

Abstract
Proximal renal tubular acidosis (RTA) (Type II RTA) is characterized by a defect in the ability to reabsorb HCO3 in the proximal tubule. This is usually manifested as bicarbonate wastage in the urine reflecting that the defect in proximal tubular transport is severe enough that the capacity for bicarbonate reabsorption in the thick ascending limb of Henle's loop and more distal nephron segments is overwhelmed. More subtle defects in proximal bicarbonate transport likely go clinically unrecognized owing to compensatory reabsorption of bicarbonate distally. Inherited proximal RTA is more commonly autosomal recessive and has been associated with mutations in the basolateral sodium-bicarbonate cotransporter (NBCe1). Mutations in this transporter lead to reduced activity and/or trafficking, thus disrupting the normal bicarbonate reabsorption process of the proximal tubules. As an isolated defect for bicarbonate transport, proximal RTA is rare and is more often associated with the Fanconi syndrome characterized by urinary wastage of solutes like phosphate, uric acid, glucose, amino acids, low-molecular-weight proteins as well as bicarbonate. A vast array of rare tubular disorders may cause proximal RTA but most commonly it is induced by drugs. With the exception of carbonic anhydrase inhibitors which cause isolated proximal RTA, drug-induced proximal RTA is associated with Fanconi syndrome. Drugs that have been recently recognized to cause severe proximal RTA with Fanconi syndrome include ifosfamide, valproic acid and various antiretrovirals such as Tenofovir particularly when given to human immunodeficiency virus patients receiving concomitantly protease inhibitors such as ritonavir or reverse transcriptase inhibitors such as didanosine.
Keywords: drug-induced pRTA, hereditary pRTA, proximal RTA, renal tubular acidosis
Introduction
Bicarbonate is freely filtered, and its concentration in the glomerular filtrate is equal to that in plasma (∼25 mEq/L). The majority of the filtered HCO3 (∼80%) is reabsorbed in the proximal tubule and the remaining 20% is reclaimed by the loop of Henle, distal tubules and collecting tubules. In an individual with a glomerular filtration rate (GFR) of 100 mL/min, ∼2500 mEq of HCO3 is filtered daily, and virtually all of this HCO3 is reabsorbed so that essentially none appears in the urine [1]. Because the vast majority of HCO3 is normally reclaimed in the proximal tubule, the finding of urinary HCO3 wastage is usually taken as evidence of a defect in proximal tubular HCO3 reabsorption [1]. When there is a major defect in HCO3 reabsorption in the proximal tubule, a larger quantity of filtered HCO3 is delivered to the distal segments, including the thick ascending limb, overwhelming the distal system and thereby causing urinary HCO3 wastage, which is the hallmark of the derangement underlying proximal (or Type II) RTA [1].
In recent years, there has been significant progress in the understanding of the mechanisms causing genetic proximal renal tubular acidosis (RTA) [2–8]. Moreover, new causes of drug-induced RTA have been recognized [9, 10]. In this review, we will discuss the mechanisms involved in the causation of hereditary proximal RTA as well as acquired causes with an emphasis on drug-induced proximal RTA.
Isolated proximal RTA versus Fanconi syndrome
Proximal RTA as an isolated defect in HCO3 transport is rare. It is characterized by a decreased rate of HCO3 reabsorption in the proximal tubule in the absence of alterations in the transport of other solutes. The impairment in HCO3 reabsorption was initially characterized as a decrease in the renal threshold for HCO3 reabsorption [11, 12].
More commonly, proximal RTA is associated with generalized dysfunction of the proximal tubule as part of the Fanconi syndrome [1, 9]. Isolated proximal RTA can be autosomal dominant, autosomal recessive and sporadic (Table 1). The autosomal recessive type is associated with severe growth retardation, ocular abnormalities such as glaucoma, cataracts and band keratopathy, and mental retardation [13]. Autosomal dominant proximal RTA, to our knowledge, has been reported in a single Costa Rican family [14]. The clinical features include mild growth retardation and reduced bone density. Sporadic isolated proximal RTA is a non-familial transient disorder that has been reported during infancy. Patients with this disorder have defective renal and intestinal bicarbonate reabsorption [13].
Table 1.
Causes of isolated proximal RTA
| Genetic causes |
|
| Acquired causes |
|
Autosomal dominant and autosomal recessive proximal RTA is usually permanent and requires life-long alkali therapy. In contrast, sporadic isolated proximal RTA is transient and alkali therapy can be discontinued after several years. After discontinuation of the alkali therapy, the condition does not return. This defect has been ascribed to proximal tubular immaturity in some infants [12].
An isolated defect of proximal tubular HCO3 reabsorption is also caused by the use of carbonic anhydrase inhibitors (see drug-induced proximal RTA). The kidney has at least two forms of carbonic anhydrase, CA II and CA IV [15]. CA II is found in the cell cytoplasm of both proximal and distal tubules and is also present in red blood cells, whereas CA IV is mainly found in the brush border of the proximal tubule. The latter is the isoform of CA involved in facilitating apical HCO3 reabsorption by preventing the luminal accumulation of H2CO3 and thus creating a more favorable pH gradient for H+ secretion [16] (Figure 1). A derangement in CA IV expression or function could specifically lead to impairment of proximal HCO3 reabsorption, but to our knowledge, CA IV deficiency has not been demonstrated in patients with hereditary proximal RTA. By contrast, familial RTA has been long recognized in patients with inherited deficiency of CA II in red blood cells [17–20]. These patients had features of both proximal and distal RTA (Type III RTA) as well as osteopetrosis, cerebral calcification and mental retardation [17, 19–22]. Hereditary distal RTA with associated features of proximal tubular dysfunction (low molecular weight proteinuria, generalized hyperaminoaciduria, hypophosphatemia with hyperphospaturia, and hypouricemia with hyperuricosuria) was described in two siblings with ATP6V1B1 mutation [23]. This rare association has never been fully understood although may go away after correction of the hypokalemia.
Fig. 1.
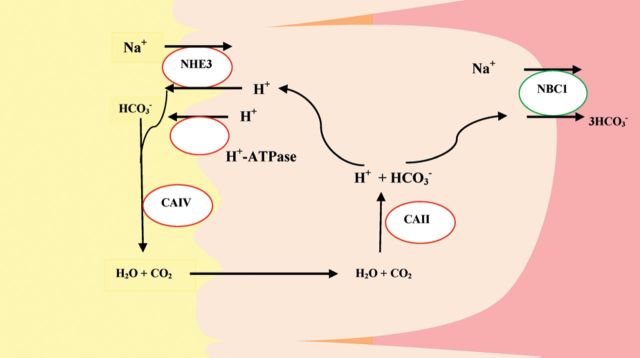
Proximal tubule bicarbonate reabsorption. CAII/IV, carbonic anhydrase II/IV; NHE3, Na+/H+ exchanger 3; NBC1, Na+/HCO3− cotransporter.
Fanconi syndrome is caused by a generalized dysfunction of proximal tubules causing loss of solutes like phosphate, uric acid, glucose, amino acids, low-molecular-weight proteins and bicarbonate [8, 24]. The genetic causes of Fanconi syndrome can be primary or secondary to systemic diseases. Primary Fanconi syndrome is caused by a missense mutation in Na phosphate cotransporter (NaPi-II) of the proximal tubular apical membrane [25]. The secondary causes of Fanconi syndrome include inherited cystinosis, galactosemia, hereditary fructose intolerance, tyrosinemia, Lowe syndrome, Alport syndrome, Wilson disease and mitochondrial disorders [8, 24, 25]. The most common inherited cause is cystinosis [26].
The acquired causes of Fanconi syndromes with proximal RTA include amyloidosis, multiple myeloma, paroxysmal nocturnal hemoglobinuria, renal transplantation, antiretroviral drugs, ifosfamide, cadmium and lead [27–30]. Of these, light-chain associated Fanconi syndrome is the most common [28]. Other causes of Fanconi syndrome are discussed under drug-induced proximal RTA (Table 2).
Table 2:
Proximal RTA with Fanconi Syndrome
| Genetic | NaPi-II cotransporter mutation |
| Inherited systemic diseases |
|
| Acquired causes | |
| Drug-induced | |
| Nucleotide Reverse Transcriptase Inhibitors | Tenofovir, Adefovir |
| Nucleoside Reverse Transcriptase Inhibitors | Didanosine, Lamivudine, Stavudine |
| Anticancer drugs | Ifosfamide, Oxaplatin, Cisplatin, |
| Anti-convulsant drugs | Valproic acid |
| Antibiotics, and | Aminoglycosides, expired tetracyclines |
| Antivirals | Cidofovir |
| Others | Streptozocin |
| Miscellaneous conditions | |
| Heavy Metals | Lead, Cadmium, Mercury, Copper |
| Vitamin D deficiency | |
| Multiple myeloma | |
| Amyloidosis | |
| Renal transplantation | |
| Paroxysmal nocturnal hemoglobinuria | |
| Aristolochic acid (Chinese herb nephropathy) | |
| Fumaric acid | |
| Suramin | |
| Paraquat | |
| L-Lysine and L-arginine | |
| Tubulo-intersticial nephritis | |
| Membranous nephropathy with anti-tubular antibodies |
Pathophysiology and clinical diagnosis
HCO3 is transported across into the peritubular space by a basolateral sodium-bicarbonate cotransporter (NBCe1), while H+ re-enters the tubular lumen mainly via a sodium-hydrogen exchanger (NHE) and to a lesser extent via H+-ATPase-driven H+ secretion (Figure 1). The secreted H+ then reacts with filtered HCO3 to form H2CO3 [31, 32]. In the proximal tubule lumen, H2CO3 is catalyzed by membrane-anchored CA IV and CA XIV to H2O and CO2 [31–33]. Conversion to CO2 facilitates reabsorption since HCO3 is relatively impermeable to the apical membrane of proximal tubule. In contrast, CO2 freely diffuses across the apical membrane into the cytosol. In the cytosol, CA II catalyzes the hydration of CO2 to form H+ and HCO3− which are then transported out of the cell via specific transporters (Figure 1).
Theoretically, proximal RTA could result from a defect in the basolateral NBCe1, the apical NHE, the apical H+-ATPase or carbonic anhydrase deficiency of either carbonic anhydrase II (cytosolic) or IV (brush border bound) (see Figure 1). So far, mutations have only been identified in NBCe1 and carbonic anhydrase genes [5]. The diagnosis of proximal RTA is established by the finding of urinary HCO3 wastage usually manifested by very alkaline urine. Patients with proximal RTA, however, have an intact ability to lower urine pH < 5.5 when the plasma HCO3 is less than the renal threshold for tubular HCO3− reabsorption (see Figure 2).
Fig. 2.
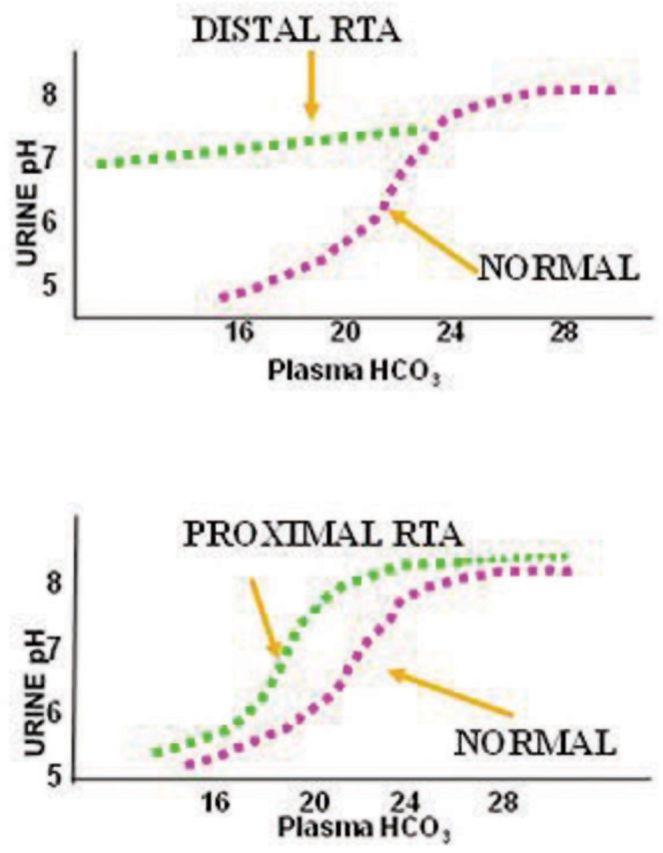
Differences in urinary pH and plasma bicarbonate in proximal and distal RTA. Adapted from Rodriguez-Soriano and Edelman [144].
The most elegant and comprehensive way of confirming the diagnosis of proximal RTA is to assess HCO3 reabsorption over a wide range of plasma HCO3. This can be done when the plasma HCO3 is increased from low levels slowly by the administration of NaHCO3 (HCO3 titration test). A marked increase in urinary HCO3 excretion occurs normally and this is reflected by an increase in urine pH as plasma HCO3 rises above the renal threshold for bicarbonate reabsorption. Below a certain threshold, however, bicarbonate excretion in patients with proximal RTA ceases and the urine pH can fall almost normally (Figure 2) [34]. This is a distinctive feature from patients with distal RTA (or Type 1 RTA) in whom the urine pH cannot fall normally regardless of the degree of acidosis (Figure 2). In normal infancy, the renal threshold is ∼22 mEq/L, whereas in older children and adults it is around 26 mEq/L [35]. In the steady state, the level of plasma HCO3 in patients with proximal RTA is close to the renal threshold. For instance, a patient with renal threshold of 18 mEq/L has a metabolic acidosis with a level of plasma bicarbonate 18 mEq/L. In this steady state, there is no bicarbonate wasting and the urine pH can be reduced close to normal levels [12].
A fractional HCO3 excretion of ≥15% clearly establishes the diagnosis of proximal RTA [36, 37]. Even lesser degrees of HCO3 wastage (i.e. fractional HCO3 excretion above 5%) are, in our opinion, sufficient for the diagnosis of less severe types of proximal RTA [36–38]. When fractional bicarbonate excretion is >15%, this reflects massive bicarbonate wastage as a result of a severe defect in proximal tubular transport which is severe enough that the capacity for bicarbonate reabsorption in the thick ascending limb of Henle's loop and more distal nephron segment is overwhelmed. More subtle defects in proximal bicarbonate transport likely go clinically unrecognized owing to compensatory reabsorption of bicarbonate in the distal nephron segment.
Glucosuria in the face of normal blood glucose, aminoaciduria, hyperphosphaturia and hyperuricosuria characterize the presence of Fanconi syndrome [39, 40]. Hypokalemia and renal potassium wasting can also be present in patients with proximal RTA associated with Fanconi syndrome [12]. Unlike patients with distal RTA, urine PCO2, measured after HCO3 infusion, should be normal in patients with proximal RTA (i.e. >70 mmHg), indicating that their distal H+ secretion is intact [41].
Hereditary proximal renal tubular acidosis
Inherited proximal RTA has been described as autosomal dominant, autosomal recessive or sporadic. Autosomal recessive inheritance in isolated proximal RTA has been linked to mutations in NBCe1 [2–7, 42]. Although the Na+/H+ exchanger 3 (NHE3) has been suggested as a candidate gene [43], to date there has been no reports of NHE3 mutations in proximal RTA patients [7]. It is of note, however, that isolated proximal RTA is very rare [2].
Autosomal dominant inheritance has been reported in a single Costa Rican family, but the gene involved has not yet been identified [14]. Lemann et al. studied some features of pRTA in two brothers from this family [14]. One brother was 20 years old, with short stature, bilateral coloboma and idiopathic subaortic stenosis. The other was 25 years old and asymptomatic. When untreated with bicarbonate, both brothers were acidotic with a urine pH of ≤5.0 consistent with proximal RTA. The asymptomatic brother had serum bicarbonate ranging from 17 to 19 mEq/L, while the other brother had bicarbonate in the range of 11.5–14 mEq/L. Radiological investigation revealed reduced bone density in both brothers.
Katzir et al. described the only other family with autosomal dominant isolated proximal RTA wherein the father and all four children had a very high fractional excretion of bicarbonate (40–60%) following bicarbonate loading [2]. They did not find mutations in any of the nine candidate genes they studied including carbonic anhydrases, NBC1 and NHE3.
Most of the proximal RTA associated with inherited causes of Fanconi syndrome result from metabolites accumulating in the proximal tubules overtime, and preventing its proper functioning [10]. In the case of inherited cystinosis, there is evidence of ATP depletion in proximal tubular cells [26]. It has been suggested that ATP depletion leads to an increase in intracellular sodium concentration which would impair all transporters that depend on it for energy [10].
Na-HCO3 cotransporter (NBCe1) mutations
The gene that encodes NBCe1 is SLC4A4 [13]. Three isoforms of NBCe1 have been described. NBCe1-A is expressed in the kidney and eye, NBCe1-B is expressed in pancreas, duodenum, colon and several other tissues, and NBCe1-C is predominantly expressed in the brain [44]. Mutations in the kidney isoform (NBCe1-A) cause proximal RTA and associated ocular abnormalities such as band karatopathy (Figure 3). Almost all cases described till now have shown autosomal recessive inheritance [3–6, 45–48].
Fig. 3.

Band keratopathy of the cornea in the left eye in a patient with autosomal recessive proximal RTA and the R298S mutation in kNBC cDNA. (Source: Igarashi et al. [13]).
Igarashi et al. were the first to report NBCe1 mutations in two unrelated patients having isolated proximal RTA with ocular abnormalities [5]. Both the patients were female, one presented at age 16 while the other presented at age 2. Their parents were normal. The common features were proximal RTA, short stature, bilateral glaucoma, cataracts and band keratopathy. Intellectual impairment was also reported in the 16-year-old patient. Serum amylase was interestingly elevated in both of them without signs of pancreatitis. This interesting association was attributed to the overlap between the kidney and pancreatic isoform of NBCe1 [5]. Plasma bicarbonate was low and urine was acidic. Genomic analysis revealed homozygous mutations in each patient, and heterozygous mutation in their asymptomatic parents. Missense mutations, R289S and R510, were identified [5].
Mutations leading to reduced activity and/or trafficking of the NBCe1 disrupt the normal bicarbonate reabsorption process of the proximal tubules leading to proximal RTA. Functional analysis has revealed reduced activity of R289S and R510 mutant NBCe1 [4]. Later studies have identified several other types of mutations in the NBCe1 gene [48]. Most of these mutations show abnormalities in trafficking as the likely mechanism of proximal RTA caused by NBCe1 mutations (see Figure 4).
Fig. 4.
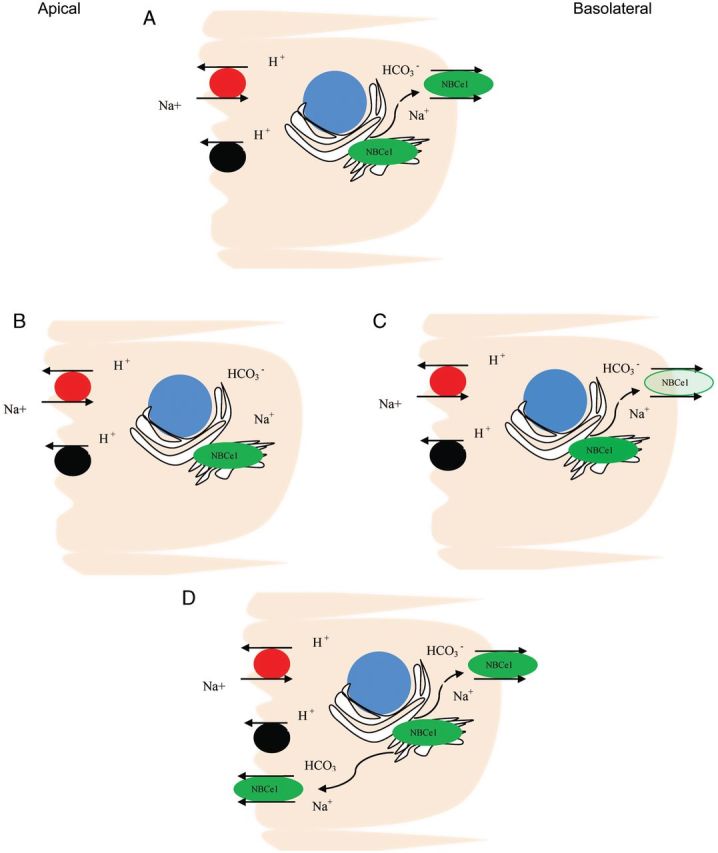
Schematic model of various mechanisms whereby NBCe1 mutations result in abnormal Na+/HCO3− transport in proximal RTA. (A) Normal, (B) Internal sequestration (R410H, R510H, E 91R,R342S, R881C), (C) decreased function(T485S, G486R), (D) internal sequestration and some mistargeting to both apical membrane and basolateral membrane(S427L).
Trafficking of mutant NBCE1 has been studied in Xenopus oocytes, ECV304 cells and polarized MDCK cells. Studies showed intracellular sequestration of mutants R410H, R510H, E91R and R342S [8]. Mutant S427L, while showing increased intracellular retention, also expressed on both apical and basolateral membranes [49]. Endoplasmic sequestration without cell surface expression was seen with mutant R881C [48]. Normal basolateral localization was observed with mutants T485S and G486R. Further investigation, however, revealed 50% reduction in transport activity [46, 47].
Using the substituted cysteine accessibility method, Zhu et al. showed that cysteine-substituted constructs R298C, S427C, T485C, G486C, L522C and A799C were processed to the plasma membrane, whereas R510C and R881C failed to process to the plasma membrane [44] (see Figure 4). In their experiment, a functional assay showed that constructs R298C, S427C, T485C, L522C and V799C have >50%, and R486C has 30–40% the transport function of NBCe1-A-5C− (human NBCe1-A construct with five endogenous cysteines substituted with serines).
Carbonic anhydrase gene mutations
Deficiency of CA II is also the primary defect underlying the autosomal recessive syndrome of osteopetrosis, RTA and cerebral calcification [17, 22]. In this disorder, it causes a mixed pattern of proximal and distal RTA (Type III RTA). Early onset of hypokalemia, paroxysmal muscle weakness, moderate-to-severe mental retardation and growth retardation are the other associated manifestations.
The CA II gene is located at q22 on chromosome 8. At least 23 different mutations have been identified so far in different kindreds with mixed RTA [8, 19, 50–52]. The syndrome of CA II deficiency, caused by these mutations, has a varied phenotypic presentation and has been diagnosed in a variety of ethnic backgrounds. It is particularly common in Arab populations of the Middle East. More than 70% of the reported cases of CA II deficiency syndrome are from these populations, probably the result of both a high rate of consanguineous marriages and an increased frequency of the CA II deficiency allele [19]. Patients of Arabic origin have a unique splice junction mutation at the junction of exon 2–intron 2 of the CA II gene (c.232+1 G>A) [53]. A study done in patients from Tunisia and Algeria traced the ancestry of all affected patients studied under an old Arab tribe of Helal who had settled there in the 10th century. Clinically, Arabic patients have a very severe phenotype. Unlike American and Belgian patients (H107Y mutations), severe cognitive impairment is a consistent feature [54]. This was suggested to be because missense mutations are phenotypically less severe than splice site mutations and frameshift mutations [19, 20]. However, later studies found frameshift mutation in an American patient with only mild learning disabilities [20].
Carbonic anhydrase II deficiencies are recessive mixed proximal–distal (Type 3) RTA [18]. Predominance of distal type of RTA (dRTA) has been reported in some cases [55, 56]. The characteristic biochemical findings are bicarbonate wasting, inability to lower urine pH <5.5, a low urine-to-blood pCO2 difference in an alkaline urine and decreased NH4 excretion [8].
Studies in mice
Gawanes et al. described that a null NBCe1−/− mice, prepared through disrupting the SLC4A4 gene, exhibited severe metabolic acidosis, growth retardation, reduced plasma sodium, hyperaldosteronism, splenomegaly, abnormal dentition, intestinal obstruction and death before weaning [57]. Blood pH and bicarbonate were very low in the mutants. Impaired transepithelial bicarbonate secretion in the colon was reported, but there was no histopathological evidence of any pancreatic abnormalities [57].
Nakamura et al. reported proximal RTA in NHE3 (NHe3−/−) mutant mice wherein the mutant mice had reduced bicarbonate and fluid reabsorption in the proximal convoluted tubules by 61% and 69%, respectively [58]. Impaired intestinal absorption with diarrhea, lower blood pressure and increased aldosterone was also noted.
TWIK-related acid-sensitive K+ channel 2 (TASK2) in the renal proximal tubules is involved in volume regulation [59]. Warth et al. produced Task−/− mutant mice, and reported a reduction in blood pH, and bicarbonate concentration in the mutant mice, whereas urine pH and bicarbonate were increased [60]. These findings were consistent with proximal RTA, and further studies suggested coupling of TASK2 activity to bicarbonate transport through external alkalinization [60].
Bicarbonate wastage and RTA have been well documented with Fanconi syndrome secondary to cystinosis in humans. A Ctns (cystinosin) (−/−) knock-out mouse model, however, did not show any significant difference in urinary bicarbonate excretion or urinary pH when compared with control wild-type mice at 10 months of age [61]. Increased urinary excretion of other biochemical parameters such as glucose, phosphate and potassium were noted in the knock-out mice consistent with Fanconi syndrome.
There has been a report of glucosuria and mild proximal RTA in patients with maturity onset diabetes of the young(MODY) Type 3, a condition resulting from a defect in a transcription factor called hepatocytes nuclear factor 1α (HNF1α). A knock-out mouse model of HNF1α, which developed glucosuria with evidence of Fanconi syndrome, suggested its important role in renal transport [62]. In a later study, knock-out mice had reduced expression of sodium/phospate cotransporters 1 and 4 (NaPi-I and NaPi-IV). Interestingly, no effect could be shown on the major renal phosphate transporter NaPi-II [63].
Other inherited proximal tubulopathies with acidosis
Lowe syndrome (oculocerebrorenal syndrome, OCRL) is an X-linked disease characterized by eye anomalies (mostly cataracts), mental retardation and Fanconi-like proximal tubulopathy [64]. The syndrome is attributed to mutations in the gene OCRL, encoding alpha-phosphatidylinositol (4,5)- biphosphate phosphatase (PIP2P) [65]. Most patients with Lowe syndrome develop significant metabolic acidosis and the disease should be distinguished from other hereditary renal acidosis disorders.
Dent's disease is an X-linked recessive proximal tubulopathy, characterized by low-molecular-weight proteinuria and hypercalciuria with nephrocalcinosis and nephrolithiasis, as well as progressive renal failure [66]. Mutations of two different genes produce Dent's disease: CLCN5 gene in two-thirds of cases [67], and OCRL, the same gene that causes Lowe's syndrome in one-third of the patients [68]. Metabolic acidosis can be found in those patients with Dent's disease due to OCRL gene mutations, and therefore should be considered in the differential diagnosis of proximal RTA, whereas those with CLCN5 mutations commonly do not develop metabolic acidosis. Patients with genetic Fanconi syndrome of Dent's disease, Lowe syndrome and autosomal dominant idiopathic Fanconi can be differentiated on the basis of their distinct urinary proteomes and metabonomes using mass spectrometry and H-NMR spectroscopy [69].
Fanconi–Bickel syndrome is a rare but well-defined autosomal recessive entity, characterized by hepatorenal glycogen accumulation, proximal renal tubular dysfunction and impaired utilization of glucose and galactose, possibly as a result of a primary defect in monosaccharide transport across the tubular membranes. RTA is part of the clinical manifestations in the affected patients [70].
Drug-induced proximal RTA
Carbonic anhydrase inhibitors cause isolated proximal RTA, whereas several drugs have been linked to the development of proximal RTA associated with Fanconi syndrome. Those include ifosfamide, oxaplatin, aminoglycosides, tenofovir, cidofovir, adefovir, didanosine, topiramate, valproic acid and others [9, 10] (Table 2).
Carbonic anhydrase inhibitors
It has been long recognized that carbonic anhydrase inhibitors, such as acetazolamide used to manage conditions such as glaucoma or increased intracranial pressure, cause isolated proximal RTA [9]. The main isoform of carbonic anhydrase found in the kidney is the membrane-bound and cytoplasmic carbonic anhydrase isoform, namely CA IV and CA II, respectively [71, 72]. CA II is more widespread and is present in almost all cells of the nephron, whereas the membrane-bound carbonic anhydrase, CA IV, has limited expression and is found mainly in the proximal tubule and is absent or expressed weakly in most segments of the collecting duct and the final segment of the proximal tubule [72]. Both isoforms of carbonic anhydrase play an important role in acid–base transport throughout the nephron [72]. In the tubular lumen, CA IV catalyzes the formation of CO2 and H2O from H2CO3. In the cytosol of tubular cells, CA II favors the formation of bicarbonate and hydrogen ion from CO2 and H2O that enter the cell (Figure 1) [72].
CA inhibitors have been used in clinical practice mainly to reduce elevated intraocular pressure in glaucoma or to treat mountain sickness [73]. Three of them (acetazolamide, methazolamide and dichlorphenamide) can be administered systemically. The other two CA inhibitors (brinzolamide and dorzolamide) are applied topically. All CA inhibitors are sulfonamide derivatives and have the potential to cause proximal RTA [73]. The defect in bicarbonate reabsorption with CA inhibitors can be explained by inhibition of CA IV located in the apical membrane of the proximal tubule cells. It has been demonstrated that some CA inhibitors, such as acetazolamide and benzolamide, are less membrane-permeable than others and as such are not as effective inhibitors of cytosolic CA as membrane-bound CA [74]. Typically CA inhibitors, therefore, cause a pure proximal RTA as a result of inhibition of the membrane-bound CA IV isoform resulting in the isolated inhibition of bicarbonate reabsorption without any associated features of Fanconi syndrome [75].
Ifosfamide
Ifosfamide is an alkylating agent, which is used in the treatment of various cancers such as bone sarcomas, soft tissue sarcomas and testicular cancer [76, 77]. It is a synthetic analog of cyclophosphamide and, like cyclophosphamide, can cause hemorrhagic cystitis. The use of Mesna has reduced the occurrence of hemorrhagic cystitis but has shown no preventive effect on its tubular toxicity which leads to Fanconi syndrome [78].
The incidence of Fanconi syndrome in treated patients has been reported between 1.4 and 5% [78]. The toxicity of ifosfamide and its late onset following discontinuation have been well recognized in several studies [78–81].
Rossi et al. did a follow-up study of 75 patients who had received ifosfamide for various malignancies [78]. Over 31 months of follow-up, five patients developed renal Fanconi syndrome as demonstrated by the presence of hyperaminoaciduria, phosphaturia, glucosuria and low serum bicarbonate [78]. Seven patients developed generalized subclinical tubulopathy which was defined as an impairment of three or all four parameters of proximal tubular solute transport (amino acids, phosphate, glucose and sodium) in the absence of acidosis or metabolic bone disease [78]. They reported that generalized subclinical tubulopathy occurred before the development of Fanconi syndrome in all five cases and moderate reduction in creatinine clearance was also reported in them [78]. Most information on ifosfamide nephrotoxicity comes from children since the use of ifosfamide in pediatric oncology is common [79–81]. By contrast, studies reporting ifosfamide-related Fanconi syndrome in adult patients are rare [82, 83]. In a recent report, Farry et al. did a long-term assessment of ifosfamide-related renal toxicity in adult patients and reported a steady decline in the estimated GFR although none of the patients progressed to end-stage renal disease [84]. Given the development of renal toxicity after completion of ifosfamide therapy, it has been suggested that patients should be followed for the development of proximal RTA [85, 86].
Nissim et al., based on their studies on rats, demonstrated that the active metabolites of ifosfamide, chloroacetaldehyde (CAA), causes renal injury by inhibiting nicotinamide adenine dinucleotide (reduced) (NADH):ubiquinone oxidoreductase (C-I), one of the enzymes in the oxidative phosphorylation pathway [87] (Figure 5). These authors showed for the first time that CAA accumulates in the renal cortex following ifosfamide treatment. They further showed that the inhibition of (C-1) led to increased NADH and decreased nicotinamide adenine dinucleotide (NAD). Furthermore, administration of agmatine (AGM), a metabolite of arginine decarboxylation, with ifosfamide prevented these changes and raised cAMP level. AGM, therefore, has been suggested as one of the potential therapies for prevention against ifosfamide-induced tubular dysfunction including Fanconi syndrome [87]. Later studies by Yaseen et al. examined the adverse effect of CAA and found that it inhibited endocytosis in the rat proximal kidney tubules which was attributed to a CAA-induced decrease in ATP levels and inhibition of V-ATPase [88] (Figure 5).
Fig. 5.

Schematic of IFO-induced nephrotoxicity. CAA, a metabolite of IFO, may inhibit NADH:oxidoreductase following dephosphorylation of AQDQ, an 18 kDa subunit of C-I. The inhibition of C-I disrupts oxidative phosphorylation, leading to multiple metabolic abnormalities, including elevation of NADH, decreased pyruvate dehydrogenase (PDH) and tricarboxylic acid cycle activity. (i) PDH, (ii) PC, (iii) glutamate-aspartate aminotransferase, (iv) glutamate dehydrogenase; C-I, C-II, C-III, C-IV, respiratory chain complexes; C-V, ATP synthetase; Cyt. C, cytochrome c, α-kg, α-ketoglutarate (source: Nissim [87]).
Oxaplatin and cisplatin
Oxaplatin-induced proximal RTA has been described as both isolated proximal RTA and as part of Fanconi syndrome. It was first described in a patient who was being treated with it for adenocarcinoma of the colon and subsequently developed hypokalemic, hyperchloremic metabolic acidosis with a normal anion gap. Evidence of Fanconi syndrome was based on glycosuria and low serum phosphate level [89]. In another report, in a similar clinical setting of oxaplatin use for adenocarcinoma of colon, the patient developed bicarbonate wasting and severe hypokalemic, hyperchloremic metabolic acidosis with a normal anion gap but no other abnormalities, thus suggesting isolated proximal RTA [90].
Fanconi syndrome has also been described with cisplatin [91]. In mice, a marked increase in urinary concentrations of glucose, amino acids such as alanine, valine, leucine, methionine and trichloroacetic acid cycle metabolites such as pyruvate and lactate was found within the first 48 h of administration of cisplatin [92]. The amino acid profile present in the urine of cisplatin-treated mice preceded the elevations in serum creatinine. Electron microscopy data at Day 3 revealed a cytotoxic effect on the S3 segment of the proximal tubules. Aminoaciduria was explained by the toxic effect of cisplatin on amino acid transporters in the proximal convoluted tubule as almost 90% of amino acid transport occurs in the proximal convoluted tubule. It has been suggested that one of the possible mechanisms of cisplatin-induced proximal tubule nephrotoxicity is reduced expression and function of sodium-dependent glucose transporters (Figure 6) [92].
Fig. 6.

Schematic representation of the proximal tubule reabsorption processes perturbed in cisplatin- or gentamcin-induced renal Fanconi-like syndromes that are manifested by the urinary elevation of glucose, NAAs and monocarboxylates. The uptake of glucose, NAA and monocarboxylate into the epithelial cells is mediated by luminal sodium-dependent transporters such as SLC5A1, SLC5A2, SLC6A18 and SLC16A7. The sodium electrochemical gradient across the luminal membrane is provided by the activity of the basolateral sodium/potassium ATPase. The entry of cisplatin (denoted as ‘C’) or gentamicin (denoted as ‘G’) into the tubular epithelial cells results in the transcriptional down-regulation of HNF1R and HNF1, which in turn leads to the reduction of mRNA levels of SLC5A1, SLC5A2 and collectrin. Cisplatin or gentamicin treatment also leads to the reduction of SLC6A18 and SLC16A7 mRNA through unknown transcription factors. Both nephrotoxicants also induce hypoxia in renal tubular cells, which leads to the transcriptional up-regulation and post-translational stabilization of hypoxia-inducible factor HIF1R. An increased amount of HIF1 up-regulates the transcription of basolateral GLUT transporters as one of the adaptive responses to proximal tubule injury. Up-regulated gene names are shown in red color, while down-regulated ones are shown in blue color. For schematic convenience, these regulated genes are shown next to each other, although they are located on different chromosomes inside the nucleus (source: Xu et al. [94]).
Portilla et al. have reported in previous studies that cisplatin inhibits peroxisome proliferator-activated receptor-alpha activity and consequently fatty acid oxidation resulting in proximal tubule cell death [92, 93]. Fibrates such as bezafibrate prevents this inhibition and thus, may be protective against cisplatin-induced proximal tubule cell death [93].
The uptake of glucose into the epithelial cells is mediated by luminal sodium-dependent transporters such as SLC5A1, SLC5A2, SLC6A18 and SLC16A7 [94]. Xu et al. reported that the entry of cisplatin into the tubular epithelial cells results in the transcriptional down-regulation of HNF1R and HNF1, which, in turn, leads to the reduction of messenger RNA (mRNA) levels of SLC5A1, SLC5A2 and collectrin [94]. Cisplatin also leads to the reduction of SLC6A18 and SLC16A7 mRNA. Collectrin, an analog of ACE2, which lacks ACE2 activity is involved in proximal tubule absorption of amino acids [95]. Inhibition of collectrin may be part of the proximal tubule defect causing proximal RTA but this, to our knowledge, has not been studied.
Antiretrovirals
Fanconi syndrome has been reported in human immunodeficiency virus (HIV)-positive patients on antiretroviral therapy [96]. Earle et al. described three cases of Fanconi in such patients. Each of these patients showed generalized tubular dysfunction with hypophosphatemia, metabolic acidosis, phosphaturia, glucosuria and generalized aminoaciduria. All three patients had glucosuria, two had proteinuria (1.6 g/24 h and 2.6 g/24 h), urinary phosphorus was high in all and aminoaciduria was present in two of them [96]. Serum analysis revealed hypophosphatemia in all, but serum bicarbonate was low normal. Two of them recovered on discontinuation of their respective antiretroviral drugs: tenofovir and adefovir. The third patient, who had been treated with cidofovir, an acyclic nucleoside phosphonate, for cytomegalovirus (CMV) retinitis 9 months before presentation, continued to require electrolyte replacement. Of note, all these antiretroviral drugs were nucleotide reverse transcriptase inhibitors. Proximal RTA has also been reported with two other nucleoside reverse transcriptase inhibitors lamivudine and stavudine [97].
In a review of Food and Drug Administration (FDA) Adverse Event Report System of Fanconi syndrome associated with Tenofovir use, it was found that the protease inhibitor, ritonovir (74%), and the nucleoside reverse transcriptase inhibitor, didanosine (43%), were most commonly used in combination with tenofovir, a nucleotide reverse transcriptase inhibitor [98]. In view of a previous drug interaction wherein the coadministration of lopinavir/ritonavir with tenofovir increased the systemic levels of tenofovir, it was suggested that these drug combinations are a risk for the development of Fanconi syndrome [99]. Therefore, patients receiving these combination drugs should be closely monitored for renal toxicity [98, 100]. It has also been suggested that factors other than drug combination may also potentiate tenofovir toxicity as 17% of the patients who did not take these combination drugs with tenofovir still developed Fanconi syndrome [98]. It is interesting to note that although tenofovir is used both to treat HIV and hepatitis B infection, Fanconi syndrome has been described more commonly in HIV patients [101]. This again points to the potentiation of tenofovir toxicity in HIV patients either through other concomitant drugs and/or HIV-associated renal damage.
Three mechanisms of tenofovir nephrotoxicity have been suggested including drug excretion in proximal tubules, genetic association and mitochondrial toxicity [101]. Proximal tubules are involved in the excretion of several drugs including tenofovir which enters the proximal tubule cells through basolateral organic anion transporters and exits using apical transporter multidrug resistance-associated protein 4 (MRP 4) [102]. Didanosine uses the same organic anion transporters to enter the proximal tubular cells, and therefore combination with tenofovir leads to a competition for the same transporters causing an increase in renal toxicity [101]. Similarly, ritonovir exits the tubular cells using multidrug resistance-associated protein 2 (MRP 2), and it is believed that somehow this interferes with tenofovir exit through MRP4 causing increased intracellular concentration of tenofovir and tubular dysfunction but the exact mechanism is unknown [101].
Pharmacogenomics has offered another mechanism as to why tenofovir may cause Fanconi syndrome. In a study of 115 HIV-infected patients who were on tenofovir, 19 (16.5%) had developed kidney tubular dysfunction, and of these, 16 had genotype CC at position-24 of ABCC2. It has been suggested that this genotype would help us to identify and then monitor patients at greater risk of developing tenofovir-associated tubulopathy [103].
There have been few reports wherein didanosine has been implicated in the development of Fanconi's syndrome in HIV patients [104–106]. D'Ythurbide et al. reported Fanconi syndrome and diabetes insipidus in an HIV patient being treated with didanosine, lamivudine, atazanavir and ritonavir [104]. On admission, the patient had hypophosphatemia, hypouricemia, hyperchloremic metabolic acidosis with a normal anion gap, normoglycemic glycosuria and low-molecular-weight proteinuria. High urinary phosphate excretion and high fractional excretion of phosphate indicated renal phosphate wasting. One month following discontinuation of didanosine, while continuing to treat with other drugs, the patient recovered completely from glycosuria, tubular proteinuria, metabolic acidosis and hypernatremia [104].
Anti-convulsant therapies
Topiramate is an anti-epileptic drug which is used mainly for seizure disorders and migraine prophylaxis [107]. Several studies have described the development of metabolic acidosis following treatment with topiramate [107–109]. Sacre et al. reported a 47-year-old woman treated with topiramate for 12 months for migraine prophylaxis who developed hyperchloremic metabolic acidosis with a normal GFR and positive urinary anion gap suggesting both proximal and distal RTA [110].
Studies have suggested inhibition of CA II by topiramate as the cause for the development of mixed RTA [73, 111]. Winum et al. found that topiramate is a very potent inhibitor of human CA Types II and XII and a medium potency inhibitor of Type IV CA [73]. Maryanoff et al. by contrast, found topiramate to have low activity against CA II [112]. However, X-ray crystallography studies have revealed a very tight association between bound topiramate and the active site of CA II consistent with topiramate's very potent inhibitory activity against CA II [111]. This inhibition of cytosolic CA could explain very well the development of mixed RTA.
Rarely, long-term treatment with valproic acid, another anticonvulsant drug, can also cause Fanconi syndrome. Knorr et al. reported that an 8-year-old boy on valproic acid for 7 years was admitted to the hospital for status epilepticus with laboratory findings of Fanconi syndrome. On discontinuation of valproate, laboratory findings returned to normal 2 months later [113]. More recent reports have described similar findings with laboratory values returning to normal following discontinuation of valproate [114]. The mechanism is not clear but a direct effect of valproic acid on the mitochondria of the proximal tubules has been implicated [115, 116].
Aminoglycosides
There are a few case reports of aminoglycoside-induced Fanconi syndrome [117–119]. Ghiculescu et al. reported the development of Fanconi syndrome in a 53-year-old man treated for respiratory infections with gentamicin [117]. The patient was profoundly hypophosphatemic and moderately hypocalcemic, and discontinuation of gentamicin resulted in recovery.
The presence of glucosuria and aminoaciduria after exposure to gentamicin has also been reported [120]. In the case of gentamicin, recent in vitro and in vivo studies performed in LLCPK1 cells, as well as in mouse kidney tissue, have shown that aminoglycoside antibiotics reduce glucose reabsorption in kidney tissue by reducing mRNA, protein expression and function of the sodium-dependent glucose transporter, which is located in the apical membrane of the proximal tubule (Figure 6) [121].
Other drugs
Other antivirals used for opportunistic infections in HIV have also been implicated in the development of Fanconi's syndrome [122]. Vittecoq et al. [122] reported the development of tubular dysfunction in HIV patients treated for CMV retinitis with cidofovir. On the fifth day of cedofovir treatment, a patient developed low serum bicarbonate, low serum phosphorous, nonselective proteinuria and glycosuria. Fanconi syndrome was diagnosed and a renal biopsy revealed degeneration and necrosis of proximal tubular cells [122]. Fanconi syndrome has also been reported after the administration of capecitabine, irinotecan and bevacizumab [123]. l-Cationic amino acids, such as lysine and l-arginine, have a profound inhibitory effect on proximal bicarbonate reabsorption and can potentially cause proximal RTA [124].
Heavy metals
Heavy metals such as lead, cadmium and mercury have been reported to be associated with proximal RTA [125]. Chronic cadmium exposure has been reported to cause Fanconi syndrome [126]. Cadmium accumulates in the proximal tubular cells through receptor-mediated endocytosis of metallothionein-bound Cd (Cd–MT). Cd–MT complexes are degraded in endosomes and lysosomes which release free Cd2+ into the cytosol. In the cytosol, it generates reactive oxygen species which leads to a cascade of damaging cellular events that can cause generalized proximal tubular dysfunction [126].
Miscellaneous causes
Proximal RTA as part of Fanconi's syndrome has been reported with several conditions including vitamin D deficiency, multiple myeloma, amyloidosis, renal transplantation and paroxysmal nocturnal hemoglobinuria [127].
There have been several reports of proximal RTA, with or without Fanconi's syndrome, in children with nutritional vitamin D deficiency or resistance to vitamin D action [128, 129]. There have also been reports of Fanconi syndrome in adult patients with vitamin D deficiency [127]. Taylor et al. reported that a 33-year-old African American woman with nutritional vitamin D deficiency, possibly as a result of various medical problems including paraparesis, developed Fanconi syndrome [127]. The patient was acidotic with hypocalcemia and aminoaciduria. Using an ammonium chloride loading test and a bicarbonate infusion test, proximal RTA was diagnosed which resolved following 2 years of vitamin D and calcium therapy [127]. To our knowledge, the exact mechanism by which vitamin D deficiency leads to Fanconi syndrome is unknown.
Messiaen et al. have reported several cases of Fanconi syndrome as a result of multiple myeloma [28]. Although the exact pathophysiology of Fanconi syndrome in multiple myeloma has not been elucidated, it has been shown in several studies that kappa light chain accumulates in the lysosomes of the proximal tubular cells and is not degradable [28, 130, 131]. Possible mechanisms could be disruption of apical membrane recyling, and/or impaired ATP production [28]. In a more recent report, accumulation of kappa light chains in the absence of crystals was demonstrated in a case of Fanconi syndrome in a patient with Waldenstrom macroglobulinemia [131]. Rochman et al. reported development of Fanconi syndrome in a patient with chronic lymphocytic leukemia who presented with proximal RTA with hypokalemia, phosphaturia and glycosuria [27]. Kappa light chains in the urine, peritubular deposits and amyloid casts were found [27]. In the light of these studies, kappa light chain has been implicated as the key factor causing Fanconi syndrome in these patients [28, 130, 131]. Rikitake et al., however, reported lambda light chain proteinuria in a 57-year-old female who developed Fanconi syndrome and had renal amyloidosis with nephrotic syndrome for the last 5 years [132].
Both proximal and distal RTA have been reported after kidney transplantation, the distal type being much more frequently found [133, 134]. Friedman et al. reported the development of Fanconi syndrome alongside rejection episode 4.5 years following a renal transplant [134]. An immunologically mediated tubular dysfunction was suggested as the possible cause [134].
Hsiao et al. reported Fanconi syndrome and chronic kidney disease in a patient with paroxysmal nocturnal hemoglobinuria (PNH) and hemosiderosis [135]. The patient was a 57-year-old female who had PNH for the last 17 years and history of multiple blood transfusion. She had the following features of Fanconi syndrome: proximal RTA, hyperphosphaturia, glucosuria and aminoaciduria. A renal biopsy revealed heavy hemosiderin deposits in the proximal tubular cells and that explained the signs of the generalized proximal tubular dysfunctions in this patient [135].
Diagnosis of proximal RTA
The diagnosis of proximal RTA requires the demonstration of urinary HCO3− wastage. This is evident whenever urine pH is high when the plasma bicarbonate is normal or slightly reduced. Patients with proximal RTA, however, have an intact ability to lower urine pH <5.5 when the plasma HCO3− is lower than the renal threshold for bicarbonate reabsorption. This is in contrast to patients with classic or Type I RTA who cannot lower urine pH maximally regardless of the degree of the acidosis (Figure 2). Skeletal abnormalities and rickets are less common with proximal RTA than in classic, distal RTA [136]. Nephrocalcinosis and nephrolithiasis are usually absent in proximal RTA but are prominent features of distal RTA [31]. Growth retardation is also seen in proximal RTA as a result of metabolic acidosis [31].
Traditionally, a fractional HCO3− excretion of ≥15% is said to be needed to establish the diagnosis of proximal RTA [36]. Given the large capacity of the distal nephron to reabsorb HCO3−, however, even minor degrees of HCO3− wastage (i.e. fractional HCO3− excretion ∼5%) are, in our opinion, sufficient for the diagnosis. The most definitive way to diagnose proximal RTA is to assess HCO3− excretion when plasma HCO3− is increased by administration of NaHCO3 (HCO3− titration test). When proximal RTA is present, a marked increase in urinary HCO3− excretion and urine pH occurs as plasma HCO3− rises above the renal threshold, whereas HCO3 excretion decreases and the urine pH falls when the plasma HCO3 is reduced. Hypokalemia and renal potassium wasting are characteristic of distal RTA [137]. In proximal RTA, hypokalemia can also occur particularly during bicarbonate therapy as discussed under treatment. Glucosuria in the face of normal blood glucose, aminoaciduria, hyperphosphaturia and hyperuricosuria characterize the presence of Fanconi's syndrome [39]. Additionally, the urinary proteomes and metabonomeses can be examined using mass spectrometry and 1H-NMR spectroscopy as it has been done in genetic forms of the renal Fanconi syndrome [69]. Ifosfamide nephrotoxicity results in tubular proteinuria, whereas the urine of a Fanconi syndrome patient contains mainly albumin. Under both the conditions, excretion of amino acids is greatly augmented [69].
Patients with a distal acidification defect (e.g. distal RTA) typically have a positive urine anion gap because of low NH4+ excretion, whereas in diarrheal states associated with metabolic acidosis, the urine anion gap is negative, reflecting the fact that NH4+ excretion is appropriately increased [138]. Information regarding NH4+ excretion from subjects with proximal RTA is limited. The excretion rates of NH4+ may not be reduced when compared with those of control subjects [11, 139, 140]. For instance, in a study by Brenes and Sanchez, the response to a 3-day acid loading test with NH4Cl was evaluated in 8 patients with isolated proximal RTA and in 10 healthy control subjects [141]. In the basal state, all subjects with proximal RTA had rates of NH4+ excretion similar to those of the control subjects, suggesting a normal pattern of ammonium renal handling. On the third day of acid loading, however, their NH4+ excretion rates were significantly lower than those of controls, demonstrating impairment in maximal urinary NH4+ excretion [14, 141]. Given this latter finding, it is likely that the urinary anion gap in proximal RTA may not be as negative as in normal people with metabolic acidosis. Although reduced, the amount of urinary ammonium may still be sufficient to result in a negative urine anion gap. Since these previous studies did not provide information on the urinary anion gap (UAG) in proximal RTA, additional information on this parameter is needed for the proper interpretation of UAG in proximal RTA. In distal RTA, the urine anion gap is consistently positive without exceptions [138].
Therapy of proximal RTA
Drug-induced proximal RTA caused by CA inhibitors is usually mild and readily reversible. One has to be very careful in patients with compromised kidney function because in this setting, metabolic acidosis can be severe with the use of CA inhibitors. Drug-induced proximal RTA associated with Fanconi syndrome usually reflects severe proximal tubular nephrotoxicity. In some cases, there are profound defects in bicarbonate and phosphate transport, such that the degree of metabolic acidosis and hypophosphatemia is severe requiring drug discontinuation. Discontinuation of the offending drugs such as antiretrovirals usually leads to recovery, but the defect can be persistent after discontinuation with some agents such as ifosfamide.
Patients with inherited and acquired proximal RTA should be given HCO3− supplements [140, 142]. In children, in particular, HCO3− replacement therapy is critical for the prevention of growth retardation due to acidosis [142]. The magnitude of the bicarbonaturia that occurs when serum HCO3− is normalized requires that large amounts of HCO3− be administered (5–15 mEq/kg body weight). By adding a thiazide diuretic (e.g. hydrochlorothiazide 25–50 mg daily), the amount of bicarbonate can be reduced. Thiazides enhance proximal tubule and loop HCO3− reabsorption by reducing the extracellular volume. A reduction in extracellular volume is sensed by the kidneys as a stimulus for tubular reabsorption of Na+ and HCO3−. This way the use of thiazide diuretics by producing mild volume depletion allows for a reduction in the amount of bicarbonate to be given daily [143].
The combination of sodium bicarbonate and thiazide administration can unfortunately aggravate hypokalemia by promoting K+ secretion in the cortical collecting tubule (CCT) as a result of enhanced distal HCO3− delivery. By being relatively poorly reabsorbed in the CCT, the presence of bicarbonate in large amounts generates a negative transepithelial voltage that promotes K+ secretion and urinary potassium wastage. Therefore, plasma K+ must be carefully monitored when these therapies are used. A mixture of sodium and potassium salts is usually needed. We recommend Polycitra (K-Shohl solution, a mixture of sodium and potassium citrate). This oral solution contains 1 mEq of K+ and 1 mEq of Na+ per mL, and the citrate, when metabolized, is equivalent to 2 mEq of HCO3− per mL.
Funding
D.B. has grant support from the National Institute of Diabetes and Digestive and Kidney Diseases (grant 1R01DK080089-01A2).
Acknowledgements
This article is dedicated in the memory of Professor Juan Rodriguez-Soriano (1933-2010) for his important contributions to the understanding of the syndromes of renal tubular acidosis and the first description of the physiological and clinical features of proximal renal tubular acidosis.
Conflict of interest statement. None declared.
References
- 1.Moorthi K, Batlle D. Renal Tubular Acidosis. In: Gennari J, Adrogue HJ, Galla JH, Madias NE, editors. Acid–Base Disorders and Their Treatment. Boca Raton: Taylor and Francis; 2005. pp. 417–459. [Google Scholar]
- 2.Katzir Z, Dinour D, Reznik-Wolf H, Nissenkorn A, Holtzman E. Familial pure proximal renal tubular acidosis–a clinical and genetic study. Nephrol Dial Transplant. 2008;23:1211–1215. doi: 10.1093/ndt/gfm583. doi:10.1093/ndt/gfm583. [DOI] [PubMed] [Google Scholar]
- 3.Demirci FY, Chang MH, Mah TS, Romero MF, Gorin MB. Proximal renal tubular acidosis and ocular pathology: a novel missense mutation in the gene (SLC4A4) for sodium bicarbonate cotransporter protein (NBCe1) Mol Vis. 2006;12:324–330. [PubMed] [Google Scholar]
- 4.Dinour D, Chang MH, Satoh J, et al. A novel missense mutation in the sodium bicarbonate cotransporter (NBCe1/SLC4A4) causes proximal tubular acidosis and glaucoma through ion transport defects. J Biol Chem. 2004;279:52238–52246. doi: 10.1074/jbc.M406591200. doi:10.1074/jbc.M406591200. [DOI] [PubMed] [Google Scholar]
- 5.Igarashi T, Inatomi J, Sekine T, et al. Mutations in SLC4A4 cause permanent isolated proximal renal tubular acidosis with ocular abnormalities. Nat Genet. 1999;23:264–266. doi: 10.1038/15440. doi:10.1038/15440. [DOI] [PubMed] [Google Scholar]
- 6.Inatomi J, Horita S, Braverman N, et al. Mutational and functional analysis of SLC4A4 in a patient with proximal renal tubular acidosis. Pflugers Arch. 2004;448:438–444. doi: 10.1007/s00424-004-1278-1. [DOI] [PubMed] [Google Scholar]
- 7.Lo YF, Yang SS, Seki G, et al. Severe metabolic acidosis causes early lethality in NBC1 W516X knock-in mice as a model of human isolated proximal renal tubular acidosis. Kidney Int. 2011;79:730–741. doi: 10.1038/ki.2010.523. doi:10.1038/ki.2010.523. [DOI] [PubMed] [Google Scholar]
- 8.Alper SL. Familial renal tubular acidosis. J Nephrol. 2010;23:S57–S76. [PubMed] [Google Scholar]
- 9.Izzedine H, Launay-Vacher V, Isnard-Bagnis C, Deray G. Drug-induced Fanconi's syndrome. Am J Kidney Dis. 2003;41:292–309. doi: 10.1053/ajkd.2003.50037. doi:10.1053/ajkd.2003.50037. [DOI] [PubMed] [Google Scholar]
- 10.Quigley R. Proximal renal tubular acidosis. J Nephrol. 2006;19:S41–S45. [PubMed] [Google Scholar]
- 11.Rodriguez Soriano J, Boichis H, Stark H, Edelmann CM., Jr Proximal renal tubular acidosis. A defect in bicarbonate reabsorption with normal urinary acidification. Pediatr Res. 1967;1:81–98. doi: 10.1203/00006450-196703000-00001. doi:10.1203/00006450-196703000-00001. [DOI] [PubMed] [Google Scholar]
- 12.Rodriguez Soriano J. Renal tubular acidosis: the clinical entity. J Am Soc Nephrol. 2002;13:2160–2170. doi: 10.1097/01.asn.0000023430.92674.e5. doi:10.1097/01.ASN.0000023430.92674.E5. [DOI] [PubMed] [Google Scholar]
- 13.Igarashi T, Sekine T, Inatomi J, Seki G. Unraveling the molecular pathogenesis of isolated proximal renal tubular acidosis. J Am Soc Nephrol. 2002;13:2171–2177. doi: 10.1097/01.asn.0000025281.70901.30. doi:10.1097/01.ASN.0000025281.70901.30. [DOI] [PubMed] [Google Scholar]
- 14.Lemann J, Jr, Adams ND, Wilz DR, Brenes LG. Acid and mineral balances and bone in familial proximal renal tubular acidosis. Kidney Int. 2000;58:1267–1277. doi: 10.1046/j.1523-1755.2000.00282.x. doi:10.1046/j.1523-1755.2000.00282.x. [DOI] [PubMed] [Google Scholar]
- 15.Purkerson JM, Schwartz GJ. The role of carbonic anhydrases in renal physiology. Kidney Int. 2007;71:103–115. doi: 10.1038/sj.ki.5002020. doi:10.1038/sj.ki.5002020. [DOI] [PubMed] [Google Scholar]
- 16.Batlle D, Haque SK. Genetic causes and mechanisms of distal renal tubular acidosis. Nephrol Dial Transplant. 2012;27:3691–3704. doi: 10.1093/ndt/gfs442. [DOI] [PubMed] [Google Scholar]
- 17.Shapira E, Ben-Yoseph Y, Eyal FG, Russell A. Enzymatically inactive red cell carbonic anhydrase B in a family with renal tubular acidosis. J Clin Invest. 1974;53:59–63. doi: 10.1172/JCI107559. doi:10.1172/JCI107559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bolt RJ, Wennink JM, Verbeke JI, Shah GN, Sly WS, Bokenkamp A. Carbonic anhydrase type II deficiency. Am J Kidney Dis. 2005;46:e71–e73. A50 doi:10.1053/j.ajkd.2005.08.023. [PubMed] [Google Scholar]
- 19.Fathallah DM, Bejaoui M, Lepaslier D, Chater K, Sly WS, Dellagi K. Carbonic anhydrase II (CA II) deficiency in Maghrebian patients: evidence for founder effect and genomic recombination at the CA II locus. Hum Genet. 1997;99:634–637. doi: 10.1007/s004390050419. doi:10.1007/s004390050419. [DOI] [PubMed] [Google Scholar]
- 20.Shah GN, Bonapace G, Hu PY, Strisciuglio P, Sly WS. Carbonic anhydrase II deficiency syndrome (osteopetrosis with renal tubular acidosis and brain calcification): novel mutations in CA2 identified by direct sequencing expand the opportunity for genotype–phenotype correlation. Hum Mutat. 2004;24:272. doi: 10.1002/humu.9266. doi:10.1002/humu.9266. [DOI] [PubMed] [Google Scholar]
- 21.Sly WS, Hewett-Emmett D, Whyte MP, Yu YS, Tashian RE. Carbonic anhydrase II deficiency identified as the primary defect in the autosomal recessive syndrome of osteopetrosis with renal tubular acidosis and cerebral calcification. Proc Natl Acad Sci USA. 1983;80:2752–2756. doi: 10.1073/pnas.80.9.2752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sly WS, Whyte MP, Sundaram V, et al. Carbonic anhydrase II deficiency identified as the primary defect in the autosomal recessive syndrome of osteopetrosis with renal tubular acidosis and cerebral calcification. N Engl J Med. 1985;313:139–145. doi: 10.1056/NEJM198507183130302. [DOI] [PubMed] [Google Scholar]
- 23.Tasic V, Korneti P, Gucev Z, Hoppe B, Blau N, Cheong HI. Atypical presentation of distal renal tubular acidosis in two siblings. Pediatr Nephrol. 2008;23(7):1177–81. doi: 10.1007/s00467-008-0796-z. doi:10.1016/0002-9343(72)90075-7. [DOI] [PubMed] [Google Scholar]
- 24.Sirac C, Bridoux F, Essig M, Devuyst O, Touchard G, Cogne M. Toward understanding renal Fanconi syndrome: step by step advances through experimental models. Contrib Nephrol. 2011;169:247–261. doi: 10.1159/000313962. doi:10.1159/000313962. [DOI] [PubMed] [Google Scholar]
- 25.Magen D, Berger L, Coady MJ, et al. A loss-of-function mutation in NaPi-IIa and renal Fanconi's syndrome. N Engl J Med. 2010;362:1102–1109. doi: 10.1056/NEJMoa0905647. doi:10.1056/NEJMoa0905647. [DOI] [PubMed] [Google Scholar]
- 26.Baum M. The Fanconi syndrome of cystinosis: insights into the pathophysiology. Pediatr Nephrol. 1998;12:492–497. doi: 10.1007/s004670050495. doi:10.1007/s004670050495. [DOI] [PubMed] [Google Scholar]
- 27.Rochman J, Lichtig C, Osterweill D, Tatarsky I, Edelman S. Adult Fanconi's syndrome with renal tubular acidosis in association with renal amyloidosis: occurrence in a patient with chronic lymphocytic leukemia. Arch Intern Med. 1980;140:1361–1363. doi:10.1001/archinte.1980.00330210109033. [PubMed] [Google Scholar]
- 28.Messiaen T, Deret S, Mougenot B, et al. Adult Fanconi syndrome secondary to light chain gammopathy. Clinicopathologic heterogeneity and unusual features in 11 patients. Medicine (Baltimore) 2000;79:135–154. doi: 10.1097/00005792-200005000-00002. doi:10.1097/00005792-200005000-00002. [DOI] [PubMed] [Google Scholar]
- 29.Riley AL, Ryan LM, Roth DA. Renal proximal tubular dysfunction and paroxysmal nocturnal hemoglobinuria. Am J Med. 1977;62:125–129. doi: 10.1016/0002-9343(77)90357-6. doi:10.1016/0002-9343(77)90357-6. [DOI] [PubMed] [Google Scholar]
- 30.Irizarry-Alvarado JM, Dwyer JP, Brumble LM, Alvarez S, Mendez JC. Proximal tubular dysfunction associated with tenofovir and didanosine causing Fanconi syndrome and diabetes insipidus: a report of 3 cases. AIDS Read. 2009;19:114–121. [PubMed] [Google Scholar]
- 31.Fry AC, Karet FE. Inherited renal acidoses. Physiology (Bethesda) 2007;22:202–211. doi: 10.1152/physiol.00044.2006. doi:10.1152/physiol.00044.2006. [DOI] [PubMed] [Google Scholar]
- 32.Batlle D, Ghanekar H, Jain S, Mitra A. Hereditary distal renal tubular acidosis: new understandings. Annu Rev Med. 2001;52:471–484. doi: 10.1146/annurev.med.52.1.471. doi:10.1146/annurev.med.52.1.471. [DOI] [PubMed] [Google Scholar]
- 33.Kaunisto K, Parkkila S, Rajaniemi H, Waheed A, Grubb J, Sly WS. Carbonic anhydrase XIV: luminal expression suggests key role in renal acidification. Kidney Int. 2002;61:2111–2118. doi: 10.1046/j.1523-1755.2002.00371.x. doi:10.1046/j.1523-1755.2002.00371.x. [DOI] [PubMed] [Google Scholar]
- 34.Soriano JR, Boichis H, Edelmann CM., Jr Bicarbonate reabsorption and hydrogen ion excretion in children with renal tubular acidosis. J Pediatr. 1967;71:802–813. doi: 10.1016/s0022-3476(67)80005-2. doi:10.1016/S0022-3476(67)80005-2. [DOI] [PubMed] [Google Scholar]
- 35.Edelmann CM, Soriano JR, Boichis H, Gruskin AB, Acosta MI. Renal bicarbonate reabsorption and hydrogen ion excretion in normal infants. J Clin Invest. 1967;46:1309–1317. doi: 10.1172/JCI105623. doi:10.1172/JCI105623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.McSherry E, Sebastian A, Morris RC., Jr Renal tubular acidosis in infants: the several kinds, including bicarbonate-wasting, classic renal tubular acidosis. J Clin Invest. 1972;51:499–514. doi: 10.1172/JCI106838. doi:10.1172/JCI106838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Edelman CMJR, Rodriguez-Soriano J, Boichis H. An isolated defect in renal bicarbonate reabsorption as a cause of hyperchloremic acidosis. J Pediatr. 1965;67:946. [Google Scholar]
- 38.Batlle DC, Chan YL. Effect of l-arginine on renal tubular bicarbonate reabsorption by the rat kidney. Miner Electr Metab. 1989;15:187–194. [PubMed] [Google Scholar]
- 39.Roth KS, Foreman JW, Segal S. The Fanconi syndrome and mechanisms of tubular transport dysfunction. Kidney Int. 1981;20:705–716. doi: 10.1038/ki.1981.200. doi:10.1038/ki.1981.200. [DOI] [PubMed] [Google Scholar]
- 40.Morris RCJ, Sebastian A. Renal tubular acidosis and the Fanconi syndrome. In: Stanbury JB, Wyngaarden JB, Frederickson DS, editors. The Metabolic Basis of Inherited Disease. New York: 1983. McGraw-Hill, [Google Scholar]
- 41.Batlle DC. Renal tubular acidosis. In: Seldin DW, Giebisch G, editors. The Regulation of Acid–Base Balance. New York: 1989. pp. 353–390. Raven Press, [Google Scholar]
- 42.Laing CM, Toye AM, Capasso G, Unwin RJ. Renal tubular acidosis: developments in our understanding of the molecular basis. Int J Biochem Cell Biol. 2005;37:1151–1161. doi: 10.1016/j.biocel.2005.01.002. doi:10.1016/j.biocel.2005.01.002. [DOI] [PubMed] [Google Scholar]
- 43.Borensztein P, Laghmani K, Froissard M, Paillard M. Molecular aspects of Na+/H+ exchange in the renal tubule: localization and adaptation to the acid–base state. Nephrologie. 1996;17:377–381. [PubMed] [Google Scholar]
- 44.Zhu Q, Kao L, Azimov R, et al. Topological location and structural importance of the NBCe1-A residues mutated in proximal renal tubular acidosis. J Biol Chem. 2010;285:13416–13426. doi: 10.1074/jbc.M109.093286. doi:10.1074/jbc.M109.093286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Igarashi T, Inatomi J, Sekine T, et al. Novel nonsense mutation in the Na+/HCO3− cotransporter gene (SLC4A4) in a patient with permanent isolated proximal renal tubular acidosis and bilateral glaucoma. J Am Soc Nephrol. 2001;12:713–718. doi: 10.1681/ASN.V124713. [DOI] [PubMed] [Google Scholar]
- 46.Horita S, Yamada H, Inatomi J, et al. Functional analysis of NBC1 mutants associated with proximal renal tubular acidosis and ocular abnormalities. J Am Soc Nephrol. 2005;16:2270–2278. doi: 10.1681/ASN.2004080667. doi:10.1681/ASN.2004080667. [DOI] [PubMed] [Google Scholar]
- 47.Suzuki M, Vaisbich MH, Yamada H, et al. Functional analysis of a novel missense NBC1 mutation and of other mutations causing proximal renal tubular acidosis. Pflugers Arch. 2008;455:583–593. doi: 10.1007/s00424-007-0319-y. doi:10.1007/s00424-007-0319-y. [DOI] [PubMed] [Google Scholar]
- 48.Toye AM, Parker MD, Daly CM, et al. The human NBCe1-A mutant R881C, associated with proximal renal tubular acidosis, retains function but is mistargeted in polarized renal epithelia. Am J Physiol Cell Physiol. 2006;291:C788–C801. doi: 10.1152/ajpcell.00094.2006. doi:10.1152/ajpcell.00094.2006. [DOI] [PubMed] [Google Scholar]
- 49.Li HC, Szigligeti P, Worrell RT, Matthews JB, Conforti L, Soleimani M. Missense mutations in Na+:HCO3− cotransporter NBC1 show abnormal trafficking in polarized kidney cells: a basis of proximal renal tubular acidosis. Am J Physiol Renal Physiol. 2005;289:F61–F71. doi: 10.1152/ajprenal.00032.2005. doi:10.1152/ajprenal.00032.2005. [DOI] [PubMed] [Google Scholar]
- 50.Aramaki S, Yoshida I, Yoshino M, et al. Carbonic anhydrase II deficiency in three unrelated Japanese patients. J Inherit Metab Dis. 1993;16:982–990. doi: 10.1007/BF00711514. doi:10.1007/BF00711514. [DOI] [PubMed] [Google Scholar]
- 51.Hu PY, Ernst AR, Sly WS, Venta PJ, Skaggs LA, Tashian RE. Carbonic anhydrase II deficiency: single-base deletion in exon 7 is the predominant mutation in Caribbean Hispanic patients. Am J Hum Genet. 1994;54:602–608. [PMC free article] [PubMed] [Google Scholar]
- 52.Hu PY, Lim EJ, Ciccolella J, Strisciuglio P, Sly WS. Seven novel mutations in carbonic anhydrase II deficiency syndrome identified by SSCP and direct sequencing analysis. Hum Mutat. 1997;9:383–387. doi: 10.1002/(SICI)1098-1004(1997)9:5<383::AID-HUMU1>3.0.CO;2-5. doi:10.1002/(SICI)1098-1004(1997)9:5<383::AID-HUMU1>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 53.Fathallah DM, Bejaoui M, Sly WS, Lakhoua R, Dellagi K. A unique mutation underlying carbonic anhydrase II deficiency syndrome in patients of Arab descent. Hum Genet. 1994;94:581–582. doi: 10.1007/BF00211035. [DOI] [PubMed] [Google Scholar]
- 54.Hu PY, Roth DE, Skaggs LA, et al. A splice junction mutation in intron 2 of the carbonic anhydrase II gene of osteopetrosis patients from Arabic countries. Hum Mutat. 1992;1:288–292. doi: 10.1002/humu.1380010404. doi:10.1002/humu.1380010404. [DOI] [PubMed] [Google Scholar]
- 55.Whyte MP, Murphy WA, Fallon MD, et al. Osteopetrosis, renal tubular acidosis and basal ganglia calcification in three sisters. Am J Med. 1980;69:64–74. doi: 10.1016/0002-9343(80)90501-x. doi:10.1016/0002-9343(80)90501-X. [DOI] [PubMed] [Google Scholar]
- 56.Strisciuglio P, Hu PY, Lim EJ, Ciccolella J, Sly WS. Clinical and molecular heterogeneity in carbonic anhydrase II deficiency and prenatal diagnosis in an Italian family. J Pediatr. 1998;132:717–720. doi: 10.1016/s0022-3476(98)70367-1. doi:10.1016/S0022-3476(98)70367-1. [DOI] [PubMed] [Google Scholar]
- 57.Gawenis LR, Bradford EM, Prasad V, et al. Colonic anion secretory defects and metabolic acidosis in mice lacking the NBC1 Na+/HCO3− cotransporter. J Biol Chem. 2007;282:9042–9052. doi: 10.1074/jbc.M607041200. doi:10.1074/jbc.M607041200. [DOI] [PubMed] [Google Scholar]
- 58.Nakamura S, Amlal H, Schultheis PJ, Galla JH, Shull GE, Soleimani M. HCO3− reabsorption in renal collecting duct of NHE-3-deficient mouse: a compensatory response. Am J Physiol. 1999;276:F914–F921. doi: 10.1152/ajprenal.1999.276.6.F914. [DOI] [PubMed] [Google Scholar]
- 59.Barriere H, Belfodil R, Rubera I, et al. Role of TASK2 potassium channels regarding volume regulation in primary cultures of mouse proximal tubules. J Gen Physiol. 2003;122:177–190. doi: 10.1085/jgp.200308820. doi:10.1085/jgp.200308820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Warth R, Barriere H, Meneton P, et al. Proximal renal tubular acidosis in TASK2 K+ channel-deficient mice reveals a mechanism for stabilizing bicarbonate transport. Proc Natl Acad Sci USA. 2004;101:8215–8220. doi: 10.1073/pnas.0400081101. doi:10.1073/pnas.0400081101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Nevo N, Chol M, Bailleux A, et al. Renal phenotype of the cystinosis mouse model is dependent upon genetic background. Nephrol Dial Transplant. 25:1059–1066. doi: 10.1093/ndt/gfp553. doi:10.1093/ndt/gfp553. [DOI] [PubMed] [Google Scholar]
- 62.Pontoglio M, Prie D, Cheret C, et al. HNF1alpha controls renal glucose reabsorption in mouse and man. EMBO Rep. 2000;1:359–365. doi: 10.1093/embo-reports/kvd071. doi:10.1093/embo-reports/kvd071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Cheret C, Doyen A, Yaniv M, Pontoglio M. Hepatocyte nuclear factor 1 alpha controls renal expression of the Npt1-Npt4 anionic transporter locus. J Mol Biol. 2002;322:929–941. doi: 10.1016/s0022-2836(02)00816-1. doi:10.1016/S0022-2836(02)00816-1. [DOI] [PubMed] [Google Scholar]
- 64.Lowe CU, Terrey M, Mac LE. Organic-aciduria, decreased renal ammonia production, hydrophthalmos, and mental retardation; a clinical entity. AMA Am J Dis Child. 1952;83:164–184. doi: 10.1001/archpedi.1952.02040060030004. [DOI] [PubMed] [Google Scholar]
- 65.Attree O, Olivos IM, Okabe I, et al. The Lowe's oculocerebrorenal syndrome gene encodes a protein highly homologous to inositol polyphosphate-5-phosphatase. Nature. 1992;358:239–242. doi: 10.1038/358239a0. doi:10.1038/358239a0. [DOI] [PubMed] [Google Scholar]
- 66.Wrong OM, Norden AG, Feest TG. Dent's disease; a familial proximal renal tubular syndrome with low-molecular-weight proteinuria, hypercalciuria, nephrocalcinosis, metabolic bone disease, progressive renal failure and a marked male predominance. QJM. 1994;87:473–493. [PubMed] [Google Scholar]
- 67.Lloyd SE, Pearce SH, Fisher SE, et al. A common molecular basis for three inherited kidney stone diseases. Nature. 1996;379:445–449. doi: 10.1038/379445a0. doi:10.1038/379445a0. [DOI] [PubMed] [Google Scholar]
- 68.Hoopes RR, Jr, Shrimpton AE, Knohl SJ, et al. Dent disease with mutations in OCRL1. Am J Hum Genet. 2005;76:260–267. doi: 10.1086/427887. doi:10.1086/427887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Vilasi A, Cutillas PR, Maher AD, et al. Combined proteomic and metabonomic studies in three genetic forms of the renal Fanconi syndrome. Am J Physiol Renal Physiol. 2007;293:F456–F467. doi: 10.1152/ajprenal.00095.2007. doi:10.1152/ajprenal.00095.2007. [DOI] [PubMed] [Google Scholar]
- 70.Manz F, Bickel H, Brodehl J, et al. Fanconi–Bickel syndrome. Pediatr Nephrol. 1987;1:509–518. doi: 10.1007/BF00849262. doi:10.1007/BF00849262. [DOI] [PubMed] [Google Scholar]
- 71.Karlmark B, Agerup B, Wistrand PJ. Renal proximal tubular acidification. Role of brush-border and cytoplasmic carbonic anhydrase. Acta Physiol Scand. 1979;106:145–150. doi: 10.1111/j.1748-1716.1979.tb06383.x. doi:10.1111/j.1748-1716.1979.tb06383.x. [DOI] [PubMed] [Google Scholar]
- 72.Schwartz GJ. Physiology and molecular biology of renal carbonic anhydrase. J Nephrol. 2002;15:S61–S74. [PubMed] [Google Scholar]
- 73.Winum JY, Poulsen SA, Supuran CT. Therapeutic applications of glycosidic carbonic anhydrase inhibitors. Med Res Rev. 2009;29:419–435. doi: 10.1002/med.20141. doi:10.1002/med.20141. [DOI] [PubMed] [Google Scholar]
- 74.Supuran CT, Scozzafava A. Benzolamide is not a membrane-impermeant carbonic anhydrase inhibitor. J Enzyme Inhib Med Chem. 2004;19:269–273. doi: 10.1080/14756360410001689559. doi:10.1080/14756360410001689559. [DOI] [PubMed] [Google Scholar]
- 75.Keilani T, Segal R, Esparaz D, Winchell G, Tanaka W, Connor J, Lippa E, Levine B, Batlle D. Effect of Dorzolamide, a novel topical carbonic anhydrase inhibitor, on urinary acidification in subjects with moderate renal impairment (clinical research meeting) J Investig Med. 1995 [Google Scholar]
- 76.Voute PA, van den Berg H, Behrendt H, Michiels E, de Kraker J. Ifosfamide in the treatment of pediatric malignancies. Semin Oncol. 1996;23:8–11. [PubMed] [Google Scholar]
- 77.Bokemeyer C, Harstrick A, Beyer J, et al. The use of dose-intensified chemotherapy in the treatment of metastatic nonseminomatous testicular germ cell tumors. German Testicular Cancer Study Group. Semin Oncol. 1998;25:24–32. discussion 45-8. [PubMed] [Google Scholar]
- 78.Rossi R, Pleyer J, Schafers P, et al. Development of ifosfamide-induced nephrotoxicity: prospective follow-up in 75 patients. Med Pediatr Oncol. 1999;32:177–182. doi: 10.1002/(sici)1096-911x(199903)32:3<177::aid-mpo3>3.0.co;2-h. doi:10.1002/(SICI)1096-911X(199903)32:3<177::AID-MPO3>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 79.Loebstein R, Atanackovic G, Bishai R, et al. Risk factors for long-term outcome of ifosfamide-induced nephrotoxicity in children. J Clin Pharmacol. 1999;39:454–461. [PubMed] [Google Scholar]
- 80.Oberlin O, Fawaz O, Rey A, et al. Long-term evaluation of Ifosfamide-related nephrotoxicity in children. J Clin Oncol. 2009;27:5350–5355. doi: 10.1200/JCO.2008.17.5257. doi:10.1200/JCO.2008.17.5257. [DOI] [PubMed] [Google Scholar]
- 81.Stohr W, Paulides M, Bielack S, et al. Ifosfamide-induced nephrotoxicity in 593 sarcoma patients: a report from the Late Effects Surveillance System. Pediatr Blood Cancer. 2007;48:447–452. doi: 10.1002/pbc.20858. doi:10.1002/pbc.20858. [DOI] [PubMed] [Google Scholar]
- 82.Negro A, Regolisti G, Perazzoli F, Davoli S, Sani C, Rossi E. Ifosfamide-induced renal Fanconi syndrome with associated nephrogenic diabetes insipidus in an adult patient. Nephrol Dial Transplant. 1998;13:1547–1549. doi: 10.1093/ndt/13.6.1547. doi:10.1093/ndt/13.6.1547. [DOI] [PubMed] [Google Scholar]
- 83.Beckwith C, Flaharty KK, Cheung AK, Beatty PG. Fanconi's syndrome due to ifosfamide. Bone Marrow Transplant. 1993;11:71–73. [PubMed] [Google Scholar]
- 84.Farry JK, Flombaum CD, Latcha S. Long term renal toxicity of ifosfamide in adult patients—5 year data. Eur J Cancer. 2012;48:1326–1331. doi: 10.1016/j.ejca.2012.03.009. doi:10.1016/j.ejca.2012.03.009. [DOI] [PubMed] [Google Scholar]
- 85.Kollataj B, Zajaczkowska M, Katski K, et al. [Acquired Fanconi-de Toni-Debre syndrome due to therapy for Ewing's sarcoma in 5-years old boy] Przegl Lek. 2006;63:220–222. [PubMed] [Google Scholar]
- 86.Rogowska E, Wozniak W. [Nephrotoxicity of ifosfamide with special reference to Fanconi Syndrome] Med Wieku Rozwoj. 2004;8:289–295. [PubMed] [Google Scholar]
- 87.Nissim I, Horyn O, Daikhin Y, Luhovyy B, Phillips PC, Yudkoff M. Ifosfamide-induced nephrotoxicity: mechanism and prevention. Cancer Res. 2006;66:7824–7831. doi: 10.1158/0008-5472.CAN-06-1043. doi:10.1158/0008-5472.CAN-06-1043. [DOI] [PubMed] [Google Scholar]
- 88.Yaseen Z, Michoudet C, Baverel G, Dubourg L. Mechanisms of the ifosfamide-induced inhibition of endocytosis in the rat proximal kidney tubule. Arch Toxicol. 2008;82:607–614. doi: 10.1007/s00204-007-0275-5. doi:10.1007/s00204-007-0275-5. [DOI] [PubMed] [Google Scholar]
- 89.Linch M, Cunningham D, Mingo O, Stiles A, Farquhar-Smith WP. Renal tubular acidosis due to oxaliplatin. Ann Oncol. 2007;18:805–806. doi: 10.1093/annonc/mdm080. doi:10.1093/annonc/mdm080. [DOI] [PubMed] [Google Scholar]
- 90.Negro A, Grasselli C, Galli P. Oxaliplatin-induced proximal renal tubular acidosis. Intern Emerg Med. 2010;5:267–268. doi: 10.1007/s11739-009-0331-7. doi:10.1007/s11739-009-0331-7. [DOI] [PubMed] [Google Scholar]
- 91.Cachat F, Nenadov-Beck M, Guignard JP. Occurrence of an acute Fanconi syndrome following cisplatin chemotherapy. Med Pediatr Oncol. 1998;31:40–41. doi: 10.1002/(sici)1096-911x(199807)31:1<40::aid-mpo11>3.0.co;2-6. doi:10.1002/(SICI)1096-911X(199807)31:1<40::AID-MPO11>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 92.Portilla D, Li S, Nagothu KK, et al. Metabolomic study of cisplatin-induced nephrotoxicity. Kidney Int. 2006;69:2194–2204. doi: 10.1038/sj.ki.5000433. doi:10.1038/sj.ki.5000433. [DOI] [PubMed] [Google Scholar]
- 93.Nagothu KK, Bhatt R, Kaushal GP, Portilla D. Fibrate prevents cisplatin-induced proximal tubule cell death. Kidney Int. 2005;68:2680–2693. doi: 10.1111/j.1523-1755.2005.00739.x. doi:10.1111/j.1523-1755.2005.00739.x. [DOI] [PubMed] [Google Scholar]
- 94.Xu EY, Perlina A, Vu H, et al. Integrated pathway analysis of rat urine metabolic profiles and kidney transcriptomic profiles to elucidate the systems toxicology of model nephrotoxicants. Chem Res Toxicol. 2008;21:1548–1561. doi: 10.1021/tx800061w. doi:10.1021/tx800061w. [DOI] [PubMed] [Google Scholar]
- 95.Kuba K, Imai Y, Ohto-Nakanishi T, Penninger JM. Trilogy of ACE2: a peptidase in the renin–angiotensin system, a SARS receptor, and a partner for amino acid transporters. Pharmacol Ther. 2010;128:119–128. doi: 10.1016/j.pharmthera.2010.06.003. doi:10.1016/j.pharmthera.2010.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Earle KE, Seneviratne T, Shaker J, Shoback D. Fanconi's syndrome in HIV+ adults: report of three cases and literature review. J Bone Miner Res. 2004;19:714–721. doi: 10.1359/jbmr.2004.19.5.714. doi:10.1359/jbmr.2004.19.5.714. [DOI] [PubMed] [Google Scholar]
- 97.Nelson M, Azwa A, Sokwala A, Harania RS, Stebbing J. Fanconi syndrome and lactic acidosis associated with stavudine and lamivudine therapy. AIDS. 2008;22:1374–1376. doi: 10.1097/QAD.0b013e328303be50. doi:10.1097/QAD.0b013e328303be50. [DOI] [PubMed] [Google Scholar]
- 98.Gupta SK. Tenofovir-associated Fanconi syndrome: review of the FDA adverse event reporting system. AIDS Patient Care STDS. 2008;22:99–103. doi: 10.1089/apc.2007.0052. doi:10.1089/apc.2007.0052. [DOI] [PubMed] [Google Scholar]
- 99.Kearney BP, Mathias A, Mittan A, Sayre J, Ebrahimi R, Cheng AK. Pharmacokinetics and safety of tenofovir disoproxil fumarate on coadministration with lopinavir/ritonavir. J Acquir Immune Defic Syndr. 2006;43:278–283. doi: 10.1097/01.qai.0000243103.03265.2b. doi:10.1097/01.qai.0000243103.03265.2b. [DOI] [PubMed] [Google Scholar]
- 100.Gupta SK, Eustace JA, Winston JA, et al. Guidelines for the management of chronic kidney disease in HIV-infected patients: recommendations of the HIV Medicine Association of the Infectious Diseases Society of America. Clin Infect Dis. 2005;40:1559–1585. doi: 10.1086/430257. doi:10.1086/430257. [DOI] [PubMed] [Google Scholar]
- 101.Hall AM, Hendry BM, Nitsch D, Connolly JO. Tenofovir-associated kidney toxicity in HIV-infected patients: a review of the evidence. Am J Kidney Dis. 2011;57:773–780. doi: 10.1053/j.ajkd.2011.01.022. doi:10.1053/j.ajkd.2011.01.022. [DOI] [PubMed] [Google Scholar]
- 102.Imaoka T, Kusuhara H, Adachi M, Schuetz JD, Takeuchi K, Sugiyama Y. Functional involvement of multidrug resistance-associated protein 4 (MRP4/ABCC4) in the renal elimination of the antiviral drugs adefovir and tenofovir. Mol Pharmacol. 2007;71:619–627. doi: 10.1124/mol.106.028233. doi:10.1124/mol.106.028233. [DOI] [PubMed] [Google Scholar]
- 103.Rodriguez-Novoa S, Labarga P, Soriano V, et al. Predictors of kidney tubular dysfunction in HIV-infected patients treated with tenofovir: a pharmacogenetic study. Clin Infect Dis. 2009;48:e108–e116. doi: 10.1086/598507. doi:10.1086/598507. [DOI] [PubMed] [Google Scholar]
- 104.D'Ythurbide G, Goujard C, Mechai F, Blanc A, Charpentier B, Snanoudj R. Fanconi syndrome and nephrogenic diabetes insipidus associated with didanosine therapy in HIV infection: a case report and literature review. Nephrol Dial Transplant. 2007;22:3656–3659. doi: 10.1093/ndt/gfm467. doi:10.1093/ndt/gfm467. [DOI] [PubMed] [Google Scholar]
- 105.Crowther MA, Callaghan W, Hodsman AB, Mackie ID. Dideoxyinosine-associated nephrotoxicity. AIDS. 1993;7:131–132. doi:10.1097/00002030-199301000-00024. [PubMed] [Google Scholar]
- 106.Izzedine H, Launay-Vacher V, Deray G. Fanconi syndrome associated with didanosine therapy. AIDS. 2005;19:844–845. doi: 10.1097/01.aids.0000168985.05209.b8. doi:10.1097/01.aids.0000168985.05209.b8. [DOI] [PubMed] [Google Scholar]
- 107.Mirza N, Marson AG, Pirmohamed M. Effect of topiramate on acid-base balance: extent, mechanism and effects. Br J Clin Pharmacol. 2009;68:655–661. doi: 10.1111/j.1365-2125.2009.03521.x. doi:10.1111/j.1365-2125.2009.03521.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Ozer Y, Altunkaya H. Topiramate induced metabolic acidosis. Anaesthesia. 2004;59:830. doi: 10.1111/j.1365-2044.2004.03884.x. doi:10.1111/j.1365-2044.2004.03884.x. [DOI] [PubMed] [Google Scholar]
- 109.Philippi H, Boor R, Reitter B. Topiramate and metabolic acidosis in infants and toddlers. Epilepsia. 2002;43:744–747. doi: 10.1046/j.1528-1157.2002.37201.x. doi:10.1046/j.1528-1157.2002.37201.x. [DOI] [PubMed] [Google Scholar]
- 110.Sacre A, Jouret F, Manicourt D, Devuyst O. Topiramate induces type 3 renal tubular acidosis by inhibiting renal carbonic anhydrase. Nephrol Dial Transplant. 2006;21:2995–2996. doi: 10.1093/ndt/gfl251. doi:10.1093/ndt/gfl251. [DOI] [PubMed] [Google Scholar]
- 111.Recacha R, Costanzo MJ, Maryanoff BE, Chattopadhyay D. Crystal structure of human carbonic anhydrase II complexed with an anti-convulsant sugar sulphamate. Biochem J. 2002;361:437–441. doi: 10.1042/0264-6021:3610437. doi:10.1042/0264-6021:3610437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Maryanoff BE, McComsey DF, Costanzo MJ, Hochman C, Smith-Swintosky V, Shank RP. Comparison of sulfamate and sulfamide groups for the inhibition of carbonic anhydrase-II by using topiramate as a structural platform. J Med Chem. 2005;48:1941–1947. doi: 10.1021/jm040124c. doi:10.1021/jm040124c. [DOI] [PubMed] [Google Scholar]
- 113.Knorr M, Schaper J, Harjes M, Mayatepek E, Rosenbaum T. Fanconi syndrome caused by antiepileptic therapy with valproic acid. Epilepsia. 2004;45:868–871. doi: 10.1111/j.0013-9580.2004.05504.x. doi:10.1111/j.0013-9580.2004.05504.x. [DOI] [PubMed] [Google Scholar]
- 114.Endo A, Fujita Y, Fuchigami T, Takahashi S, Mugishima H. Fanconi syndrome caused by valproic acid. Pediatr Neurol. 42:287–290. doi: 10.1016/j.pediatrneurol.2009.12.003. doi:10.1016/j.pediatrneurol.2009.12.003. [DOI] [PubMed] [Google Scholar]
- 115.Hawkins E, Brewer E. Renal toxicity induced by valproic acid (Depakene) Pediatr Pathol. 1993;13:863–868. doi: 10.3109/15513819309048273. doi:10.3109/15513819309048273. [DOI] [PubMed] [Google Scholar]
- 116.Lenoir GR, Perignon JL, Gubler MC, Broyer M. Valproic acid: a possible cause of proximal tubular renal syndrome. J Pediatr. 1981;98:503–504. doi: 10.1016/s0022-3476(81)80736-6. [DOI] [PubMed] [Google Scholar]
- 117.Ghiculescu RA, Kubler PA. Aminoglycoside-associated Fanconi syndrome. Am J Kidney Dis. 2006;48:e89–e93. doi: 10.1053/j.ajkd.2006.08.009. doi:10.1053/j.ajkd.2006.08.009. [DOI] [PubMed] [Google Scholar]
- 118.Gainza FJ, Minguela JI, Lampreabe I. Aminoglycoside-associated Fanconi's syndrome: an underrecognized entity. Nephron. 1997;77:205–211. doi: 10.1159/000190274. doi:10.1159/000190274. [DOI] [PubMed] [Google Scholar]
- 119.Melnick JZ, Baum M, Thompson JR. Aminoglycoside-induced Fanconi's syndrome. Am J Kidney Dis. 1994;23:118–122. doi: 10.1016/s0272-6386(12)80820-1. [DOI] [PubMed] [Google Scholar]
- 120.Lenz EM, Bright J, Knight R, et al. Metabonomics with 1H-NMR spectroscopy and liquid chromatography-mass spectrometry applied to the investigation of metabolic changes caused by gentamicin-induced nephrotoxicity in the rat. Biomarkers. 2005;10:173–187. doi: 10.1080/13547500500094034. doi:10.1080/13547500500094034. [DOI] [PubMed] [Google Scholar]
- 121.Fleck C, Schwertfeger M, Taylor PM. Regulation of renal amino acid (AA) transport by hormones, drugs and xenobiotics - a review. Amino Acids. 2003;24:347–374. doi: 10.1007/s00726-002-0316-6. doi:10.1007/s00726-002-0316-6. [DOI] [PubMed] [Google Scholar]
- 122.Vittecoq D, Dumitrescu L, Beaufils H, Deray G. Fanconi syndrome associated with cidofovir therapy. Antimicrob Agents Chemother. 1997;41:1846. doi: 10.1128/aac.41.8.1846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Shaikh A, Wiisanen ME, Gunderson HD, Leung N. Acquired Fanconi syndrome after treatment with capecitabine, irinotecan, and bevacizumab. Ann Pharmacother. 2009;43:1370–1373. doi: 10.1345/aph.1M120. doi:10.1345/aph.1M120. [DOI] [PubMed] [Google Scholar]
- 124.Batlle D, Hays S, Foley R, Chan Y, Arruda JA, Kurtzman NA. Proximal renal tubular acidosis and hypophosphatemia induced by arginine. Adv Exp Med Biol. 1982;151:239–249. doi: 10.1007/978-1-4684-4259-5_30. doi:10.1007/978-1-4684-4259-5_30. [DOI] [PubMed] [Google Scholar]
- 125.Garcia Rodriguez JF, Sanchez-Guisande D, Novoa D, Romero R, Arcocha V. Fanconi syndrome caused by mercury chloride poisoning. Rev Clin Esp. 1989;184:111–112. [PubMed] [Google Scholar]
- 126.Johri N, Jacquillet G, Unwin R. Heavy metal poisoning: the effects of cadmium on the kidney. Biometals. 2010;23:783–792. doi: 10.1007/s10534-010-9328-y. doi:10.1007/s10534-010-9328-y. [DOI] [PubMed] [Google Scholar]
- 127.Taylor HC, Elbadawy EH. Renal tubular acidosis type 2 with Fanconi's syndrome, osteomalacia, osteoporosis, and secondary hyperaldosteronism in an adult consequent to vitamin D and calcium deficiency: effect of vitamin D and calcium citrate therapy. Endocr Pract. 2006;12:559–567. doi: 10.4158/EP.12.5.559. [DOI] [PubMed] [Google Scholar]
- 128.Guignard JP, Torrado A. Proximal renal tubular acidosis in vitamin D deficiency rickets. Acta Paediatr Scand. 1973;62:543–546. doi: 10.1111/j.1651-2227.1973.tb08154.x. doi:10.1111/j.1651-2227.1973.tb08154.x. [DOI] [PubMed] [Google Scholar]
- 129.Vainsel M, Manderlier T, Vis HL. Proximal renal tubular acidosis in vitamin D deficiency rickets. Biomedicine. 1975;22:35–40. [PubMed] [Google Scholar]
- 130.Maldonado JE, Velosa JA, Kyle RA, Wagoner RD, Holley KE, Salassa RM. Fanconi syndrome in adults. A manifestation of a latent form of myeloma. Am J Med. 1975;58:354–364. doi: 10.1016/0002-9343(75)90601-4. doi:10.1016/0002-9343(75)90601-4. [DOI] [PubMed] [Google Scholar]
- 131.Bridoux F, Sirac C, Hugue V, et al. Fanconi's syndrome induced by a monoclonal Vkappa3 light chain in Waldenstrom's macroglobulinemia. Am J Kidney Dis. 2005;45:749–757. doi: 10.1053/j.ajkd.2004.12.020. doi:10.1053/j.ajkd.2004.12.020. [DOI] [PubMed] [Google Scholar]
- 132.Rikitake O, Sakemi T, Yoshikawa Y, Nagano Y, Watanabe T. Adult Fanconi syndrome in primary amyloidosis with lambda light-chain proteinuria. Jpn J Med. 1989;28:523–526. doi: 10.2169/internalmedicine1962.28.523. doi:10.2169/internalmedicine1962.28.523. [DOI] [PubMed] [Google Scholar]
- 133.Batlle DC, Mozes MF, Manaligod J, Arruda JA, Kurtzman NA. The pathogenesis of hyperchloremic metabolic acidosis associated with kidney transplantation. Am J Med. 1981;70:786–796. doi: 10.1016/0002-9343(81)90534-9. doi:10.1016/0002-9343(81)90534-9. [DOI] [PubMed] [Google Scholar]
- 134.Friedman A, Chesney R. Fanconi's syndrome in renal transplantation. Am J Nephrol. 1981;1:45–47. doi: 10.1159/000166488. doi:10.1159/000166488. [DOI] [PubMed] [Google Scholar]
- 135.Hsiao PJ, Wang SC, Wen MC, Diang LK, Lin SH. Fanconi syndrome and CKD in a patient with paroxysmal nocturnal hemoglobinuria and hemosiderosis. Am J Kidney Dis. 55:e1–e5. doi: 10.1053/j.ajkd.2009.07.022. doi:10.1053/j.ajkd.2009.07.022. [DOI] [PubMed] [Google Scholar]
- 136.Brenner RJ, Spring DB, Sebastian A, et al. Incidence of radiographically evident bone disease, nephrocalcinosis, and nephrolithiasis in various types of renal tubular acidosis. N Engl J Med. 1982;307:217–221. doi: 10.1056/NEJM198207223070403. doi:10.1056/NEJM198207223070403. [DOI] [PubMed] [Google Scholar]
- 137.Batlle D, Moorthi KM, Schlueter W, Kurtzman N. Distal renal tubular acidosis and the potassium enigma. Semin Nephrol. 2006;26:471–478. doi: 10.1016/j.semnephrol.2006.12.001. doi:10.1016/j.semnephrol.2006.12.001. [DOI] [PubMed] [Google Scholar]
- 138.Batlle DC, Hizon M, Cohen E, Gutterman C, Gupta R. The use of the urinary anion gap in the diagnosis of hyperchloremic metabolic acidosis. N Engl J Med. 1988;318:594–599. doi: 10.1056/NEJM198803103181002. doi:10.1056/NEJM198803103181002. [DOI] [PubMed] [Google Scholar]
- 139.Brenes LG, Brenes JN, Hernandez MM. Familial proximal renal tubular acidosis. A distinct clinical entity. Am J Med. 1977;63:244–252. doi: 10.1016/0002-9343(77)90238-8. doi:10.1016/0002-9343(77)90238-8. [DOI] [PubMed] [Google Scholar]
- 140.Nash MA, Torrado AD, Greifer I, Spitzer A, Edelmann CM., Jr Renal tubular acidosis in infants and children. Clinical course, response to treatment, and prognosis. J Pediatr. 1972;80:738–748. doi: 10.1016/s0022-3476(72)80124-0. doi:10.1016/S0022-3476(72)80124-0. [DOI] [PubMed] [Google Scholar]
- 141.Brenes LG, Sanchez MI. Impaired urinary ammonium excretion in patients with isolated proximal renal tubular acidosis. J Am Soc Nephrol. 1993;4:1073–1078. doi: 10.1681/ASN.V441073. [DOI] [PubMed] [Google Scholar]
- 142.McSherry E, Morris RC., Jr Attainment and maintenance of normal stature with alkali therapy in infants and children with classic renal tubular acidosis. J Clin Invest. 1978;61:509–527. doi: 10.1172/JCI108962. doi:10.1172/JCI108962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Rampini S, Fanconi A, Illig R, Prader A. Effect of hydrochlorothiazide on proximal renal tubular acidosis in a patient with idiopathic ‘de toni-debre-fanconi syndrome. Helv Paediatr Acta. 1968;23:13–21. [PubMed] [Google Scholar]
- 144.Rodriguez-Soriano J, Edelmann CM., Jr Renal tubular acidosis. Annu Rev Med. 1969;20:363–382. doi: 10.1146/annurev.me.20.020169.002051. doi:10.1146/annurev.me.20.020169.002051. [DOI] [PubMed] [Google Scholar]