


Abstract
A key step in proliferation of retroviruses is the conversion of their RNA genome to double-stranded DNA, a process catalysed by multifunctional reverse transcriptases (RTs). Dimeric and monomeric RTs have been described, the latter exemplified by the enzyme of Moloney murine leukaemia virus. However, structural information is lacking that describes the substrate binding mechanism for a monomeric RT. We report here the first crystal structure of a complex between an RNA/DNA hybrid substrate and polymerase-connection fragment of the single-subunit RT from xenotropic murine leukaemia virus-related virus, a close relative of Moloney murine leukaemia virus. A comparison with p66/p51 human immunodeficiency virus-1 RT shows that substrate binding around the polymerase active site is conserved but differs in the thumb and connection subdomains. Small-angle X-ray scattering was used to model full-length xenotropic murine leukaemia virus-related virus RT, demonstrating that its mobile RNase H domain becomes ordered in the presence of a substrate—a key difference between monomeric and dimeric RTs.
INTRODUCTION
To proliferate, retroviruses must integrate their genetic information into the genome of the infected cell. As the retroviral genome is encoded in single-stranded RNA, it is converted to double-stranded DNA (dsDNA) through a multi-step process (1) using the RNA- and DNA-dependent DNA polymerase and ribonuclease H (RNase H) activities of the viral reverse transcriptase (RT). Reverse transcription initiates from host-derived tRNA hybridized to the primer binding site near the 5′-end of the viral genome and proceeds until RT reaches the extreme 5′ terminus of the genome, thereby creating (−) strand strong-stop DNA. RNase H activity degrades the RNA strand of the resulting RNA/DNA hybrid, liberating the nascent strand of (−) DNA and allowing it to hybridize with the 3′ end of the genome through a process designated (−) strand transfer. As (−) DNA synthesis resumes, RNase H activity continues to degrade the RNA strand in the resulting RNA/DNA hybrid, with the exception of one or two short polypurine tracts (PPTs) that prime synthesis of (+) strand strong-stop DNA. After a second strand transfer event and release of the tRNA and PPT primers, bidirectional DNA synthesis produces the integration-competent double-stranded viral DNA.
The N-terminal DNA polymerase domains of RTs resemble other nucleic acid polymerases and have been likened to a right hand with subdomains designated fingers, palm and thumb (2). A fourth subdomain, the connection, links the DNA polymerase and C-terminal RNase H domains. Both dimeric and monomeric retroviral RTs have been described, the former exemplified by the human immunodeficiency virus-1 (HIV-1) enzyme. HIV-1 RT is an asymmetric heterodimer of 66 and 51 kDa subunits (p66 and p51) that are proteolytically cleaved from the gag-pol precursor during virus maturation (3). The p66 subunit contains the DNA polymerase and RNase H domains, whereas p51 lacks an RNase H domain, has an altered conformation relative to the equivalent segment of its p66 counterpart and provides a structural platform that serves to support and activate the larger subunit. A similar subunit organization has been described for the RTs of related lentiviruses (4,5). The best-characterized monomeric RT is the ∼75 kDa enzyme from the gammaretrovirus Moloney murine leukaemia virus (Mo-MLV) (6–8). One important consequence of the dimeric versus monomeric architecture of RTs is the placement of their RNase H domain. The domain of HIV-1 RT is rigidly placed on the p51 subunit platform (2), whereas the Mo-MLV RNase H counterpart is connected to the rest of the enzyme via a flexible linker and assumed to be mobile (9).
HIV-1 RT has been extensively characterized structurally and to this point is the only RT for which structures of complexes with productively bound substrates are available. The structures determined for HIV-1 RT include its complexes with (i) dsDNA (10); (ii) dsDNA and the incoming nucleotide (11); and (iii) an RNA/DNA hybrid in which the RNA strand contains the sequence of the HIV-1 3′ PPT and flanking regions (12). Mo-MLV RT is the only monomeric RT for which structural information is available. Several structures of an N-terminal fragment comprising the fingers and palm subdomains have been reported, including structures containing dsDNA (13–15). In these structures, however, the duplex failed to contact critical active site residues of the DNA polymerase domain, and its position differed significantly from that in substrate complexes of HIV-1 RT and related DNA polymerases. These discrepancies were reconciled by the notion that such ternary complexes reflected an intermediate translocation state (14). The full-length Mo-MLV RT has also been crystallized, but only the DNA polymerase and connection subdomain were defined in the corresponding structure, and the RNase H domain was disordered (9).
RNase H activity is essential for retrovirus replication (16) and is responsible for several critical steps of proviral DNA synthesis, including DNA strand transfer and generation and specific removal of the tRNA and PPT primers. In contrast to HIV-1 RNase H, the isolated Mo-MLV domain retains activity but lacks specificity for some important intermediates in reverse transcription (17,18). The Mo-MLV RNase H domain contains a characteristic element designated the ‘basic protrusion’, which is absent from the HIV-1 enzyme. This motif is important for substrate binding and comprises a short helix and loop, which together form a bulge on the protein surface (19). Deleting the basic protrusion in the RNase H domain of Mo-MLV RT does not inhibit RNase H activity but blocks virus infectivity (20). In the structures of two gammaretroviral RNases H that were initially reported, the basic protrusion was removed to obtain crystals that diffracted X-rays to high resolution (21,22). Recently, a structure of the intact RNase H domain of the xenotropic murine leukaemia virus-related virus (XMRV), a close relative of Mo-MLV, has been determined (23).
XMRV was originally proposed as the aetiological agent of prostate cancer (24) and chronic fatigue syndrome (25,26), but subsequent studies have unequivocally dismissed this notion, showing that XMRV increases through recombination following passaging human tumours in mice (27,28). Nevertheless, XMRV, a close relative to Mo-MLV, remains a replication-competent gammaretrovirus capable of infecting human cells.
Existing Mo-MLV RT structures provide only limited and fragmentary knowledge about the mechanism of action of monomeric RTs, and no structures of Mo-MLV RT in a complex with productively bound nucleic acid are available. Therefore, our aim was to solve a crystal structure of a monomeric RT in complex with an RNA/DNA hybrid. We elected to work on the enzyme from the VP62 isolate of XMRV, which, excluding an unstructured N-terminus, differs in the sequence of the polymerase and connection domains in only five positions from that of Mo-MLV RT (P30L, L234Q, Q238R, D422N, L463M); hence, the two enzymes can be considered essentially identical. Here, we report the first crystal structure of a complex between the polymerase-connection region of a monomeric gammaretroviral RT and its substrate together with biochemical data that provide insights in the mechanism of substrate binding. We also used our co-crystal structure for a comprehensive comparison with HIV-1 RT to elucidate structural and mechanistic similarities and differences between these enzymes. Lastly, we present small-angle X-ray scattering (SAXS) data for the full-length enzyme that, together with modelling, provide insights about the mobility and arrangement of the RNase H domain in the context of a full-length XMRV RT monomer.
MATERIALS AND METHODS
Crystallization
Protein expression and purification is described in Supplementary Information. Briefly, RT from XMRV isolate VP62 was expressed in Escherichia coli strain BL21 (DE3) Magic and purified on Nickel, ion exchange and size exclusion columns. HPLC-purified RNA and DNA oligonucleotides were purchased from Metabion International AG. The lengths of oligonucleotides used for crystallization were based on previous DNase I footprinting data (29). Before crystallization, protein was mixed with DNA/RNA hybrid in a 1:1.2 molar ratio and a final protein concentration of 5 mg/ml. The DNA/RNA hybrid (hybrid 1) was produced by annealing an RNA oligonucleotide (5′-AACAGAGUGCGACACCUGAUUCCAU-3′) and a DNA oligonucleotide (5′-TGGAATCAGGTGTCGCACTCTG-3′). The resulting hybrid had a 22 bp duplex region and overhangs of the RNA strand: 2 nt overhang at the 5′-end of the RNA and 1 nt overhang at the 3′ end. Crystals of the nucleoprotein complex were obtained at room temperature by hanging-drop vapour diffusion. Initial crystallization conditions were identified using the INDEX screen from Hampton Research. Following optimization, the best crystals were obtained by mixing 1 μl of protein–DNA/RNA complex with 1 μl of reservoir buffer containing 0.2 M ammonium sulfate, 100 mM BisTris (pH 5.0) and 17% PEG3350 and addition of 0.2 µl 20% w/v benzamidine to the drop. Before data collection, crystals were cryoprotected by step-wise addition of 50% glycerol to the crystallization drop to a final concentration of 25% and flash frozen in liquid N2. The content of the crystals was analysed by polyacrylamide gel electrophoresis (Sodium dodecyl sulphate–polyacrylamide gel electrophoresis for protein and Tris-borate-EDTA–urea gel for nucleic acid).
Diffraction data collection, structure solution and refinement
X-ray diffraction data were collected at 14.1 beam line of Berliner Elektronenspeicherring-Gesellschaft für Synchrotronstrahlung (BESSY) (30) for selenomethionine crystals (at Se peak wavelength of 0.979 Å) and native data at the European Synchrotron Radiation Facility (ESRF) at 23-2 beamline on a Mar225 CCD detector at 100 K. Diffraction data were processed and scaled with HKL2000 (31). The statistics of diffraction data is summarized in Table 1. The structure was solved by molecular replacement, using the Mo-MLV RT structure as the search model (Protein Data Bank ID: 1RW3) (9) and PHASER program (32). Iterative building with COOT (33) was carried out, and refinement with Phenix (34) was monitored throughout using R-free, calculated with 5% of unique reflections. In the final model, 99.5% of the residues are within the allowed regions of the Ramachandran plot.
Table 1.
Data collection and refinement statistics of XMRV RT- RNA/DNA complex crystals
| Data collection | Native | SeMet (two crystals) |
|---|---|---|
| Space group | P43212 | P43212 |
| Cell dimensions | ||
| a, b, c (Å) | 98.1, 98.1, 201.8 | 97.9, 97.9, 201.3 |
| α, β, γ(°) | 90, 90, 90 | 90, 90, 90 |
| Resolution (Å) | 30-3.04 (3.09-3.04)* | 50-3.4 (3.46-3.40) |
| Rmerge | 9.9 (96.4) | 16.0 (53.9) |
| I/σI | 26.8 (2.7) | 16.5 (1.6) |
| Completeness (%) | 100 (100) | 94.5 (65.9) |
| Redundancy | 13.0 (13.3) | 13.4 (6.5) |
| Refinement | ||
| Resolution (Å) | 3.04 | |
| Number of reflections | 19172 | |
| Rwork/Rfree | 22.4/28.0 | |
| Number of atoms | 4071 | |
| Protein | 3413 | |
| Ligand/ion | 621 | |
| Water | 37 | |
| B-factors (Å2) | 75.3 | |
| Protein | 69.3 | |
| Ligand/ion | 109.6 | |
| Water | 52.4 | |
| R.m.s. deviations | ||
| Bond lengths (Å) | 0.011 | |
| Bond angles (°) | 1.01 | |
*Values in parentheses are for the highest-resolution shell.
In the DNA polymerase domain and connection subdomain, several regions could not be traced due to the lack of interpretable electron density: the extreme N terminus (the His-tag and protein residues 1–27), two loops in fingers domain (residues 104–107 and 175–181), two residues from thumb subdomain (330 and 331) and a fragment of the connection domain (449–454). The last residue of the connection domain that could be traced in our structure is Pro487.
The composite simulated annealing omit maps were calculated with Crystallography & NMR System (CNS) 1.3 (35) using the default parameters with 5% of the model omitted at each step. Anomalous difference maps for selenomethionine data set were calculated both in CNS 1.3 and in Phenix giving essentially the same results. Structural analyses, including superpositions and secondary structure assignments, were performed in Pymol (http://www.pymol.org). The same software was used to prepare the structural figures. Nucleic acid geometry was analysed by Curves+ (36). The structure was deposited in the PDB under the accession code 4HKQ.
Biochemical studies of XMRV RT variants
RNA-dependent DNA polymerase and RNase H activities were simultaneously evaluated via the ability to support DNA strand transfer (37). DNA synthesis was initiated by adding 1 μL of RT (150 ng) to 9 μL of mixture containing 50 nM donor Cy5-RNA template/Cy3-DNA primer, 250 nM acceptor RNA template and 200 μM dNTPs in 10 mM Tris–HCl, (pH 7.8), 9 mM MgCl2, 80 mM NaCl, 5 mM dithiothreitol. Samples were incubated at 37°C for 5, 10, 20 and 40 min, then quenched with equal volume of 8 M urea in TBE buffer. Polymerization and hydrolysis products were resolved by high voltage, denaturing polyacrylamide gel electrophoresis and visualized by fluorescent imaging (Typhoon Trio+, GE Healthcare).
SAXS data analysis
Synchrotron SAXS data were collected on the X33 beamline at EMBL (DESY, Hamburg, Germany) (38). Protein buffer contained 10 mM HEPES (pH 6.5), 5% glycerol, 100 mM KCl and 1 mM DTT. Samples were prepared for XMRV RT alone and for a mixture of the protein with DNA/RNA hybrids (hybrid 1, PPT-18 and PPT-19) at a 1:1.2 molar ratio and a final protein concentration of 0.9 or 1.8 mg/ml. The PPT substrates had following sequences: PPT-19 – RNA strand: 5′-UAGUCUCCAGAAAAAGGGGGGAAUG-3′, DNA strand: 5′-ATTCCCCCCTTTTTCTGGAGAC-3′. PPT-18: RNA: 5′-AGUCUCCAGAAAAAGGGGGGAAUGA-3′ and DNA 3′-CATTCCCCCCTTTTTCTGGAGA-5′.
Pilatus one-megapixel array detector (Dectris, Switzerland) was used to record 15 s exposures. The sample-to-detector distance was set to 2.7 m and covered a range of momentum transfer 0.08 nm−1 < s < 6.0 nm−1, (s = 4 π sin θ/λ, where 2θ is the scattering angle and λ = 0.15 nm is the X-ray wavelength used in measurements). No measurable radiation damage was detected by comparison of eight successive time frames with 15 s exposures.
All SAXS data manipulations were performed with the PRIMUS software suite (39). Radius of gyration Rg and forward scattering I (0) were calculated using Guinier approximation with PRIMUS’s AutoRg program. Based on p (r) distribution obtained from GNOM (40), maximum diameter values Dmax were calculated for each sample. Molecular weights estimates were calculated based on bovine serum albumin scattering profile used as a standard. A low resolution envelope was determined from the scattering data for XMRV RT – PPT-19 complex. Thirty independent ab initio reconstructions were obtained with DAMMIF (41) and averaged with DAMAVER (42). The normal spatial discrepancy parameter for PPT-19 was in the range of 0.72–0.96, which indicates high similarity of reconstructions and that unique solution was identified. This reconstruction should, however, be treated with caution, as it was generated with DAMMIF, which cannot handle multiphase (protein/nucleic acid) scattering data. Applying MONSA (43), which is dedicated to process multiphase data, was not possible for XMRV RT owing to differences between its apo and complex conformations. The final reconstruction was superimposed onto the atomic model of the complex (with PPT-19 substrate) with SUPCOMB20 (44), taking into account the enantiomers.
Inter-domain flexibility of the apo protein was initially assessed with basic parameters inferred from a scattering profile (Rg, Dmax), Kratky plot and consecutively explored with the Ensemble Optimization Method (EOM) (45). EOM consists of two separate programs: Random Chain (RANCH) and Genetic Algorithm Judging Optimization of Ensemble (GAJOE). RANCH generates a pool of random models with the target sequence while preserving structural fragments provided by the user. GAJOE selects the optimal ensemble of models with combined scattering intensities best-matching experimental data. Thus, EOM allows the coexistence of a number of conformations in solution, providing goodness of fit measure (χ) and Rg distribution for the selected ensemble.
To perform sampling of RNase H domain position in the vicinity of the RNA/DNA hybrid, an initial pool of decoys was generated using the REFINER program (46). Polymerase and RNase H domains were treated as rigid bodies, whereas the inter-domain linker covered a range of conformations. Additionally, an N-terminal fragment and 4 terminal residues at the C-termini were modelled. A final non-redundant set of 29 733 decoys was selected by clustering with the Cα root mean square deviation threshold of 3 Å. Each decoy from this set was complemented with the atomic model of the RNA/DNA hybrid. The configurations clashing with RNase H domain were filtered out. As a result, a set of 26 617 decoys was obtained. For each model, fitting to the SAXS data was conducted with CRYSOL (47). A discrepancy χ, defined as:
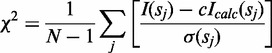 |
where, N is the number of experimental points, c is a scaling factor, Icalc (sj) and σ are the calculated intensity and experimental error at the momentum transfer sj, respectively (47), was calculated. In addition, the distance between the active site of the RNase H domain and the scissile phosphate in substrate (separation of Cα atom of Asp534 and phosphorus atom of the nucleotide located 19 bp from the active site of polymerase domain) was measured for each model and plotted against χ. Analogous analyses were performed for apo form. Owing to the absence of the RNA/DNA hybrid in the structure, RNase H domain-scissile phosphate distance was measured with the respect to the virtual point in 3D space corresponding to a phosphorus atom.
RESULTS AND DISCUSSION
Structure of XMRV RT in complex with RNA/DNA substrate
To gain insight into the mechanism of a gammaretroviral RT, we solved the crystal structure of the XMRV enzyme in complex with an RNA/DNA hybrid (Table 1 and Figure 1A). Details of the solution, refinement of the structure and its overall description can be found in Supplementary Information. The structure was solved by molecular replacement using the model of apo Mo-MLV RT (PDB code: 1RW3) (9), but significant portions of the thumb and connection subdomains required retracing (Supplementary Information, Supplementary Figures S1 and S2). The DNA polymerase and connection domains of XMRV RT could be traced, but although the RNase H domain was present in the crystal, we failed to observe its electron density and consider it disordered. Analysis of the crystal packing interactions shows that there is enough space in the crystal to accommodate the RNase H domain (Supplementary Figure S4). For the nucleic acid, we traced 16 of 25 residues of the RNA and 14 of 22 residues of the DNA strand (Supplementary Figure S3).
Figure 1.

Overall structure of XMRV RT in complex with RNA/DNA hybrid. (A) The protein is shown in cartoon representation with subdomains colour-coded pink for palm, cyan for fingers, yellow for thumb and green for connection. RNA template and DNA primer are coloured red and blue, respectively. (B) Surface representation of XMRV RT with electrostatic potential (±15 kT/e) coded in blue (positive) and red (negative). Nucleic acid is shown in cartoon representation (yellow for RNA and green for DNA).
We also corrected the apo Mo-MLV RT structure by changing the tracing in the thumb and connection domains to the one observed in our complex structure and refining the new model against 1RW3 structure factors deposited in the PDB. After those corrections, the apo structure resembles the XMRV RT protein from our complex structure—individual domains of these two RTs can be superimposed with low root mean square deviations (RMSD) for C-α atoms of 0.7–1.2 Å (Supplementary Table S1). However, considerable global conformational changes occur in the presence of substrate and are described in the Supplementary Information and Supplementary Figure S5.
Contacts with RNA/DNA
The RNA/DNA hybrid in our structure interacts with all domains of the protein (Figure 1A) and is comfortably accommodated by the substrate-binding channel, which is overall positively charged (Figure 1B). All but one protein-nucleic acid interaction involve the phosphodiester backbone, agreeing with the lack of sequence specificity (Figure 2A). Overall, the protein covers 14 nt of the primer and 16 nt of the template strands, in good agreement with the DNase I footprinting studies of Mo-MLV RT lacking the RNase H domain (29). The minor groove of the substrate has the width of ∼9 Å in the vicinity of the active site (nucleotides −3 and −4 of the template) and ∼10.5 Å around the thumb subdomain (nucleotides −5 and −6 of the template). These values indicate that both strands adopt an A-form conformation. This is in agreement with the fact that RTs are able to extend tRNA primers on RNA template—such dsRNA substrates can only adopt a pure A-form. Further, towards the connection domain, the minor groove is ∼8.5 Å wide, indicating that the hybrid adopts an intermediate conformation between A- and B-forms.
Figure 2.

Substrate binding. (A) Diagram showing interactions between XMRV RT and nucleic acid. Ovals are coloured according to protein subdomains of Figure 1. Black outline denotes residues for which equivalents can be found in HIV-1 RT (putative equivalent denoted with dashed oval). Stacking of Tyr64 with the RNA overhang is shown as parallel lines. Van der Waals interactions are shown as grey dashed lines and polar interactions as blue dashed lines. (B) Interaction of XMRV RT with the terminal portion of the RNA/DNA duplex (shown in red and blue for RNA and DNA, respectively). XMRV RT protein is in surface representation. Tyr64 and residues forming the ‘pin’ stabilizing the conformation of the template in front of the incoming nucleotide are shown as cyan sticks, and the dNTP modelled based on the HIV-1 RT structure (PDB ID: 1RTD) is shown as dark grey sticks. (C) Interactions with the 2′-OH groups of the template. The RNA strand is shown as red sticks and 2′-OH groups as spheres. Hydrogen bonds are indicated with dashed lines. (D) Binding of primer nucleotides by residues of the thumb subdomain. In panels (B) and (D), a composite simulated annealing omit map contoured at 1 σ is overlaid on the substrate (blue mesh).
A comparison of the apo structures of Mo-MLV and HIV-1 RT (6,48) showed that residues comprising the DNA polymerase active site, and participating in binding of catalytic metal ions and the incoming dNTP, are highly conserved; thus, the active site architecture of the two enzymes is very similar (Supplementary Figure S6). Our structure provides further support for this conservation by showing that the trajectory of the substrate near the DNA polymerase active site, and positioning of the 3′-OH of the DNA primer strand in particular, is superimposable. This implies that although the incoming dNTP and divalent metal ions are absent from our substrate complex, their mode of binding almost certainly parallels that of HIV-1 RT (11). A detailed comparison of the polymerase active sites of XMRV and HIV-1 RTs can be found in Supplementary Information.
The template RNA in our structure contains a 5′ overhang with terminal nucleotide +2 flipped out (Figure 2A and B). XMRV RT residues contributing to stabilization of this nucleotide are Tyr64 and Leu99. The aromatic ring of Tyr64 forms a stacking interaction with the base of the nucleotide, and this interaction is further stabilized in our crystals by a lattice contact (Supplementary Figure S4C). A characteristic feature of RTs is their ability to perform DNA synthesis concurrent with the displacement of downstream nucleic acid hybridized with the template. A Y64A substitution of Mo-MLV RT selectively reduced displacement synthesis (49), whereas a recombinant virus containing this mutation failed to replicate. KMnO4 probing showed that displacement synthesis involves melting of base pairs +1 and +2 ahead of the DNA polymerase active site (50). Stacking between the unpaired base and Tyr64 is likely a key element of this mechanism and is supported by observations that the aromatic ring of the amino acid is sufficient for displacement synthesis, as a Y64F variant exhibits wild-type enzymatic properties (49). The equivalent of Tyr64 in HIV-1 RT is Trp24, which was experimentally demonstrated to contribute towards substrate binding (51).
In the XMRV RT co-crystal, RNA nucleotide +2 also interacts through its 2′-OH group with fingers subdomain residues Asp114 and Arg116 located opposite the active site (Figure 2B). The side chain of Arg116 is located ∼3.5 Å from the phosphate of nucleotide +1 and protrudes from the protein surface to form a ‘pin’ that guides template trajectory such that the base of nucleotide +1 is positioned to pair with the incoming dNTP (Figure 2B). To fulfill this function, the pin must be rigid, and the conformation of the Arg116 side chain is stabilized by a strong ionic interaction with Asp114. Biochemical data confirm the importance of both Arg116 and Asp114. Substituting Arg116 of Mo-MLV RT with Lys or Leu reduced DNA polymerase activity on homopolymeric RNA/DNA and abolished activity on a template with random sequence (14). Similarly, substituting Asp114 with Asn reduced activity on a homopolymeric RNA template by 60% and on random sequence RNA template ∼5-fold. Mutating either residue also inhibited virus replication (52,53). Proteins with D114A or R116A substitutions are also significantly less processive, unable to resolve hairpins in the template and displayed reduced affinity for nucleic acid (52). HIV-1 RT counterparts of these residues are Asp76 and Arg78. When Arg78 was substituted with Lys, HIV-1 RT retained 50% of its DNA polymerase activity (54), and a later study showed that an R78A substitution increased fidelity and decreased affinity for DNA and RNA templates (55).
Template nucleotides +1, −1 and −3 form hydrogen bonds through their 2′-OH groups with backbone carbonyls of Gly191, Lys193 (both located at the boundary of the fingers and palm subdomains) and Pro130 from the palm (Figure 2C). These interactions could underlie a preference for RNA as the template strand. Indeed, HIV-1 RT binds RNA/DNA substrates ∼10-fold tighter than dsDNA (56). Moreover, the 5 terminal bp of RNA/DNA located at the polymerase active site were sufficient for enhanced binding (56). The HIV-1 counterparts of Gly191 and Lys193 are Gly152 and Lys154, respectively, and the carbonyl of Lys154 forms an interaction with the RNA 2′-OH in the HIV RT-RNA/DNA complex (PDB ID: 1HYS) (12). Pro130 has no clear equivalent in HIV-1 RT—perhaps its role is fulfilled by Gln91. In XMRV RT, Gly191 and Lys193 are located in a loop whose conformation is stabilized by a hydrogen bond between the side chain of Asn119 and the backbone amide of Lys193. This stabilization likely enhances substrate binding. Interestingly, an N119A substitution in Mo-MLV RT reduced DNA polymerase activity 50% on a homopolymeric RNA template and completely abolished activity on random-sequence RNA (14). Further towards 3′ end of the RNA template, the phosphate groups of nucleotides −5 to −8 interact with a positively charged patch on the surface of the connection domain formed by the side chains of Lys397, Lys398 and Lys425, which have no obvious equivalents in HIV-1 RT.
For interactions with the primer strand, the phosphate group of the penultimate DNA nucleotide is strongly bound by the guanidinium group of Arg284 located in the first helix of the thumb subdomain (Figure 2D). No clear equivalent of this residue exists in HIV-1 RT. Nucleotides −3, −4 and −5 form van der Waals interactions with the side chains and backbone of α-helix F of the thumb, which is inserted into the minor groove of the substrate. We note a minor deformation of the primer backbone in this region, although we cannot exclude the possibility that it is induced by a displacement of the thumb domain resulting from crystal packing interactions (Supplementary Figure S4B). A prominent interaction involving α-helix F is stacking of the ribose ring of DNA nucleotide −3 with Phe309. If a 2′-OH were present in the ribose ring of the nucleotide, it would impose less effective stacking, and therefore this interaction can select against ribonucleotides in the primer strand. The HIV-1 RT counterpart is Trp266, whose substitution completely abolishes DNA polymerase activity (57). RTs are known to extend RNA primers poorly, other than those of (−) and (+) strand synthesis (tRNA and the PPT, respectively) (3). Perhaps this specificity is partially conferred by Phe309 in XMRV RT and Trp266 in HIV-1 RT.
A notable interaction is made by the phosphate group of nucleotide −5 of the DNA strand with Arg298 and Arg301 of α-helix F of the thumb domain, equivalent to HIV-1 residues Gln258 and Asn255, respectively (Figure 2D). The side chains of the two residues form ‘tweezers’ that, together with Glu302, hold the backbone of the DNA strand. The following fragment of the DNA strand does not interact with the protein, but nucleotide −12 forms an interaction with Arg456 and nucleotide −13 a hydrogen bond with Trp406. Both residues are located in the connection subdomain and lack obvious equivalents in HIV-1 RT. Interestingly, when a PPT substrate is modelled into the structure of XMRV RT (see later in the text), the isolated substrate contact mediated by Trp406 and Arg456 is at the boundary between the A- and G-tracts, which has been shown to be an element critical for PPT recognition (58). It is tempting to speculate that this contact may be more efficient for PPT owing to the special structure of the A-tract, which leads to better positioning of the substrate for RNase H cleavage and a kinetic preference for hydrolysis at the PPT-U3 junction.
In summary, the protein–nucleic acid interactions can be divided in several segments. For template binding, these are the interactions with RNA overhang (mediated by Tyr64 and the ‘pin’), followed by a region of interactions with 2′-OH groups and subsequently by a positively charged patch binding the backbone of the RNA. For the primer, most of the interactions are mediated by the thumb followed by an isolated interaction with the connection domain. In the vicinity of the active site, protein–nucleic acid contacts are conserved between HIV-1 and XMRV RT. However, further towards the connection domain, substrate binding is mediated by a different set of residues.
Site-directed mutagenesis of substrate contacts
Site-directed mutagenesis was used to assess the importance of novel nucleic acid contacts that lacked equivalents in HIV-1 RT. We prepared XMRV RT variants K397A/K398A, R311A/K425A and W406A/R456A, with substitutions in the thumb and connection domains, and tested their ability to support DNA strand transfer, the activity which simultaneously monitors DNA polymerase and RNase H activities of the protein. This assay comprised a Cy5 5′-labelled donor RNA template annealed to a Cy3 5′-labelled DNA primer in the presence of a nucleotide acceptor RNA template. Donor and acceptor RNA templates shared 20 homologous nucleotides at their 5′- and 3′-termini, respectively. Initial RNA-dependent DNA synthesis produces a 40 nt strand transfer intermediate (STI) and RNase H-mediated strand transfer and subsequent DNA synthesis lead to a 60 nt product (STP) (Figure 3A).
Figure 3.

Biochemical characterization of the XMRV RT variants. The DNA strand transfer assay is outlined schematically in (A) and described in the main text. The results are shown in (B). Notations P20 and T40 indicate migration positions of the Cy3-labelled, 20 nt DNA primer and Cy5-labelled RNA template, respectively, before initiation of DNA synthesis. Full-length primer extension on the donor RNA template is evidenced by accumulation of the strand transfer intermediate, STI40, whereas RNase H hydrolysis products are defined by R21/20, R18/17 and R14/13. Transfer of nascent DNA to the acceptor RNA template and continued DNA synthesis is evidenced by accumulation of the 60 nucleotide strand transfer product, STP60. Samples were withdrawn after 5 min (Lanes a), 10 min (Lanes b), 20 min (Lanes c) and 40 min (lanes d) for analysis. Positions of Ala substitutions XMRV RT are indicated below each panel and shown in (C) with lime green for K397A/K398A, blue for R311A/K425A and purple for W406A/R456A.
Data in Figure 3B show that the 40 nt STI is efficiently synthesized by all proteins, indicating that contacts between the connection domain and the substrate do not play a major role in DNA polymerase activity. They do, however, affect RNase H function—in the case of variants K397A/K398A and W406A/R456A, we observe reduced RNase H activity as evidenced by a slower decrease in the T40 RNA template. In addition, W406A/R456A RT displayed altered cleavage specificity, generating 21 and 20 nt fragments without any shorter products. The R21/20 cleavage corresponds to the positioning of the substrate in which the blunt end of the RNA/DNA STI is stably bound at the polymerase active site. For the other cleavage sites (R18/17 and in particular R13/14), the end of the hybrid shifts from the polymerase domain and substrate binding involves only interactions with the thumb and connections domains. One of these important contacts is lost in the W406A/R456A variant (Figure 3C), which likely explains why the cleavages at R21/20 involving interactions between the hybrid and the polymerase domain are preferred. Unexpectedly, R311A/K425A variant displayed enhanced RNase H activity and consequently higher rate of strand transfer. It is not clear what causes this increase in the activity, but overall our data show that the connection domain participates in correctly positioning the substrate for RNase H cleavage.
Comparison with structures of HIV-1 RT
We compared the structures of XMRV RT and HIV-1 RT containing (i) an RNA/DNA (PDB ID: 1HYS) (12) and (ii) dsDNA and the incoming nucleotide (PDB ID: 1RTD) (11). Figure 4A shows a structure-based alignment for the two proteins. Individual subdomains of XMRV and HIV-1 RT are similar, and the palm, fingers, thumb and connection can be superimposed with RMSDs between 1.2 and 1.7 Å (Table 2). However, we noted that α-helix E (residues 282–290) of the XMRV RT thumb is replaced by an extended fragment in HIV-1 RT (Figure 4A). Consensus secondary structure predictions, calculated using Genesilico metaserver (59), suggest that an equivalent α-helix exists in RTs from gamma- and spumaretroviruses, whereas for lenti-, alpha-, beta- and deltaretroviral RTs, this fragment is predicted to be extended (Supplementary Figure S7). Therefore, the presence of this α-helix defines two classes of RTs. In XMRV, this α-helix harbours Arg284 which, as described earlier in the text, forms an important contact with the primer strand.
Figure 4.

Comparison of gammaretroviral and lentiviral RT structures. (A) Structure-based alignment of the sequences of HIV-1 and XMRV RT. Residues involved in forming the active site and in binding of the incoming dNTP are highlighted in yellow, whereas those involved in binding template and primer are highlighted in red and blue, respectively. Critical functional residues are indicated and the line between sequences colour codes the subdomains is as in Figure 1A. α-helices are indicated as tubes and β-strands as arrows and labelled. (B and D) Superposition of the palm/fingers subdomain from XMRV RT complex structure, HIV-1 RT in complex with RNA/DNA (PDB ID: 1HYS) (B) and HIV-1 RT complexed with dsDNA (PDB: 1RTD) (D). XMRV RT palm subdomain is in pink and fingers in cyan. HIV-1 RT structure is shown in orange. For clarity, only the substrate from our XMRV RT structure is presented. (C and E) palm subdomain-based superposition of XMRV RT complex structure with structures of HIV-1 RT complexed with RNA/DNA (PDB ID: 1HYS) (E) and dsDNA (PDB ID: 1RTD) (C). HIV-1 RT is shown in orange for p66 subunit, and the RNase H is shown in darker colour. The p51 subunit and XMRV RT are omitted for clarity. The RNA/DNA hybrid from XMRV RT is shown in red (RNA) and blue (DNA) and the substrates from HIV RT structures in pink (RNA) and cyan (DNA). The axes of the nucleic acid are shown as spheres (dark blue for XMRV RT complex and cyan for HIV-1 complexes).
Table 2.
Superpositions of the structures of substrate-bound XMRV and HIV-1 RTs
| Superimposed domain (Total number of traced Cα atoms in each XMRV RT domain given in parentheses) | RMSD (Å) calculated for: |
||||
|---|---|---|---|---|---|
| Substrate | Fingers | Palm | Thumb | Connection | |
| RNA/DNA complex, 1HYS | |||||
| Substrate 30 pairs of atomsa | 0.5 | 5.8 | 2.3 | 3.1 | 4.1 |
| Fingers 84 pairs of Cα atoms (of 106) | 7.3 | 1.7 | 8.2 | 14.9 | 17.1 |
| Palm 91 pairs of Cα atoms (of 136) | 1.1 | 4.6 | 1.3 | 3.1 | 3.1 |
| Thumb 53 pairs of Cα atoms (of 79) | 2.9 | 6.1 | 5.3 | 1.6 | 2.3 |
| Connection 54 pairs of Cα atoms (of 120) | 4.7 | 8.1 | 5.7 | 3.1 | 1.6 |
| dsDNA complex, 1RTD | |||||
| Substrate 24 pairs of atomsa | 0.4 | 2.4 | 1.8 | 3.0 | 3.4 |
| Fingers 84 pairs of Cα atoms (of 106) | 1.6 | 1.2 | 3.6 | 3.7 | 6.2 |
| Palm 91 pairs of Cα atoms (of 136) | 0.7 | 2.7 | 1.3 | 3.0 | 3.2 |
| Thumb 53 pairs of Cα atoms (of 79) | 2.7 | 3.5 | 4.4 | 1.5 | 2.8 |
| Connection 54 pairs of Cα atoms (of 120) | 5.2 | 7.3 | 5.8 | 3.1 | 1.6 |
The individual subdomains were superimposed [the resulting root mean square deviations (RMSDs) of pairs of C−α atoms are shown in bold] and the RMSD values for the other subdomains are given.
aPhophodiester backbone atoms of nucleotides −1, −2 and −3 of the primer and template strands were used for superposition.
A more significant difference is observed in the connection subdomain. In XMRV RT, we traced α-helix L between residues 456 and 465. This helix contains two methionines, and we could verify our tracing by anomalous difference maps for the selenomethionine data set (Supplementary Figure S2C). The corresponding region in HIV-1 RT forms an extended structure located at the p66-p51 interface, where an equivalent helix cannot be accommodated. Moreover, in the p51 subunit, owing to its altered conformation, the region corresponding to α-helix L of XMRV RT is tightly packed between the connection, palm and fingers, preventing accommodation of an α-helix. Therefore, converting the α-helical region to an extended structure likely reflects adaptation to the dimeric architecture of lentiviral RTs.
The overall structures of proteins in the substrate complexes of XMRV and HIV-1 RTs are similar. The palm, thumb and connection subdomains of XMRV RT (198 C-α atoms) can be superimposed on structures of HIV-1 RT with an RMSD of 2.0 and 1.9 Å for 1HYS and 1RTD, respectively. One key difference is positioning of their fingers subdomains. These superimpose well between XMRV RT – substrate complex and HIV-1 ternary complex (PDB ID: 1RTD), adopting a ‘half-open’ conformation (11), whereas the conformation in the HIV-1 RT – RNA/DNA complex (PDB ID: 1HYS) is more open (Figure 4B and D). Therefore, our structure may more closely resemble an elongating complex in the presence of the incoming dNTP.
We next compared nucleic acid positioning and trajectory in our structure versus RNA/DNA or dsDNA from HIV-1 RT structures. This differs between the two RTs, regardless whether the palm subdomain or the terminal region of the substrate is used for superposition (Figure 4C and E). The HIV-1 and XMRV RT substrates are superimposable for the first 3 bp going from the polymerase active site, but after this region, their trajectories differ. In our XMRV RT structure, the substrate passes closer to the connection subdomain, which would not be possible for HIV-1 RT, as it would invoke clashes with both the connection and RNase H domain. The trajectory of XMRV RT substrate results in formation of an isolated contact between the substrate and the connection domain mediated by Trp406 and Arg456 which, as described earlier in the text, may play a role in PPT recognition.
Model of full-length XMRV RT and its verification through SAXS experiments
As the RNase H domain is not visible in our structure, we prepared models of the full-length XMRV RT complex by combining our structure with those of XMRV RNase H (23) and human RNase H1 in complex with RNA/DNA (19) (described in more detail in Supplementary Information). Models with polymerase-RNase H distance of 18 or 19 bp are free of steric clashes, whereas any distance shorter than 18 bp imposes a severe clash between the RNase H and connection domains. This is in agreement with biochemical data, which show that very little 3′-end directed cleavage by Mo-MLV occurs at distances 17 bp or closer to the polymerase active site (60). The model with 19 bp separation between the polymerase and RNase H active sites is shown in Figure 5A.
Figure 5.

Full-length XMRV RT model and SAXS data. (A) Model of the full-length protein based on the XMRV RT substrate complex structure of Figure 1A, and structures of human RNase H1 and the isolated XMRV RNase H domain. The modelled RNase H domain is depicted in orange. Disordered regions are in grey (only a single modelled conformation is shown for clarity). The 19 bp distance between the DNA polymerase and RNase H active sites was assumed for model preparation. An ab initio calculated SAXS density map for XMRV RT – PPT-19 complex is overlaid on the model (grey). (B and C) Experimental (black circles and grey error bars) and theoretical (red line) scattering curves for XMRV RT – PPT-19 complex (B) and apo protein (C), computed by CRYSOL and EOM, respectively. The plots display the logarithm of the scattering intensity as a function of momentum transfer s = 4π sin θ/λ (where 2θ is the scattering angle and λ = 0.15 nm is the X-ray wavelength used in the measurements). (D) EOM analysis of the SAXS data for apo protein. The frequencies of models with particular Rg values are plotted for the pool used in EOM (red) and selected to fit the SAXS data for apo XMRV RT (blue). (E) Two examples of models selected in EOM analysis, superimposed based on the polymerase-connection domains, show different positioning of the RNase H domain (orange). The coarse-grained models of the linker and terminal regions are shown as small spheres for one model only.
To verify the correctness of the model and to provide further insights into the structure of the full-length enzyme, XMRV RT was examined in the absence and presence of RNA/DNA hybrids by SAXS. Complexes with several hybrids were examined, including the duplex used for crystallization (hybrid 1) and hybrids with strands of the same length but a sequence corresponding to the Mo-MLV PPT, which differs from the XMRV PPT by one nucleotide. Substrates with preferred RNase H cleavage sites located 18 nt (PPT-18) and 19 nt (PPT-19) from the 3′-end of the recessed DNA were used. We first calculated the radius of gyration (Rg), maximum particle dimension (Dmax) and particle volume values based on SAXS data (Table 3). For all three parameters, the values were larger for protein alone and the complex with hybrid 1 than for complexes with hybrids PPT-18 and PPT-19, implying that complexes with PPT hybrids induced a more compact structure. This may reflect multiple positions of the RNase H domain for the protein alone and hybrid 1 complex and a more ordered RNase H domain interacting with the PPT substrates. Therefore, for further analysis of the model of full-length protein in complex with the substrate described earlier in the text, we used SAXS data for PPT-19 complex. These data show very good agreement with the theoretical scattering curve calculated based on the model with χ value of 0.92 (Figure 5B). We also calculated 30 independent ab initio reconstructions for PPT-18 and PPT-19 data. PPT-19 averaged filtered reconstruction showed very good agreement with our model of the full-length protein (χ = 0.89) (Figure 5A).
Table 3.
Parameters derived from SAXS experiments
| Rg (nm) | Dmax (nm) | VP (nm3) | |
|---|---|---|---|
| apo | 3.95 ± 0.10 | 13.5 ± 0.5 | 160 ± 10 |
| hybrid 1 | 3.80 ± 0.10 | 13.0 ± 0.5 | 172 ± 10 |
| PPT-18 | 3.60 ± 0.10 | 12.0 ± 0.5 | 155 ± 10 |
| PPT-19 | 3.60 ± 0.10 | 12.0 ± 0.5 | 155 ± 10 |
Rg, radius of gyration; Dmax, maximum size of the particle; Vp, excluded volume of the hydrated particle estimated from Porod asymptotics.
We next explored further the potential mobility of the RNase H domain. We first used Normal Mode Analysis scored against SAXS data (Supplementary Information), which indicated mobility of RNase H domain in apo protein and confirmed the correctness of the full-length XMRV RT substrate complex model. We next applied EOM (45) using models with random RNase H domain positions based on coarse-grained representation of the linker between the connection and RNase H domain. The theoretical scattering curve calculated based on an ensemble selected in EOM showed very good agreement with the experimental SAXS data for apo protein (Figure 5C), with χ of 0.91. We noted a bimodal distribution of models selected from EOM with a larger fraction with compact conformation (with Rg between 35 and 40 Å) and a minor fraction of the extended conformations (with Rg between 43 and 48 Å) (Figure 5D and E). From this result, we conclude that the RNase H domain is mobile, but its positioning is not completely random with two preferred regions, between which it can easily alternate.
To better probe potential interactions between the RNase H domain and its substrate, we prepared another ensemble with shorter distances between the RNase H domain and the hybrid and with full-atom representation of the N terminus and linker region. The ensemble contained 29 733 models for apo form and 26 617 models for the complex (protein models with clashes with RNA/DNA hybrid were removed from the complex ensemble). Models were subsequently scored for their agreement with SAXS data for both the apo protein and XMRV RT – PPT-19 complex. For each model, the score was plotted versus the distance between the active site of the RNase H domain (C-α position of Asp534) and the position of phosphorus atom of the scissile phosphate of the PPT-19 substrate. SAXS data for the apo protein (Supplementary Figure S8A) show minimal correlation of the score of the model with the distance between the position of the substrate and the RNase H domain, demonstrating that different positions of RNase H domain fit equally well to the SAXS data. In contrast, for RT – PPT-19 complex data, models with shorter distance between the active site of the RNase H domain and the scissile phosphate show better χ scores than models with this domain located further from the hybrid (Supplementary Figure S8B), indicating that for the PPT-19 complex, the RNase H domain interacts with the substrate.
In conclusion, the Normal Mode Analysis, EOM and the χ versus distance plot analysis indicate mobility of the RNase H domain in the absence of the substrate and its ordering on the PPT hybrids. No such ordering is observed for hybrid 1, for which the SAXS data are similar to the apo protein, probably reflecting higher affinity of the RNase H domain for the PPT sequence over the random sequence in hybrid 1. Furthermore, the SAXS data support our model of full-length XMRV RT interacting with the substrate.
CONCLUSIONS
Our studies of XMRV RT provide the first comprehensive structural analysis of the interaction of a monomeric gammaretroviral enzyme with an RNA/DNA substrate. We show here that interactions between the DNA polymerase domains and the substrate, as well as the active site composition, are highly conserved among monomeric and dimeric RTs. However, a pronounced difference is the positioning and mobility of their RNase H domains. In HIV-1 RT, this domain is relatively rigidly positioned by the p66 connection subdomain and p51 DNA polymerase domain. In contrast, as demonstrated by our SAXS data, the XMRV RNase H domain is mobile in the absence of the substrate. RNase H activity is responsible for the mechanistically more intricate steps of reverse transcription, such as DNA strand transfer or generation and specific removal of the PPT primer. Given the structural differences between monomeric and dimeric RTs, it is interesting that some properties of the RNase H activity of the two are conserved. For example, Mo-MLV RT can use the HIV-1 PPT sequence for priming (+) strand synthesis (61), and HIV-1 RT can use Mo-MLV PPT with only slightly affected cleavage specificity (62). Further structural studies should elucidate the atomic details of RNase H function in the context of these RTs.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online: Supplementary Materials and Methods, Supplementary Results, Supplementary Table 1, Supplementary Figures 1–8 and Supplementary References [63–81].
FUNDING
Polish National Science Center [contract number N N301 439738 to M.N.]; FP7 HEALTHPROT project [contract number 229676 to M.N.]; the WeNMR project [European FP7 e-Infrastructure grant, contract No. 261572; http://www.wenmr.eu to D.I.S.], Foundation for Polish Science [grant TEAM/2009-4/2 to J.M.B], Intramural Research Program (IRP) of the National Cancer Institute, National Institutes of Health, Department of Health and Human Services [to S.F.J. Le G.], federal funds from the National Institutes of Health [Contract # HHSN261200800001E to M.K.B.]. The research of M.N. was supported in part by an International Early Career Scientist grant from the Howard Hughes Medical Institute. M.N. is also a recipient of Foundation for Polish Science ‘Ideas for Poland’ award. The access to ESRF was financed by the Polish Ministry of Science and Higher Education [project no. ESRF/73/2006]. The research leading to these results has also received funding from the European Community’s Seventh Framework Program [agreement no. 226716]. Funding for open access charge: International Institute of Molecular and Cell Biology, Polish National Science Center Grant [contract number N N301 439738 to M.N.].
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
The authors thank Alexander Wlodawer and Robert Crouch for critical reading of the manuscript and Zbigniew Dauter for the help with crystallographic data processing and analysis. They acknowledge Iwona Ptasiewicz for excellent technical assistance. They would like to thank the staff of beamline 23-2 at European Synchrotron Radiation Facility (ESRF) and beamline 14-1 at Berliner Elektronenspeicherring-Gesellschaft für Synchrotronstrahlung (BESSY) for assistance with data collection. The content of this publication does not necessarily reflect Cancer Institute, National Institutes of Health, the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the US Government.
REFERENCES
- 1.Gilboa E, Mitra SW, Goff S, Baltimore D. A detailed model of reverse transcription and tests of crucial aspects. Cell. 1979;18:93–100. doi: 10.1016/0092-8674(79)90357-x. [DOI] [PubMed] [Google Scholar]
- 2.Kohlstaedt LA, Wang J, Friedman JM, Rice PA, Steitz TA. Crystal structure at 3.5 A resolution of HIV-1 reverse transcriptase complexed with an inhibitor. Science. 1992;256:1783–1790. doi: 10.1126/science.1377403. [DOI] [PubMed] [Google Scholar]
- 3.Herschhorn A, Hizi A. Retroviral reverse transcriptases. Cell. Mol. Life Sci. 2010;67:2717–2747. doi: 10.1007/s00018-010-0346-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.North TW, Cronn RC, Remington KM, Tandberg RT, Judd RC. Characterization of reverse transcriptase from feline immunodeficiency virus. J. Biol. Chem. 1990;265:5121–5128. [PubMed] [Google Scholar]
- 5.Thomas DA, Furman PA. Purification and kinetic characterization of equine infectious anemia virus reverse transcriptase. Biochem. Biophys. Res. Commun. 1991;180:1365–1371. doi: 10.1016/s0006-291x(05)81346-4. [DOI] [PubMed] [Google Scholar]
- 6.Cote ML, Roth MJ. Murine leukemia virus reverse transcriptase: structural comparison with HIV-1 reverse transcriptase. Virus Res. 2008;134:186–202. doi: 10.1016/j.virusres.2008.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Moelling K. Characterization of reverse transcriptase and RNase H from friend-murine leukemia virus. Virology. 1974;62:46–59. doi: 10.1016/0042-6822(74)90302-x. [DOI] [PubMed] [Google Scholar]
- 8.Roth MJ, Tanese N, Goff SP. Purification and characterization of murine retroviral reverse transcriptase expressed in Escherichia coli. J. Biol. Chem. 1985;260:9326–9335. [PubMed] [Google Scholar]
- 9.Das D, Georgiadis MM. The crystal structure of the monomeric reverse transcriptase from Moloney murine leukemia virus. Structure. 2004;12:819–829. doi: 10.1016/j.str.2004.02.032. [DOI] [PubMed] [Google Scholar]
- 10.Ding J, Das K, Hsiou Y, Sarafianos SG, Clark AD, Jr, Jacobo-Molina A, Tantillo C, Hughes SH, Arnold E. Structure and functional implications of the polymerase active site region in a complex of HIV-1 RT with a double-stranded DNA template-primer and an antibody Fab fragment at 2.8 A resolution. J. Mol. Biol. 1998;284:1095–1111. doi: 10.1006/jmbi.1998.2208. [DOI] [PubMed] [Google Scholar]
- 11.Huang H, Chopra R, Verdine GL, Harrison SC. Structure of a covalently trapped catalytic complex of HIV-1 reverse transcriptase: implications for drug resistance. Science. 1998;282:1669–1675. doi: 10.1126/science.282.5394.1669. [DOI] [PubMed] [Google Scholar]
- 12.Sarafianos SG, Das K, Tantillo C, Clark AD, Jr, Ding J, Whitcomb JM, Boyer PL, Hughes SH, Arnold E. Crystal structure of HIV-1 reverse transcriptase in complex with a polypurine tract RNA:DNA. EMBO J. 2001;20:1449–1461. doi: 10.1093/emboj/20.6.1449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cote ML, Yohannan SJ, Georgiadis MM. Use of an N-terminal fragment from moloney murine leukemia virus reverse transcriptase to facilitate crystallization and analysis of a pseudo-16-mer DNA molecule containing G-A mispairs. Acta. Crystallogr. D Biol. Crystallogr. 2000;56:1120–1131. doi: 10.1107/s0907444900008246. [DOI] [PubMed] [Google Scholar]
- 14.Najmudin S, Cote ML, Sun D, Yohannan S, Montano SP, Gu J, Georgiadis MM. Crystal structures of an N-terminal fragment from Moloney murine leukemia virus reverse transcriptase complexed with nucleic acid: functional implications for template-primer binding to the fingers domain. J. Mol. Biol. 2000;296:613–632. doi: 10.1006/jmbi.1999.3477. [DOI] [PubMed] [Google Scholar]
- 15.Sun D, Jessen S, Liu C, Liu X, Najmudin S, Georgiadis MM. Cloning, expression, and purification of a catalytic fragment of Moloney murine leukemia virus reverse transcriptase: crystallization of nucleic acid complexes. Protein Sci. 1998;7:1575–1582. doi: 10.1002/pro.5560070711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tisdale M, Schulze T, Larder BA, Moelling K. Mutations within the RNase H domain of human immunodeficiency virus type 1 reverse transcriptase abolish virus infectivity. J. Gen. Virol. 1991;72(Pt 1):59–66. doi: 10.1099/0022-1317-72-1-59. [DOI] [PubMed] [Google Scholar]
- 17.Schultz SJ, Champoux JJ. RNase H domain of Moloney murine leukemia virus reverse transcriptase retains activity but requires the polymerase domain for specificity. J. Virol. 1996;70:8630–8638. doi: 10.1128/jvi.70.12.8630-8638.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhan X, Crouch RJ. The isolated RNase H domain of murine leukemia virus reverse transcriptase. Retention of activity with concomitant loss of specificity. J. Biol. Chem. 1997;272:22023–22029. doi: 10.1074/jbc.272.35.22023. [DOI] [PubMed] [Google Scholar]
- 19.Nowotny M, Gaidamakov SA, Ghirlando R, Cerritelli SM, Crouch RJ, Yang W. Structure of human RNase H1 complexed with an RNA/DNA hybrid: insight into HIV reverse transcription. Mol. Cell. 2007;28:264–276. doi: 10.1016/j.molcel.2007.08.015. [DOI] [PubMed] [Google Scholar]
- 20.Telesnitsky A, Goff SP. RNase H domain mutations affect the interaction between Moloney murine leukemia virus reverse transcriptase and its primer-template. Proc. Natl Acad. Sci. USA. 1993;90:1276–1280. doi: 10.1073/pnas.90.4.1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kirby KA, Marchand B, Ong YT, Ndongwe TP, Hachiya A, Michailidis E, Leslie MD, Sietsema DV, Fetterly TL, Dorst CA, et al. Structural and inhibition studies of the RNase H function of xenotropic murine leukemia virus-related virus reverse transcriptase. Antimicrob. Agents Chemother. 2012;56:2048–2061. doi: 10.1128/AAC.06000-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lim D, Gregorio GG, Bingman C, Martinez-Hackert E, Hendrickson WA, Goff SP. Crystal structure of the moloney murine leukemia virus RNase H domain. J. Virol. 2006;80:8379–8389. doi: 10.1128/JVI.00750-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhou D, Chung S, Miller M, Le Grice SF, Wlodawer A. Crystal structures of the reverse transcriptase-associated ribonuclease H domain of Xenotropic murine leukemia-virus related virus. J. Struct. Biol. 2012;177:638–645. doi: 10.1016/j.jsb.2012.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schlaberg R, Choe DJ, Brown KR, Thaker HM, Singh IR. XMRV is present in malignant prostatic epithelium and is associated with prostate cancer, especially high-grade tumors. Proc. Natl Acad. Sci. USA. 2009;106:16351–16356. doi: 10.1073/pnas.0906922106. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 25.Lombardi VC, Ruscetti FW, Das Gupta J, Pfost MA, Hagen KS, Peterson DL, Ruscetti SK, Bagni RK, Petrow-Sadowski C, Gold B, et al. Detection of an infectious retrovirus, XMRV, in blood cells of patients with chronic fatigue syndrome. Science. 2009;326:585–589. doi: 10.1126/science.1179052. [DOI] [PubMed] [Google Scholar]
- 26.Mikovits JA, Lombardi VC, Pfost MA, Hagen S, Ruscetti FW. Detection of an infectious retrovirus, XMRV, in blood cells of patients with chronic fatigue syndrome. Virulence. 2010;1:386–390. doi: 10.4161/viru.1.5.12486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Delviks-Frankenberry K, Cingoz O, Coffin JM, Pathak VK. Recombinant origin, contamination, and de-discovery of XMRV. Curr. Opin. Virol. 2012;2:499–507. doi: 10.1016/j.coviro.2012.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Paprotka T, Delviks-Frankenberry KA, Cingoz O, Martinez A, Kung HJ, Tepper CG, Hu WS, Fivash MJ, Jr, Coffin JM, Pathak VK. Recombinant origin of the retrovirus XMRV. Science. 2011;333:97–101. doi: 10.1126/science.1205292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wohrl BM, Georgiadis MM, Telesnitsky A, Hendrickson WA, Le Grice SF. Footprint analysis of replicating murine leukemia virus reverse transcriptase. Science. 1995;267:96–99. doi: 10.1126/science.7528942. [DOI] [PubMed] [Google Scholar]
- 30.Mueller U, Darowski N, Fuchs MR, Forster R, Hellmig M, Paithankar KS, Puhringer S, Steffien M, Zocher G, Weiss MS. Facilities for macromolecular crystallography at the Helmholtz-Zentrum Berlin. J. Synchrotron Radiat. 2012;19:442–449. doi: 10.1107/S0909049512006395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Otwinowski Z, Minor W. In: Methods in Enzymol. Carter CW, Sweet RM, editors. Vol. 276. New York: Academic Press; 1997. pp. 307–326. [Google Scholar]
- 32.McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J. Appl. Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr. D. Biol. Crystallogr. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Brunger AT. Version 1.2 of the Crystallography and NMR system. Nat. Protoc. 2007;2:2728–2733. doi: 10.1038/nprot.2007.406. [DOI] [PubMed] [Google Scholar]
- 36.Lavery R, Moakher M, Maddocks JH, Petkeviciute D, Zakrzewska K. Conformational analysis of nucleic acids revisited: Curves+ Nucleic Acids Res. 2009;37:5917–5929. doi: 10.1093/nar/gkp608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chung S, Miller JT, Johnson BC, Hughes SH, Le Grice SF. Mutagenesis of human immunodeficiency virus reverse transcriptase p51 subunit defines residues contributing to vinylogous urea inhibition of ribonuclease H activity. J. Biol. Chem. 2012;287:4066–4075. doi: 10.1074/jbc.M111.314781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Blanchet CE, Zozulya AV, Kikhney AG, Franke D, Konarev PV, Shang W, Klaering R, Robrahn B, Hermes C, Cipriani F, et al. Instrumental setup for high-throughput small- and wide-angle solution scattering at the X33 beamline of EMBL Hamburg. J. Appl. Cryst. 2012;45:489–495. [Google Scholar]
- 39.Konarev PV, Volkov VV, Sokolova AV, Koch MHJ, Svergun DI. PRIMUS: a Windows PC-based system for small-angle scattering data analysis. J. Appl. Cryst. 2003;36:1277–1282. [Google Scholar]
- 40.Svergun DI. Determination of the regularization parameter in indirect-transform methods using perceptual criteria. J. Appl. Cryst. 1992;25:495–503. [Google Scholar]
- 41.Franke D, Svergun DI. DAMMIF, a program for rapid ab-initio shape determination in small-angle scattering. J. Appl. Cryst. 2009;42:342–346. doi: 10.1107/S0021889809000338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Volkov VV, Svergun DI. Uniqueness of ab initio shape determination in small-angle scattering. J. Appl. Cryst. 2003;36:860–864. doi: 10.1107/S0021889809000338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Svergun DI. Restoring low resolution structure of biological macromolecules from solution scattering using simulated annealing. Biophys. J. 1999;76:2879–2886. doi: 10.1016/S0006-3495(99)77443-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kozin MB, Svergun DI. Automated matching of high- and low-resolution structural models. J. Appl. Cryst. 2001;34:33–41. [Google Scholar]
- 45.Bernado P, Mylonas E, Petoukhov MV, Blackledge M, Svergun DI. Structural characterization of flexible proteins using small-angle X-ray scattering. J. Am. Chem. Soc. 2007;129:5656–5664. doi: 10.1021/ja069124n. [DOI] [PubMed] [Google Scholar]
- 46.Boniecki M, Rotkiewicz P, Skolnick J, Kolinski A. Protein fragment reconstruction using various modeling techniques. J. Comput. Aided Mol. Des. 2003;17:725–738. doi: 10.1023/b:jcam.0000017486.83645.a0. [DOI] [PubMed] [Google Scholar]
- 47.Svergun D, Barberato C, Koch MHJ. CRYSOL – a Program to evaluate x-ray solution scattering of biological macromolecules from atomic coordinates. J. Appl. Cryst. 1995;28:768–773. [Google Scholar]
- 48.Georgiadis MM, Jessen SM, Ogata CM, Telesnitsky A, Goff SP, Hendrickson WA. Mechanistic implications from the structure of a catalytic fragment of Moloney murine leukemia virus reverse transcriptase. Structure. 1995;3:879–892. doi: 10.1016/S0969-2126(01)00223-4. [DOI] [PubMed] [Google Scholar]
- 49.Paulson BA, Zhang M, Schultz SJ, Champoux JJ. Substitution of alanine for tyrosine-64 in the fingers subdomain of M-MuLV reverse transcriptase impairs strand displacement synthesis and blocks viral replication in vivo. Virology. 2007;366:361–376. doi: 10.1016/j.virol.2007.04.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Winshell J, Champoux JJ. Structural alterations in the DNA ahead of the primer terminus during displacement synthesis by reverse transcriptases. J. Mol. Biol. 2001;306:931–943. doi: 10.1006/jmbi.2001.4439. [DOI] [PubMed] [Google Scholar]
- 51.Agopian A, Depollier J, Lionne C, Divita G. p66 Trp24 and Phe61 are essential for accurate association of HIV-1 reverse transcriptase with primer/template. J. Mol. Biol. 2007;373:127–140. doi: 10.1016/j.jmb.2007.07.044. [DOI] [PubMed] [Google Scholar]
- 52.Gu J, Villanueva RA, Snyder CS, Roth MJ, Georgiadis MM. Substitution of Asp114 or Arg116 in the fingers domain of moloney murine leukemia virus reverse transcriptase affects interactions with the template-primer resulting in decreased processivity. J. Mol. Biol. 2001;305:341–359. doi: 10.1006/jmbi.2000.4281. [DOI] [PubMed] [Google Scholar]
- 53.Pfeiffer JK, Georgiadis MM, Telesnitsky A. Structure-based moloney murine leukemia virus reverse transcriptase mutants with altered intracellular direct-repeat deletion frequencies. J. Virol. 2000;74:9629–9636. doi: 10.1128/jvi.74.20.9629-9636.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Boyer PL, Ferris AL, Hughes SH. Cassette mutagenesis of the reverse transcriptase of human immunodeficiency virus type 1. J. Virol. 1992;66:1031–1039. doi: 10.1128/jvi.66.2.1031-1039.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kim B, Ayran JC, Sagar SG, Adman ET, Fuller SM, Tran NH, Horrigan J. New human immunodeficiency virus, type 1 reverse transcriptase (HIV-1 RT) mutants with increased fidelity of DNA synthesis. Accuracy, template binding, and processivity. J. Biol. Chem. 1999;274:27666–27673. doi: 10.1074/jbc.274.39.27666. [DOI] [PubMed] [Google Scholar]
- 56.Bohlayer WP, DeStefano JJ. Tighter binding of HIV reverse transcriptase to RNA-DNA versus DNA-DNA results mostly from interactions in the polymerase domain and requires just a small stretch of RNA-DNA. Biochemistry. 2006;45:7628–7638. doi: 10.1021/bi051770w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gao HQ, Boyer PL, Arnold E, Hughes SH. Effects of mutations in the polymerase domain on the polymerase, RNase H and strand transfer activities of human immunodeficiency virus type 1 reverse transcriptase. J. Mol. Biol. 1998;277:559–572. doi: 10.1006/jmbi.1998.1624. [DOI] [PubMed] [Google Scholar]
- 58.Rattray AJ, Champoux JJ. Plus-strand priming by Moloney murine leukemia virus. The sequence features important for cleavage by RNase H. J. Mol. Biol. 1989;208:445–456. doi: 10.1016/0022-2836(89)90508-1. [DOI] [PubMed] [Google Scholar]
- 59.Kurowski MA, Bujnicki JM. GeneSilico protein structure prediction meta-server. Nucleic Acids Res. 2003;31:3305–3307. doi: 10.1093/nar/gkg557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Schultz SJ, Zhang M, Champoux JJ. Preferred sequences within a defined cleavage window specify DNA 3′ end-directed cleavages by retroviral RNases H. J. Biol. Chem. 2009;284:32225–32238. doi: 10.1074/jbc.M109.043158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Pullen KA, Champoux JJ. Plus-strand origin for human immunodeficiency virus type 1: implications for integration. J. Virol. 1990;64:6274–6277. doi: 10.1128/jvi.64.12.6274-6277.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Pullen KA, Rattray AJ, Champoux JJ. The sequence features important for plus strand priming by human immunodeficiency virus type 1 reverse transcriptase. J. Biol. Chem. 1993;268:6221–6227. [PubMed] [Google Scholar]
- 63.Wu N, Christendat D, Dharamsi A, Pai EF. Purification, crystallization and preliminary X-ray study of orotidine 5'-monophosphate decarboxylase. Acta Crystallogr. D Biol. Crystallogr. 2000;56:912–914. doi: 10.1107/s090744490000576x. [DOI] [PubMed] [Google Scholar]
- 64.Evans P. Scaling and assessment of data quality. Acta Crystallogr. D Biol. Crystallogr. 2006;62:72–82. doi: 10.1107/S0907444905036693. [DOI] [PubMed] [Google Scholar]
- 65.Karplus PA, Diederichs K. Linking crystallographic model and data quality. Science. 2012;336:1030–1033. doi: 10.1126/science.1218231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Goodwin KD, Long EC, Georgiadis MM. A host-guest approach for determining drug-DNA interactions: an example using netropsin. Nucleic Acids Res. 2005;33:4106–4116. doi: 10.1093/nar/gki717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Chowdhury K, Kaushik N, Pandey VN, Modak MJ. Elucidation of the role of Arg 110 of murine leukemia virus reverse transcriptase in the catalytic mechanism: biochemical characterization of its mutant enzymes. Biochemistry. 1996;35:16610–16620. doi: 10.1021/bi961462l. [DOI] [PubMed] [Google Scholar]
- 68.Basu A, Basu S, Modak MJ. Site-directed mutagenesis of Moloney murine leukemia virus reverse transcriptase. Demonstration of lysine 103 in the nucleotide binding site. J. Biol. Chem. 1990;265:17V162–17166. [PubMed] [Google Scholar]
- 69.Gao G, Orlova M, Georgiadis MM, Hendrickson WA, Goff SP. Conferring RNA polymerase activity to a DNA polymerase: a single residue in reverse transcriptase controls substrate selection. Proc. Natl Acad. Sci. USA. 1997;94:407–411. doi: 10.1073/pnas.94.2.407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Drosopoulos WC, Prasad VR. Increased polymerase fidelity of E89G, a nucleoside analog-resistant variant of human immunodeficiency virus type 1 reverse transcriptase. J. Virol. 1996;70:4834–4838. doi: 10.1128/jvi.70.7.4834-4838.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Oude Essink BB, Back NK, Berkhout B. Increased polymerase fidelity of the 3TC-resistant variants of HIV-1 reverse transcriptase. Nucleic Acids Res. 1997;25:3212–3217. doi: 10.1093/nar/25.16.3212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Pandey VN, Kaushik N, Rege N, Sarafianos SG, Yadav PN, Modak MJ. Role of methionine 184 of human immunodeficiency virus type-1 reverse transcriptase in the polymerase function and fidelity of DNA synthesis. Biochemistry. 1996;35:2168–2179. doi: 10.1021/bi9516642. [DOI] [PubMed] [Google Scholar]
- 73.Wainberg MA, Drosopoulos WC, Salomon H, Hsu M, Borkow G, Parniak M, Gu Z, Song Q, Manne J, Islam S, et al. Enhanced fidelity of 3TC-selected mutant HIV-1 reverse transcriptase. Science. 1996;271:1282–1285. doi: 10.1126/science.271.5253.1282. [DOI] [PubMed] [Google Scholar]
- 74.Roberts JD, Bebenek K, Kunkel TA. The accuracy of reverse transcriptase from HIV-1. Science. 1988;242:1171–1173. doi: 10.1126/science.2460925. [DOI] [PubMed] [Google Scholar]
- 75.Eswar N, Webb B, Marti-Renom MA, Madhusudhan MS, Eramian D, Shen MY, Pieper U, Sali A. Comparative protein structure modeling using Modeller. Curr. Protoc. Bioinformatics. 2006;(Chapter 5) doi: 10.1002/0471250953.bi0506s15. Unit 5.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Sgourakis NG, Lange OF, DiMaio F, Andre I, Fitzkee NC, Rossi P, Montelione GT, Bax A, Baker D. Determination of the structures of symmetric protein oligomers from NMR chemical shifts and residual dipolar couplings. J. Am. Chem. Soc. 2011;133:6288–6298. doi: 10.1021/ja111318m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Gorba C, Miyashita O, Tama F. Normal-mode flexible fitting of high-resolution structure of biological molecules toward one-dimensional low-resolution data. Biophys. J. 2008;94:1589–1599. doi: 10.1529/biophysj.107.122218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Xiong Y, Eickbush TH. Similarity of reverse transcriptase-like sequences of viruses, transposable elements, and mitochondrial introns. Mol. Biol. Evol. 1988;5:675–690. doi: 10.1093/oxfordjournals.molbev.a040521. [DOI] [PubMed] [Google Scholar]
- 79.Puglia J, Wang T, Smith-Snyder C, Cote M, Scher M, Pelletier JN, John S, Jonsson CB, Roth MJ. Revealing domain structure through linker-scanning analysis of the murine leukemia virus (MuLV) RNase H and MuLV and human immunodeficiency virus type 1 integrase proteins. J. Virol. 2006;80:9497–9510. doi: 10.1128/JVI.00856-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Tanese N, Telesnitsky A, Goff SP. Abortive reverse transcription by mutants of Moloney murine leukemia virus deficient in the reverse transcriptase-associated RNase H function. J. Virol. 1991;65:4387–4397. doi: 10.1128/jvi.65.8.4387-4397.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Leo B, Schweimer K, Rosch P, Hartl MJ, Wohrl BM. The solution structure of the prototype foamy virus RNase H domain indicates an important role of the basic loop in substrate binding. Retrovirology. 2012;9:73. doi: 10.1186/1742-4690-9-73. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.