


Abstract
Lens epithelium-derived growth factor (LEDGF/p75) tethers lentiviral preintegration complexes (PICs) to chromatin and is essential for effective HIV-1 replication. LEDGF/p75 interactions with lentiviral integrases are well characterized, but the structural basis for how LEDGF/p75 engages chromatin is unknown. We demonstrate that cellular LEDGF/p75 is tightly bound to mononucleosomes (MNs). Our proteomic experiments indicate that this interaction is direct and not mediated by other cellular factors. We determined the solution structure of LEDGF PWWP and monitored binding to the histone H3 tail containing trimethylated Lys36 (H3K36me3) and DNA by NMR. Results reveal two distinct functional interfaces of LEDGF PWWP: a well-defined hydrophobic cavity, which selectively interacts with the H3K36me3 peptide and adjacent basic surface, which non-specifically binds DNA. LEDGF PWWP exhibits nanomolar binding affinity to purified native MNs, but displays markedly lower affinities for the isolated H3K36me3 peptide and DNA. Furthermore, we show that LEDGF PWWP preferentially and tightly binds to in vitro reconstituted MNs containing a tri-methyl-lysine analogue at position 36 of H3 and not to their unmodified counterparts. We conclude that cooperative binding of the hydrophobic cavity and basic surface to the cognate histone peptide and DNA wrapped in MNs is essential for high-affinity binding to chromatin.
INTRODUCTION
Lens epithelium-derived growth factor (LEDGF/p75), a chromatin-binding protein, is essential for effective integration of HIV-1 cDNA into human chromosomes (1–4). As a cellular cofactor, LEDGF/p75 controls the site specificity of HIV-1 integration and preferentially navigates lentiviral preintegration complexes (PICs) to the actively transcribed genes (2). LEDGF/p75 acts as a bifunctional tether with its C-terminal fragment (termed Integrase-Binding Domain or IBD) specifically engaging lentiviral integrases (INs), and its N-terminal portion interacting with chromatin. Although the structural foundations for the LEDGF IBD:IN interaction have been characterized in detail (5,6), the molecular mechanism for how the N-terminal portion of LEDGF/p75 associates with chromatin is not understood.
The N-terminal portion of LEDGF/p75 contains a PWWP domain, which is essential for tight interactions of the full-length protein with chromatin. Deletion of the PWWP domain disrupted association of cellular LEDGF with condensed chromosomes in mitosis (7–9) and significantly compromised its ability to support HIV-1 replication (10). Experiments with deletion mutants and chimeric proteins have further revealed a role of LEDGF PWWP in integration site selectivity (7,11). For example, HIV-1 integration sites observed with full length LEDGF/p75 and its truncated counterpart, which lacked the PWWP domain, differed markedly (9). Furthermore, chimeric proteins that had the chromatin-binding part of LEDGF replaced with different chromatin-binding modules were still effective cofactors for lentiviral PICs (12) but redirected viral integration to alternative genomic regions (7,11). Taken together, the published studies have demonstrated that the PWWP domain is a major determinant for tight and site-selective binding of LEDGF/p75 to chromatin.
The LEDGF PWWP domain is a member of the Tudor domain ‘Royal Family’. PWWP domains are defined by a conserved Pro-Trp-Trp-Pro signature motif. Atomic structures for several of these PWWP domains are available, and their biochemical properties have been characterized in detail (13–19). The PWWP domains of Dnmt3a, Brpf1, MSH-6, NSD1, NSD2 and N-PAC have been shown to preferentially bind H3K36me3 (14,17,20). Recently, LEDGF PWWP has also been demonstrated to bind the H3K36me3 peptide and not its unmodified counterpart (21). Although the binding affinities of LEDGF PWWP with H3K36me3 have not been determined, the studies with other PWWP domains revealed that Kd values for these interactions are in the low millimolar range (14,18). Clearly, such low binding affinities for the PWWP:H3K36me3 interactions alone cannot explain the key chromatin-tethering role of these domains and raise a possibility that these interactions could be enhanced by additional interactions of the PWWP domains with DNA or other cellular factors.
PWWP domains from different proteins have been shown to also bind DNA (13,16,19); however, DNA-binding affinities varied markedly (16,19). For example, the Kd values for DNMT3b and HDGF PWWPs binding to DNA were reported to be ∼230 nM and ∼100 μM, respectively (16,19). Earlier studies argued against a tight interaction of LEDGF PWWP with synthetic DNA (10). Thus, the available biochemical data cannot explain how the PWWP domain contributes to tight binding of LEDGF/p75 to chromatin.
Here, we demonstrate that LEDGF PWWP directly interacts with purified mononucleosomes (MNs) with high affinity in the absence of additional cellular factors. Furthermore, we have determined the solution structure of LEDGF PWWP and identified two functionally important regions of the protein: a well-defined hydrophobic cavity that selectively interacts with H3K36me3 and the adjacent basic surface bound to DNA. We propose that the cooperative binding of these interfaces to the trimethylated H3 histone tail and DNA components of MNs is essential for the high-affinity binding of LEDGF/p75 to chromatin.
MATERIALS AND METHODS
Expression and purification of recombinant isotopically labelled LEDGF PWWP proteins for NMR studies
N-terminal hexahistidine-tagged LEDGF PWWP (amino acids 1–93) was engineered from pFT1-LEDGF (22) and expressed in Escherichia coli strain BL21(DE3) (Agilent). Cultures were grown in M9 minimal medium supplemented with 1% (v/v) Eagle Basal Vitamin Mix (Life Technologies, Gaithersburg, MD, USA), containing 1 g/l 15N-ammonium chloride (Cambridge Isotope Laboratories, Inc.) as the sole nitrogen source or 1 g/l 15N-ammonium chloride and 2 g/l 13C-glucose (Cambridge Isotope Laboratories, Inc.) as the sole carbon source. Cultures (24 l total) were grown at 37°C to an optical density of 0.6 at 600 nm and induced for 3 h by addition of 0.5 mM isopropyl-thio-β-d-galactopyranoside (IPTG) at 30°C. Tag-free PWWP was purified according to (23) with minor changes noted. Bacterial cells were disrupted by sonication in 1 M NaCl, 50 mM Hepes, pH 7.5, 2 mM β-mercaptoethanol (BME), 35 mM imidazole and 1 tablet of Complete EDTA-free Protease Inhibitor (Roche). The protein was purified with HisTrap HP (GE Healthcare) using a linear gradient from 35 mM to 500 mM imidazole in 500 mM NaCl, 50 mM Hepes, pH 7.5, 2 mM BME. The hexahistidine tag was removed using PreScission Protease (GE Healthcare), leaving four non-native N-terminal residues (Gly-Pro-Gly-Ser). The protein was then purified with HiTrap Heparin HP (GE Healthcare) using a linear gradient from 250 mM to 1 M NaCl in 50 mM Hepes, pH 7.5, 2 mM BME. Peak fractions were further purified by size-exclusion chromatography using HiLoad 16/60 Superdex 200 (GE Healthcare) in 150 mM NaCl, 50 mM Hepes, pH 7.5, 2 mM BME.
Expression and purification of recombinant proteins
Plasmids encoding N-terminal glutathione S-transferase (GST) fused to LEDGF/p75 (1–530) and LEDGF PWWP (1–100) were constructed in pGEX-4Ti and were a gift from Dr Alan Engelman (24,25). Plasmids encoding N-terminal GST LEDGF IBD (amino acids 347–469) were constructed in pGEX-4Ti (a gift from Dr Alan Engelman) (24) and a C-terminal hexahistidine tag was engineered. LEDGF/p75 (amino acids 1–530), LEDGF PWWP (amino acids 1–100) and LEDGF IBD (amino acids 347–469) were expressed in E. coli strain BL21(DE3) and purified as previously described (22,24,25).
Preparation of native MNs
The entire purification scheme was performed on ice or at 4°C. Nuclear pellets were prepared from SupT1 cells by dounce homogenization in lysis buffer (15 mM Hepes, pH 7.5, 0.5 M sucrose, 60 mM KCl, 0.25 mM EDTA, 2 mM BME and Complete EDTA-free Protease Inhibitor) supplemented with 1× FastBreak Cell Lysis Reagent (Promega). The nuclear pellet was washed and resuspended in MN buffer (15 mM HEPES, pH 7.5, 70 mM NaCl, 20 mM KCl, 5 mM MgCl2, 3 mM CaCl2, 2 mM BME and Complete EDTA-free Protease Inhibitor). Micrococcal nuclease digestion of chromatin and the removal of chromatin-bound proteins were adapted from (26) with minor changes noted. Salt-stripped chromatin was purified by sucrose gradient ultracentrifugation using a linear 5–25% gradient of sucrose in 15 mM Hepes, pH 7.5, 800 mM NaCl, 0.25 mM EDTA, 2 mM BME and Complete EDTA-free Protease Inhibitor at 100 000g for 16 hours at 4°C. Fractions were analysed by 1.5% agarose 1× Tris-acetate-EDTA electrophoresis and stained with ethidium bromide. For sucrose gradient-purified MNs, fractions were concentrated and dialysed into 15 mM HEPES, pH 7.5, 100 mM NaCl, 0.25 mM EDTA, 2 mM BME and protease inhibitor. For MNs that required additional purification, size exclusion chromatography was performed using a HiLoad 16/60 Superdex 200 column in 200 mM NaCl, 15 mM Hepes, pH 7.5, 0.25 mM EDTA and 2 mM BME. The fractions containing MNs were supplemented with Complete EDTA-free Protease Inhibitor upon elution.
Preparation of tri-methyl-lysine analogue containing MNs
Xenopus laevis core histones, H2A, H2B, H3 and H4, were expressed in E. coli and purified as previously described (27,28). The MNs containing the tri-methyl-lysine analogue at position 36 of H3 (referred here as H3KC36me3 MNs) were prepared using the previously described method (29). Briefly, the K36C mutation in H3 was introduced by site-directed mutagenesis. Purified and lyophilized mutant H3 was dissolved in 1 M HEPES, pH 7.8, 4 M guanidine chloride, 10 mM d/l-methionine and 20 mM DTT, and incubated for 1 h at 37°C. The protein was then treated with 100 mg of the alkylating agent (2-bromoethyl) trimethylammonium bromide, at 50°C for 2.5 h. Subsequently, 10 mM DTT was added to the reaction and incubated for an additional 2.5 h at 50°C. The reaction was quenched with 675 mM BME, and the modified H3 was purified from the reaction mixture using a PD-10 column. H3 containing KC36me3 was combined with the other three core histones (unmodified H2A, H2B and H4) in equimolar ratios, refolded and the resulting histone octamers were purified by size exclusion chromatography (27,28). For control experiments, unmodified histone octamers were formed using recombinant wild-type H2A, H2B, H3 and H4. MNs were reconstituted using purified histone octamers and 147 bp synthetic DNA of the Widom ‘601’ sequence (30).
Binding of cellular LEDGF/p75 to MNs
SupT1 MNs were purified as described above, omitting the addition of 800 mM NaCl to remove chromatin-bound proteins. To monitor histone H3 and LEDGF/p75, sucrose gradient fractions were subjected to SDS-PAGE and developed by Western blot using a rabbit polyclonal histone H3 antibody (Abcam) and a mouse monoclonal LEDGF antibody, respectively (BD Transduction Laboratories). To monitor DNA, sucrose gradient fractions were digested with proteinase K and analysed by 1.5% agarose gel electrophoresis stained with ethidium bromide.
Electrophoretic Mobility Shift Assay
DNA (300 nM) was incubated with increasing amounts of GST-PWWP for 30 min at room temperature in the buffer containing 25 mM Hepes (pH 7.5), 150 mM NaCl, 5 mM MgCl2, 1 mM DTT and 50 μg/ml BSA. The nucleoprotein complexes were analysed using 1% agarose gel electrophoresis and visualized with ethidium bromide. The following three DNA preparations were assayed in parallel reactions: ∼147 bp native mononucleosomal DNA, 40 bp DNA containing either SMYD1 or non-specific sequence. To obtain native DNA, purified MNs were digested with proteinase K (Invitrogen) for 2 h at 60°C, and DNA was then extracted with phenol:chloroform:isoamyl alcohol (25:24:1). An additional wash with chloroform:isoamyl alcohol (24:1) was performed, and the aqueous layer was then selected for ethanol precipitation to recover the native mononucleosomal DNA. SMYD1 DNA, used for titrations (below), was prepared by annealing the following synthetic oligonucleotides: 5′-CAGGCTGGTCTTGAACTCCTGACCTCAGATGATCCATGTG-3′ and 5′-CACATGGATCATCTGAGGTCAGGAGTTCAAGACCAGCCTG-3′. The following oligonucleotides were used to prepare non-specific DNA: 5′-CTGCAGAAGCTTGGTGCCGGGGCCGCTCAATTGGTCGTAG-3′ and 5′-CTACGACCAATTGAGCGGCCCCGGCACCAAGCTTCTGCAG-3′.
GST pull-down assays
Glutathione Sepharose 4B was equilibrated with the pull-down buffer containing 100 mM NaCl, 150 mM Hepes, pH 7.5, 0.5% (v/v) NP-40, 2 mM BME and Complete EDTA-free Protease Inhibitor cocktail. LEDGF/p75, LEDGF PWWP and LEDGF IBD (500 nM) were preincubated with equilibrated resin for 90 min at 4°C. SupT1 MNs (200 nM) were then incubated with the prebound resin for an additional 90 min at 4°C. The resin was washed three times with the pull-down buffer to remove unbound proteins. Bound proteins were analysed by SDS-PAGE. Histone H3 was detected by Western blot analysis using a histone H3 antibody (Abcam).
The following assay was used to determine the Kd for the LEDGF PWWP interaction with native, H3KC36me3-modified and unmodified MNs. GST-LEDGF PWWP (100 nM) were mixed with increasing concentrations of MNs to form a complex. The mixture was incubated with Glutathione Sepharose 4B resin equilibrated with the pull-down buffer. Unbound proteins were removed by washing three times with the pull-down buffer, and the bound proteins were analysed by SDS-PAGE. Histone H3 was detected by Western blot analysis using a rabbit polyclonal histone H3 antibody (Abcam) and quantified using ImageJ software. To estimate Kd values for the GST-LEDGF PWWP binding to MN, the data were fit to the Hill equation using Origin 8 Software (OriginLab).
Mass spectrometric analysis
To determine detailed protein composition of MN preparations prior to and after their pull-down with GST-LEDGF PWWP, the samples were subjected to denaturing SDS-PAGE, and protein bands were visualized by Microwave Blue stain (Protiga). The entire lane was excised and cut into 0.5 cm pieces. The gel pieces were destained and dehydrated according to the described procedure (31). In-gel proteolysis was performed using 0.5 μg of trypsin to generate small peptide peaks amenable to MS and MS/MS analysis. The tryptic peptides were desalted using reverse-phase Vivapure C18 micro spin column (Sartorius Stedim Biotech) and desiccated with a Speed Vac. Dried samples were dissolved in 0.2 % formic acid and analysed by nanoLC-LTQ Orbitrap MS/MS mass spectrometry as previously described (32).
Briefly mass spectrometry analyses were performed on a hybrid LTQ Orbitrap (Thermo Fisher Scientific) equipped with a nanoelectrospray ion source (Thermo Fisher Scientific) connected to an EASY-nLC. Peptide fractionation was performed on a 15 cm reversed phase analytical column packed in-house with C18 beads (ReproSil-Pur C18-AQ medium; Dr Maisch GmbH) using a linear gradient from 5% to 28% acetonitrile in 0.2% formic acid for 50 min, followed by an isocratic gradient of 80% acetonitrile in 0.2% formic acid for 10 min at a flow rate of 350 nl/min.
The mass spectrometer was operated in data-dependent mode automatically switching between full-scan MS and tandem MS acquisition. Survey full scan mass spectra were acquired after accumulation of 500 000 ions, with a resolution of 60 000 at 400 m/z. The top ten most intense ions from the survey scan were isolated and, after the accumulation of 5000 ions, fragmented in the linear ion trap by collisionally induced dissociation. Preview scan mode was enabled. Precursor ion charge state screening was enabled, and all singly charged and unassigned charge states were rejected. The dynamic exclusion list was enabled with a relative mass window of 10 ppm and early expiration turned on.
NMR structure experiments
[U-15N]- or [U-15N,13C]-labelled LEDGF PWWP was concentrated to 350 μM in buffer containing 50 mM HEPES, pH 7.6, 150 mM NaCl and 0.02% NaN3. D2O was added to 5% (v/v) and 4,4-dimethyl-4-silapentane-1-sulfonic acid (DSS) to 0.66 mM to the NMR sample. NMR spectra were collected at 25°C on 600- and 800-MHz Bruker Avance DRX spectrometers equipped with cryogenically cooled triple resonance single-axis gradient probes. Data were processed with NMRPipe (33) and analysed with NMRViewJ (34).
Triple resonance spectra [HNCO, HNCACB, CBCA(CO)NH] (35) were recorded on [U-15N,13C]-LEDGF PWWP for backbone assignments. The backbone assignments were obtained by manual inspection of the data within NMRViewJ, with assistance of the Probabilistic Interaction Network of Evidence (PINE) algorithm (36). The backbone was assigned to 95.3% completeness (81 of 85 non-proline residues). Three of the four non-native N-terminal amino acids (Gly-Pro-Gly) and the C-terminal (Ser) were unassigned.
Triple resonance spectra [HBHA(CO)NH, (H)CC(CO)NH-TOCSY, HC(C)H-TOCSY] (35) were recorded for side chain assignments. Aliphatic side chain assignments were obtained by manual inspection of the data with use of the 1H-13C HSQC to resolve overlapped regions. The aliphatic side chain protons were 79% assigned. The majority of unassigned aliphatic protons corresponded to overlapped resonances from Lys and Pro side chains. Aliphatic side chains other than Pro and Lys were assigned to 98%.
Three-dimensional 13C-edited and 15N-edited NOESY spectra were recorded at 800 MHz, with a mixing time of 200 ms, along with a 13C-edited aromatic region NOESY. The aromatic side chains were assigned based on NOE correlations to the aromatic protons from the previously assigned Hβ, Hα, HN and HN(i + 1) protons in the 15N- and 13C-edited NOESY spectra. The assignments were confirmed by finding mirror NOEs in the 13C-edited aromatic region NOESY. NOEs for neighbouring protons were also used to confirm assignments and to assign protons too far away from the backbone and aliphatic protons to have an NOE. Additional aromatic residues were assigned iteratively during structure calculations. Aromatic side chains were assigned to 80%. The majority of unassigned aromatic protons were the Hζ of Phe, due to the large distance from assigned backbone resonances, and overlap in the spectra. Excluding the Hζ of Phe, the aromatic side chains were 93% assigned.
Chemical shifts were used as input to the TALOS+ program (37), which yielded a secondary structure map for LEDGF PWWP that matched the secondary structure of the HDGF PWWP, with the exception of significantly less helical character near the C-terminus. This analysis also yielded torsion angle restraints that were used for the structure determination.
NOESY peak lists were prepared using NMRViewJ (34). The NOE signals were automatically assigned, and structural calculations were performed iteratively using CYANA 2.1 (38). The x-ray structure PDB ID: 4FU6 was used to start the structural calculation. One hundred and fifty structures were calculated, with the 20 structures having the lowest target function used for the ensemble. Structure statistics are summarized in Supplementary Table S2.
H3K36me3 titration
Synthetic peptides TKVARKSAPATGGV-K(me3)-KPHRY (termed here as H3K36Me3) and its unmodified counterpart TKVARKSAPATGGVKKPHRY (termed here as H3K36) were purchased from Biomatik. The 40 mM stock solutions of peptides were prepared in the NMR buffer (150 mM NaCl, 50 mM Hepes, pH 7.5, 2 mM BME) and titrated into 100 μM [U-15N]-LEDGF PWWP. The following peptide:LEDGF PWWP ratios were monitored: 0.25:1, 0.5:1, 1:1, 2:1, 4:1, 8:1, 16:1, 24:1, 32:1, 40:1, 48:1, 56:1, 64:1, 72:1 and 80:1. Each addition slightly diluted the sample with the final titration point containing 81.8 μM [U-15N]-LEGDF PWWP and 6.5 mM H3K36me3 peptide. The 1H-15N HSQC spectra were recorded after each addition at 25°C, and the data were analysed using NMRViewJ (34). The chemical shift perturbations for the largest titration point were normalized using the following equation: 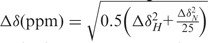 (39). The chemical shift perturbations were mapped onto the structure of LEDGF PWWP using a colour gradient from white to red, with white indicating small or no perturbations and red indicating large perturbations.
(39). The chemical shift perturbations were mapped onto the structure of LEDGF PWWP using a colour gradient from white to red, with white indicating small or no perturbations and red indicating large perturbations.
DNA Titration
The 40 bp SMYD1 DNA was dissolved in the NMR buffer (150 mM NaCl, 50 mM Hepes, pH 7.5, 2 mM BME) and titrated into 100 μM [U-15N]-LEDGF PWWP. The following DNA:LEDGF PWWP were monitored 0.1:1, 0.2:1, 0.4:1, 0.8:1, 1.6:1 and 3.2:1. The final titration point contained 78.5 μM [U-15N]-LEDGF PWWP and 251.2 μM DNA. The 1H-15N HSQC spectra were recorded and analysed identical to the peptide titration experiments. Chemical shift perturbations were mapped onto the structure using a gradient from white to green, with white indicating small or no perturbations and green indicating large perturbations.
Molecular modelling of the LEDGF PWWP binding to the MN
The solution NMR structure of LEDGF PWWP was docked onto the crystal structure of nucleosome core particle (PDB # 1AOI) (40). The chemical shift perturbations (CSP) data were exploited to position LEDGF PWWP onto the nucleosome. The histone tail of H3 was modelled using the Coot software (41) showing interactions with the LEDGF PWWP domain in a manner consistent with CSP results obtained from H3K36Me3 titration. Energy minimization was applied to the modelled histone tail with Insight II software package (Accelrys Inc., San Diego, CA, USA). The CSP data from the DNA titration experiments were exploited to guide LEDGF PWWP interactions with the wrapped DNA.
RESULTS
LEDGF/p75 and its PWWP domain tightly interact with MNs
In vitro experiments with recombinant LEDGF/p75 have shown that the full-length protein tightly interacts with both free DNA and nucleosomes (10,24,42). However, it is not clear whether cellular LEDGF/p75 associates with chromatin through its binding to nucleosomal or naked DNA sites. To address this question, we subjected chromatin isolated from SupT1 cells to micrococcal nuclease treatment and fractionated the digestion products by sucrose gradient. MNs are composed of ∼147-bp DNA fragments wrapped around an octamer of core histones and are resistant to nuclease digestion, while the linker DNA and free DNA segments are susceptible to nuclease cleavages. Therefore, the elution profiles of H3 and ∼147-nt dsDNA fragments were monitored as markers for MNs. Figure 1 shows clearly that all of the detectable cellular LEDGF/p75 co-eluted with these markers indicating that the protein is tightly associated with MNs.
Figure 1.

Cellular LEDGF/p75 is associated with MNs. SupT1 chromatin was digested by micrococcal nuclease and subjected to sucrose, equilibrium density gradient ultracentrifugation. Twenty-two 0.5 ml fractions were collected from the top of the gradient and analysed. (A) and (B) depict SDS-PAGE analysis of the fractions detected by Western blotting with LEDGF/p75 and H3 antibodies, respectively. The first lanes in each SDS-PAGE image contain protein markers. Subsequent lanes contain sucrose gradient fractions with respective fraction number indicated in the top of each image. (C) Sucrose gradient fractions 1 to 22 were treated with proteinase K and analysed with agarose gel electrophoresis. The first lane of the left agarose gel image shows the migration of 147-mer synthetic double-stranded DNA. The second lane of the left and the first lane of the right images show the DNA markers (Bioline Quanti-Marker 1 kb).
Previous studies (24,25) have shown that recombinant LEDGF/p75 and LEDGF PWWP interact with nucleosomes reconstituted with synthetic DNA and HeLa histones. Since the published studies used synthetic DNA, we repeated the binding experiments using purified native MNs (Figure 2A). GST-tagged full length LEDGF/p75, LEDGF PWWP and LEDGF IBD (Supplementary Figure S1) were incubated with native MNs to monitor their binding. As expected full-length LEDGF/p75 and LEDGF PWWP but not LEDGF IBD bound to MNs (Figure 2B). These experiments confirm that LEDGF PWWP is sufficient for binding to MNs. Therefore, our further studies have focused on investigating only the LEDGF PWWP contribution to the interaction with MNs.
Figure 2.
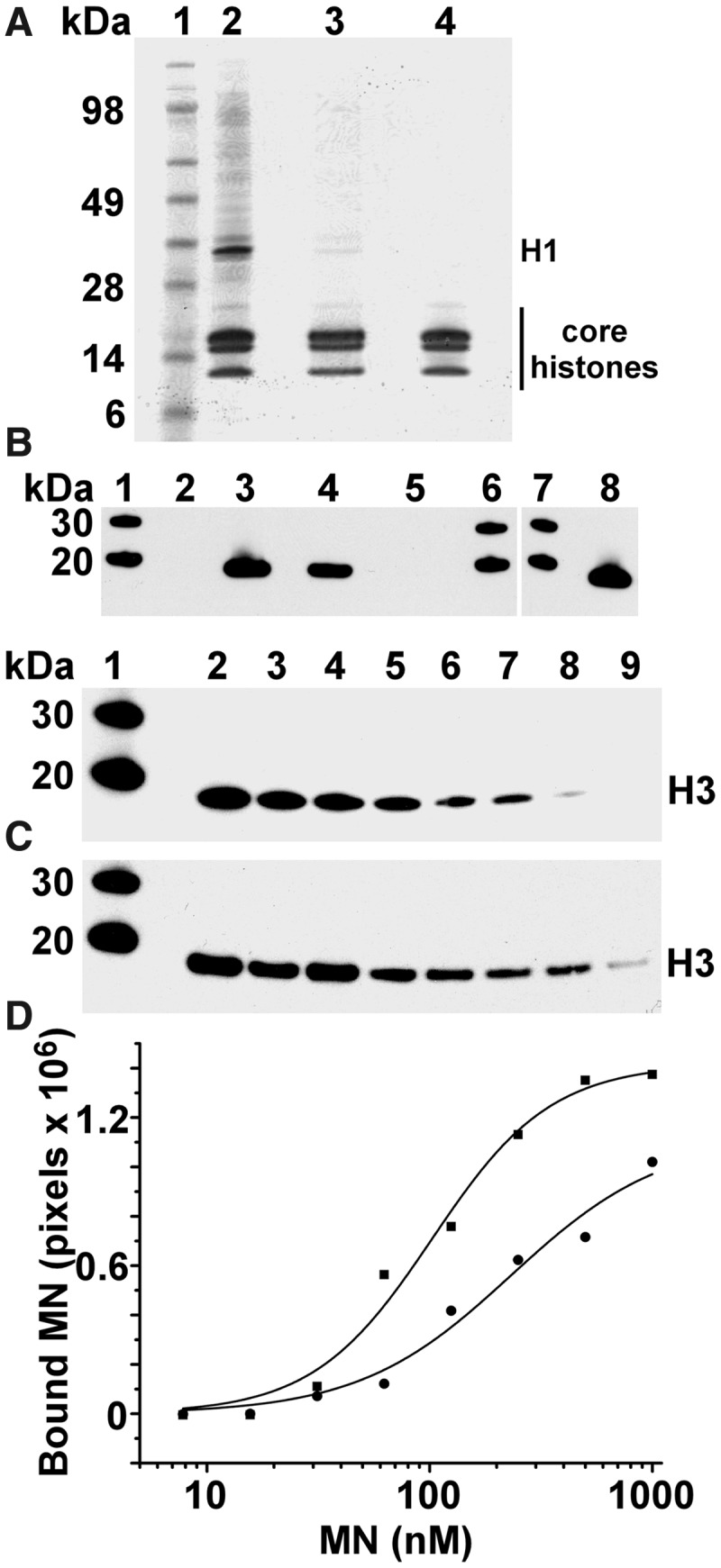
LEDGF PWWP tightly binds purified MNs. (A) Coomassie-stained SDS-PAGE analysis of MNs purified from SupT1 cells. Lane 1: molecular weight markers; lane 2: micrococcal nuclease-digested chromatin; lane 3: MN fractions recovered from sucrose gradient ultracentrifugation of the digested chromatin; lane 4: MN fractions that were purified further with size exclusion chromatography. (B) Pull-down experiments to probe the interactions of GST LEDGF/p75, GST LEDGF PWWP and GST LEDGF IBD with purified MNs. Lane contents are as follows: lanes 1, 6 and 7 contain protein molecular weight markers; lane 2: negative control with MNs plus the beads; lane 3: GST LEDGF/p75 plus MNs; lane 4: GST LEDGF PWWP plus MNs; lane 5: GST LEDGF IBD plus MNs; lane 8: 1/10th loading of MNs. Histone H3 antibody was used to monitor the interactions. (C) Western blot analysis of GST LEDGF PWWP interactions with MN fractions from sucrose gradient (top image) and size exclusion chromatography (bottom image) purification steps. Lane 1: protein standards; lanes 2–9: 100 nM GST LEDGF PWWP was incubated with decreasing concentrations of MNs followed by pull-down using GST-beads. (D) The intensities of GST LEDGF PWWP-bound histone H3 bands were quantified using ImageJ software and fit the data to the Hill equation. Circles and squares show GST LEDGF PWWP binding to MNs purified by sucrose gradient and size exclusion chromatography, respectively.
Next, we examined whether cellular proteins other than core histones can mediate the LEDGF PWWP binding to MNs. Nuclease-treated chromatin (lane 2 in Figure 2A) pelleted upon centrifugation at 10 000g for 10 min and could not be used for binding assays. In contrast, the MNs recovered by NaCl treatment of the digested chromatin and their subsequent fractionation by sucrose, equilibrium density gradient centrifugation (lane 3 in Figure 2A) were readily soluble and used for binding assays. Coomassie-stained SDS-PAGE analysis of sucrose gradient fractions revealed that the majority of the proteins were core histones with additional faint bands of other proteins being also present. To determine the full composition of the sample, we cut out the entire lane 3 in Figure 2A and subjected it to MS analysis. The data in Supplementary Table S1 show that a number of cellular proteins were present in the sample in addition to core histones. We next performed the pull-down assays using GST LEDGF PWWP and sucrose gradient-purified MNs. The starting input and pull-down samples were normalized for total protein and subjected to mass spectrometry (MS)-based proteomic analysis in parallel experiments. The data in Supplementary Table S1 show ‘Unweighted Spectrum Count’ for the identified proteins, which provide a semi-quantitative comparison of relative abundance of a given protein in either the pulled-down or input samples. As expected, LEDGF PWWP and histone proteins in both the pulled-down fractions and the input were at comparable levels due to the fact that the samples were adjusted based on total protein loaded. However, relative quantities of other cellular proteins present in the input sample were markedly decreased after pull-down of MNs with GST LEDGF PWWP (Supplementary Table S1). For example, although significant quantities of Heterochromatin protein-1-β and topoisomerase peptide peaks were detected in the partially purified MN preparations, these proteins were absent in the pull-down fractions. These results show that LEDGF PWWP disfavoured MNs associated with other chromatin-binding partners, possibly due to their masking of interacting interfaces on the MNs that were therefore unavailable for LEDGF PWWP.
Supplementary Table S1 shows that besides core histones, two proteins, H1 and ubiquinone biosynthesis monooxygenase COQ6, were also present in the pulled-down samples. To determine whether these proteins contributed to binding or if they remained associated with MNs without affecting the interactions with LEDGF PWWP, we further purified MNs using size exclusion chromatography. Visual inspection of the Coomassie-stained gel indicated that the faint bands corresponding to H1 and other high molecular weight cellular proteins present in sucrose gradient fractions were effectively removed from MNs by size exclusion chromatography (compare lanes 3 and 4 in Figure 2A). Furthermore, MS analysis of the GST LEDGF PWWP pull-down of highly purified MNs revealed only core histones; H1 or other cellular proteins were not detected in this sample (data not shown). Proteomic experiments were repeated three times, with only core histones being consistently pulled-down by GST LEDGF PWWP.
We next compared the binding affinities of LEDGF PWWP with partially purified (lane 3, Figure 2A) and highly purified (lane 4, Figure 2A) MNs. GST-tagged PWWP was incubated with increasing amounts of MNs, and the amount of bound MN was determined for each titration point. The apparent Kd values estimated from these experiments were 232 ± 23 nM and 102 ± 8 nM for LEDGF PWWP binding to MNs from sucrose gradient and size exclusion chromatography fractions, respectively (Figures 2C and D). The slightly higher apparent Kd value observed with the sucrose gradient fractions is consistent with our MS data demonstrating that residual amounts of chromatin-associated proteins interfere rather than facilitate LEDGF PWWP interaction with MNs. Taken together, our results in Figure 2 and Supplementary Table S1 argue strongly that LEDGF PWWP directly interacts with purified MNs.
Solution structure of LEDGF PWWP
To understand the structural basis for the recognition of MNs by LEDGF PWWP, we first determined the solution structure of the apo-protein using NMR (PDB ID: 2M16, Figure 3A and Supplementary Table S2). The protein has the characteristic PWWP domain fold (13,14,16,18,19,43) with a five-strand anti-parallel β-barrel (β1, 10–14; β2, 20–25; β3, 40–44; β4, 49–53; β5, 58–60) forming the protein core (Figure 3B). The protein also has a 310 helix (α1, 55–57) between β4 and β5. The C-terminal portion consists of two α-helices (α2, 61–67; α3, 81–86) (Figure 3B). The structure is well defined in the structured regions with a RMSD of 0.4 Å for backbone and 0.9 Å for all heavy atoms (Supplementary Table S2). The first four N-terminal residues, the last two C-terminal residues and loop between β2 and β3 were not well defined by the NMR data and appear to be relatively flexible. In addition, four non-native residues remaining at the protein N-terminus after proteolytic cleavage of the affinity tag were unstructured and not included in the structure calculation.
Figure 3.

The solution structure of LEDGF PWWP. (A) Superposition of the 20 structures that best fit the NMR data (PDB ID: 2M16). (B) The representative structure of LEDGF PWWP domain, with β sheets coloured blue, α helixes coloured red and loops coloured grey. (C) The electrostatic potential mapped to the surface of the protein. The neutral hydrophobic pocket and positively charged surface (blue) are indicated. Labels highlight residues flanking the hydrophobic pocket.
In parallel with these studies, we learned of unpublished coordinates 4FU6 deposited in the PDB by the Structural Genomics consortium (http://www.thesgc.org/). These coordinates represent the same domain as studied here, though it has additional N-terminal residues incorporated for recombinant expression and purification. The NMR-derived and x-ray crystal structures are highly similar (Supplementary Figure S2). However, a significant difference is observed beginning in the loop following helix α2 through several residues of helix α3 (residues 68–80). These differences are manifested in the geometry of helix α3, and in the packing of Tyr68 and Phe77 relative to the hydrophobic core. Careful inspection of the NOE pattern for residues 76–80, which are helical in 4FU6, showed that they were missing the Hα-HN (i,i + 3) NOEs characteristic of an α-helix (Supplementary Figure S2). These data, together with the fact that this region is in a contact point between monomers in the crystal (Supplementary Figure S3), indicate that the solution and crystal structures differ in this region, and highlight a region of protein flexibility that may be important for ligand binding and recognition.
A key feature of the LEDGF PWWP solution structure is a well-defined hydrophobic pocket (Figure 3C). The loops connecting β1 with β2, β3 with β4 and α1 with α2 as well as β4 strand contribute to the cavity. The pocket has an extended ‘horseshoe’ shape with well-defined walls on its three sides (top, left and right) and an opening at its bottom end. Side chains of Trp21 and Phe44 are at the base of the cavity. The aliphatic side chains of Arg74 and Lys75 define the top wall. Side chains of Met15, Tyr18 and Pro19 form the left wall, whereas the methyl of Thr47 and the aliphatic groups of the side chain of Glu49 form the right side of the pocket. There is an opening between Tyr18 and Glu49 at the bottom end of the cavity with the methyl group of Ala51 lying at its floor. The dimensions of the LEDGF PWWP pocket (width ∼4 Å, depth ∼6 Å and length ∼10.5 Å) are suitable to readily accommodate a post-translationally modified Lys side chain with the opening at the bottom of the cavity enabling the side chain to extend from the histone peptide backbone into the cavity.
An electrostatic potential map of the protein reveals a highly basic protein surface adjacent to the left of the hydrophobic pocket (Figure 3C). This surface comprises the loops between β1 and β2, β4 and β5 and α2 and α3 as well as helices α1 and α2. The following basic residues are a part of the surface: Lys14, Lys16, Lys56, Lys67, Lys70, Lys73, Arg74 and Lys75. Mutagenesis studies have previously highlighted the functional importance of several of these residues in LEDGF PWWP (25).
The LEDGF PWWP hydrophobic pocket interacts with H3K36me3
To determine LEDGF PWWP interfaces interacting with MNs, we have exploited the NMR amide resonance assignments and monitored CSP upon complex formation. Titration of purified MNs into LEDGF PWWP was accompanied by significant broadening and disappearance of signals, instead of detectable shift perturbations. This could be explained by motional broadening induced by a tight binding of the significantly larger molecular weight MNs to LEDGF PWWP. Therefore, we next investigated contributions of individual components of MNs by monitoring LEDGF PWWP interactions with small histone peptides and DNA.
LEDGF PWWP has recently been shown to preferentially interact with H3K36me3 (21,44). To identify the peptide-binding site, we examined CSPs upon complex formation. Figure 4 and Supplementary Figures S4 and S5 show that the largest perturbations occurred in the region directly surrounding the hydrophobic cavity. These include the backbone amides from Lys14, Met15, Tyr18 and Trp21, which form the left face of the pocket; the side chain amide from Trp21, located in the base of the cavity; and Arg24, Phe43, Phe44, Phe45, Thr47, His48, Glu49, Thr50 and Ala51 located to the right of the cavity and forming the β4 sheet. Interestingly, these perturbations extend on both sides of this β sheet, suggesting the tail of the peptide wraps around β4 and reaches the opposite side of the hydrophobic cavity. Such a binding mode is also supported by large perturbations in residues Phe5, Gly8, Lys91 and Phe92, which are located along the backside of LEDGF PWWP. Saturation during the titration was not reached at the highest concentration (6.5 mM) of the peptide tested (Supplementary Figures S6A and B), indicating that the binding affinity is in the low mM range. It should be noted that Brpf1 PWWP binds H3K36me3 with a similar Kd of ∼2.7 mM (14).
Figure 4.

Identification of LEDGF PWWP residues interacting with H3K36me3 and DNA. (A) Overlay of a small region of the 1H-15N HSQCs of the titration of H3K36me3 into LEDGF PWWP (full spectra are shown in Supplementary Figure S4A). Free LEDGF PWWP is in black and the 80:1 titration point is in red. (B) Overlay of a small region of the 1H-15N HSQCs of the titration of DNA into LEDGF PWWP (full spectra are in Supplementary Figure S4C). Free LEDGF PWWP is in black and the 3.2:1 titration point is in green. (C) The CSP caused by H3K36me3 mapped onto LEDGF PWWP. White indicates small or no perturbation, and red indicates large perturbation (see the quantitative results in Supplementary Figure S5). (D) The CSP caused by DNA mapped to LEDGF PWWP. White indicates small or no perturbation, and green indicates large perturbation (see the quantitative results in Supplementary Figure S5).
In control experiments, we used NMR to monitor PWWP binding to unmodified peptides. Although similar CSPs were observed for residues flanking the hydrophobic pocket (i.e., Gly8, His48, Lys91 and Phe92; Supplementary Figures S4 and S5), no significant CSPs were observed within the hydrophobic pocket (compare Supplementary Figures S4A and B; also Supplementary Figures S5, S6A and B). These data indicate that although the binding modes for unmethylated and trimethylated peptides are similar, the additional interaction between LEDGF PWWP hydrophobic pocket and the modified peptide provides enhanced affinity.
The LEDGF PWWP basic surface non-specifically binds DNA
To delineate contributions of DNA for the high affinity LEDGF PWWP:MN interactions, we isolated native ∼147-mer DNAs by protease treatment of purified MNs followed by phenol–chloroform extraction of DNA. Since HDGF PWWP, a close homologue of LEDGF PWWP, has been shown to preferentially bind SMYD1 sequences (15), this DNA was also analysed in parallel experiments. The control experiments contained non-specific synthetic DNA. The electrophoretic mobility shift assay (EMSA) data in Supplementary Figure S7 have revealed very similar Kd values of ∼1.5 μM for the LEDGF PWWP binding to all three DNA preparations, indicating that LEDGF PWWP binds DNA non-specifically.
To determine the LEDGF PWWP interfaces that bind DNA, we monitored CSPs upon titrating DNA into the protein (Figure 4 and Supplementary Figures S4 and S5). The most perturbed amides correspond to residues distributed across the basic face, and include backbone amides of Thr2, Arg3, Asp4, Lys14, Lys16, Tyr18, His20, Asn38, Gly54, Lys56, Asp57, Ile58, Phe59, Lys67, Tyr68, Lys70, Arg74, Lys75, Gly76 and Glu79, and side chain of Asn78. This region is highly positively charged (compare Figures 3C and 4D) and thus likely to establish favourable electrostatic interactions with the phosphate backbone of DNA. Residues Asp30 and Gly31 (at the bottom right of LEDGF PWWP in Figure 4D) are also perturbed. These residues may not directly interact with DNA but instead undergo a conformational change due to the higher flexibility in this region. The quantitative analysis of CSPs (Supplementary Figures S6C and D) suggests a low micromolar binding affinity and is in good agreement with the EMSA experiments (Supplementary Figure S7).
Comparative analysis of the binding results (Figure 4 and Supplementary Figures S4 and S5) reveals that 17 and 23 residues were significantly perturbed by H3K36me3 and DNA, respectively. Of these, only two residues Lys14 and Tyr18 were affected by both ligands. It should be noted that the backbones of these residues directly contribute to the hydrophobic cavity while their side chains are extended to the basic surface. Thus, the hydrophobic cavity and the basic interface provide adjacent and distinct interfaces to engage H3K36me3 and DNA, respectively.
LEDGF PWWP tightly binds H3KC36me3 MNs
Purified native MNs contain a variety of histone post-translational modifications, including H3K36me3. To directly test the significance of H3K36 tri-methylation for the high-affinity interaction of LEDGF PWWP with MNs, we used the method (29) of site-specific incorporation of the tri-methyl-lysine analogue in recombinant histones. In vitro reconstituted MNs containing tri-methyl-lysine analogues have been shown to be functionally similar to their natural counterparts (29). We incorporated a tri-methyl-lysine analogue at position 36 of H3 and mixed the modified protein with unmodified, recombinant H2A, H2B and H4, and 147-bp synthetic DNA to reconstitute H3KC36me3 MNs. For control experiments, MNs containing unmodified core histones were prepared. We next monitored GST LEDGF PWWP binding to H3KC36me3-modified and unmodified MNs using a GST pull-down assay (Figure 5B). GST LEDGF PWWP tightly interacted with H3KC36me3 MNs (Kd of ∼48 ± 11 nM), whereas only residual binding was observed with unmodified MNs at 500 nM (Figure 5B and C). The Kd value for unmodified MNs could not be determined from these assays, as at concentrations exceeding 500 nM, we observed significant non-specific interactions of MNs with the glutathione resin. Since the only difference between H3KC36me3 MNs and their unmodified counterparts (Figure 5) is the tri-methylation of residue 36 of H3, we conclude that this modification plays a critical role in high-affinity binding of LEDGF PWWP to MNs.
Figure 5.

LEDGF PWWP tightly and preferentially binds H3KC36me3 MNs. (A) Western blot analysis of H3KC36me3-modified and unmodified MNs. Lane 1: molecular weight markers; lanes 2 and 3: inputs (1/20th) for H3KC36me3-modified and unmodified MNs, respectively. (B) Western blot analysis of GST PWWP interaction with increasing concentrations of H3KC36me3-modified or unmodified MNs. Lane 1: molecular weight markers; lanes 2 to 8: the GST PWWP pull-down of decreasing concentrations (500 nM to 7.8 nM) of H3KC36me3-modified (top) or unmodified (bottom) MNs. (C) The histone H3 bands were detected by the histone H3 antibody and quantified using ImageJ software and fit to the Hill equation. H3 band intensities were normalized using the 30 kDa band intensity from the molecular weight marker in each gel. Squares correspond to the H3KC36me3-modified MN bound and circles correspond to the unmodified MN bound.
A model for LEDGF PWWP binding to MN
Our NMR data together with the available structure of MN (40) allowed us to construct an atomic model for the LEDGF PWWP interaction with MN. Figure 6 and Supplementary Figure S8 show the basic surface of LEDGF PWWP engaging the DNA wrapped around the histone core. It should be noted that while nucleosomal DNA is significantly bent, we used a free 40-bp DNA for mapping binding sites by NMR (Figure 4 and Supplementary Figures S4–S6). Despite these differences, the majority of residues identified by CSPs (including Lys14, Lys16, Tyr18, His20, Lys56, Asp57, Phe59, Lys67, Tyr68, Lys70, Arg74 and Lys75) were also found to interact with the nucleosomal DNA in the proposed model (Figure 6 and Supplementary Figure S8).
Figure 6.

Close-up of the model for the LEDGF PWWP binding to the MN (see Supplementary Figure S8 for the full view of the complex).
Our molecular model agrees well with the NMR results for the H3K36me3 peptide binding to LEDGF PWWP (Figure 4 and Supplementary Figures S4–S6). The H3 histone tail extends from the core structure of the MN and follows its binding path on LEDGF PWWP, which passes directly underneath the hydrophobic pocket and then curls around the β4 sheet (Figure 6 and Supplementary Figure S8). The opening at the bottom of the ‘horseshoe’-shaped cavity allows the trimethylated Lys36 side chain of the H3 histone tail to extend from the peptide backbone into the hydrophobic pocket to establish specific interactions.
DISCUSSION
Cooperative binding of LEDGF PWWP to H3K36me3 and DNA is essential for its high-affinity interaction with MNs
The present studies reveal a direct high-affinity binding of LEDGF PWWP with purified native MNs in a manner that is not mediated by other cellular factors. The NMR solution structure of LEDGF PWWP combined with titration experiments enabled us to identify the key interacting interfaces: the hydrophobic cavity selectively coordinates trimethylated Lys36 of the H3 histone tail and the basic surface non-specifically binds DNA. Although our NMR titration data agree with the recent reports (21,44) that LEDGF PWWP preferentially binds H3K36me3, the data in Supplementary Figure S6 importantly show that the LEDGF PWWP:H3K36me3 interaction exhibits a very low affinity (Kd is in the low millimolar range). Therefore, these protein–protein interactions alone are not likely to be sufficient for the tight binding observed for LEDGF PWWP to chromatin. Furthermore, we demonstrate that LEDGF PWWP binds DNA (Supplementary Figures S6 and S7). Since the binding affinities of LEDGF PWWP with H3K36me3 or DNA are significantly lower than with MNs (Figure 2 and Supplementary Figures S6 and S7), we conclude that the hydrophobic pocket and the basic surface act synergistically to ensure high-affinity binding of LEDGF PWWP with MNs. This notion is further supported by our observations (Figure 5) that LEDGF PWWP preferentially and tightly binds in vitro reconstituted MNs containing H3KC36me3 compared with their unmodified counterparts.
Such a cooperative mode of binding is likely to be a general mechanism of the PWWP modules. For example, Brpf1 PWWP displayed selective, but weak, binding (Kd of ∼2.7 mM) to the isolated H3K36me3 peptide (14). At the same time, a number of PWWP domains have been shown to exhibit DNA-binding activities (13,16,19). It should be noted that Lys36 is located in proximity to the DNA wrapped around the histone core [(40) and Figure 6]. Therefore, it is logical to propose that PWWP modules have evolved to exploit this feature to stabilize their interactions with chromatin by also directly engaging the DNA and do not solely rely on protein–protein interactions.
Importantly, there is an excellent correlation between our findings and published mutagenesis results (25). Based on amino acid sequence similarities between LEDGF PWWP and its homologues, Engelman and coworkers selected 24 residues for mutagenesis experiments. These mutations were introduced in the context of ectopically expressed full-length LEDGF, and their ability to rescue HIV-1 infection in LEDGF/p75 knock out cells was examined (25). Of particular interest are the substitutions of residues forming the hydrophobic pocket. Trp21 and Ala51 were found to be the most critical for LEDGF/p75 function, with important contributions of Met15, Thr47 and Glu49 also being observed (25). Our data in Figure 4 and Supplementary Figures S4–S6 show that these residues interact with H3K36me3 and not with unmodified peptide or DNA.
The mutational analyses have also revealed the importance of the basic surface for LEDGF/p75-dependent stimulation of HIV-1 integration. For example, substitution of Lys14 with Glu supported ∼16% of wild-type LEDGF/p75 levels of HIV-1 infectivity. Furthermore, the mutant with substitutions at six basic residues (K56A, K67A, K70A, K73A, R74A, K75A) failed to stimulate HIV-1 infectivity (25). Our data in Figure 4 and Supplementary Figures S4–S6 show that these residues directly interact with DNA. Taken together, the mutagenesis results and our findings collectively indicate the functional significance of the hydrophobic cavity and basic interface for LEDGF/p75 binding to chromatin.
The cooperative mechanism of LEDGF PWWP engagement with H3K36me3 and mononucleosomal DNA is likely to play a key role in directing LEDGF/p75 to cognate chromatin sites and accordingly navigating HIV-1 integration into the actively transcribed genes. Indeed, the trimethylated Lys36 is an epigenetic marker for active transcription units. At the same time, LEDGF PWWP is likely to have greater access to nucleosomal DNA in the context of actively transcribed genes. These two factors of the trimethylation of Lys36 and more accessible DNA being available together would contribute to preferential integration of viral cDNA at these sites.
In addition, the following correlation is particularly noteworthy. In vitro, purified IN favours integration into DNA wrapped in nucleosomes compared with free DNA (45,46), and in infected cells, HIV-1 integration sites in chromosomal DNA are periodically distributed on the nucleosome surface (47). Our findings that cellular LEDGF/p75 primarily binds MNs (Figure 1) with LEDGF PWWP tightly engaging the outer surface of MNs (Figure 6 and Supplementary Figure S8) provide structural clues for how the cellular cofactor tethers HIV-1 PICs at nucleosomal sites, which in turn are the preferred substrates for the IN-mediated pair-wise integration of viral cDNA.
Distinct structural features of LEDGF PWWP
The five β strand core of the LEDGF PWWP domain is highly similar to other solved PWWP domain structures [Supplementary Figure S9 and (13,14,16,18,19,43)]. This similarity extends to the loop between β1 and β2, and the loop between β3 and β4, as well as strand β4 (coloured green in Supplementary Figure S9), which define the base and side walls of the hydrophobic pocket. However, there are important differences between LEDGF PWWP and its homologues (Supplementary Figure S9). For example, the C-terminal portion of LEDGF PWWP located at the upper wall of the pocket (coloured red in Supplementary Figure S9) has less alpha helical character than other homologues. The PWWP domains from HDGF, HDGF-2 (or HRP-2), Brpf1 and Pdp1 each have a helix–loop–helix structure in this region that directly contributes to the hydrophobic pocket. In contrast, the respective α3-helix in LEDGF PWWP is significantly shorter (81–86) and thus a longer loop (68–80) forms the top of the hydrophobic pocket, with Arg74 and Lys75 directly forming the top wall. Significantly, both NMR titration (Figure 4 and Supplementary Figures S4 and S5) and site-directed mutagenesis experiments (25) confirm the functional importance of these residues. Moreover, this is the region of the protein that exhibits conformational differences between the solution and deposited crystal structures, implicating them in induced-fit recognition.
Comparison of the LEDGF PWWP structure with the Brpf1 PWWP complexed with the cognate H3K36me3 peptide reveals further striking differences involving the protein regions that interact with the extended histone peptide tail (Supplementary Figure S10). Although the hydrophobic cavities in Brpf1 PWWP and LEDGF PWWP similarly engage the trimethylated Lys36, the N-terminal portion of the histone tail is oriented differently in these two complexes. The peptide extends from the hydrophobic cavity along the front bottom surface of Brpf1 PWWP engaging the loop between β2 and β3, residues from an α-helix (1125–1139), two short β barrels (1113–1115 and 1118–1120) and their connecting loop (1121–1124) (coloured red in Supplementary Figure S10). These Brpf1 PWWP structural elements differ significantly from the LEDGF PWWP structure, which contains a small flexible loop in this region (coloured red in Supplementary Figure S10). Our NMR titration results indicate the N-terminal portion of the histone tail curls around LEDGF PWWP to interact with the back side of the hydrophobic cavity.
These distinct structural features of LEDGF PWWP could help to develop a new class of inhibitors that would selectively interfere with LEDGF/p75 binding with chromatin. LEDGF/p75 is essential for effective HIV-1 integration into human chromosomes, and small molecules that impair LEDGF/p75 association with chromatin would consequently inhibit HIV-1 replication. Furthermore, LEDGF/p75 is over-expressed in many solid tumours and plays a key role in cancer cell survival (48,49). These cellular activities of LEDGF/p75 have been linked to the role of LEDGF PWWP in DNA repair (44). Therefore, the selective inhibitors of LEDGF PWWP interaction with chromatin could offer a new avenue for anti-cancer drug discovery as well.
ACCESSION NUMBERS
PDB ID: 2M16.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online: Supplementary Tables 1 and 2, Supplementary Figures 1–10 and Supplementary References [13,14,18,19,50–52].
FUNDING
National Institutes of Health [AI062520 and GM103368 to M.K., GM077234 to M.P.F.]; National Cancer Institute, National Institutes of Health [under contract HHSN261200800001E with SAIC-Frederick, Inc. (to R.J.G.)]. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U. S. Government. Funding for open access charge: National Institutes of Health [GM103368].
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
We wish to thank Julian W. Bess, Jr., Rodman B. Smith, William H. Bohn and David B. Westcott of the AIDS and Cancer Virus Program, Biological Products Core, for producing the SupT1 cells for this study. We also thank Jacques Kessl, Ross Larue and Amit Sharma for critical reading of the manuscript.
REFERENCES
- 1.Cherepanov P, Maertens G, Proost P, Devreese B, Van Beeumen J, Engelborghs Y, De Clercq E, Debyser Z. HIV-1 integrase forms stable tetramers and associates with LEDGF/p75 protein in human cells. J. Biol. Chem. 2003;278:372–381. doi: 10.1074/jbc.M209278200. [DOI] [PubMed] [Google Scholar]
- 2.Ciuffi A, Llano M, Poeschla E, Hoffmann C, Leipzig J, Shinn P, Ecker JR, Bushman F. A role for LEDGF/p75 in targeting HIV DNA integration. Nat. Med. 2005;11:1287–1289. doi: 10.1038/nm1329. [DOI] [PubMed] [Google Scholar]
- 3.Llano M, Saenz DT, Meehan A, Wongthida P, Peretz M, Walker WH, Teo W, Poeschla EM. An essential role for LEDGF/p75 in HIV integration. Science. 2006;314:461–464. doi: 10.1126/science.1132319. [DOI] [PubMed] [Google Scholar]
- 4.Shun MC, Raghavendra NK, Vandegraaff N, Daigle JE, Hughes S, Kellam P, Cherepanov P, Engelman A. LEDGF/p75 functions downstream from preintegration complex formation to effect gene-specific HIV-1 integration. Genes Dev. 2007;21:1767–1778. doi: 10.1101/gad.1565107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cherepanov P, Ambrosio AL, Rahman S, Ellenberger T, Engelman A. Structural basis for the recognition between HIV-1 integrase and transcriptional coactivator p75. Proc. Natl Acad. Sci. USA. 2005;102:17308–17313. doi: 10.1073/pnas.0506924102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hare S, Di Nunzio F, Labeja A, Wang J, Engelman A, Cherepanov P. Structural basis for functional tetramerization of lentiviral integrase. PLoS Pathog. 2009;5:e1000515. doi: 10.1371/journal.ppat.1000515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gijsbers R, Ronen K, Vets S, Malani N, De Rijck J, McNeely M, Bushman FD, Debyser Z. LEDGF hybrids efficiently retarget lentiviral integration into heterochromatin. Mol. Ther. 2010;18:552–560. doi: 10.1038/mt.2010.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Llano M, Vanegas M, Hutchins N, Thompson D, Delgado S, Poeschla EM. Identification and characterization of the chromatin-binding domains of the HIV-1 integrase interactor LEDGF/p75. J. Mol. Biol. 2006;360:760–773. doi: 10.1016/j.jmb.2006.04.073. [DOI] [PubMed] [Google Scholar]
- 9.Gijsbers R, Vets S, De Rijck J, Ocwieja KE, Ronen K, Malani N, Bushman FD, Debyser Z. Role of the PWWP domain of lens epithelium-derived growth factor (LEDGF)/p75 cofactor in lentiviral integration targeting. J. Biol. Chem. 2011;286:41812–41825. doi: 10.1074/jbc.M111.255711. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 10.Turlure F, Maertens G, Rahman S, Cherepanov P, Engelman A. A tripartite DNA-binding element, comprised of the nuclear localization signal and two AT-hook motifs, mediates the association of LEDGF/p75 with chromatin in vivo. Nucleic Acids Res. 2006;34:1653–1675. doi: 10.1093/nar/gkl052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ferris AL, Wu X, Hughes CM, Stewart C, Smith SJ, Milne TA, Wang GG, Shun MC, Allis CD, Engelman A, et al. Lens epithelium-derived growth factor fusion proteins redirect HIV-1 DNA integration. Proc. Natl Acad. Sci. USA. 2010;107:3135–3140. doi: 10.1073/pnas.0914142107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Meehan AM, Saenz DT, Morrison JH, Garcia-Rivera JA, Peretz M, Llano M, Poeschla EM. LEDGF/p75 proteins with alternative chromatin tethers are functional HIV-1 cofactors. PLoS Pathog. 2009;5:e1000522. doi: 10.1371/journal.ppat.1000522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Qiu Y, Zhang W, Zhao C, Wang Y, Wang W, Zhang J, Zhang Z, Li G, Shi Y, Tu X, et al. Solution structure of the Pdp1 PWWP domain reveals its unique binding sites for methylated H4K20 and DNA. Biochem. J. 2012;442:527–538. doi: 10.1042/BJ20111885. [DOI] [PubMed] [Google Scholar]
- 14.Vezzoli A, Bonadies N, Allen MD, Freund SM, Santiveri CM, Kvinlaug BT, Huntly BJ, Gottgens B, Bycroft M. Molecular basis of histone H3K36me3 recognition by the PWWP domain of Brpf1. Nat. Struct. Mol. Biol. 2010;17:617–619. doi: 10.1038/nsmb.1797. [DOI] [PubMed] [Google Scholar]
- 15.Yang J, Everett AD. Hepatoma-derived growth factor binds DNA through the N-terminal PWWP domain. BMC Mol. Biol. 2007;8:101. doi: 10.1186/1471-2199-8-101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Qiu C, Sawada K, Zhang X, Cheng X. The PWWP domain of mammalian DNA methyltransferase Dnmt3b defines a new family of DNA-binding folds. Nat. Struct. Biol. 2002;9:217–224. doi: 10.1038/nsb759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dhayalan A, Rajavelu A, Rathert P, Tamas R, Jurkowska RZ, Ragozin S, Jeltsch A. The Dnmt3a PWWP domain reads histone 3 lysine 36 trimethylation and guides DNA methylation. J. Biol. Chem. 2010;285:26114–26120. doi: 10.1074/jbc.M109.089433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wu H, Zeng H, Lam R, Tempel W, Amaya MF, Xu C, Dombrovski L, Qiu W, Wang Y, Min J. Structural and histone binding ability characterizations of human PWWP domains. PloS One. 2011;6:e18919. doi: 10.1371/journal.pone.0018919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lukasik SM, Cierpicki T, Borloz M, Grembecka J, Everett A, Bushweller JH. High resolution structure of the HDGF PWWP domain: a potential DNA binding domain. Protein Sci. 2006;15:314–323. doi: 10.1110/ps.051751706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vermeulen M, Eberl HC, Matarese F, Marks H, Denissov S, Butter F, Lee KK, Olsen JV, Hyman AA, Stunnenberg HG, et al. Quantitative interaction proteomics and genome-wide profiling of epigenetic histone marks and their readers. Cell. 2010;142:967–980. doi: 10.1016/j.cell.2010.08.020. [DOI] [PubMed] [Google Scholar]
- 21.Pradeepa MM, Sutherland HG, Ule J, Grimes GR, Bickmore WA. Psip1/Ledgf p52 binds methylated histone H3K36 and splicing factors and contributes to the regulation of alternative splicing. PLoS Genet. 2012;8:e1002717. doi: 10.1371/journal.pgen.1002717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vandegraaff N, Devroe E, Turlure F, Silver PA, Engelman A. Biochemical and genetic analyses of integrase-interacting proteins lens epithelium-derived growth factor (LEDGF)/p75 and hepatoma-derived growth factor related protein 2 (HRP2) in preintegration complex function and HIV-1 replication. Virology. 2006;346:415–426. doi: 10.1016/j.virol.2005.11.022. [DOI] [PubMed] [Google Scholar]
- 23.Hare S, Shun MC, Gupta SS, Valkov E, Engelman A, Cherepanov P. A novel co-crystal structure affords the design of gain-of-function lentiviral integrase mutants in the presence of modified PSIP1/LEDGF/p75. PLoS Pathog. 2009;5:e1000259. doi: 10.1371/journal.ppat.1000259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Botbol Y, Raghavendra NK, Rahman S, Engelman A, Lavigne M. Chromatinized templates reveal the requirement for the LEDGF/p75 PWWP domain during HIV-1 integration in vitro. Nucleic Acids Res. 2008;36:1237–1246. doi: 10.1093/nar/gkm1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shun MC, Botbol Y, Li X, Di Nunzio F, Daigle JE, Yan N, Lieberman J, Lavigne M, Engelman A. Identification and characterization of PWWP domain residues critical for LEDGF/p75 chromatin binding and human immunodeficiency virus type 1 infectivity. J. Virol. 2008;82:11555–11567. doi: 10.1128/JVI.01561-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schnitzler GR. Isolation of Histones and Nucleosome Cores from Mammalian Cells. Curr. Protoc. Mol. Biol. 2000:21.25.21–21.25.12. doi: 10.1002/0471142727.mb2105s50. [DOI] [PubMed] [Google Scholar]
- 27.Luger K, Rechsteiner TJ, Richmond TJ. Expression and purification of recombinant histones and nucleosome reconstitution. Methods Mol. Biol. 1999;119:1–16. doi: 10.1385/1-59259-681-9:1. [DOI] [PubMed] [Google Scholar]
- 28.Manohar M, Mooney AM, North JA, Nakkula RJ, Picking JW, Edon A, Fishel R, Poirier MG, Ottesen JJ. Acetylation of histone H3 at the nucleosome dyad alters DNA-histone binding. J. Biol. Chem. 2009;284:23312–23321. doi: 10.1074/jbc.M109.003202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Simon MD, Chu F, Racki LR, de la Cruz CC, Burlingame AL, Panning B, Narlikar GJ, Shokat KM. The site-specific installation of methyl-lysine analogs into recombinant histones. Cell. 2007;128:1003–1012. doi: 10.1016/j.cell.2006.12.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lowary PT, Widom J. New DNA sequence rules for high affinity binding to histone octamer and sequence-directed nucleosome positioning. J. Mol. Biol. 1998;276:19–42. doi: 10.1006/jmbi.1997.1494. [DOI] [PubMed] [Google Scholar]
- 31.McKee CJ, Kessl JJ, Norris JO, Shkriabai N, Kvaratskhelia M. Mass spectrometry-based footprinting of protein-protein interactions. Methods. 2009;47:304–307. doi: 10.1016/j.ymeth.2008.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lee JE, Sweredoski MJ, Graham RL, Kolawa NJ, Smith GT, Hess S, Deshaies RJ. The steady-state repertoire of human SCF ubiquitin ligase complexes does not require ongoing Nedd8 conjugation. Mol. Cell Proteomics. 2011;10:M110 006460. doi: 10.1074/mcp.M110.006460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 34.Johnson BA. Using NMRView to visualize and analyze the NMR spectra of macromolecules. Methods Mol. Biol. 2004;278:313–352. doi: 10.1385/1-59259-809-9:313. [DOI] [PubMed] [Google Scholar]
- 35.Sattler M, Schleucher J, Griesinger C. Heteronuclear multidimensional NMR experiments for the structure determination of proteins in solution employing pulsed field gradients. Prog. Nucl. Mag. Res. Sp. 1999;34:93–158. [Google Scholar]
- 36.Bahrami A, Assadi AH, Markley JL, Eghbalnia HR. Probabilistic interaction network of evidence algorithm and its application to complete labeling of peak lists from protein NMR spectroscopy. PLoS Comput. Biol. 2009;5:e1000307. doi: 10.1371/journal.pcbi.1000307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shen Y, Delaglio F, Cornilescu G, Bax A. TALOS+: a hybrid method for predicting protein backbone torsion angles from NMR chemical shifts. J. Biomol. NMR. 2009;44:213–223. doi: 10.1007/s10858-009-9333-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Guntert P. Automated NMR structure calculation with CYANA. Methods Mol. Biol. 2004;278:353–378. doi: 10.1385/1-59259-809-9:353. [DOI] [PubMed] [Google Scholar]
- 39.Garrett DS, Seok YJ, Peterkofsky A, Clore GM, Gronenborn AM. Identification by NMR of the binding surface for the histidine-containing phosphocarrier protein HPr on the N-terminal domain of enzyme I of the Escherichia coli phosphotransferase system. Biochemistry. 1997;36:4393–4398. doi: 10.1021/bi970221q. [DOI] [PubMed] [Google Scholar]
- 40.Luger K, Mader AW, Richmond RK, Sargent DF, Richmond TJ. Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature. 1997;389:251–260. doi: 10.1038/38444. [DOI] [PubMed] [Google Scholar]
- 41.Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta. Crystallogr. D Biol. Crystallogr. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.McNeely M, Hendrix J, Busschots K, Boons E, Deleersnijder A, Gerard M, Christ F, Debyser Z. In vitro DNA tethering of HIV-1 integrase by the transcriptional coactivator LEDGF/p75. J. Mol. Biol. 2011;410:811–830. doi: 10.1016/j.jmb.2011.03.073. [DOI] [PubMed] [Google Scholar]
- 43.Nameki N, Tochio N, Koshiba S, Inoue M, Yabuki T, Aoki M, Seki E, Matsuda T, Fujikura Y, Saito M, et al. Solution structure of the PWWP domain of the hepatoma-derived growth factor family. Protein Sci. 2005;14:756–764. doi: 10.1110/ps.04975305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Daugaard M, Baude A, Fugger K, Povlsen LK, Beck H, Sorensen CS, Petersen NH, Sorensen PH, Lukas C, Bartek J, et al. LEDGF (p75) promotes DNA-end resection and homologous recombination. Nat. Struct. Mol. Biol. 2012;19:803–810. doi: 10.1038/nsmb.2314. [DOI] [PubMed] [Google Scholar]
- 45.Pryciak PM, Sil A, Varmus HE. Retroviral integration into minichromosomes in vitro. EMBO J. 1992;11:291–303. doi: 10.1002/j.1460-2075.1992.tb05052.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pruss D, Reeves R, Bushman FD, Wolffe AP. The influence of DNA and nucleosome structure on integration events directed by HIV integrase. J. Biol. Chem. 1994;269:25031–25041. [PubMed] [Google Scholar]
- 47.Wang GP, Ciuffi A, Leipzig J, Berry CC, Bushman FD. HIV integration site selection: analysis by massively parallel pyrosequencing reveals association with epigenetic modifications. Genome Res. 2007;17:1186–1194. doi: 10.1101/gr.6286907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Daugaard M, Kirkegaard-Sorensen T, Ostenfeld MS, Aaboe M, Hoyer-Hansen M, Orntoft TF, Rohde M, Jaattela M. Lens epithelium-derived growth factor is an Hsp70-2 regulated guardian of lysosomal stability in human cancer. Cancer Res. 2007;67:2559–2567. doi: 10.1158/0008-5472.CAN-06-4121. [DOI] [PubMed] [Google Scholar]
- 49.Daniels T, Zhang J, Gutierrez I, Elliot ML, Yamada B, Heeb MJ, Sheets SM, Wu X, Casiano CA. Antinuclear autoantibodies in prostate cancer: immunity to LEDGF/p75, a survival protein highly expressed in prostate tumors and cleaved during apoptosis. Prostate. 2005;62:14–26. doi: 10.1002/pros.20112. [DOI] [PubMed] [Google Scholar]
- 50.Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Risler JL, Delorme MO, Delacroix H, Henaut A. Amino acid substitutions in structurally related proteins. A pattern recognition approach. Determination of a new and efficient scoring matrix. J. Mol. Biol. 1988;204:1019–1029. doi: 10.1016/0022-2836(88)90058-7. [DOI] [PubMed] [Google Scholar]
- 52.Gouet P, Courcelle E, Stuart DI, Metoz F. ESPript: analysis of multiple sequence alignments in PostScript. Bioinformatics. 1999;15:305–308. doi: 10.1093/bioinformatics/15.4.305. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.