

Abstract
Renal hypodysplasia (RHD) is characterized by small and/or disorganized kidneys following abnormal organogenesis. Mutations in several genes have been identified recently to be associated with RHD in humans, including BMP4, a member of the transforming growth factor (TGF)-β family of growth factors. DACH1 has been proposed as a candidate gene for RHD because of its involvement in the EYA-SIX-DACH network of renal developmental genes. Here, we present a patient with renal dysplasia carrying homozygous missense mutations in both BMP4 (p.N150K) and DACH1 (p.R684C). The genotype–phenotype correlation in the family hints at an oligogenic mode of inheritance of the disease in this kindred. Functional analyses of the identified DACH1 mutation in HEK293T cells demonstrated enhanced suppression of the TGF-β pathway suggesting that both mutations could act synergistically in the development of the phenotype in this patient. This finding provides a model for RHD as an oligo-/polygenic disorder and supports a role for DACH1 in the development of RHD in humans.
Keywords: kidney dysplasia, molecular genetics, oligogenic inheritance
Background
Renal hypodysplasia (RHD) is characterized by disturbed renal parenchymal architecture with or without formation of cysts and constitutes a major cause of chronic renal failure in children [1]. A number of genes have been identified to cause hereditary RHD in a subset of patients, including HNF1B (TCF2), PAX2, EYA1 and SIX1 [2]. Generally, mutations in these genes are inherited in an autosomal-dominant manner. However, the majority of cases of non-syndromic RHD occur sporadically or follow non-mendelian inheritance. The latter are thought to be complex, oligogenic or polygenic disorders and identification of disease-causing genes is challenging [3]. Few genes have been identified so far to play a role in non-mendelian RHD, including BMP4, a member of the transforming growth factor (TGF)-β family of growth factors involved in key steps of murine and human nephrogenesis including regulation of ureteric budding and branching morphogenesis [4]. Mutations in BMP4 have been shown to be associated with the development of RHD in humans [5].
DACH1, the human homologue of the Drosophila dachshund gene, encodes a transcription cofactor participating in the EYA-SIX-DACH network of developmental genes regulating cell differentiation during human nephrogenesis [6–8].
Both BMP4 and DACH1 are expressed in the developing murine kidney at the site of mesenchymal–epithelial interaction. While BMP4 is expressed mainly in the mesenchyme surrounding the branching ureter, DACH1 mRNA is expressed in the tip and trunk of the branching ureter as well as the comma-shaped bodies and convoluted tubules [4, 9]. In the EYA-SIX-DACH network, Six1 and Eya1 have been shown to initiate ureteric budding by transcriptional activation of glial cell line derived neurotropic factor, Gdnf (Figure 1) and human mutations in either of the encoding genes lead to Branchio-Oto-Renal (BOR) syndrome. To date, no human mutations in DACH1 have been described and Dach1-deficient mice die postnatally without gross abnormalities of the kidneys or other organs [10]. However, DACH1 expression is down-regulated in human dysplastic kidneys compared with normal kidneys [11] and results of deletion mapping studies have shown DACH1 to be located in a chromosomic region associated with congenital anomalies of the kidney and the urinary tract [12]. We, therefore, believed DACH1 to be an interesting candidate implicated in the development of RHD in humans.
Fig. 1.

Molecular regulation of ureteric budding and branching morphogenesis. Eya1 (indirectly), Six1 and Pax2 (directly) activate transcription of GDNF, initializing budding and consequent branching of the ureter via Ret and Gfra1. In vitro studies have demonstrated a role of Dach1 as a co-repressor or co-activator of Six1, with Eya1 acting as a molecular switch. In vitro Bmp4 inhibits ectopic budding and branching of the ureter by inhibition of Gdnf signalling. In addition to its function in the Six-Dach-Eya network, Dach1 inhibits TGF-β/BMP4 signalling directly in human breast cancer cell lines (dashed line).
Methods
DNA was extracted from peripheral blood leucocytes by standard methods. Screening for sequence alterations was performed by single-strand conformation analysis (SSCA; Multiphor II, Amersham Biosciences, Piscataway, NJ) following individual PCR-amplification of all protein-coding exons and flanking intronic sequences of the DACH1 gene (NCBI RefSeq NG_011849.2). Primers were designed using the Primer3 software (http://frodo.wi.mit.edu/cgi-bin/primer3/primer3_www.cgi). All primer sequences are available upon request. Samples showing altered patterns in SSCA were analysed by bidirectional sequencing using the Big-Dye Terminator Cycle Sequencing Kit (Applied Biosystems, Foster City, USA). The sequencing products were analysed in an Applied Biosystems 3100 Genetic Analyser. Pathogenic mutations were excluded in 200 control chromosomes. Nonsynonymous sequence variants were analysed for functional effects on protein level by multiple prediction tools [13–15]
Patient
The patient was identified as part of a study of genetic variations in renal developmental genes in an unselected cohort of 250 patients with RHD, including patients of the ESCAPE trial (EU QLGI-CT-2002-00908). Pathogenic mutations or deletions in HNF1B and PKHD1 have been excluded in our index patient I.S. prior to the study of BMP4 and DACH1. I.S. is a non-ESCAPE patient, who is in medical care at the University-Children's Hospital Heidelberg, Germany. The study was approved by the local ethics committee and written informed consent was obtained from the family.
Plasmid construction
The expression plasmids that include an N-terminal FLAG peptide for DACH1 and DACH1-DS domain deleted (ΔDS) were constructed as described by Wu et al. [16]. Flag-DACH1-p.R684C was generated by site-directed mutagenesis (GenScript, Piscataway, NJ, USA) according to the manufacturer's protocol. The reporter plasmids 3-TP Lux and SBE-4 Luc were described in Wu et al. [16]. AP-1 Luc, c-Jun Luc and c-Fos Luc have also been described previously [17].
Cell culture, DNA transfection and luciferase assays
Cell Culture, DNA Transfection and Luciferase Assays were performed as described previously [17]. HEK293T and MDA-MB-231 cell lines were cultured in DMEM supplemented with 10% fetal calf serum and 1% penicillin/1% streptomycin. Transfections were performed using Superfect transfection reagent (Qiagen, Valencia, CA, USA) according to the manufacturer's protocol. For reporter gene assays, cells were transiently transfected with an appropriate combination of the reporter, expression plasmids and control vector. In some experiments, cells were serum-starved for 36 h and stimulated with or without TGF-β (1 ng/mL) for 12 h before collecting cells for luciferase assay. Transfection efficiency was normalized by cotransfection with 0.2 mg of pRL-CMV plasmid (Promega, Madison, WI, USA) and was measured with the Promega dual-luciferase reporter assay system according to the manufacturer's protocol. Statistical analyses were performed using Student's t test.
Case report
I.S. was born at term after an uneventful pregnancy. At the age of 4 years, the boy presented with anaemia, dystrophy and renal insufficiency. Ultrasound revealed enlarged kidneys (>97.P) with bilateral multiple cyst formation and hyperechogenic parenchyma. Kidney function deteriorated over a period of 1 year leading to end-stage renal disease at the age of 5 4/12 years. Seven months later, the patient received an allogenic kidney graft. Besides recurrent urinary tract infections and a CMV infection, there were no major complications. The patient is currently 19 years old and has a good graft function with a creatinine baseline of 0.8 mg/dL (71 µmol/L). Apart from congenital unilateral ptosis, no extrarenal anomalies were identified.
The patient is the first child of consanguineous parents. The parents are double cousins, sharing both sets of grandparents as ancestors. A second cousin had died shortly after birth from undefined cystic kidney disease. No other family member was known to be affected by renal disease. However, ultrasound examination revealed bilateral medullary renal cysts in the father without impairment of renal function. The patient's mother (III-4), his uncles (III-2, III-3) as well as all four siblings had a normal renal ultrasound and normal kidney function.
The patient was found to carry two constitutional homozygous missense mutations in BMP4 and DACH1. The BMP4 mutation p.N150K has been described previously in the same patient and has been shown to impair protein function in a zebrafish overexpression assay [5]. In addition to the mutation in BMP4, our index patient carries a homozygous missense mutation in exon 10 of the DACH1 gene, (NM_080759.4:c.2050C> T; NP_542937.2:p.R684C) leading to an amino acid substitution of arginine to cysteine at position 684 of the DACH1 protein. This amino acid position is highly conserved among vertebrates and zebrafish, and p.R684C is not annotated as an SNP in human genetic variation databases (www.ncbi.nlm.nih.gov/projects/SNP, www.1000genomes.org). In silico analysis of DACH1-p.R684C was performed using multiple prediction tools [13–15]. All the algorithms predicted an alteration of protein function with high scores.
Genotyping of the family revealed both alterations to be germline mutations. The parents as well as another sibling, one uncle and one grandmother carried the heterozygous BMP4 mutation (Figure 2). Two siblings, both parents, two grandparents and one uncle were carriers of the heterozygous sequence variation in DACH1. As both paternal grandparents are heterozygous for DACH1 p.R684C consanguinitiy is possible in this marriage. One uncle (III-2) was homozygous for p.R684C in DACH1 and one sister (IV-2) homozygous for p.N150K in BMP4. However, no other family member except the index patient I.S. was affected by both mutations in a double homozygous state.
Fig. 2.

Pedigree of the family showing a high degree of consanguinity. The severely affected index patient (IV-1) carries homozygous mutations in BMP4 and DACH1. Two relatives carrying single homozygous mutations are clinically unaffected (III-2, IV-2) while one of the double heterozygous mutation carriers (III-5, father to the index patient) shows a mild phenotype. The genotypes of the deceased second cousins are unknown. For identification of individuals roman numerals are used to indicate the generation followed by arabic numerals for each individual. Individuals are consecutively numbered starting from the left in each generation. Arabic numbers in the pedigree itself indicate multiple individuals of the same sex and phenotype.
To asses further the functional impact of DACH1-p.R684C, cell culture transfection assays were performed with DACH1 mutants generated by site-directed mutagenesis. Luciferase reporter assays in HEK293T and MDA-231 cells demonstrated similar repression of AP-1, c-Jun and c-Fos promoter activity by wild-type DACH1 and DACH1-p.R684C in response to serum stimulation but enhanced repression of TGF-β signalling by the DACH1-p.R684C mutant compared with wild-type DACH1 (Figure 3E and F). A DACH1 construct deleted for the Dach DNA binding domain served as a negative control (DACH1ΔDS) [18]. TGF-β signalling plays a key role in the inhibition of epithelial cell proliferation, production of extracellular matrix components, regulation of differentiation and cellular apoptosis. TGF-β-induced apoptosis is inhibited by DACH1 [16] and we demonstrate here the enhanced repressor activity of the DACH1-p.R684C mutant.
Fig. 3.
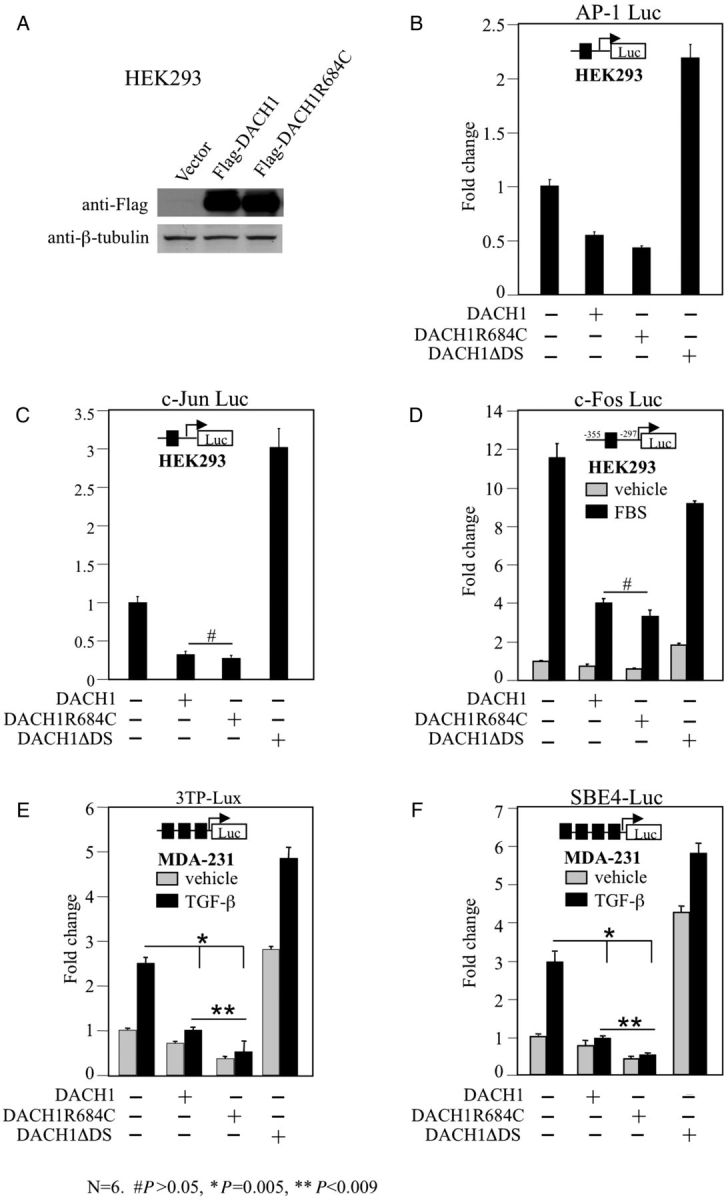
Repression of TGF-β signalling by DACH1-p.R684C. Western blot analysis of HEK293T cells transiently transfected with Flag-tagged DACH1 and DACH1-p.R684C expression plasmids demonstrating equal protein abundance of wild-type and mutant DACH1 (A). Repression levels of AP-1 (B), c-Jun (C) and c-fos promoter (D, in response to serum stimulation) by wild-type DACH1 and DACH1-p.R684C in HEK293T cells. DACH1 protein with deleted DS domain (DACH1ΔDS) served as negative control. Strong repressor activity of DACH1-p. R684C in MDA-231 cells after co-transfection with 3TP-Lux (E) and SBE-4 Luc (F) reporter and stimulation with or without TGF-β for 12 h. DACH1 protein with deleted DS domain (DACH1ΔDS) served as negative control. Data are shown as mean ± SE for n = 6 separate transfections.
Discussion
The patient presented here is carrying double homozygous mutations in two renal developmental genes, BMP4 and DACH1. The two unaffected single homozygous mutation carriers (III-2 and IV-2) practically rule out the possibility of a monogenic recessive trait for either of the genes involved. Strikingly, the index patient is the only family member homozygous for both mutations (Figure 2). This strongly supports the notion that synergistic digenic effects underlie the complex pattern of inheritance in this kindred. An unequivocal digenic tetraallelic mode of inheritance as suggested by Katsanis et al. for a subset of patients with Bardet–Biedl Syndrome [19] cannot be established by this data because of the unknown genotype of the deceased second cousin (III-9) as well as the occurrence of renal cysts in the double heterozygous father (III-5). These findings favour the assumption of BMP4-/DACH1-gene dosage effects and the presence of additional genetic or non-genetic modifiers of the disease in the family.
BMP4 has been shown previously to be associated with renal dysplasia in humans and the mutation p.N150K described in this patient impairs protein function in an experimental setting involving protein overexperession in zebrafish embryos [5]. Moreover, in vitro studies show that the mutation leads to reduced mRNA abundance in cultured human cell lines [20]. Evidence for a role of DACH1 in the development of human renal disease comes from deletion mapping studies [12] and has recently been strengthened by the results of genome-wide association studies. In a population-based study involving 67 093 individuals (including 5807 individuals with chronic renal insufficiency), Köttgen et al. identified a single-nucleotide polymorphism in DACH1 to be associated with reduced kidney function in adulthood, suggesting that nearby common sequence variants or even rare alleles in DACH1 can contribute to the development of renal dysfunction in humans [21]. Furthermore, interactions between the BMP4 and the DACH1 pathways have been demonstrated in vitro. BMP4 utilizes the TGF-ß signal transduction pathway via SMAD4 and Wu et al. have shown that DACH1 antagonizes TGF-β signalling in human breast cancer cell lines by binding SMAD4 [16]. Here we show that the DACH1 mutation p.R684C enhances the potential for suppression of the TGF-β signalling pathway in vitro compared with wild-type DACH1. We, therefore, postulate that the functional BMP4 defect caused by the mutation p.N150K may be aggravated by gain of function in DACH1 leading to further disruption of the TGF-β/BMP4 pathway in this patient. The interaction between the two pathways during kidney development has not been studied extensively so far. BMP4 is expressed in periureteric stroma cells and exerts its effects as a secretory growth factor by binding on Alk3/Alk6-receptors on the surface of ureteric epithelial cells, preventing ureteric budding/branching by inhibiting Gdnf-signalling in these cell lines [4]. Dach1 has also been shown to be expressed in ureteric epithelial cells [9] and could therefore directly influence the key steps in kidney development by inhibiting the TGF-β/BMP4 pathway in this lineage, thus acting as a modifier of BMP4 function.
This finding is in line with the emerging understanding of RHD as an oligo-/polygenic disorder in which the accumulation of multiple subtle mutations or polymorphisms (mutational load) leads to dysfunction of the corresponding developmental programme [22]. Further functional studies as well as supplemental data on human DACH1 genotypes will be required to establish a consistent model for the interaction between BMP4 and DACH1 and to clarify the role of DACH1 for the development of human RHD.
Acknowledgements
Financial support for this study was obtained from the Thyssen Foundation (AZ-10.07.2.173) and The Dietmar Hopp Foundation (S. Weber, F. Schaefer) as well as R01CA70896, R01CA75503, R01CA86072 (R.G. Pestell), The Kimmel Cancer Center NIH Cancer Center Core Grant P30CA56036 (R.G. Pestell), a generous grant from the Dr. Ralph AND Marian C. Falk Medical Research Trust (R.G. Pestell) and the Department of Defense Concept Award W81XWH-11-1-0303 (K. Wu), a grant from The Pennsylvania Department Of Health (R.G. Pestell) and support from The National Natural Science Foundation Of China (81072169, 81172422 TO K. WU). The Department disclaims responsibility for any analysis, interpretations or conclusions. R.G.P. holds owner interests (Value Unknown) for The Submitted US Provisional Application No. 61/292,749 (Georgetown University Ref: 2008-038), ‘methods And Compositions For The Diagnosis, Prognosis And Treatment Of Cancer.’ The authors thank the patient and his family for kindly participating in this study.
References
- 1.Woolf AS. A molecular and genetic view of human renal and urinary tract malformations. Kidney Int. 2000;58:500–512. doi: 10.1046/j.1523-1755.2000.00196.x. doi:10.1046/j.1523-1755.2000.00196.x. [DOI] [PubMed] [Google Scholar]
- 2.Weber S, Moriniere V, Knüppe T, et al. Prevalence of mutations in renal developmental genes in children with renal hypodysplasia: results of the ESCAPE Study. J Am Soc Nephrol. 2006;17:2864–2870. doi: 10.1681/ASN.2006030277. [DOI] [PubMed] [Google Scholar]
- 3.Sanna-Cherchi S, Caridi G, Weng PL, et al. Genetic approaches to human renal agenesis/hypoplasia and dysplasia. Pediatr Nephrol. 2007;22:1675–1684. doi: 10.1007/s00467-007-0479-1. doi:10.1007/s00467-007-0479-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Miyazaki Y, Oshima K, Fogo A, et al. Bone morphogenetic protein 4 regulates the budding site and elongation of the mouse ureter. J Clin Invest. 2000;105:863–873. doi: 10.1172/JCI8256. doi:10.1172/JCI8256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Weber S, Taylor JC, Winyard P, et al. SIX2 and BMP4 mutations associate with anomalous kidney development. J Am Soc Nephrol. 2008;19:891–903. doi: 10.1681/ASN.2006111282. doi:10.1681/ASN.2006111282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ikeda K, Watanabe Y, Ohto H, et al. Molecular interaction and synergistic activation of a promoter by Six, Eya, and Dach proteins mediated through CREB binding protein. Mol Cell Biol. 2002;22:6759–6766. doi: 10.1128/MCB.22.19.6759-6766.2002. doi:10.1128/MCB.22.19.6759-6766.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li X, Oghi KA, Zhang J, et al. Eya protein phosphatase activity regulates Six1-Dach-Eya transcriptional effects in mammalian organogenesis. Nature. 2003;426:247–254. doi: 10.1038/nature02083. doi:10.1038/nature02083. [DOI] [PubMed] [Google Scholar]
- 8.Popov VM, Wu K, Powel MJ, et al. The Dachshund gene in development and hormone-responsive tumorigenesis. Trends Endocrinol Metab. 2010;21:41–49. doi: 10.1016/j.tem.2009.08.002. doi:10.1016/j.str.2010.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ayres JA, Shum L, Akarsu AN, et al. DACH: genomic characterization, evaluation as a candidate for postaxial polydactyly type A2, and developmental expression pattern of the mouse homologue. Genomics. 2001;77:18–26. doi: 10.1006/geno.2001.6618. doi:10.1006/geno.2001.6618. [DOI] [PubMed] [Google Scholar]
- 10.Davis RJ, Shen W, Sandler YI, et al. Dach1 mutant mice bear no gross abnormalities in eye, limb, and brain development and exhibit postnatal lethality. Mol Cell Biol. 2001;21:1484–1490. doi: 10.1128/MCB.21.5.1484-1490.2001. doi:10.1128/MCB.21.5.1484-1490.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jain S, Suarez AA, McGuire J, et al. Expression profiles of congenital renal dysplasia reveal new insights into renal development and disease. Pediatr Nephrol. 2007;22:962–974. doi: 10.1007/s00467-007-0466-6. doi:10.1007/s00467-007-0466-6. [DOI] [PubMed] [Google Scholar]
- 12.Vats AN, Ishwad C, Vats KR, et al. Steroid-resistant nephrotic syndrome and congenital anomalies of kidneys: evidence of locus on chromosome 13q. Kidney Int. 2003;64:17–24. doi: 10.1046/j.1523-1755.2003.00066.x. doi:10.1046/j.1523-1755.2003.00066.x. [DOI] [PubMed] [Google Scholar]
- 13.Schwarz JM, Rodelsperger C, Schuelke M, et al. MutationTaster evaluates disease-causing potential of sequence alterations. Nat Methods. 2010;7:575–576. doi: 10.1038/nmeth0810-575. doi:10.1038/nmeth0810-575. [DOI] [PubMed] [Google Scholar]
- 14.Ramensky V, Bork P, Sunyaev S, et al. Human non-synonymous SNPs: server and survey. Nucleic Acids Res. 2002;30:3894–3900. doi: 10.1093/nar/gkf493. doi:10.1093/nar/gkf493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Adzhubei IA, Schmidt S, Peshkin L, et al. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7:248–249. doi: 10.1038/nmeth0410-248. doi:10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wu K, Yang Y, Wang C, et al. DACH1 inhibits transforming growth factor-beta signaling through binding Smad4. J Biol Chem. 2003;278:51673–51684. doi: 10.1074/jbc.M310021200. doi:10.1074/jbc.M310021200. [DOI] [PubMed] [Google Scholar]
- 17.Wu K, Liu M, Li A, et al. Cell fate determination factor DACH1 inhibits c-Jun-induced contact-independent growth. Mol Biol Cell. 2007;18:755–767. doi: 10.1091/mbc.E06-09-0793. doi:10.1091/mbc.E06-09-0793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhou J, Wang C, Wang Z, et al. Attenuation of Forkhead signaling by the retinal determination factor DACH1. Proc Natl Acad Sci USA. 2010;107:6864–6869. doi: 10.1073/pnas.1002746107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Katsanis N, Eichers ER, Ansley SJ, et al. BBS4 is a minor contributor to Bardet–Biedl syndrome and may also participate in triallelic inheritance. Am J Hum Genet. 2002;71:22–29. doi: 10.1086/341031. doi:10.1086/341031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tabatabaeifar M, Schlingmann KP, Litwin M, et al. Functional analysis of BMP4 mutations identified in pediatric CAKUT patients. Pediatr Nephrol. 2009;24:2361–2368. doi: 10.1007/s00467-009-1287-6. doi:10.1007/s00467-009-1287-6. [DOI] [PubMed] [Google Scholar]
- 21.Kottgen A, Pattaro C, Boger CA, et al. New loci associated with kidney function and chronic kidney disease. Nat Genet. 2010;42:376–384. doi: 10.1038/ng.568. doi:10.1038/ng.568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ichikawa I, Kuwayama F, Pope JC, et al. Paradigm shift from classic anatomic theories to contemporary cell biological views of CAKUT. Kidney Int. 2002;61:889–898. doi: 10.1046/j.1523-1755.2002.00188.x. doi:10.1046/j.1523-1755.2002.00188.x. [DOI] [PubMed] [Google Scholar]