

Abstract
Background
Renovascular hypertension (RVH) is characterized by chronic inflammation of the stenotic kidney and progressive renal dysfunction. Neutrophil gelatinase-associated lipocalin (NGAL), an acute phase protein induced in inflammatory conditions and ischemia, is a novel biomarker for acute kidney injury. We hypothesized that chronic RVH would be associated with increased renal and circulating NGAL levels.
Methods
We prospectively measured renal vein and inferior vena cava (IVC) levels of NGAL and inflammatory cytokines in essential hypertensive (EH) and RVH patients, during constant sodium intake and anti-hypertensive regimens, and compared them with systemic levels in age-matched normotensive subjects (n = 22 each). In addition, we measured urinary NGAL and kidney injury molecule (KIM)-1 in all patients.
Results
Blood pressure, serum creatinine, estimated glomerular filtration rate (eGFR), lipid panels and medications were similar in RVH and EH. Systemic, stenotic and contralateral renal vein levels of NGAL were all similarly elevated in RVH versus normal hypertension and EH (P < 0.05), as were renal vein levels of inflammatory markers like tumor necrosis factor-α. Furthermore, renal vein NGAL levels inversely correlated with eGFR, and directly with renal vein (but not systemic) levels of inflammatory markers. Urinary levels of NGAL and KIM-1 were elevated in both EH and RVH, as were systemic levels of C-reactive protein.
Conclusions
Chronic RVH is associated with elevated NGAL levels, likely due to ongoing kidney and systemic inflammation and ischemia. These findings may also imply the occurrence of the inflammation process in chronic RVH, which might contribute to the poorer outcomes of RVH compared with EH patients.
Keywords: cytokines, inflammation, KIM-1, NGAL, renovascular hypertension
Introduction
Renovascular hypertension (RVH), a common disorder in patients with atherosclerotic vascular occlusive disease of the kidney, is characterized by increased activity of the renin–angiotensin system, reduced renal perfusion pressure and progressive renal dysfunction [1]. Moreover, the combination of RVH and decreased glomerular filtration rate (GFR) predisposes to several complications such as coronary artery disease, cerebrovascular disease and peripheral vascular disease, leading to an increase in cardiovascular mortality [2].
Recent studies have suggested a role for renal inflammation in aggravating renal dysfunction in experimental RVH [3]. We have previously shown in swine RVH that the post-stenotic kidney up-regulates the expression of the proinflammatory chemokine monocyte chemoattractant protein (MCP-1) [4] and NFκB [5], as well as an influx of inflammatory cells [6], which are associated with decreased GFR and impaired endothelial function. Furthermore, a recent clinical study showed elevated systemic C-reactive protein (CRP) levels and peripheral leukocytes in patients with atherosclerotic renal artery stenosis versus EH and healthy controls [7].
Neutrophil gelatinase-associated lipocalin (NGAL), a 25 kDa amino acid protein, is a member of the lipocalin family largely expressed in activated neutrophils and tubular epithelial cells in response to inflammation [8]. NGAL plays an important role in innate immunity. Accumulating evidence also has established both urinary and plasma NGAL levels as predictive biomarkers for acute kidney injury (AKI) and its outcomes in varied groups of patients including pediatric [9] and adult [10] populations, critically ill patients [11] and contrast-induced nephropathy [12]. NGAL may originate from both renal and extra-renal sites. In ischemic murine AKI models, NGAL mRNA expression is initially up-regulated in the post-ischemic kidney, followed by increased urinary NGAL levels within 2 h after acute ischemia [13]. However, whether NGAL is released from the post-stenotic kidney of chronic human RVH has not been determined.
Similarly, kidney injury molecule (KIM)-1, a transmembrane type 1 glycoprotein expressed at very low levels in the normal kidney, has been proposed as a promising biomarker for the early prediction of AKI. KIM-1 is up-regulated in tubular epithelial cells of rats in response to ischemia [14] and in urine of patients with AKI, suggesting that KIM-1 is a sensitive indicator of acute tubular injury [15, 16]. Its expression is also similarly elevated in human renal biopsies of EH and patients with renal diseases compared with normal controls [17–19]. However, whether its expression is up-regulated in RVH patients remains unknown.
This study tested the hypothesis that atherosclerotic RVH would be associated with increased renal and circulating NGAL levels, as well as urinary NGAL and KIM-1 levels. In addition, we hypothesized that NGAL levels would correlate with the elevation of inflammatory biomarkers levels in essential hypertensive (EH) and RVH patients.
Materials and methods
Patient population
The study was approved by the Institutional Review Board of the Mayo Clinic in accordance with the Declaration of Helsinki and the Health Insurance Portability and Accountability Act (HIPAA) guidelines. All patients provided written informed consent before enrollment.
From 2008 to present, we prospectively enrolled patients with EH (n = 22) or unilateral renal artery stenosis (n = 22), participating in previously described inpatient protocol studies [20]. In addition, sex– and age-matched normotensive controls (n = 22) were recruited and serum samples collected through the Mayo Clinic Biobank. Urine samples were also collected from 16 consenting healthy age-matched potential kidney donors. See the Supplementary data for detailed methods of subject recruitment through the Mayo Clinic Biobank.
RVH patients included in the study met the entry criteria analogous to those required for enrollment in the Cardiovascular Outcomes for Renal Atherosclerotic Lesions (CORAL trial) [21]. The severity of renal artery stenosis was determined by renal artery Doppler ultrasound velocity acceleration (peak systolic velocity >200 cm/s), or MR/CT angiography with evident stenosis >60% and/or post-stenotic dilation. Exclusion criteria included patients with uncontrolled hypertension [systolic blood pressure (SBP) >180 mmHg, despite antihypertensive therapy], diabetes requiring insulin or oral hypoglycemic medications, recent cardiovascular event (myocardial infarction, stroke, congestive heart failure within 6 months), pregnancy and kidney transplant. In addition, patients with AKI, defined in accordance with criteria established by the Acute Kidney Injury Network (AKIN) [22], were excluded. In EH and RVH patients, controlled sodium intake (150 mEq of sodium) was maintained through the duration of the study. In addition, treatment with diuretics, angiotensin-converting enzyme inhibitors or angiotensin receptor blockers was continued or initiated at the usual recommended daily dose. A single dose of furosemide (20 mg) had been administered to EH and RVH patients for unrelated protocol studies [20] 1 day before renal vein sampling.
Clinical and laboratory parameters including age, sex, height, weight, body mass index, systolic, diastolic, mean arterial pressure (MAP), serum creatinine, proteinuria, plasma renin activity (PRA), uric acid, troponin I, low-density lipoprotein, high-density lipoprotein (HDL), total cholesterol and triglyceride levels were evaluated at study entry by standard procedures. eGFR was calculated using the chronic kidney disease epidemiology collaboration (CKD-EPI) formula [23]. In addition, comorbidities and medication intake were recorded, as well as cardiac echocardiography-derived left ventricular ejection fraction (LVEF) and left ventricular mass index (LVMI) assessed within 6 months before the study. Single-kidney renal blood flow (RBF) was measured in RVH and EH patients using multi-detector CT, as previously described [20].
Renal vein and urine sampling
Blood samples for NGAL and inflammatory cytokine analysis were obtained from the right and left renal vein and infra-renal inferior vena cava (IVC) of all hypertensive patients by placing a catheter via the femoral or internal jugular vein, as previously described [20]. For EH patients, individual kidney (right and left) renal vein levels were averaged. In normotensive controls, only peripheral (antecubital) blood samples were collected. Samples were stored at −80°C until measurement. Urine samples were collected in all hypertensive patients 1 day before blood sampling, and in potential kidney donors from September 2010 to January 2011. Collected samples were centrifuged, and the supernatant was stored.
Measurements
Aliquoted plasma samples were centrifuged at 3000 g/5 min and samples (25 μL) incubated overnight at 4°C. Renal vein and IVC levels of interferon (IF)-γ, tumor necrosis factor (TNF)-α, TNF receptor (sTNFR)-1 and macrophage inflammatory protein (MIP)-1δ were measured by Luminex (Millipore, cat No: MPXHCYTO-60K; MPXHCYP2-62K and HSCR-32K). Signals were read by the Bio-plex 200 systems (BIO-RAD). In addition, to assess the severity of atherosclerotic disease, we measured systemic levels of CRP by enzyme-linked immunosorbent assay (ELISA) (R&D systems, Cat# DCRP00), and MCP-1, interleukin (IL)-6, adiponectin and soluble receptor for advanced glycation end-products (sRAGE) levels by Luminex.
NGAL (ng/mL) was tested by ELISA according to manufacturer's protocol (BioPorto Diagnostics, Cat# KIT 036) in 10 μL of renal vein, IVC plasma and urine samples diluted 1:500. Final volumes of 100 μL were used for assay. Urinary KIM-1 levels were evaluated by ELISA using a commercially available kit (R&D systems, Cat# DKM100). Urinary NGAL and KIM-1 were expressed as nanogram/milligram of urinary creatinine.
All measurements were performed by a single investigator blinded to the clinical data.
Statistical analysis
Normally distributed data were expressed as mean ± SD, while non-normally distributed was expressed as medium (range). The Shapiro–Wilk test was used to test for deviation from normality. Group comparisons were accomplished using parametric analysis of variance followed by unpaired Student's t-test and non-parametric (Wilcoxon and Kruskal–Wallis) statistics as appropriate. Plasma levels of NGAL and inflammatory markers were corrected for serum creatinine levels. Plasma and urinary NGAL, as well as urinary KIM-1 levels were also corrected for eGFR. Regressions were calculated by the least-squares fit to compare renal function, injury markers and levels of inflammatory cytokines. Statistical significance was accepted for P ≤ 0.05. Statistical analysis was performed using JMP software package version 8.0 (SAS Institute Inc., Cary, NC).
Results
Table 1 summarizes the clinical, laboratory and demographic characteristics of the patients included in the study. SBP was elevated in EH and RVH patients compared with normal (P < 0.05), with no significant differences found in antihypertensive regimens between the groups. LVMI was similarly higher in RVH and EH compared with normal (P < 0.05 both), but troponin I levels and LVEF did not differ among the groups. Lipid profile was similar in all patients, except that HDL levels were lower in RVH compared with normal and EH patients.
Table 1.
Clinical, laboratory and demographic data (mean ± SD) of healthy volunteers, EH and RVH patients
| Healthy volunteers | EH | RVH | |
|---|---|---|---|
| Demographics | |||
| Number of patients | 22 | 22 | 22 |
| Age (years) | 69.1 ± 6.7 | 67.3 ± 12.7 | 65.6 ± 9.5 |
| Gender (male/female) | 11/22 | 11/22 | 11/22 |
| Body mass index | 25.6 ± 3.6 | 27.4 ± 3.4 | 27.4 ± 4.3 |
| Duration of hypertension (years) | 18.2 ± 7.3 | 15.2 ± 13.3 | |
| Related laboratory measures | |||
| Systolic blood pressure (mmHg) | 120.4 ± 8.9 | 137.3 ± 21.6* | 131.3 ± 20.3* |
| Diastolic blood pressure (mmHg) | 70.6 ± 8.1 | 70.7 ± 15.3 | 70.3 ± 8.2 |
| Mean blood pressure (mmHg) | 87.2 ± 7.3 | 92.9 ± 13.9 | 90.6 ± 10.3 |
| Total cholesterol (mg/dL) | 178.3 ± 31.2 | 185.7 ± 33.1 | 171.1 ± 23.2 |
| High-density lipoprotein (mg/dL) | 57.9 ± 12.7 | 57.2 ± 16.5 | 43.1 ± 13.6*,† |
| Low-density lipoprotein (mg/dL) | 95.2 ± 26.8 | 102.1 ± 26.2 | 99.2 ± 25.5 |
| Triglycerides (mg/dL) | 126.1 ± 43.0 | 146.3 ± 68.4 | 143.8 ± 89.3 |
| Leukocytes (×109/L) | 6.1 ± 1.7 | 6.4 ± 1.4 | 6.4 ± 1.4 |
| Troponin I (ng/mL) | 0.86 ± 0.18 | 0.94 ± 0.17 | 1.46 ± 0.48 |
| Left ventricular EF (%) | 64.0 ± 2.8 | 63.3 ± 3.5 | 61.1 ± 5.4 |
| Left ventricular mass index (g/m2) | 74.8 ± 23.3 | 109.0 ± 40.0* | 105.9 ± 21.8* |
| Concomitant medication | |||
| No. of Antihypertensive drugs (median) | 0 | 3 (1–4) | 3 (1–4) |
| Lipid-lowering drugs (No. of patients) | 0 | 18/22 | 17/22 |
| Renal function | |||
| Serum creatinine (mg/dL) | 0.9 ± 0.1 | 1.0 ± 0.3 | 1.2 ± 0.4* |
| eGFR-CDK-EPI (mL/min/1.73/m2) | 75.8 ± 12.1 | 72.2 ± 20.3 | 62.2 ± 22.0* |
| Urine protein (mg/24 h) | 88.1 ± 82.4 | 90.1 ± 75.1 | 82.8 ± 49.2 |
| STK or single-kidney RBF (mL/min) | — | 368.3 ± 142.3 | 213.6 ± 123.9† |
| CLK-RBF (mL/min) | 417.7 ± 128.9‡ | ||
| Albumin/creatinine ratio (mg/g) | 23.9 ± 15.4 | 18.2 ± 23.9 | 22.0 ± 36.3 |
| Uric acid (mg/dL) | 5.1 ± 1.1 | 6.1 ± 1.3* | 7.4 ± 1.6*,† |
EF, ejection fraction; eGFR-CKD-EPI, estimated glomerular filtration rate-chronic kidney disease epidemiology collaboration; STK, stenotic kidney; CLK, contralateral kidney; RBF, renal blood flow.
*P ≤ 0.05 versus normal.
†P ≤ 0.05 versus EH.
‡P ≤ 0.05 versus RVH (STK).
Serum creatinine levels were higher and eGFR lower in RVH compared with normal controls (both, P < 0.05 versus normal), but were not different from those in EH patients (both P > 0.05). Systemic PRA levels were elevated in both hypertensive groups (P = 0.008 and <0.0001 versus normal, respectively), but not different between them (P = 0.12), although renal vein PRA levels were higher in RVH compared with EH (P = 0.04). RBF was lower in the stenotic kidney than in the contralateral kidney or in EH (Table 1, P < 0.0001 and 0.0006, respectively), underscoring the hemodynamic significance of the stenoses. Urinary protein excretion and albumin/creatinine ratio did not differ among the groups. Finally, serum uric acid levels were significantly elevated in both hypertensive groups, but higher in RVH patients compared with EH (P = 0.04).
Plasma NGAL
Systemic, stenotic kidney vein and contralateral kidney vein levels of NGAL were all similarly elevated in RVH compared with both EH and healthy volunteers (Figure 1A, all P < 0.05), whereas neither systemic nor renal vein NGAL levels in EH were different from normal subjects. A direct correlation was found between systemic and renal vein NGAL levels (P < 0.0001) and both also correlated inversely with eGFR and directly with serum creatinine levels in RVH patients (Figure 1B and C). Moreover, stenotic kidney venous NGAL levels correlated directly with stenotic kidney venous levels of all measured inflammatory markers (Figure 2A–D), but not with their systemic levels (Figure 2E–H). Neither systemic nor RV levels of NGAL correlated with any measure of blood pressure or LVMI.
Fig. 1.

Systemic and renal vein (RV) levels of NGAL in normal, EH, stenotic (STK) and contralateral (CLK) kidneys of RVH patients (A). The top and bottom of the boxes are the estimated 75th and 25th percentiles, respectively. The vertical lines extend from the 75th percentile to the highest and from the 25th percentile to the lowest data points. RV and systemic NGAL levels inversely correlated with eGFR (B), but directly with serum creatinine (C, Scr) levels in RVH patients. *P < 0.05 versus normal, #P < 0.05 versus EH.
Fig. 2.

Renal vein (RV) NGAL levels directly correlated with RV levels of MIP-1δ (A), IF-γ (B), TNF-α (C) and TNF receptor (TNFR-1) (D) in renovascular hypertensive patients, whereas their systemic levels did not correlate (E–H).
Urinary biomarker levels
Urine NGAL levels were similarly elevated in EH and RVH patients compared with normal volunteers (Figure 3A, both P < 0.05 versus normal), and did not correlate with either renal vein (R2 = 0.01, P = 0.41) or IVC (R2 = 0.02, P = 0.24) NGAL levels in hypertensive patients.
Fig. 3.
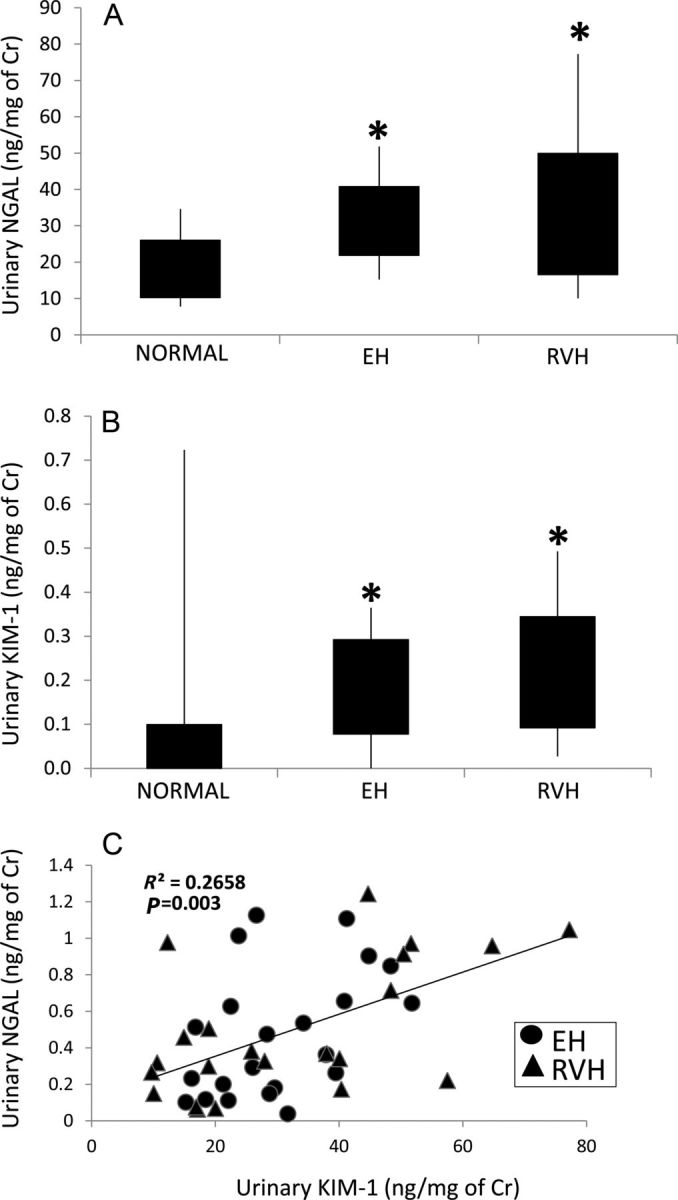
Urinary NGAL (A) and KIM-1 (B) levels in EH and RVH patients. A direct correlation was found between urinary KIM-1 and urinary NGAL levels (C). *P < 0.05 versus normal.
Urinary KIM-1 levels were also elevated in EH and RVH compared with normal, but were not different between these hypertensive groups (Figure 3B, P = 0.35). Finally, a statistically significant direct correlation was found between urinary KIM-1 and urinary NGAL levels (Figure 3C).
Renal inflammatory markers
Renal vein levels of IF-γ, TNF-α, TNFR-1 and MIP-1δ were higher only in the stenotic kidney vein compared with normal and EH (Table 2, P < 0.05 for all). In addition, levels of IF-γ and TNF-α were higher in the stenotic kidney vein compared with systemic and contralateral kidney venous levels in RVH (Table 2, P < 0.05 for all).
Table 2.
Systemic and renal vein (RV) levels (mean ± SD) of PRA, NGAL and cytokines in healthy volunteers, EH and RVH patients
| Healthy volunteers |
EH |
RVH |
||||
|---|---|---|---|---|---|---|
| Systemic | Systemic | RV | Systemic | CLK RV | STK RV | |
| PRA (ng/mL/h) | 0.5 ± 0.5 | 7.9 ± 8.1* | 10.3 ± 10.4 | 13.9 ± 13.7* | 15.3 ± 14.5 | 20.0 ± 16.4† |
| NGAL (ng/mL) | 65.6 ± 24.0 | 84.2 ± 44.4 | 66.4 ± 36.7 | 117.0 ± 53.3*,† | 128.3 ± 45.1*,† | 110.5 ± 50.2*,† |
| IF-γ (pg/mL) | 4.8 ± 2.8 | 6.5 ± 4.1 | 5.7 ± 4.7 | 5.8 ± 2.0 | 6.1 ± 3.5 | 9.7 ± 3.4*,†,#,‡ |
| TNF-α (pg/mL) | 3.6 ± 1.6 | 3.9 ± 1.9 | 4.1 ± 2.0 | 4.7 ± 2.1 | 4.9 ± 1.2 | 6.8 ± 2.3*,†,#,‡ |
| TNFR-1 (pg/mL) | 964.1 ± 272.6 | 1104.5 ± 498.4 | 1016.5 ± 449.2 | 1160.0 ± 387.8 | 1115.0 ± 449.4 | 1457.8 ± 859.8*,† |
| MIP-1δ (pg/mL) | 2146.3 ± 2324.4 | 2619.2 ± 1474.5 | 2343.8 ± 1300.8 | 3573.0 ± 2553.6 | 3520.6 ± 3103.2 | 4235.8 ± 3528.0*,†,‡ |
CLK, contralateral kidney; STK, stenotic kidney; IF, interferon; TNF, tumor necrosis factor-α; TNFR, TNF receptor; MIP, macrophage inflammatory protein.
*P ≤ 0.05 versus normal.
†P ≤ 0.05 versus EH (RV).
#P ≤ 0.05 versus RVH (systemic).
‡P ≤ 0.05 versus RVH (CLK).
Markers of atherosclerotic disease and systemic inflammation
White blood cell count did not differ among the groups (Table 1). Systemic CRP levels were significantly and similarly (P = 0.67) elevated in EH and RVH patients compared with normal (Figure 4A, P = 0.05 and 0.007, respectively). However, systemic levels of MCP-1, IL-6, adiponectin and sRAGE did not differ among the groups (data not shown). Finally, no significant correlations were found between systemic NGAL levels and CRP (Figure 4B, R2 = 0.01, P = 0.55), MCP-1 (R2 = 0.001, P = 0.80), IL-6 (R2 = 0.003, P = 0.73), adiponectin (R2 = 0.0009, P = 0.85) or sRAGE (R2 = 0.05, P = 0.13) levels.
Fig. 4.
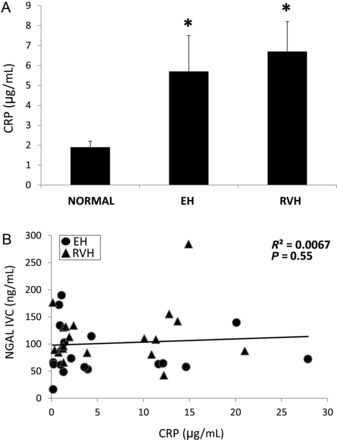
Systemic levels of CRP in normal, EH and RVH patients (A). Systemic NGAL levels did not correlate with CRP levels in EH and RVH patients (B).
Statistical differences in plasma and urinary markers between the groups persisted after adjustment for serum creatinine and eGFR (Supplementary data, Tables S1–S3).
Discussion
This study demonstrates, for the first time, elevated NGAL levels in both the systemic circulation and the stenotic kidney vein of RVH compared with EH patients. The comparable systemic and renal venous levels of NGAL may imply extra-renal production, yet their correlations with GFR, serum creatinine and with increased renal vein levels of inflammatory cytokines implicates NGAL as a marker of renal inflammation and dysfunction in renovascular disease. Taken together, our findings support a role for renal inflammatory pathways as a component of kidney injury beyond a stenotic lesion in human RVH.
RVH represents a common complication in patients with atherosclerotic renal arterial disease, associated with high rates of target organ injury [24]. Despite extensive research, the mechanisms responsible for irreversible parenchymal injury and occasionally progression to end-stage renal disease from RVH remain obscure. Yet, experimental studies attribute a cardinal role for inflammation and ischemia in eliciting kidney damage.
This study assessed, for the first time, concomitant circulating, renal vein and urinary levels of NGAL. NGAL is an acute-phase protein induced in inflammatory conditions and ischemia, and a reliable biomarker for predicting AKI in various clinical settings [9–12] under standardized conditions in stable hypertensive patients. Our results show that systemic and renal vein NGAL levels were elevated in chronic RVH patients compared with EH and healthy volunteers, which may imply ongoing inflammatory processes or acute ischemic events in the post-ischemic kidney.
In humans, both systemic and renal NGAL pools have been described [25], both of which may reflect extra-renal disease. Systemic NGAL, usually found in low concentrations in the plasma, is up-regulated in response to a variety of conditions, including inflammation, ischemia, infections, neoplasia and atherosclerosis [26, 27]. Once circulating NGAL reaches the kidney, it is filtered and binds with high affinity to megalin, which is expressed in proximal tubular cells. As a result, only a small proportion of systemic NGAL normally is excreted in the urine. On the other hand, the renal pool represents NGAL locally produced at the thick ascending limb of Henle and collecting ducts in response to acute processes linked to kidney injury [25, 28].
Previous studies show that NGAL levels may facilitate differentiating chronic kidney disease (CKD) and prerenal reduction in GFR from intrinsic AKI patients [29, 30], stipulating that NGAL is a marker of active kidney damage. Nevertheless, its levels are also elevated in patients with Stages 2–4 CKD and inversely correlate with eGFR [31]. Studies in mice have shown a direct relationship between NGAL expression and CKD progression [32], suggesting NGAL as a biomarker of AKI-to-CKD transition [33]. Remarkably, plasma and urinary NGAL levels in our study population were substantially lower than those typically observed in patients with AKI (Supplementary data, Table S4), arguing against the presence of AKI. However, CKD alone was unlikely to account for the increase in NGAL levels in our study, because eGFR and serum creatinine levels did not differ between EH and RVH patients and the differences between these groups persisted after adjustment for both measures of renal function. Importantly, elevated NGAL levels in our RVH patients might imply greater tissue damage in the post-stenotic compared with EH kidney [20]. Furthermore, the relationship between eGFR and serum creatinine with plasma NGAL supports a role of this biomarker as a predictor of kidney injury. However, we cannot exclude a role for intermittent acute changes in post-stenotic renal perfusion pressure that may induce recurrent hemodynamic injury to the affected kidney.
Notably, NGAL levels are also elevated in patients with cardiovascular diseases such as heart failure [34] and left ventricular hypertrophy [17]. However, our enrolled patients were free of symptomatic vascular disease other than renovascular, and LVEF, LVMI, blood pressure and troponin I levels did not differ between EH and RVH patients, arguing against cardiovascular disease as the primary determinant of NGAL elevation. Yet, a number of studies documented a positive association between NGAL levels and atherosclerotic disease, implicating NGAL in the development of atherosclerosis [35, 36]. NGAL is also increased in conditions linked to atherosclerosis such as coronary artery disease [37] and abdominal aortic aneurysm [38]. Furthermore, NGAL levels are elevated in patients with vascular remodeling and plaque instability, in association with increased mortality rates [39]. However, systemic levels of markers of active atherosclerotic disease such as CPR, MCP-1, IL-6, adiponectin and sRAGE were not different between RVH and EH patients, arguing against a markedly greater diffuse atherosclerotic burden in RVH, despite their renal artery atherosclerotic lesions. Pertinently, we found lower HDL levels in RVH patients compared with EH and normal controls, which may imply increased cardiovascular risk in this group of patients [40]. Similarly, previous studies in patients with atherosclerotic renal artery stenosis found that a drop in HDL was associated with intra-renal atherosclerotic vessel disease and global glomerulosclerosis [41]. Hence, we cannot exclude a link between mild atherosclerosis or low HDL and elevated NGAL.
Importantly, our study showed that renal vein levels of several inflammatory biomarkers were higher in the post-stenotic kidney of RVH compared with EH patients and normal controls, consistent with an important inflammatory component beyond the stenotic lesion. This association between renal ischemia and inflammation is increasingly being identified in experimental and human RVH [42]. Rats with unilateral renal artery stenosis show an interstitial chronic inflammatory infiltrate in the ischemic kidney consisting of B lymphocytes, T lymphocytes and macrophages [3, 43], suggesting that inflammation triggers interstitial extracellular matrix accumulation and fibrosis in the clipped kidney. Furthermore, plasma levels of soluble CD40 ligand, a transmembrane protein of the TNF superfamily involved in inflammation and thrombosis, are increased in RVH patients compared with normal controls [44]. These findings corroborate and extend data from our group showing that chronic swine RVH is associated with renal and systemic inflammation, leading to kidney functional deterioration [4]. Our results indicated that the stenotic human kidney in RVH is also a source of inflammation. These pro-inflammatory cytokines, possibly released by parenchymal or inflammatory cells that populate the kidney, might impair reparative processes and induce kidney deterioration.
Some of the inflammatory markers up-regulated in the stenotic RVH kidney are capable of stimulating epithelial cells to synthesize NGAL [45]. In support of this notion, we found that renal vein NGAL levels directly correlated with renal vein levels of all measured inflammatory biomarkers, while their systemic levels did not. Nevertheless, unlike other inflammatory biomarkers, NGAL levels from the stenotic and contralateral kidneys, in addition to systemic NGAL levels, were all similarly elevated in RVH patients, implying a more generalized level of systemic NGAL production, possibly in circulating leukocytes. These observations suggest that NGAL might be produced both within and outside the kidney as a component or consequence of renal inflammation. Nevertheless, the low NGAL gradient across the stenotic kidney may also reflect a continuous rate of renal NGAL production combined with a long plasma half-life, although renal production alone might not suffice to sustain equivalent concentration of NGAL in the infra-renal IVC, which represents the entire systemic venous circulation.
In order to evaluate its origin, we measured urinary levels of both NGAL and KIM-1, which were elevated similarly in RVH and EH patients, implying some degree of kidney damage in both groups. Incidentally, urinary levels in RVH reflect combined contribution from both the stenotic and contralateral kidneys. Thus, the elevated urinary but unaltered plasma NGAL levels in EH patients, in contrast to the increase in both in RVH, may support the contention that systemic NGAL in RVH was primarily produced in response to renal inflammation observed only in this group.
This study is limited by its relatively small study population and cross-sectional nature. The specific criteria used in our patient selection may limit the translation power of our findings to the more complex ARAS patients encountered in clinical practice. Pertinently, the majority of patients were treated with blockers of the renin–angiotensin system and statins, which may mask atherosclerotic disease activity. In addition, our studies involved measurements at a single time point; the predictive value of NGAL on clinical kidney outcomes should be addressed in future investigations. Furthermore, the lack of association between either systemic or RV NGAL levels with any measure of blood pressure or LVMI limits the potential role of NGAL as a predictive marker. We also did not examine the independent contribution of atherosclerosis on NGAL levels, which warrants independent and thorough future studies. Nevertheless, our results demonstrate that RVH is associated with concurrent parenchymal inflammation and elevated NGAL levels.
Conclusions
RVH is associated with chronic hypoperfusion of the renal parenchyma, which compromises renal hemodynamics and function. Therefore, it is critical to identify the mechanisms underlying renal tissue injury to prevent progression to renal failure. We observed that elevated renal vein levels of inflammatory biomarkers in the post-stenotic kidney of RVH patients correlated with NGAL levels, indicating active renal inflammation. However, because renal venous and systemic plasma NGAL levels were all similarly elevated, yet correlated specifically with renal release of inflammatory signals, a considerable proportion of circulating NGAL might have been produced outside the kidney, but conceivably in response to renal inflammation and dysfunction. The association between stenotic kidney vein NGAL levels and GFR might reflect both acute and chronic inflammatory processes of renal injury, which represent important contributors toward kidney functional deterioration in RVH patients. Future studies will need to dissect and identify the specific mechanisms eliciting NGAL production in patients with RVH and renal inflammation. Recognition of components related to acute injury and inflammation in the post-stenotic kidney, as well as their relationship to systemic inflammation and GFR, may constitute an important step for formulating new endpoints and designing novel therapeutic approaches for this disorder.
Supplementary data
Supplementary data are available online at http://ndt.oxfordjournals.org.
Conflict of interest statement
The results presented in this paper have not been published previously in whole or part, except in an abstract format.
Acknowledgements
The authors are grateful to Mr. Marshall Behrens, Mayo Clinic Immunology, for his technical support, and Dr. Keith Knutson for the use of the Luminex machine. Partly supported by the NIH grant numbers: HL085307, DK73608, DK090358, HL77131 and UL1-RR024150, and by Mayo Clinic Center for Individualized Medicine.
References
- 1.Preston RA, Epstein M. Ischemic renal disease: an emerging cause of chronic renal failure and end-stage renal disease. J Hypertens. 1997;15:1365–1377. doi: 10.1097/00004872-199715120-00001. doi:10.1097/00004872-199715120-00001. [DOI] [PubMed] [Google Scholar]
- 2.Kalra PA, Guo H, Kausz AT, et al. Atherosclerotic renovascular disease in United States patients aged 67 years or older: risk factors, revascularization, and prognosis. Kidney Int. 2005;68:293–301. doi: 10.1111/j.1523-1755.2005.00406.x. doi:10.1111/j.1523-1755.2005.00406.x. [DOI] [PubMed] [Google Scholar]
- 3.Matavelli LC, Huang J, Siragy HM. Angiotensin at receptor stimulation inhibits early renal inflammation in renovascular hypertension. Hypertension. 2011;57:308–313. doi: 10.1161/HYPERTENSIONAHA.110.164202. doi:10.1161/HYPERTENSIONAHA.110.164202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhu XY, Chade AR, Krier JD, et al. The chemokine monocyte chemoattractant protein-1 contributes to renal dysfunction in swine renovascular hypertension. J Hypertens. 2009;27:2063–2073. doi: 10.1097/HJH.0b013e3283300192. doi:10.1097/HJH.0b013e3283300192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chade AR, Rodriguez-Porcel M, Herrmann J, et al. Beneficial effects of antioxidant vitamins on the stenotic kidney. Hypertension. 2003;42:605–612. doi: 10.1161/01.HYP.0000089880.32275.7C. doi:10.1161/01.HYP.0000089880.32275.7C. [DOI] [PubMed] [Google Scholar]
- 6.Chade AR, Rodriguez-Porcel M, Herrmann J, et al. Antioxidant intervention blunts renal injury in experimental renovascular disease. J Am Soc Nephrol. 2004;15:958–966. doi: 10.1097/01.asn.0000117774.83396.e9. doi:10.1097/01.ASN.0000117774.83396.E9. [DOI] [PubMed] [Google Scholar]
- 7.Saeed A, Herlitz H, Nowakowska-Fortuna E, et al. Oxidative stress and endothelin-1 in atherosclerotic renal artery stenosis and effects of renal angioplasty. Kidney Blood Press Res. 2011;34:396–403. doi: 10.1159/000328732. doi:10.1159/000328732. [DOI] [PubMed] [Google Scholar]
- 8.Cowland JB, Sorensen OE, Sehested M, et al. Neutrophil gelatinase-associated lipocalin is up-regulated in human epithelial cells by IL-1 beta, but not by TNF-alpha. J Immunol. 2003;171:6630–6639. doi: 10.4049/jimmunol.171.12.6630. [DOI] [PubMed] [Google Scholar]
- 9.Mishra J, Dent C, Tarabishi R, et al. Neutrophil gelatinase-associated lipocalin (NGAL) as a biomarker for acute renal injury after cardiac surgery. Lancet. 2005;365:1231–1238. doi: 10.1016/S0140-6736(05)74811-X. doi:10.1016/S0140-6736(05)74811-X. [DOI] [PubMed] [Google Scholar]
- 10.Wagener G, Jan M, Kim M, et al. Association between increases in urinary neutrophil gelatinase-associated lipocalin and acute renal dysfunction after adult cardiac surgery. Anesthesiology. 2006;105:485–491. doi: 10.1097/00000542-200609000-00011. doi:10.1097/00000542-200609000-00011. [DOI] [PubMed] [Google Scholar]
- 11.Kumpers P, Hafer C, Lukasz A, et al. Serum neutrophil gelatinase-associated lipocalin at inception of renal replacement therapy predicts survival in critically ill patients with acute kidney injury. Crit Care. 2010;14:R9. doi: 10.1186/cc8861. doi:10.1186/cc8861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hirsch R, Dent C, Pfriem H, et al. NGAL is an early predictive biomarker of contrast-induced nephropathy in children. Pediatr Nephrol. 2007;22:2089–2095. doi: 10.1007/s00467-007-0601-4. doi:10.1007/s00467-007-0601-4. [DOI] [PubMed] [Google Scholar]
- 13.Mishra J, Ma Q, Prada A, et al. Identification of neutrophil gelatinase-associated lipocalin as a novel early urinary biomarker for ischemic renal injury. J Am Soc Nephrol. 2003;14:2534–2543. doi: 10.1097/01.asn.0000088027.54400.c6. doi:10.1097/01.ASN.0000088027.54400.C6. [DOI] [PubMed] [Google Scholar]
- 14.Ichimura T, Bonventre JV, Bailly V, et al. Kidney injury molecule-1 (KIM-1), a putative epithelial cell adhesion molecule containing a novel immunoglobulin domain, is up-regulated in renal cells after injury. J Biol Chem. 1998;273:4135–4142. doi: 10.1074/jbc.273.7.4135. doi:10.1074/jbc.273.7.4135. [DOI] [PubMed] [Google Scholar]
- 15.Liangos O, Tighiouart H, Perianayagam MC, et al. Comparative analysis of urinary biomarkers for early detection of acute kidney injury following cardiopulmonary bypass. Biomarkers. 2009;14:423–431. doi: 10.1080/13547500903067744. doi:10.1080/13547500903067744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Han WK, Waikar SS, Johnson A, et al. Urinary biomarkers in the early diagnosis of acute kidney injury. Kidney Int. 2008;73:863–869. doi: 10.1038/sj.ki.5002715. doi:10.1038/sj.ki.5002715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Leoncini G, Mussap M, Viazzi F, et al. Combined use of urinary neutrophil gelatinase-associated lipocalin (UNGAL) and albumin as markers of early cardiac damage in primary hypertension. Clin Chim Acta. 2011;412:1951–1956. doi: 10.1016/j.cca.2011.06.043. doi:10.1016/j.cca.2011.06.043. [DOI] [PubMed] [Google Scholar]
- 18.Malyszko J, Bachorzewska-Gajewska H, Malyszko JS, et al. Serum neutrophil gelatinase-associated lipocalin as a marker of renal function in hypertensive and normotensive patients with coronary artery disease. Nephrology (Carlton) 2008;13:153–156. doi: 10.1111/j.1440-1797.2007.00899.x. doi:10.1111/j.1440-1797.2007.00899.x. [DOI] [PubMed] [Google Scholar]
- 19.van Timmeren MM, van den Heuvel MC, Bailly V, et al. Tubular kidney injury molecule-1 (KIM-1) in human renal disease. J Pathol. 2007;212:209–217. doi: 10.1002/path.2175. doi:10.1002/path.2175. [DOI] [PubMed] [Google Scholar]
- 20.Gloviczki ML, Glockner JF, Lerman LO, et al. Preserved oxygenation despite reduced blood flow in poststenotic kidneys in human atherosclerotic renal artery stenosis. Hypertension. 2010;55:961–966. doi: 10.1161/HYPERTENSIONAHA.109.145227. doi:10.1161/HYPERTENSIONAHA.109.145227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Murphy TP, Cooper CJ, Dworkin LD, et al. The Cardiovascular Outcomes with Renal Atherosclerotic Lesions (CORAL) study: rationale and methods. J Vasc Interv Radiol. 2005;16:1295–1300. doi: 10.1097/01.RVI.0000176301.69756.28. doi:10.1097/01.RVI.0000176301.69756.28. [DOI] [PubMed] [Google Scholar]
- 22.Mehta RL, Kellum JA, Shah SV, et al. Acute kidney injury network: report of an initiative to improve outcomes in acute kidney injury. Crit Care. 2007;11:R31. doi: 10.1186/cc5713. doi:10.1186/cc5713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Levey AS, Stevens LA, Schmid CH, et al. A new equation to estimate glomerular filtration rate. Ann Intern Med. 2009;150:604–612. doi: 10.7326/0003-4819-150-9-200905050-00006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Losito A, Fagugli RM, Zampi I, et al. Comparison of target organ damage in renovascular and essential hypertension. Am J Hypertens. 1996;9:1062–1067. doi: 10.1016/0895-7061(96)00199-9. doi:10.1016/0895-7061(96)00199-9. [DOI] [PubMed] [Google Scholar]
- 25.Schmidt-Ott KM, Mori K, Li JY, et al. Dual action of neutrophil gelatinase-associated lipocalin. J Am Soc Nephrol. 2007;18:407–413. doi: 10.1681/ASN.2006080882. doi:10.1681/ASN.2006080882. [DOI] [PubMed] [Google Scholar]
- 26.Bolignano D, Coppolino G, Lacquaniti A, et al. From kidney to cardiovascular diseases: NGAL as a biomarker beyond the confines of nephrology. Eur J Clin Invest. 2010;40:273–276. doi: 10.1111/j.1365-2362.2010.02258.x. doi:10.1111/j.1365-2362.2010.02258.x. [DOI] [PubMed] [Google Scholar]
- 27.Bolignano D, Donato V, Lacquaniti A, et al. Neutrophil gelatinase-associated lipocalin (NGAL) in human neoplasias: a new protein enters the scene. Cancer Lett. 2010;288:10–16. doi: 10.1016/j.canlet.2009.05.027. doi:10.1016/j.canlet.2009.05.027. [DOI] [PubMed] [Google Scholar]
- 28.Haase M, Haase-Fielitz A, Bellomo R, et al. Neutrophil gelatinase-associated lipocalin as a marker of acute renal disease. Curr Opin Hematol. 2011;18:11–18. doi: 10.1097/MOH.0b013e3283411517. [DOI] [PubMed] [Google Scholar]
- 29.Singer E, Elger A, Elitok S, et al. Urinary neutrophil gelatinase-associated lipocalin distinguishes pre-renal from intrinsic renal failure and predicts outcomes. Kidney Int. 2011;80:405–414. doi: 10.1038/ki.2011.41. doi:10.1038/ki.2011.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nickolas TL, O'Rourke MJ, Yang J, et al. Sensitivity and specificity of a single emergency department measurement of urinary neutrophil gelatinase-associated lipocalin for diagnosing acute kidney injury. Ann Intern Med. 2008;148:810–819. doi: 10.7326/0003-4819-148-11-200806030-00003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mitsnefes MM, Kathman TS, Mishra J, et al. Serum neutrophil gelatinase-associated lipocalin as a marker of renal function in children with chronic kidney disease. Pediatr Nephrol. 2007;22:101–108. doi: 10.1007/s00467-006-0244-x. doi:10.1007/s00467-006-0244-x. [DOI] [PubMed] [Google Scholar]
- 32.Viau A, El Karoui K, Laouari D, et al. Lipocalin 2 is essential for chronic kidney disease progression in mice and humans. J Clin Invest. 2010;120:4065–4076. doi: 10.1172/JCI42004. doi:10.1172/JCI42004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ko GJ, Grigoryev DN, Linfert D, et al. Transcriptional analysis of kidneys during repair from AKI reveals possible roles for NGAL and KIM-1 as biomarkers of AKI-to-CKD transition. Am J Physiol Renal Physiol. 2010;298:F1472–F1483. doi: 10.1152/ajprenal.00619.2009. doi:10.1152/ajprenal.00619.2009. [DOI] [PubMed] [Google Scholar]
- 34.Bolignano D, Basile G, Parisi P, et al. Increased plasma neutrophil gelatinase-associated lipocalin levels predict mortality in elderly patients with chronic heart failure. Rejuvenation Res. 2009;12:7–14. doi: 10.1089/rej.2008.0803. doi:10.1089/rej.2008.0803. [DOI] [PubMed] [Google Scholar]
- 35.Giaginis C, Zira A, Katsargyris A, et al. Clinical implication of plasma neutrophil gelatinase-associated lipocalin (ngal) concentrations in patients with advanced carotid atherosclerosis. Clin Chem Lab Med. 2010;48:1035–1041. doi: 10.1515/CCLM.2010.211. doi:10.1515/cclm.2010.211. [DOI] [PubMed] [Google Scholar]
- 36.Forsblad J, Gottsater A, Persson K, et al. Clinical manifestations of atherosclerosis in an elderly population are related to plasma neopterin, NGAL and endothelin-1, but not to Chlamydia pneumoniae serology. Int Angiol. 2002;21:173–179. [PubMed] [Google Scholar]
- 37.Paulsson J, Dadfar E, Held C, et al. Activation of peripheral and in vivo transmigrated neutrophils in patients with stable coronary artery disease. Atherosclerosis. 2007;192:328–334. doi: 10.1016/j.atherosclerosis.2006.08.003. doi:10.1016/j.atherosclerosis.2006.08.003. [DOI] [PubMed] [Google Scholar]
- 38.Ramos-Mozo P, Madrigal-Matute J, Vega de Ceniga M, et al. Increased plasma levels of NGAL, a marker of neutrophil activation, in patients with abdominal aortic aneurysm. Atherosclerosis. 2012;220:552–556. doi: 10.1016/j.atherosclerosis.2011.11.023. doi:10.1016/j.atherosclerosis.2011.11.023. [DOI] [PubMed] [Google Scholar]
- 39.Falke P, Elneihoum AM, Ohlsson K. Leukocyte activation: relation to cardiovascular mortality after cerebrovascular ischemia. Cerebrovasc Dis. 2000;10:97–101. doi: 10.1159/000016037. doi:10.1159/000016037. [DOI] [PubMed] [Google Scholar]
- 40.Camont L, Chapman MJ, Kontush A. Biological activities of HDL subpopulations and their relevance to cardiovascular disease. Trends Mol Med. 2011;17:594–603. doi: 10.1016/j.molmed.2011.05.013. [DOI] [PubMed] [Google Scholar]
- 41.Keddis MT, Garovic VD, Bailey KR, et al. Ischaemic nephropathy secondary to atherosclerotic renal artery stenosis: clinical and histopathological correlates. Nephrol Dial Transplant. 2010;25:3615–3622. doi: 10.1093/ndt/gfq269. doi:10.1093/ndt/gfq269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Textor SC, Lerman LO. Inflammatory cell markers as indicators of atherosclerotic renovascular disease. Clin J Am Soc Nephrol. 2012;7:193–195. doi: 10.2215/CJN.12641211. doi:10.2215/CJN.12641211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Truong LD, Farhood A, Tasby J, et al. Experimental chronic renal ischemia: morphologic and immunologic studies. Kidney Int. 1992;41:1676–1689. doi: 10.1038/ki.1992.241. doi:10.1038/ki.1992.241. [DOI] [PubMed] [Google Scholar]
- 44.Haller S, Adlakha S, Reed G, et al. Platelet activation in patients with atherosclerotic renal artery stenosis undergoing stent revascularization. Clin J Am Soc Nephrol. 2011;6:2185–2191. doi: 10.2215/CJN.03140411. doi:10.2215/CJN.03140411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Arena A, Stassi G, Iannello D, et al. Both IL-1beta and TNF-alpha regulate NGAL expression in polymorphonuclear granulocytes of chronic hemodialysis patients. Mediators Inflamm. 2010;2010:613937. doi: 10.1155/2010/613937. doi:10.1155/2010/613937. [DOI] [PMC free article] [PubMed] [Google Scholar]