

Abstract
In this work, addition of OH− to one-electron oxidized thymidine (dThd) and thymine nucleotides in basic aqueous glasses is investigated. At pHs ca. 9–10 where the thymine base is largely deprotonated at N3, one-electron oxidation of the thymine base by Cl2•− at ca. 155 K results in formation of a neutral thyminyl radical, T(−H)•. Assignment to T(−H)• is confirmed by employing 15N substituted 5'-TMP. At pH ≥ ca. 11.5, formation of the 5-hydroxythymin-6-yl radical, T(5OH)•, is identified as a metastable intermediate produced by OH− addition to T(−H)• at C5 at ca. 155 K. Upon further annealing to ca. 170 K, T(5OH)• readily converts to the 6-hydroxythymin-5-yl radical, T(6OH)•. One-electron oxidation of N3-methyl-thymidine (N3-Me-dThd) by Cl2•− at ca. 155 K produces the cation radical (N3-Me-dThd•+) for which we find a pH dependent competition between deprotonation from the methyl group at C5 and addition of OH− to C5. At pH 7 the 5-methyl deprotonated species is found; however, at pH ca. 9, N3-Me-dThd•+ produces T(5OH)• that on annealing up to 180 K forms T(6OH)•. Through use of deuterium substitution at C5' and on the thymine base, i.e., specifically employing [5',5”-D,D]-5'-dThd, [5',5”-D,D]-5'-TMP, [CD3]-dThd and [CD3,6D]-dThd, we find unequivocal evidence for T(5OH)• formation and its conversion to T(6OH)•. The addition of OH− to the C5 position in T(−H)• and N3-Me-dThd•+ is governed by spin and charge localization. DFT calculations predict that the conversion of the “reducing” T(5OH)• to the “oxidizing” T(6OH)• occurs by a unimolecular OH group transfer from C5 to C6 in the thymine base. The T(5OH)• to T(6OH)• conversion is found to occur more readily for deprotonated dThd and its nucleotides than for N3-Me-dThd. In agreement, calculations predict that the deprotonated thymine base has a lower energy barrier (ca. 6 kcal/mol) for OH transfer than its corresponding N3-protonated thymine base (14 kcal/mol).
Introduction
The reactions of hydroxyl radical (•OH) with thymine (Thy), its nucleoside and nucleotide derivatives have been extensively investigated by pulse radiolysis in aqueous solution at ambient temperature.1 – 12 The hydroxyl radical has been shown to add predominantly (ca. 90%) to the C5-C6 double bond of the thymine base with a diffusion-controlled rate producing 5-hydroxythyminyl-C6 (C5-OH adduct) radical (TNH(5OH)•) (30%) and 6-hydroxythyminyl-C5 (C6-OH adduct) radical (TNH(6OH)•) (60%) (scheme 1). In addition, a small extent (ca. 10%) of H-atom abstraction from the methyl group at the C5 of thymine base moiety results in the formation of UN3HCH2• (scheme 1).1, 7, 9, 11 The high reduction potential of •OH (2.3 V at pH 7),13 should in principle, cause one-electron-oxidation of all the four nucleobases.14,15 However, experimentally, •OH is found to be not as oxidizing as its high reduction potential suggests.1 Recent theoretical calculations have shown that most of the reduction potential of •OH derives from the solvation of the OH− that is formed after electron transfer and this makes the initial electron transfer step of the one-electron oxidation by •OH a slow process.16 Consequently, the addition and H-atom abstraction reactions of •OH become kinetically favored in comparison to slower one-electron oxidation. Moreover, the high electrophilicity of •OH17 makes its addition to the electron rich C5-C6 double bond of thymine base favored over H-atom abstraction.1, 2, 6–8,11,12
Scheme 1.

The electrophilic addition and H-atom abstraction reactions of •OH with Thy and its derivatives and the addition of water or OH− to the one-electron oxidized Thy and its derivatives reported in the literature1 – 12 are summarized in this scheme.
The electrophilic addition of •OH to the C5-C6 double bond in thymine base has been modeled by DFT (B3LYP/6–31G**) using the COSMO solvation model.18 The reaction free energies are predicted to be: ΔG = −10.2 kcal/mol for addition at C5 (i.e., T(5OH)• formation) and ΔG = −20.4 kcal/mol for addition at C6 (i.e., T(6OH)• formation).18 H-atom abstraction from the methyl group at C5 in the thymine base (UN3HCH2•) is found to be the most exergonic with ΔG = −27.0 kcal/mol. Thus, formation of UN3HCH2• via H-atom abstraction by •OH is thermodynamically favored over the electrophilic addition of •OH to the C5-C6 double bond of thymine base.2 However, as mentioned above, UN3HCH2• is experimentally found to be a minor product via •OH attack at about 10% yield.1 Thus, the reactions of •OH with thymine and its derivatives are clearly kinetically controlled.1, 2, 6–8, 11, 12
Pulse radiolysis1,4,19 and continuous wave (CW) electron spin resonance (ESR) spectroscopy20–22 studies, proposed that one-electron oxidation of Thy and in its various derivatives by SO4•− results in transient formation of thymine π-cation radical (T•+) at pH 7 which quickly undergoes addition of water (or of OH−) at C6 to form an OH adduct, TNH(6OH)• (scheme 1). ESR spectroscopic studies of photoionized thymine and N1-substituted thymine compounds in frozen aqueous solutions at 77 K show that under acid, neutral, or basic conditions the one-electron oxidized thymine base radical (T(−H)•) is produced.23–25 For one-electron oxidized thymine base itself in 8 M NaOH, formation of the allylic UN3HCH2• via deprotonation from the methyl group of T(−H)• is observed upon annealing to higher temperatures (170 to 190 K).23,24 However, for dThd and other N1-substituted thymines photoionized in very basic conditions (8 M NaOH) at 77 K, formation of T(6OH)• via addition of OH− to T(−H)• is observed even at this low temperature.26,27 Substitution at N1 favors addition of OH− to the C5-C6 double bond of one-electron oxidized thymine systems. Later studies in other DNA systems show both OH− addition and methyl deprotonation processes are competitive.28,29
Addition of water (or of OH−) to one-electron oxidized thymine derivatives and reaction of •OH with thymine derivatives thus lead to the formation of identical radical intermediates – TNH(5OH)•, TNH(6OH)•, and UN3HCH2• (scheme 1). However, there is some evidence from pulse radiolysis investigations that the kinetically formed initial OH-adduct radicals can rearrange to thermodynamically more stable OH-adduct radicals. For example, a combination of pulse radiolysis, ESR with flow systems, and product analyses study has reported that in aqueous solution, under acidic conditions (pH 3) at room temperature the 5-hydroxy-1,3-dimethyluracilyl-C6 (C5-OH adduct) radical of 1,3-dimethyluracil converts to the corresponding 6-hydroxy-1,3-dimethyluracilyl-C5 (C6-OH adduct) radical.1, 2, 5–11,12
For thymine systems, a rearrangement reaction of C5-OH adduct radical to its corresponding C6-OH adduct radical, i.e., TNH(5OH)• to TNH(6OH)•, has not been reported till date (scheme 1). In this work, investigation of one-electron oxidation of dThd, L-dThd, α-dThd, N3-Me-dThd, and its nucleotides is conducted using ESR spectroscopy. Employing deuterated derivatives [5',5”-D,D]-5'-dThd, [5',5”-D,D]-5'-TMP, [CD3]-dThd, and [CD3,6D]-dThd, unequivocal evidence for the formation of T(5OH)• via OH− addition to the one-electron oxidized thymine base moiety and the base catalyzed unimolecular conversion of T(5OH)• to its corresponding T(6OH)• are presented in this work. Using N3-Me-dThd, the results at pH values ca. 7 to 12 clearly establish the competition between deprotonation from the methyl group at C5 and addition of OH− to C5 in one-electron oxidized thymine. T(5OH)• to T(6OH)• conversion for N3-Me-dThd has a higher activation barrier than that in the T(5OH)• to T(6OH)• conversion found in dThd and in its nucleotides where the thymine base is N3-deprotonated. Theoretical calculations have been carried out to illustrate the role of the protonation state of the N3 atom of thymine base in the rearrangement of T(5OH)• to its corresponding T(6OH)•. Theoretically, we find T(5OH)• to T(6OH)• rearrangement has a lower energy barrier (ca. 6 kcal/mol) for the N3-deprotonated thymine base in comparison to that (14 kcal/mol) for the N3-protonated thymine base. The theoretically calculated values of reduction potential predict that T(5OH)• is “reducing” whereas T(6OH)• is “oxidizing” in nature. These predictions are in agreement with the pulse radiolysis studies in aqueous solutions.1–12
Materials and Methods
Compounds purchased
Thymidine (dThd), thymidine-5'-monophosphate (5'-TMP), thymidine-5'-diphosphate (5'-TDP), thymidine-5'-triphosphate (5'-TTP), thymidine-3',5'-cyclic monophosphate (3',5'-cTMP), and lithium chloride (99% anhydrous, Sigma Ultra) were purchased from Sigma Chemical Company (St. Louis, MO, USA). N3-methyl-thymidine (N3-Me]-dThd), L-thymidine (L-dThd), alpha-thymidine (α-dThd), [Methyl-D3]-thymidine ([CD3]-dThd), thymidine-3',5'-bisphosphate (T-3',5'-BP) were obtained from Carbosynth Ltd (Berkshire, UK). 2',3'-Dideoxythymidine-5'-triphosphate (2',3'-dd-5'-TTP) was procured from TriLink Biotechnologies (San Diego, CA, USA). 15N-5'-TMP was procured from Cambridge Isotope Laboratories (Andover, MA, USA). Potassium persulfate (crystal) was obtained from Mallinckrodt, Inc. (Paris, KY, USA). Deuterium oxide (99.9 atom % D) was obtained from Aldrich Chemical Company Inc. (Milwaukee, WI, USA). 5',5”-Dideuterothymidine ([5',5”-D,D]-dThd) was purchased from Omicron Biochemicals, Inc. (South Bend, IN, USA). Thymidine-α,α,α,6-d4 ([CD3,6D]-dThd) was purchased from CDN Isotopes (Quebec, Canada). Following our earlier works,15,30–34 all compounds were used without any further purification.
Compound synthesized
5',5”-Dideutero-thymidine-5'-monophosphate ([5',5”-D,D]-5'-TMP
[5',5”-D,D]-dThd, available from Omicron Biochemicals, was converted to [5',5”-D,D]-3'-O-acetylthymidine by standard literature procedure.35 The latter was phosphorylated using Yoshikawa protocol36 and purified on DEAD Sephadex A-25 column to give [5',5”-D,D]-thymidine-5'-monophosphate ([5',5”-D,D]-5'-TMP) as triethylammonium salt (see supporting information pages S2 and S3).
Sample preparation
-
(a)
Preparations of Solutions of dThd and its Derivatives: Following our previous work with the monomers of DNA and RNA,15,30–34 0.5 to 10 mg of the solute (each of dThd, its deuterated derivatives, and its nucleotides) was dissolved in 1 ml of 7.5 M LiCl in D2O (or in H2O, if required). 6 to 20 mg K2S2O8 was added as the electron scavenger so that only the formation of one-electron oxidized radicals and their subsequent reactions could be studied.
-
(b)
pH adjustments: As per our previous work,15,30–34,37 the pHs of the solutions of dThd and its derivatives were adjusted from ca. 8 to ca. 12 by quick addition of appropriate micromolar amounts of 0.1 to 1 M NaOH in D2O (or in H2O) under ice-cooled conditions. We note here that all the pH values reported in this work are approximate owing to the high ionic strength (7.5 M LiCl) of these solutions and due to the use of pH papers.15,30–34,37 These homogenous solutions were degassed by bubbling thoroughly with the nitrogen gas at room temperature.15,30–34,37
-
(c)
Preparation of glassy samples: These pH-adjusted homogeneous solutions were drawn into 4 mm Suprasil quartz tubes (Catalog no. 734-PQ-8, WILMAD Glass Co., Inc., Buena, NJ, USA). The tubes containing these solutions were then rapidly immersed into liquid nitrogen (77 K). Rapid cooling of these solutions to 77 K resulted in transparent glassy solutions.15,30–34,37 These glassy (7.5 M LiCl / D2O (or H2O) samples are supercooled homogeneous solutions in the temperature range 77 to 175 K.
-
(d)
Storage of these glassy samples: Following our works,15,30–34,37 samples were always stored in Teflon containers at 77 K in the dark before and after irradiation and also after annealing.
γ-Irradiation of glassy samples
Following our previous studies,15,30–34,37 these transparent glassy samples were γ irradiated (absorbed dose = 1.4 kGy) at 77 K in Teflon containers.
Annealing of the glassy samples
As per our earlier work,15, 38 progressive annealing of these γ-irradiated glassy samples of dThd and its various derivatives were carried out in 5°C steps from 140 to 175 K for 15 to 20 min at each temperature. Employing a variable temperature assembly (Air products), annealing of each sample was carried out in the dark via cooled nitrogen gas which regulated the gas temperature within ±4°C.15 Using a copper–constantan thermocouple which is in direct contact with the sample, we have monitored the temperature of the sample during its annealing. Annealing of the sample in the temperature range 140 to 160 K causes softening of the glassy phase thereby allowing the matrix radical e.g. Cl2•− to migrate and to cause one-electron oxidation of the solute. Further annealing (160 to 175 K) causes more softening of the glass thereby allowing us to study solution phase chemistry of the one-electron oxidized thymine with the aid of ESR. K2S2O8 is not found to react with radicals formed by one-electron oxidization of the thymine base for temperatures up to 180K.30
Electron Spin Resonance (ESR)
After γ-irradiation at 77 K, the ESR spectrum of the sample was recorded also at 77 K. After annealing, the glassy samples were immersed in liquid nitrogen (77 K), and an ESR spectrum was recorded at 40 dB (20 μW). A Varian Century Series ESR spectrometer operating at 9.3 GHz with an E-4531 dual cavity, 9-inch magnet and with a 200 mW klystron was used to record the ESR spectra of these samples.15,30–34,37,38 Fremy's salt, g(center) = 2.0056, AN = 13.09 G, was used for field calibration.15,30–34,37,38
All ESR spectra reported in this work were recorded at 77 K.
Calculations based on Density Functional Theory (DFT)
We used Becke's three parameter exchange functionals (B3)39,40 with Lee, Yang, and Parr's correlation functional (LYP)41 and 6-31G** basis set designated as B3LYP/6-31G** method. Our works15,30–34,37,38 along with the works of others42–45 have shown that the density functional B3LYP method with compact basis set 6-31G* or 6-31G** is quite appropriate to obtain the reliable hyperfine coupling constant (HFCC) value of radicals which are comparable to the experimentally determined HFCCs. Thus, B3LYP/6-31G** method has been used in this work to study the reaction of •OH with Thy and the reaction of OH− with neutral N3-deprotonated thyminyl radical (T(−H)•, scheme 2) in the presence of six water molecules. The polarized continuum model (PCM), based on the integral equation formalism (IEF) model,46–49 was also used to consider the effect of bulk solution on these reactions. The calculations were performed using the Gaussian 09 suite of programs50 and GaussView51 and JMOL52 programs were used to plot the spin densities and molecular structures. The spin densities and distribution of charges were obtained employing–the Spartan'10 program set53 at the B3LYP/6-311++G**//B3LYP/6-31G* level of theory in the gas phase. The structures of the sugar and base radicals derived from dThd and its derivatives including nucleotides studied in this work are shown in scheme 2.
Scheme 2.

The structures of the thymine base radicals used in this work. The atom numbering scheme is shown here. Representative structural formula of the adduct radical (T(5OH)•) formed via addition of OH−/ OD− at C5 in T(−H)• and that of T(6OH)• formed via reorganization of T(5OH)• are presented in this scheme.
Results and Discussion
-
(A)Experimental:
-
Formation of T(−H)• in dThd and thymine nucleotides: confirmed by 15N incorporated 5'-TMP:At pH ca. 9 and above, the thymine base moiety is deprotonated at N3 in dThd and its nucleotides.1 As a result, the redox potential of N3-deprotonated thymine in dThd and in its nucleotides is significantly lower than that of its N3-protonated form.1 Therefore, the one-electron oxidation of dThd and various thymine nucleotides investigated in this work, i.e., 5'-TMP, 5'-TDP, 5'-TTP, 3',5'-cTMP, and T-3',5'-BP by Cl2•− at pHs ca. ≥9 should occur only at the thymine base (scheme 3) leading to formation of the neutral N3-deprotonated thyminyl radical (T(−H)•, scheme 2, and also Figure 10 in section B). One-electron oxidation of N3-Me-dThd by Cl2•− at pHs ca. ≥9 results in the formation of “pristine” base cation radical (N3-Me-dThd•+ (schemes 2, 3, and also supporting information Figure S15)).The ESR spectrum (green) from 5'-TMP obtained via annealing at 160 K at pH ca. 10 is presented in Figure 1A. The spectrum from a similarly prepared sample of 5'-TMP at pH ca. 8 and those from samples of 3',5'-cTMP, and T-3',5'-BP at pH ca. 10 were found to have the same hyperfine structure as in spectrum 1A. On the basis of the previous work,23–29 the spectrum 1A has been assigned to N3-deprotonated thyminyl radical (T(−H)•).To add support to this assignment, experiments were carried out using 15N incorporated 5'-TMP (where all the N-atoms in thymine ring were 15N labeled) and the results are presented in Figure 1B. The ESR spectrum (red) of 15N incorporated 5'-TMP at pH ca. 10 in Figure 1B was obtained from identical procedures as the 14N sample in Figure 1A. This spectrum is assigned to [15N]-T(−H)•. T(−H)• and N3-Me-dThd•+ were produced following our method of preparation of one-electron oxidized species in LiCl glasses.30–34 ESR spectra showing the formation of N3-Me-dThd•+ in irradiated glassy samples of N3-Me-dThd are presented in supporting information, Figure S17.The [14N]-T(−H)• (green) spectrum and [15N]-T(−H)• (red) spectrum are simulated using known parameters: isotropic β hyperfine coupling constant (HFCC) values of 3 methyl protons at 21 G, isotropic β HFCC value of C1'-β-H atom is 7 G, a mixed Lorentzian/Gaussian (1/1) linewidth of 6.4 G, with g-values of (2.0025, 2.0043, 2.0043). For [14N]-T(−H)• (green) spectrum 1A, the 14N HFCC values are (13.2, 1.0, 1.0) G, whereas the corresponding 15N HFCC values for [15N]-T(−H)• (red) spectrum 1B are (18.2, 1.0, 1.0) G. These 14N and proton HFCC values are in very good agreement with the corresponding HFCC values for T(−H)• reported in the literature.23–29The three reference markers (open triangles) in this Figure and in other Figures show the position of Fremy's salt resonance with the central marker at g = 2.0056. Each of these markers is separated from each other by 13.09 G.
-
Formation of T(5OH)• via addition of OH−to C5 of T(−H)• in 5'-TMP and the subsequent conversion of T(5OH)• to T(6OH)•:In Figure 2A, the ESR spectrum of a γ-irradiated glassy sample of 5'-TMP (0.5 mg/ml) at pH ca. 12 is shown in black. Comparison of the central part of the total 880 G wide multiplet Cl2•− spectrum (brown)15 with the black spectrum (both 220 G scans) shows that the black spectrum has the expected two low field resonances from Cl2•− and a sharp singlet from SO4•−. The Cl2•− is formed from radiation-produced holes, and SO4•− is produced by reaction of radiation-produced electrons with S2O8−2.15,30 Upon annealing to 140 K, SO4•− reacts with Cl− to form additional Cl2•− (brown spectrum in A) as found in our earlier work.30Annealing of this sample at 155 K for 15 min resulted in the black spectrum in Figure 2B. Comparison of this black spectrum in 2B with the Cl2•− spectrum (brown) in 2A shows that the line components of Cl2•− are still present at 155 K. For 5'-TMP at pH ca.12 the phosphate group as well as the N3 atom in the thymine moiety in 5'-TMP are fully deprotonated, thereby making the one-electron oxidation of the thymine base by Cl2•− in 5'-TMP quite facile.25–29 Overlap of the green spectrum of T(−H)• on the black spectrum in Figure 2B shows some indications of the presence of weak line components of T(−H)• in the black spectrum. As the concentration of 5'-TMP is raised from 0.5 mg/ml to 10 mg/ml, the line components of T(−H)• become more prominent (supporting information Figure S3).Further annealing of this sample at 160 K for 15 min results in the loss of Cl2•− and the growth of a doublet spectrum, Figure 2C. This ca. 20 G doublet is assigned to T(5OH)• (scheme 4). This radical has a localized spin at C6 which couples to the C6 alpha-H atom resulting in such an anisotropic doublet15,30,31,33,34,37,54,55 (see section 4). The spectra in 2B and 2C suggest that T(5OH)• formation occurs via addition of OH− to C5 at the C5–C6 double bond of T(−H)• (scheme 4). However, additional small line components (indicated by arrows) that are assigned to T(6OH)• (spectrum 2D, vide infra) appear in spectrum 2C. A more isolated doublet spectrum for T(5OH)• is shown in spectrum 2E (black) by subtraction of 30% of spectrum 2D from spectrum 2C.Subsequent annealing of this sample at 175 K for 20 min resulted in the loss of T(5OH)• with concomitant formation of spectrum 2D (black) which is assigned to T(6OH)• (scheme 4). We note that annealing up to 190 K of various Thy and dThd derivatives in 8 M NaOD glasses which were photoionized at 77 K, resulted in spectra that are similar to the spectrum 2D.26,27 These spectra were also assigned to T(6OH)• that is formed via addition of OH− at C6 in thymine moiety of T(−H)• (scheme 4). Annealing at 175 K results in conversion of T(5OH)• to T(6OH)•. These observations suggest that the initial formation of T(5OH)• via addition of OH− at C5 in the thymine moiety of T(−H)• is kinetically controlled.The theoretical calculations (see section (B) predict that the spin density distribution in the optimized structure of T(−H)• has the spin predominantly localized at the C5 in the thymine ring. Localization of positive charge is also found in the C5/C6 region in the thymine ring (see supporting information table T1). It is the spin localization that leads to the kinetically-controlled addition of OH− at C5 leading to the formation of T(5OH)•. Furthermore, our theoretical calculations predict that the subsequent formation of T(6OH)• is thermodynamically favored over the initially produced T(5OH)•. Owing to structural similarity of the N3-H moiety in thymine ring to the N-H moiety of succinimide, their pKa values are quite similar (9 to 9.5).1 Thus, for dThd and its various derivatives including nucleotides under our experimental conditions (pH≥ ca. 11.5), one-electron oxidized thymine (T(−H)•) as well as T(5OH)•, and T(6OH)• are N3-deprotonated (schemes 2 to 4). For N3-Me-dThd, T(5OH)• and T(6OH)• which have an N3-methyl group, N3-Me-dThd•+ formation is observed instead of T(−H)• (see section 3).Analyses of spectrum 2D (black) shows that T(6OH)• spectrum results from a composite spectra of two conformers. In one conformer of T(6OH)•, the C6-H atom is axial (see scheme 4) with a dihedral angle between the C6-H bond and the z-axis of the radical site p-orbital that approaches 0° and which results in a large β-C6-H HFCC value of 45 G. The other conformer of T(6OH)• has the C6-H atom in a more equatorial position (see scheme 4), with the C6-H atom nearer the node of C5-p orbital containing the unpaired spin. From the McConnell relationship56 (aiso ≅ Bcos2θ, B = 45 G) the β-C6-H HFCC of 9 G coupling suggests the dihedral angle (θ) between the C6-H bond and the z-axis of the radical site-p-orbital is about 117°. Both conformers have identical HFCC value of 21 G for the three methyl β-protons. The simulation shows that the abundance of the two different conformers of T(6OH)•, are 20% for the axial conformer of 45G β-coupling and 80% for the near equatorial conformer of 9 G β-coupling (scheme 4). The simulated spectrum (red, Figure 2D) which was obtained based on these couplings and abundances, matches the experimental spectrum (black, Figure 2D) very well. For further details of the simulation parameters, see supporting information Figure S2. In previous studies, authentic T(6OH)• spectrum was obtained unambiguously via debromination of 5-bromo-6-hydroxythymine on dissociative electron attachment.27 Line components due to the less abundant C6-H equatorial isomer of T(6OH)• found in this work27 are also apparent in the spectra of this earlier work but were ignored and not assigned to C6-H equatorial isomer of T(6OH)•. Thus, these T(6OH)• conformers with C6-H-axial or C6-H equatorial, are obtained via two different pathways - addition of OH− at C5–C6 double bond in thymine moiety of T(−H)• (this work along with previous work26 – 29) and debromination of 5-bromo-6-hydroxythymine27.The effects of concentration of 5'-TMP and number of phosphate substitution at the 5'-site in 5'-TMP on the formation of T(5OH)• along with the extent of subsequent conversion of T(5OH)• to the conformers of T(6OH)• are reported in section 5.
-
Addition of OH−to C5 of N3-Me-dThd•+ at pH 7 to 12:One-electron oxidation of N3-Me-dThd in our system (LiCl glass, pH ca. ≥9) leads to the formation of the N3-Me-dThd•+ (see section 1, scheme 3, and supporting information Figure S17). Therefore, to investigate the effect of concentration of OH− on the formation of T(5OH)• along with the extent of subsequent conversion of T(5OH)• to the conformers of T(6OH)•, addition of OH− to N3-Me-dThd•+ in matched samples of N3-Me-dThd (2 to 2.5 mg/ml) prepared at pHs ca. 7, 9, and 12) have been studied and these results are shown in Figures 3 (pH ca. 9), 4 (pH ca. 12) to 5 (pH ca. 7).Analyses of Figures 3 to 5 suggest the following salient points:
-
(a)Formation of N3-Me-dThd•+ and its reactivity: N3-Me-dThd•+ formation on one-electron oxidation by Cl2•− at 155 K is observed at pH 9 (Figure 3A). Progressive annealing of this sample from 77 to 155 K is shown in supporting information Figure S17. On raising the OH− concentration to pH 12, we find for an otherwise identical sample that a prominent ca. 20 G doublet due to T(5OH)• is observed at 155 K (see Figure 4A). The comparison of spectrum in Figure 3A with that in Figure 4A clearly shows that at higher pH (9 vs 12), formation of T(5OH)• via addition of OH− to C5 in N3-Me-dThd•+ is observed owing to the higher concentration of OH−. For the sample at pH ca. 9, the corresponding formation of T(5OH)• is observed only by annealing to higher temperatures (160 to 165 K).Figure 5 shows that N3-Me-dThd•+ at pH 7 readily deprotonates from the methyl group at C5 leading to the formation of UCH2• (70%) (analogous to scheme 1, for N3-Me-dThd) in completion with OH− (or water) addition to form T(6OH)• (30%). Thus, deprotonation of N3-Me-dThd•+ is competitive with addition of OH−/H2O to C5 in N3-Me-dThd•+ at pH 7; however, at higher pHs ca. 9 and 12, formation of T(5OH)• (scheme 4) via addition of OH− to C5 in N3-Me-dThd•+ is observed. Addition of water to N3-Me-dThd•+ likely has a slightly higher activation energy than OH− addition and explains the competitive formation of UCH2•. We note that pulse radiolysis studies of various one-electron oxidized thymine derivatives at room temperature under alkaline conditions (pH ≤ 10) show predominant formation of T(6OH)• reportedly as a result of OH− addition at C6 in T(−H)•.19
-
(b)Formation of T(6OH)• in N3-Me-dThd: As already observed with 5'-TMP (see section 2), conversion of T(5OH)• to T(6OH)• is observed via progressively annealing the sample in the range 160 to 180 K (see spectra 4B to 4E). The intensity of the spectra remains constant with annealing temperature (see spectra 4B to 4E). This is consistent with the conversion of T(5OH)• to T(6OH)• being unimolecular in nature. A simulation (red) of the experimental (black) spectrum in Figure 4E shows that the abundance of only conformer of T(6OH)• with C6-H axial (scheme 4) having 33 G C6-H β-coupling, 19 G β-H coupling of each of the three methyl proton. The simulated spectrum (red, Figure 4E) which has been obtained based on these couplings, 8 G linewidth, and giso = 2.0035, matches the experimental spectrum (black, Figure 4E) very well. The comparison of spectrum in Figure 3E with that in Figure 4E (supporting information Figure S19) clearly shows that even at pH ca. 9, formation of the same T(6OH)• with C6-H axial conformation occurs from T(5OH)• via progressively annealing the sample in the range 160 to 180 K. The fact that the spectra at pH 9 and 12 are the same for T(6OH)• shows deprotonation from OH does not occur.
T(−H)• produced in 5'-TMP at pH ca. 8–10, does not add OH− even on annealing to 170 K (see supporting information Figure S18), and neither the formation of T(5OH)• nor its subsequent conversion to T(6OH)• is observed. This combined with the fact that addition of OH− to N3-Me-dThd•+ is found pH ca. 9 clearly shows that both the spin density and charge distribution in the oxidized base (see part (B), section 1) determine the site and rate of reaction. -
(a)
-
Confirmation of formation of T(5OH)• and subsequent production of T(6OH)• in dThd and thymine nucleotides by using deuterated derivatives of dThd and thymine nucleotides:The ca. 20 G anisotropic doublet that is found in the samples annealed at 155 to 160 K is assigned to T(5OH)•. However, a ca. 20 G doublet from an anisotropic αH atom is expected to be generated for any radical species with a single αH hyperfine coupling. In our previous works,15,30,31,33,34,37,54,55 C5'• spectrum shows such an anisotropic doublet. To distinguish between T(5OH)• and C5'•, we have employed a number of deuterated compounds. [5',5”-D,D]-dThd and [5',5”-D,D]-5'-TMP were used to provide an unequivocal evidence of C5'• formation. For the [CD3]-dThd sample, owing to deuteration of the C5-methyl group, an anisotropic doublet spectrum from the C6-H α-proton coupling in T(5OH)• should be observed. But, the T(6OH)• spectrum in [CD3]-dThd sample should be affected by the loss of methyl group hyperfine couplings. On the other hand, in [CD3,6D]-dThd, owing to the deuteration of the C5-methyl group and C6-H, the loss of all couplings is expected due to the formation of T(5OH)• and T(6OH)•. Therefore, [CD3]-dThd and [CD3,6D]-dThd were used to confirm the formation of T(5OH)• and T(6OH)•. The results are presented in Figure 6.The detailed annealing studies of these samples are shown in supporting information Figures S4, S5, S6, and S10.We have found that the results of [5',5”-D,D]-5'-TMP (supporting information Figure S6) are similar to its nucleoside [5',5”-D,D]-dThd (supporting information Figure S4) and hence the results of the nucleosides are presented here.In Figure 6A, collapse of the central anisotropic ca. 15 to 20 G doublet is observed in [CD3,6D]-dThd but not in [5',5”-D,D]-dThd, [CD3]-dThd, and [5',5”-D,D]-5'-TMP. Since this anisotropic doublet is found in the [CD3]-dThd (ca. 15 G), [5',5”-D,D]-dThd (ca. 20 G) and [5',5”-D,D]-5'-TMP (ca. 20 G) samples, the radical site is clearly not at C5'. The collapse of doublet in [CD3,6D]-dThd sample owing to deuteration of all the ring hydrogen atoms and the appearance of the doublet in [CD3]-dThd sample is consistent only with the assignment of T(5OH)• with the radical site at C6 in the thymine base. Thus, the initial kinetically controlled formation of T(5OH)• by addition of OH− at C5 of T(−H)• is confirmed.On further annealing to 170 K for 15 min, the full line components of T(6OH)• formed are observed in [5',5”-D,D]-dThd and [5',5”-D,D]-5'-TMP samples (see Figure 6B). The loss of the line components in the [CD3]-dThd sample arising from the methyl group and the loss all line components for [CD3,6D]-dThd (Figure 6B) clearly show that the hyperfine couplings in non-deuterated dThd arise from the three methyl β-protons at C5 and the β-C6-proton in the thymine base. This is only consistent with the assignment of the radical as T(6OH)•.Analysis of the T(6OH)• spectrum from Figure 6B (thick sky blue) for the [CD3]-dThd sample is shown in Figure 7. This spectrum is a composite of two spectra– 60% of singlet spectrum C (red) and 40 % of doublet spectrum D (violet). The singlet spectrum 6C is assigned to the near equatorial conformer of T(6OH)• whereas the doublet spectrum 6D is assigned to the axial conformer T(6OH)• (C6-H β HFCC ca. 39 G). The simulated spectrum (green, Figure 7B) which was obtained based on these couplings and abundances, matches the experimental spectrum (Figure 6B and Figure 7A, thick sky blue) very well.Therefore, these results clearly establish the conversion of the kinetically favored T(5OH)• to the two different conformers of the thermodynamically favored radical, T(6OH)• (scheme 4).
-
Factors affecting the formation of T(5OH)• and subsequent production of T(6OH)• in dThd and thymine nucleotides:
-
(a)Effect of concentration on formation T(5OH)• and T(6OH)• :With increasing concentration of 5'-TMP (section 2), T(−H)• became more prominent at low temperatures (supporting information Figure S3A). At higher concentrations of 5'-TMP (10 mg/ml, supporting information Figure S3), on annealing, near identical T(5OH)• spectrum is found to that shown in Figure 2E (black). However, with increasing 5'-TMP concentration from 0.5 to 10 mg/ml (Figure 8), the conversion of T(5OH)• to T(6OH)• is found to more greatly favor the T(6OH)• conformer with C6-H in the equatorial position. Thus, the outer line components resulting from T(6OH)• with C6-H axial conformer (β C6-H HFCC ca. 45 G) are decreased substantially in the spectrum. Although an increase in concentration of 5'-TMP does not affect the conversion of T(5OH)• to T(6OH)• (supporting information Figure S3), the decrease of line components of T(6OH)• with C6-H axial conformer is likely a result of stacking of 5'-TMP57,58 at higher concentrations that prefers the formation of T(6OH)• with C6-H equatorial conformer i.e., low β C6-H coupling (ca. 9 G).Unlike the results of increase in concentration of 5'-TMP (section 2(a)), increase of concentration of dThd samples from 3 mg/ml to 10 mg/ml did not affect (supporting information Figures S7 and S8) the relative abundance of the conformers of T(6OH)• (C6-H axial and C6-H equatorial).
-
(b)Effect of the number of phosphate groups at the 5'-site i.e., dThd, 5'-TMP, 5'-TDP, and 5'-TTP:For dThd (3 mg/ml and 10 mg/ml (supporting information Figures S7, S8)), 5'-TDP or 5'-TTP (3 mg/ml each, supporting information Figure S9), the T(5OH)• spectrum is also found to be an doublet of ca. 20 G with lineshape, and g value very similar to that of the T(5OH)• spectrum for 5'-TMP as found in Figure 2E. These findings show that phosphate groups at the 5'-site in thymine nucleotides (zero, mono, di-, or tri) do not affect the production of T(5OH)•.Results shown in Figure 2D for 5'-TMP (0.5 mg/ml) as well as in Figure 8B for 5'-TMP (3 mg/ml) present evidence of formation of for both T(6OH)• conformers (scheme 4, C6-H axial or C6- equatorial). However, nearly complete formation of the T(6OH)• with C6-H equatorial conformer is observed in 5'-TDP and in 5'-TTP (see supporting information Figure S9). These results show that T(5OH)• to T(6OH)• conversion is not affected in case of 5'-TDP and 5'-TTP. But, the apparent coupling ranges and abundances of C6-H axial and C6-H equatorial conformers of T(6OH)• appear to be affected by the number of 5'-phosphate groups in these samples (supporting information Figure S16).
-
(c)Nature of the solvent (D2O vs. H2O):Formation of T(5OH)• and its subsequent conversion to T(6OH)• has been studied employing dThd (0.8 mg/ml) in H2O glasses (7.5 M LiCl / H2O (supporting information Figure S11)). These results have been compared to the corresponding results obtained using dThd in D2O glasses (7.5 M LiCl / D2O). No observable difference in spectra other than a small line-broadening has been observed on formation of T(5OH)• and its subsequent conversion to T(6OH)• in H2O glasses vs. D2O glasses. Since T(5OH)• and T(6OH)• do not have exchangeable protons with expected hyperfine couplings, the ESR spectra found on formation of T(5OH)• and its subsequent conversion to T(6OH)• is not influenced by change of the solvent from D2O to H2O.
-
(d)Orientation of the sugar relative to the base in dThd e.g., L-dThd, α-dThd:We examine the effect of D-L isomerism using L-dThd and structural isomerism at C1' using α-dThd on the formation of T(5OH)• and T(6OH)• as well as on the relative abundances of the C6-H axial and C6-H equatorial conformers (see supporting information Figures S12 to S14).The center of each spectrum, lineshape, and total hyperfine splitting regarding formation of T(5OH)• and its subsequent conversion to T(6OH)• in L-dThd (supporting information Figure S12) as well in α-dThd (supporting information Figures S13 and S14) are found to be nearly identical to the corresponding spectra found in dThd. Thus, these results establish that the formation of T(5OH)• and T(6OH)•, and their interconversion are not affected by changing from D (dThd (β-anomer)) to L (L-dThd (β-anomer)) or changing the anomeric nature at C1' from dThd (D- sugar, β-anomer) to α-dThd (D- sugar, α-anomer). The fact that the relative abundances of the axial and equatorial conformers are unaffected is good evidence that the preference for each conformer is a function of the energetics of the thymine ring not on interactions with the sugar.
-
(a)
-
Formation of T(5OH)• and subsequent production of T(6OH)• in other thymine nucleotides:One-electron oxidation of several other thymine nucleotides (3',5'-cTMP, T-3',5'-BP, 2',3'-dd-5'-TTP) at pH ca. 12 by Cl2•− (see Figure 9) were also investigated. These results show that
-
(i)the T(5OH)• spectrum in each of these nucleotides is an anisotropic doublet with total hyperfine splitting, g value at the center, and the lineshape are similar to those of the T(5OH)• spectrum found in dThd, 5'-TMP, 5'-TDP, and in 5'-TTP samples. This finding shows that phosphate substitution in 3',5'-cTMP, T-3',5'-BP as well as substitution of 3'-OH by H in 2',3'-dd-5'-TTP did not affect formation of T(5OH)•.
-
(ii)As found for 5'-TDP, 5'-TTP (see section 5(b)), and in high concentration (10 mg/ml) of 5'-TMP samples (see section 5(a)), upon annealing to 170 – 175 K, the T(6OH)• conformer with C6-H equatorial (β C6-H HFCC ca. 9 G) spectrum is predominantly observed in these nucleotides.
-
(i)
-
Is back conversion of T(6OH)• to T(5OH)• possible?In addition, it is found that photoexcitation of T(6OH)• at 143 K for 1.5 h employing a 405 nm laser (30 mW), does not lead to the back conversion of T(6OH)• to T(5OH)• (see supporting information Figure S11) due to the lack of OH group transfer. These results are in contrast to the already reported59 heat or light-induced interconversion of TNH(6H)• to TNH(5H)• in single crystals. Proton transfers are expected to be more facile.
-
Assignment of axial and equatorial conformers of T(5OH)• and T(6OH)• from hyperfine couplings:The hyperfine couplings and g-values for T(5OH)• and T(6OH)• observed in dThd and its various derivatives studied in this work are shown in Table 1. For T(5OH)•, only a 20 G HFCC from the C6 α-hydrogen which is lost on deuteration is observed. For T(6OH)• it is evident from Table 1 that one less abundant (0 to 30%) conformer which has C6-H axial with a small dihedral angle to the radical p-orbital (ca. 0 to 20°) that results in a large β C6-H HFCC of ca. 35 to 45 G (scheme 4), and one more dominant (70 to 80%) conformer has the C6-H equatorial with a larger dihedral angle to the radical p-orbital (ca. 115°) resulting in a small β C6-H HFCC of ca. 6 to 9 G (scheme 4). We note here that, only T(6OH)• in the C6-H axial conformer is observed in N3-Me-dThd (Figure 4). Only the T(6OH)• C6-H equitorial conformer is found in high concentration of 5'-TMP (10 mg/ml, Figure 8) and in 3',5'-cTMP, T-3',5'-BP, 2',3'-dd-5'-TTP (Figure 9). Thus, on the basis of C6-H hyperfine couplings, only these two conformers of T(6OH)• are found. ESR spectra cannot attribute these conformers to specific stereoisomers (6R or 6S) though they are shown in scheme 4. We recognize that a variety of diasteroisomers are also possible (scheme 4); however, ESR spectra have provided no evidence that they influence the hyperfine couplings of the conformers. Even when the structural isomer α-dThd is employed, ESR spectra of T(6OH)• show one large and one small β C6-H HFCC of identical couplings to the β-anomer, dThd.We note here that employing ESR studies with flow systems at room temperature one-electron oxidation by SO4•− of Thy and its derivatives at pH 7 results in water addition and the formation of only the C6-H equatorial conformer of T(6OH)• with small β C6-H HFCC of 11.25 G in Thy, of 15.1 G in 1-methylthymine, and of 12.7 G in 5'-TMP.20–22
-
- (B) DFT calculations:
-
Spin density and charge distribution in optimized structure of T(−H)•:Images of the spin densities and distribution of charges in the optimized structure of T(−H)• of thymidine (see Figure 10) and that in the optimized structure of N3-Me-dThd•+ (see supporting information Figure S15) were obtained using the Spartan'10 program set53 at the B3LYP/6-311++G**//B3LYP/6-31G* level of theory in the gas phase. These structures of T(−H)• and N3-Me-dThd•+ are represented in scheme 2 and have been used throughout this work.The spin density and charge distributions in the optimized structures of T(−H)• and N3-Me-dThd•+ clearly show that in both T(−H)• and N3-Me-dThd•+, the spin is localized mainly at C5 and N1 with no significant spin on N3. The N3 and oxygen atoms of the thymine base in both T(−H)• and N3-Me-dThd•+ have net negative charge whereas the N1, C6, C5, and C4 atoms of the thymine base have net positive charge. Thus, it is expected that this charge separation in both T(−H)• and N3-Me-dThd•+ would be further stabilized in a polar solvent (for example, water). The spin distributions in one-electron oxidized 5'-TMP in solution (supporting information Figure S1) also show similar distribution and clearly represent that even at pH ca. 10 for T(−H)•, the spin densities are still found in the thymine base. This is supported by our ESR spectroscopic studies (see Figure 1). Both spin density and charge distributions in both T(−H)• and N3-Me-dThd•+ contribute to the kinetically-controlled addition of −OH at C5 leading to the formation of T(5OH)• (see Figures 2 to 5).
-
Potential energy surface (PES) of the reaction of OH− with T(−H)•:We have investigated the addition of OH− to T(−H)• at C5 and its subsequent transfer to C6 (scheme 4) in the presence of six water molecules using the B3LYP/6-31G** method. The initial addition of OH− occurs from the top at C5 on the thymine ring in T(−H)• (forming C5-OH adduct radical (T(5OH)•, schemes 1 and 4)) as this is the site of highest spin density in the one-electron oxidized thymine ring (Figures 9 and 10). The initially produced T(5OH)• is found to be unstable to C6-OH adduct radical (T(6OH)•, schemes 1 and 4) formation as the calculation shows that T(6OH)• is favored over T(5OH)• and the process of conversion T(5OH)• to T(6OH)• is found to be exothermic (Figure 11). The T(6OH)• was found to be more stable than T(5OH)• by ca. 4 kcal/mol. We have calculated the PES (Figure 11) for the transfer of OH group from C5 (for T(5OH)•) to C6 (for T(6OH)•) with respect to the change of dihedral angle Φ (C5-C4-C6-C5') employing the B3LYP/6-31G** method (Figures 10 and 11). At each point on the PES, the geometries were fully optimized in the gas phase and in solution (PCM) by constraining the specified dihedral angle only. The calculated PES (Figure 11) for the transfer of OH group from C5 (for T(5OH)•) to C6 (for T(6OH)•) involves relatively small barrier ca. 6 kcal/mol in gas phase and also in solution. This result, along with the results shown in Figures 2 to 8 clearly establishes that though the formation of T(5OH)• is initially favorable, T(6OH)• is the thermodynamically stable radical. Experimentally, we find that in our supercooled homogeneous aqueous solutions, T(5OH)• formed at ca. 155 K is converted to T(6OH)• upon annealing to 170 K and above. Moreover, a detailed analysis of Mulliken charge and spin density distribution in thymine, OH and water molecules are presented in the supporting information Table T1. From Figure 11 and supporting information Table T1 it is evident that T(5OH)• and T(6OH)• have the spin density localized at C6 and C5 sites, respectively; however, during OH group transfer (at the transition state, see Figure 11) from C5 to C6, about 60% of the spin remains on the thymine ring and the remaining ca. 40% spin is on the OH group.
-
PES of the reaction of OH−with T•+:The chemistry after addition of OH− to T•+, (one-electron oxidized thymine with N3 protonated as expected at pH 7) is also treated theoretically in this work. Since the resultant species, T(5OH)• with H atom at N3 (TNH(5OH)•) and T(6OH)• with H atom at N3 (TNH(6OH)•), formed between the reaction of OH− with T•+ are the same (scheme 1) as found for the corresponding addition of •OH to C5–C6 double bond of pyrimidines, previous experimental studies of reactions •OH with pyrimidines1 – 11 are of interest.Our ESR studies have established that the initially preferred site of OH− addition to T(−H)• is at C5 thereby producing T(5OH)• just as found for addition of •OH to thymine. Thus in our calculation of OH− addition to T•+, we have initially optimized the geometries of TNH(5OH)• in the presence of six water molecules. Subsequently, we have calculated the PES for the transfer of the OH moiety from C5 to C6 with respect to the change of dihedral angle Φ (C5-C4-C6-C5') of the thymine ring using the B3LYP/6-31G** method (Figure 12). At each point on the PES, the geometries were fully optimized in the gas phase and in solution (PCM) by constraining the specified dihedral angle only. The B3LYP/6-31G** calculated spin density distribution shows that TNH(5OH)• has ca. 80% spin density localized on the C6 atom with a small delocalization on N1, C2, O2 atoms of thymine and on OH moiety. The geometry of the TNH(5OH)• is quite non-planar and the Φ (C5-C4-C6-C5') is ca. −32°. From the PES (Figure 12), it is evident that transfer of OH from C5 to C6 involves a barrier of ca. 14 kcal/mol in both gas phase and in solution and the process is exothermic in nature and TNH(6OH)• is more stable than TNH(5OH)• by ca. 7 kcal/mol. In the case of TNH(6OH)• formation, the spin density is largely localized (ca. 80%) on the C5 atom.Comparison of the PES for the reaction of OH−with T(−H)• (Figure 11) with that of the reaction of OH− with T•+ (or, the reaction of •OH with T, Figure 12) clearly shows that the transfer of OH from C5 to C6 is more favored when the one-electron oxidized thymine is N3-deprotonated (i.e., for T(−H)•) both in the gas phase and in solution due to the lower barrier (ca. 6 kcal/mol) than for T•+ (ca. 14 kcal/mol) with the N3-proton in place. This explains our experimental finding that T(5OH)• formed from T(−H)• readily converts to T(6OH)• at 170K and the corresponding conversion for T(5OH)• produced from N3-Me-dThd•+ occurs at a higher temperature, 180 K. Moreover, pulse radiolysis studies1–11 at pH 7 of OH radical attack where the N3 proton is present indicate only ca. 30% formation of TNH(5OH)• as expected from the higher barrier for conversion of TNH(5OH)• to TNH(6OH)•.
-
Theoretically predicted relative stabilities of T(6OH)• in axial and equatorial conformations and HFCC values:The B3LYP/6-31G** fully optimized conformations (C6-H equatorial and C6-H axial, shown in scheme 4) of T(6OH)• in the gas phase are shown in Figure 13. These calculations show that (i) the C6-H equatorial conformation of T(6OH)• is more stable than its C6-H axial conformation by 1.09 kcal/mol and (ii) the calculated HFCC value of β C6-H axial in T(6OH)• is higher (21.7 G) than the corresponding value of 7.5 G when it is equatorial. Therefore, it is expected that both conformers of T(6OH)• should be observed and the C6-H equatorial conformer would be the more favorable one in agreement with experiment (see Table 1).
-
Calculated values of reduction potential of the radicals studied in this work:The reduction potential values of T•+, T(−H)•, T(5OH)•, and T(6OH)• were calculated using the ωB97x/6-31++G(d) method. This method has recently provided quite accurate values of ionization potentials of the DNA bases.60 Comparison of the calculated values of reduction potential of these radicals with the corresponding values reported in the literature is presented in Table 2 below.Using uracil and its derivatives, pulse radiolysis studies have established that T(6OH)• oxidizes N,N,N',N'-tetramethylphenylene diamine (TMPD (E7 = 0.27 V))4 and thus, the reduction potential of T(6OH)• at pH 7 must be higher than 0.27 V. On the other hand, it has been unequivocally established that T(5OH)• has reducing properties (quickly reduces tetranitromethane (TNM) (E7 = 0.31 V61) and hence the reduction potential of T(5OH)• at pH 7 must be higher than 0.31 V. It has been observed previously that theoretically calculated oxidation-reduction potentials of the various nucleobases and nucleosides62,63 are closer to redox potential values obtained by cyclic voltametry in non-aqueous solvents (e.g. DMF)64 than those obtained using pulse radiolysis in aqueous solution at pH 7.1, 14 But, the trend of the theoretically calculated reduction potential values shown in Table 2 matches with the corresponding trend found using pulse radiolysis in aqueous solution at pH 71, 14. Therefore, on the basis of reactions of TMPD and TNM with T(6OH)• and T(5OH)• respectively, it is clear that T(5OH)• is a more “reducing” radical than T(6OH)• as predicted by theoretical calculations. T(6OH)• is known to be a oxidizing radical.4
-
Scheme 3.
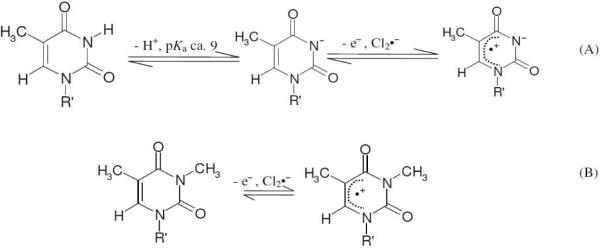
(A) Formation of T(−H)• via one-electron oxidation of N3-deprotonated thymine anion in dThd (R' = 2'-deoxyribose) and its various derivatives (e.g., L-dThd, α-dThd) and nucleotides (e.g., for 5'-TMP, R' = 2'-deoxyribose-5'-phosphate). (B) The production of N3-Me-dThd•+ by one-electron oxidation of N3-Me-dThd.
Figure 10.
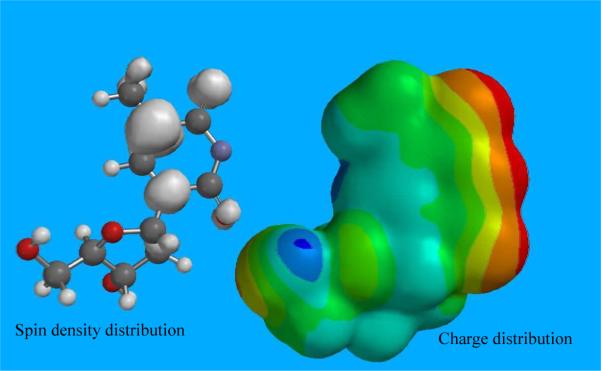
The optimized structure with spin densities and distribution of charge in T(−H)• of dThd calculated at the B3LYP/6-311++G**//B3LYP/6-31G* level in the gas phase. Red color region represents negative charge whereas the blue color region represents positive charge distributions. The radical has considerable charge separation and is near zwitterionic in nature.
Figure 1.
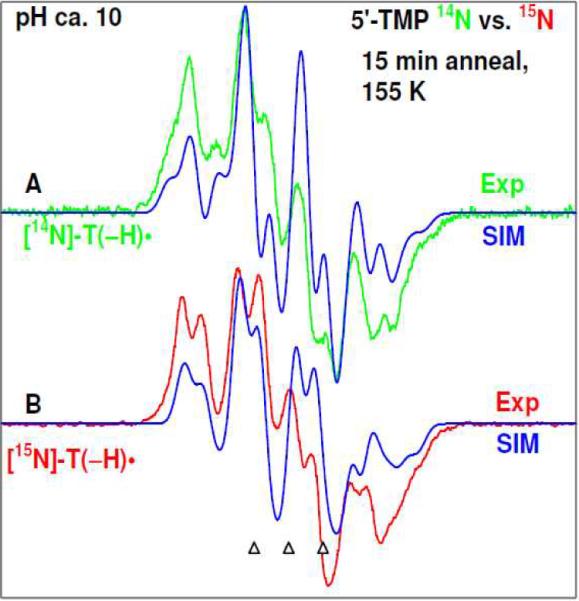
ESR spectrum of T(−H)• formed via annealing at 155 – 160 K after one-electron oxidation of the thymine base by Cl2•− in aqueous glassy sample of (A) unlabeled 5'-TMP, green color, and of (B) 15N labeled 5'-TMP, red color. The simulated spectra (blue) (for simulation parameters, see text) are superimposed on the top of the each experimentally recorded spectrum.
Figure 2.
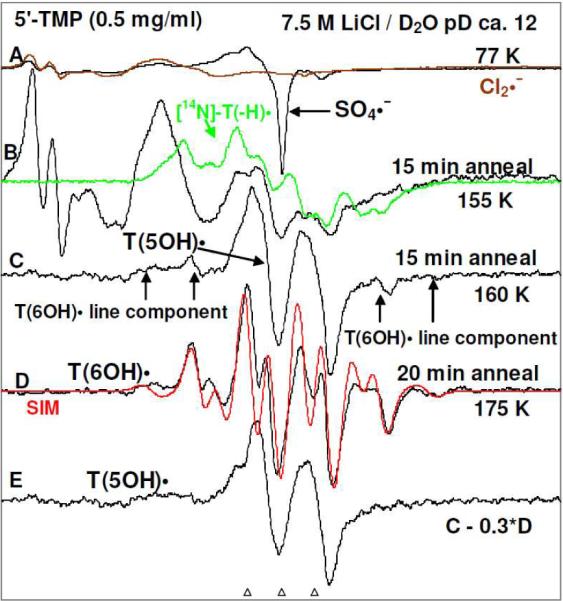
ESR spectra of (A) γ-irradiated glassy sample of 5'-TMP (0.5 mg/ml in 7.5 M LiCl/D2O) in the presence of electron scavenger K2S2O8 at pH ca. 12 (black); partial spectrum (brown, 220 G scan) of a pure Cl2•− spectrum is overlapped on the black spectrum in A to show the Cl2•− line components in the black spectrum. (B–D) Spectra (black) found after annealing to (B) 155 K for 15 min, (C) 160 K for 15 min, (D) 175 K for 20 min. The spectrum of T(−H)• (green, Figure 1A) is overlapped on spectrum (B). The red spectrum in (D) is the simulated spectrum of T(6OH)• which is a mixture of T(6OH)• with β C6-H HFCC of 9 G (80%) and with β C6-H HFCC of 45 G (20%). See text and supporting information Figure S2 for details of simulation. (E) The black spectrum, assigned to T(5OH)•, is obtained by 30% subtraction of spectrum (D) from spectrum (C). All spectra are recorded at 77 K.
Scheme 4.
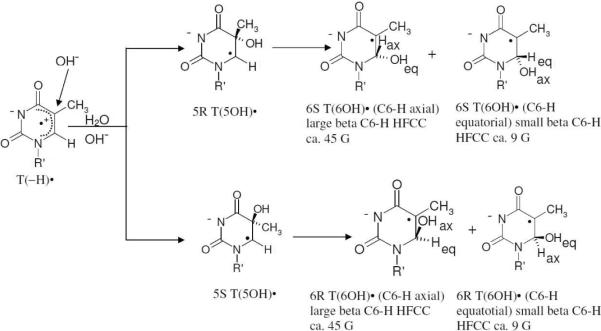
Addition of OH− to C5 of T(−H)• yields T(5OH)• (5R and/or 5S). Subsequently, T(5OH)• is converted to various diastereomers of T(6OH)•. The R and S steroisomers of T(6OH)• can not be distinguished by ESR spectroscopy and only the axial and equatorial conformers can be distinguished on the basis of β C6-H HFCC values whereas for T(5OH)• only C6αH hyperfine coupling is experimentally observable and it is same (ca. 20 G) for R or S.
Figure 3.
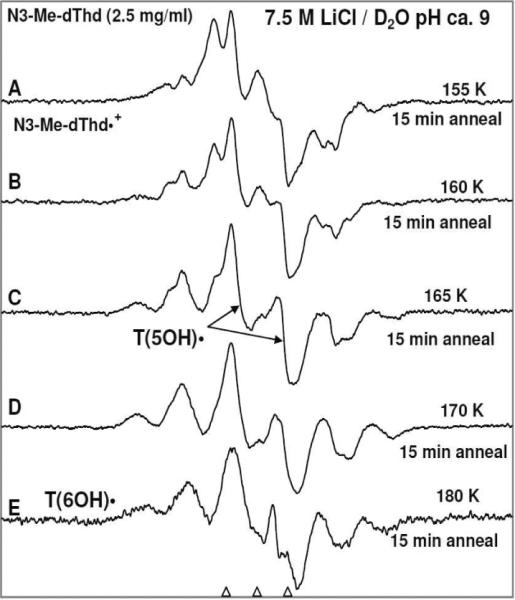
ESR spectra of (A) γ-irradiated glassy sample of N3-Me-dThd (2.5 mg/ml in 7.5 M LiCl/D2O) in the presence of electron scavenger K2S2O8 at pH ca. 9 and annealed to 155 K for 15 min. This spectrum is assigned to chiefly N3-Me-dThd•+ (see supporting information Figure S17). (B) 160 K for 15 min, (C) 165 K for 15 min (the central doublet is assigned to T(5OH)•), (D) 170 K for 15 min. (E) 180 K for 15 min. This spectrum is predominantly due to T(6OH)• with C6-H axial conformation (scheme 4) along with a small (ca. 15%) central doublet due to T(5OH)•.
Figure 4.
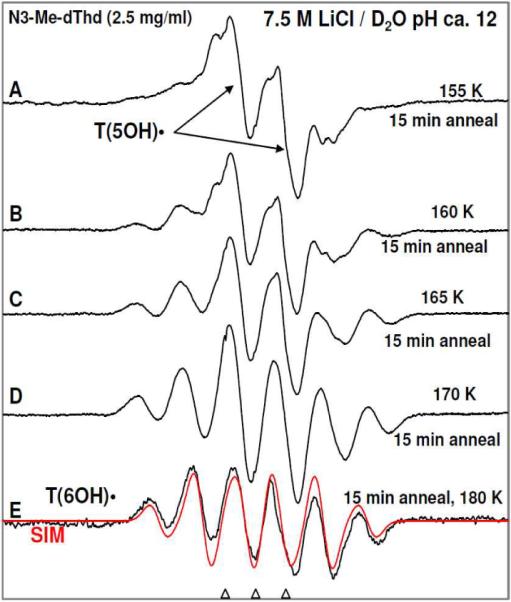
ESR spectra of (A) γ-irradiated glassy sample of N3-Me-dThd (2.5 mg/ml in 7.5 M LiCl/D2O) in the presence of electron scavenger K2S2O8 at pH ca. 12 and annealed to 155 K for 15 min and the central doublet is assigned to T(5OH)•. (B) 160 K for 15 min, (C) 165 K for 15 min, (D) 170 K for 15 min. (E) 180 K for 15 min. This spectrum is assigned to T(6OH)• in the C6-H axial conformation (scheme 4). The red spectrum is the simulated spectrum of T(6OH)• using HFCC values of each of the 3 β methyl protons (19 G) with β C6-H HFCC of 33 G, linewidth 8 G, and g = 2.0035.
Figure 5.
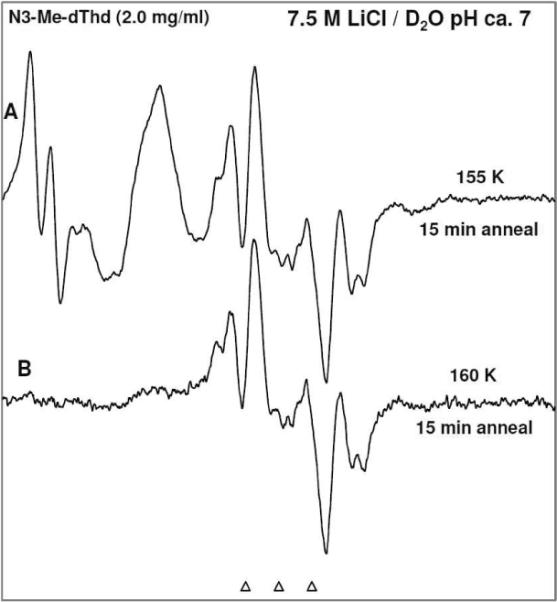
ESR spectra of (A) γ-irradiated glassy sample of N3-Me-dThd (2.5 mg/ml in 7.5 M LiCl/D2O) in the presence of electron scavenger K2S2O8 at pH ca. 12 and annealed to 155 K for 15 min showing line components due to Cl2•−, and UCH2• with some T(6OH)•. (B) After annealing at 160 K for 15 min showing 70% UCH2• and 30% T(6OH)•.
Figure 6.

ESR spectra obtained from dThd (black), [5',5”-D,D]-dThd (pink), [CD3]-dThd (thick sky blue), and [CD3,6D]-dThd (blue). These samples were prepared identically [concentration = 2 to 3 mg/ml, in the presence of the electron scavenger K2S2O8 at pH ca. 12], γ-irradiated to a dose of 1.4 kGy. (A) Spectra of T(5OH)• obtained via annealing to 155 K for 15 min. (B) Spectra of T(6OH)• obtained after annealing each sample to 170 K for 15 min.
Figure 7.
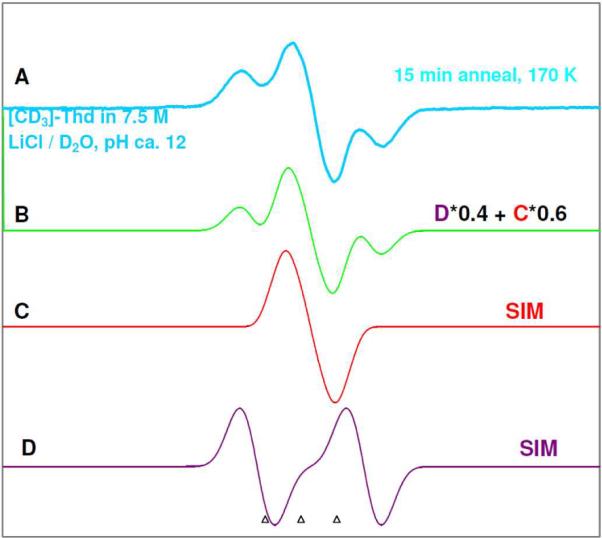
(A) The experimentally recorded (77 K) T(6OH)• spectrum (thick sky blue, also in Figure 7B) obtained in the glassy (7.5 M LiCl/D2O) sample of [CD3]-dThd sample (3 mg/ml) in the presence of the electron scavenger K2S2O8 at pH ca. 12, γ-irradiated to a dose of 1.4 kGy and progressively annealed to 170 K for 15 min. (B) Spectrum T(6OH)• (green) due to addition of 60% of spectrum (C) to 40% of spectrum (D). (C) Simulated T(6OH)• spectrum (red) using the parameters: HFCC values of 3 methyl deuterons 3.2 G and of C6-H 9 G, g = 2.0035 along with a mixed Lorentzian/Gaussian (1/1) linewidth of 6 G. These couplings are those expected from Table 1 and are unresolved. They only affect the lineshape. (D) Simulated T(6OH)• spectrum (violet) using the parameters: HFCC values of 3 methyl deuterons 3.2 G and of C6-H 39 G, g = 2.0035 along with a mixed Lorentzian/Gaussian (1/1) linewidth of 8 G. Here only the 39 G coupling is resolved.
Figure 8.

Dependence of 5'-TMP concentration in mg/ml on the type of conformer (C6-H axial or C6-H equatorial) of T(6OH)• formation via annealing at 175 K. ESR spectra of one-electron oxidized 5'-TMP (A) 5'-TMP (0.5 mg/ml). This spectrum is shown in Figure 2D in the manuscript, (B) 5'-TMP (3 mg/ml). (C) 5'-TMP (10 mg/ml) and this spectrum is shown in Figure S3D.
Figure 9.
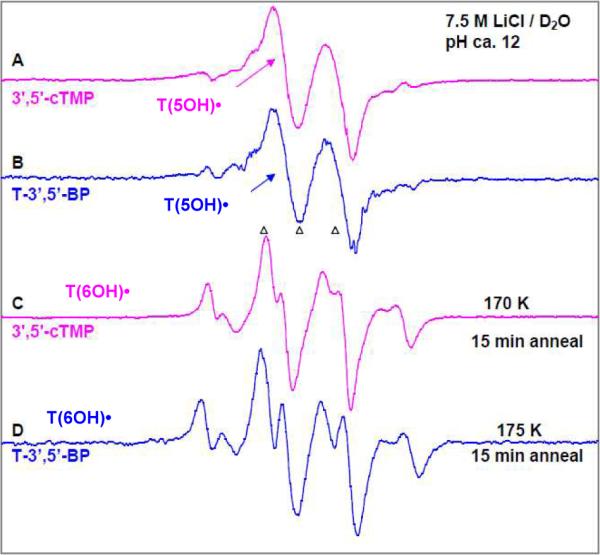
ESR spectra of T(5OH)• (the central anisotropic doublet) isolated from glassy sample of (A) 3',5'-cTMP (Pink) and (B) T-3',5'-BP (blue). Both these samples were prepared identically [concentration =3 mg/ml, in the presence of the electron scavenger K2S2O8 at pH ca. 12], γ-irradiated to a dose of 1.4 kGy, and annealed to 160 K for 15 min. ESR spectra of T(6OH)• in (C) 3',5'-cTMP (Pink) sample was annealed to 170 K for 15 min, and in (D) T-3',5'-BP sample annealed to 175 K for 15 min (Blue). The spectra obtained using 2',3'-dd-5'-TTP samples are found to be very similar to spectra (A) to (D) and hence are not shown here.
Table 1.
Hyperfine couplings and g-values for T(5OH)• and T(6OH)•.a
| Compound (3 mg/ml) | Hyperfine coupling constants (G) |
g-value | ||
|---|---|---|---|---|
| T(5OH)• | T(6OH)• | |||
|
| ||||
| αH(C6) | Me(3βH) | lpH(C6)/%b | ||
| dThd, L-dThd, α-dThd, [5',5"-D,D]-dThd | ca. 20 (1αH) | 2.0023 | ||
|
| ||||
| 21 | 9 (70%) | 2.0035 | ||
|
| ||||
| 21 | 39 (30%) | 2.0035 | ||
| N3-Me-dThd | ca. 20 (1αH) | 2.0023 | ||
|
|
||||
| 19c | 41c |
2.0035 | ||
| 19d | 33d | |||
| 5'-TMPe, [5',5"-D,D]-5'-TMP | ca. 20 (1αH) | 2.0023 | ||
|
|
||||
| 21 | 9 (80%) | 2.0035 | ||
|
|
||||
| 21 | 45 (20%) | 2.0035 | ||
| 3',5'-cTMP, T-3',5'-BP, 2',3'-dd-5'-TTP | ca. 20 (1αH) | 2.0023 | ||
|
|
||||
| 21 | 9 | 2.0035 | ||
For T(5OH)• on annealing (155–160K) recorded at 77 K and for T(6OH)• on annealing (175–180 K) recorded at 77 K; both at pH (ca. 12), and concentration (2.5 to 3 mg/ml) unless otherwise stated.
The experimentally determined percentage composition of C6-H equatorial and axial conformers of T(6OH)• are in parenthesis.
On annealing at 175 K recorded at 77 K (see supporting information Figure S20).
On annealing at 180 K recorded at 77 K (see Figure 4).
The C6-H β-proton hyperfine couplings are found to be concentration dependent (see Figure 8).
Figure 11.

B3LYP/6-31G** calculated potential energy surface for the OH transfer from C5 atom to the C6 atom in the thymine base of hydrated T(5OH)• with respect to the change of dihedral angle Φ (C5-C4-C6-C5') of thymine ring. The structures at the beginning (ca. -32°) and end (ca. 16°) are fully optimized. The up and down arrows at C5 show the movement of C5 atom with respect to C4, C6, and C5' atoms. The energies (kcal/mol) were calculated in gas phase ( ) and in PCM (
) and in PCM ( ). The variation of spin density distribution along the PES is also shown at some chosen points (see supporting information Table T1).
). The variation of spin density distribution along the PES is also shown at some chosen points (see supporting information Table T1).
Figure 12.

B3LYP/6-31G** calculated potential energy surface for the OH transfer from C5 to C6 atoms of hydrated TNH(5OH)• with respect to the change of dihedral angle Φ (C5-C4-C6-C5') of thymine ring. The structures at the beginning (ca.-32°) and end (ca. 15°) are fully optimized. The up and down arrows at C5 show the movement of C5 atom with respect to C4, C6, and C5' atoms. The energies (kcal/mol) were calculated in the gas phase ( ) and in PCM (
) and in PCM ( ). The variation of spin density distribution along the PES is also shown at some chosen points, for details see supporting information Table T1.
). The variation of spin density distribution along the PES is also shown at some chosen points, for details see supporting information Table T1.
Figure 13.
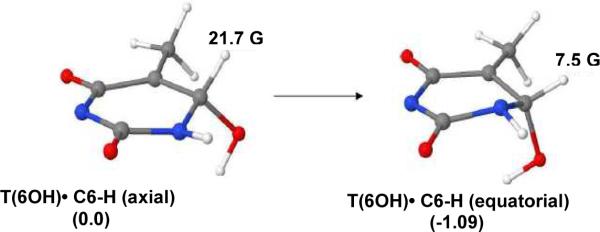
B3LYP/6-31G** fully optimized conformations of T(6OH)• (C6-H axial) and T(6OH)• (C6-H equatorial) in the gas phase with thymine base deprotonated at N3. The relative stabilities of the two conformers of T(6OH)• in kcal/mol are provided in parenthesis.
Table 2.
Comparison of the calculated values of reduction potential of T•+ , T(−H)•, T(5OH)• , and T(6OH)•, with the corresponding values reported in the literature.
| Redox couple | Calculated reduction potential (V), this work | Experimental reduction potential (V) |
|---|---|---|
| T•+ / T | E0 = 2.13 | E7 = 1.7a E0 = 2.09b |
| T(−H)• / T(−H)− | E0 = 1.22 | - |
| T(5OH)+ / T(5OH)• | E0 = 0.38 | - |
| T(OH)+c / T(6OH)• | E0 = 0.73 | - |
Ref. 1 pp 216: This value was obtained employing pulse radiolysis of aqueous solution of dThd at room temperature.
Ref. 64: This value was obtained employing cyclic voltametry of solution of dThd in dimemthylformamide (DMF) at room temperature.
After one-electron oxidation of T(6OH)• the OH moiety immediately transfers from C6 to C5 forming T(5OH)+.
Conclusions
The following salient findings are drawn from the present work:
-
The Spin and Charge Distribution in T(−H)•:
Our DFT calculated charge distribution in T(−H)• is near zwitterion in character with the sites of OH− addition occurring at the positive sites. However, for both T(−H)• and N3-Me-dThd•+, the spin density is largely localized to the C5 and this is the initial site of OH− addition. Thus, while both spin and charge distributions in T(−H)• and N3-Me-dThd•+ are contributors to the formation of T(5OH)•, the spin density directs the OH− attack site likely through initial weak three electron bond formation of a σσ* type between the radical site and a lone pair on the OH−.
-
Thermodynamics of the Site of OH−addition to one electron oxidized thymine derivatives:
The experimental studies in this work have clearly shown that C5 is the favored site of initial addition of OH− to both T(−H)• and N3-Me-dThd•+. However, theoretical calculations (this work, and previous efforts1, 11, 65) predict a greater thermodynamic stability of T(6OH)• over that of T(5OH)•. This clearly shows that the addition of OH− to T(−H)• is kinetically and not thermodynamically controlled.
We note that formation of the allylic UN3HCH2• (or UCH2•)1,2,19–29 via deprotonation of one-electron oxidized thymine derivatives is a reaction that competes with the addition of OH− (scheme 1) and is found to be the case here for N3-Me-dThd at pH 7. However, pulse radiolysis studies as well as ESR studies with flow systems of thymine derivatives in aqueous solution at room temperatures have established that at alkaline pHs (pH ≥ 10), OH− addition to C5–C6 double bond of one-electron oxidized thymine is favored over deprotonation from the methyl group at C5 to form UCH2•.1,19,22 In agreement our work with T(−H)• clearly shows that the rate of formation of T(5OH)• is dependent on the OH− concentration with no observable formation at pH ca. 8–10 with rapid formation at pH ca.12. For N3-Me-dThd•+ OH− addition to C5 does occur at pH ca. 9 but only at higher annealing temperatures whereas at pH ca.12 OH− addition occurs more readily as expected.
-
Oxidizing and reducing nature of T(5OH)• and T(6OH)•:
Using uracil and its derivatives, the work of Fujita and Steenken4 and Fujita and co-workers10 have unequivocally established that the T(5OH)• has reducing properties (quickly reduces tetranitromethane (TNM)) while T(6OH)• has oxidizing properties (quickly oxidizes N,N,N',N'-tetramethylphylene diamine (TMPD)). The theoretically calculated reduction potential values of T(5OH)• and T(6OH)• (Table 2) along with the predictions of relative reducing and oxidizing properties based on theoretically calculated ionization potentials and electron affinities65 clearly support the “reducing” nature of T(5OH)• and “oxidizing” nature of T(6OH)•. Furthermore, β-oxoalkyl type radicals such as T(6OH)• have been shown to have oxidizing properties.4
-
The conversion of T(5OH)• to T(6OH)• is thermodynamically controlled:
The experimentally observed conversion of T(5OH)• to T(6OH)• via annealing at 170 to 175 K clearly show that kinetically favored T(5OH)• intermediate is converted to a more stable species T(6OH)•. Theoretical calculations reported in this work also show that T(5OH)• produced via addition of OH− to the C5–C6 double bond of one-electron oxidized thymine (T(−H)• and T•+) is thermodynamically less stable than T(6OH)•.
This conclusion is supported by the literature. For example, pulse radiolysis with conductivity detection studies of aqueous solution (pH 7) of 1,3-dimethyluracil at room temperature have shown predominant (≥ 90%) formation of 5-hydroxy-1,3-dimethyluracilyl-C6 (C5-OH adduct) radical as confirmed by the quick reduction of tetranitromethane.1,2,4–12 However, under acidic conditions (pH 3), 5-hydroxy-1,3-dimethyluracilyl-C6 (C5-OH adduct) radical has been reported to convert to the corresponding 6-hydroxy-1,3-dimethyluracilyl-C5 (C6-OH adduct) radical.1,5–9,11,12 Thus, the conversion of T(5OH)• to T(6OH)• observed in this work appears to be thermodynamically controlled.
We note that ESR studies of addition of water molecules to one-electron oxidized halopyrimidines in 5.3 M H2SO4 glasses on annealing at 155 to 165 K show formation of C6-OH adduct radical most likely from C5-OH adduct radical to C6-OH adduct radical conversion.66
-
The conversion of T(5OH)• to T(6OH)• is unimolecular:
The conversion of T(5OH)• to T(6OH)• at 165 – 175 K is found to be independent of the concentrations (0.5 to 10 mg/ml) in dThd and in thymine nucleotides. For all concentrations during the conversion of T(5OH)• to T(6OH)•, the total spectral intensity of the ESR spectra in the range of 165 to 175 K remained unchanged within experimental uncertainty. For N3-Me-dThd the T(5OH)• adduct initially formed converts to T(6OH)• as quickly at pH 9 as at pH 12. This is in accord with a unimolecular conversion of T(5OH)• to T(6OH)• as proposed in this work.
-
Is the conversion of T(5OH)• to T(6OH)• also acid and base catalyzed?
Previous work in aqueous solution, under acidic conditions (pH 3) at room temperature reports that the 5-hydroxy-1,3-dimethyluracil-6-yl radical of 1,3-dimethyluracil (C5-OH adduct) converts to its corresponding 6-hydroxy-1,3-dimethyluracilyl-C5 (C6-OH adduct) radical by a acid catalyzed mechanism.1,5–9,11,12 Our theoretical and experimental work with dThd and thymine nucleotides suggests unimolecular conversion of T(5OH)• to T(6OH)• occurs under alkaline conditions at 170 K. However, we cannot also eliminate some contribution of a catalytic action of OH− by concerted addition to T(5OH)• and elimination to form T(6OH)•.
Our results suggest that alkaline conditions at room temperature, conversion of T(5OH)• to T(6OH)• would be fast and perhaps too short to be detected by pulse radiolysis (scheme 1). Indeed, pulse radiolysis studies of various one-electron oxidized thymine derivatives at room temperature under alkaline conditions (pH ≥ 10) show predominant formation of T(6OH)• reportedly as a result of OH− addition at C6 in T(−H)• (scheme 1).19 Our experimental work and low energy barrier predicted by our theoretical calculations for the rearrangement of T(5OH)• to T(6OH)• clearly shows that formation of T(6OH)• results from the metastable intermediate T(5OH)•.
Supplementary Material
Acknowledgment
AA, AK, ANH, BJP, VP, and MDS thank the National Cancer Institute of the National Institutes of Health (Grant RO1CA45424) for support. YL and SFW thank the National Cancer Institute of the National Institutes of Health (Grant SC1CA138176) for support. AA and MDS thank Prof. A. W. Bull for helpful suggestions.
Footnotes
Supporting Information available: Supporting information contains the following: (1) Synthesis of [5',5"-D,D]-Thymidine 5'-monophosphate as triethylammonium salt; (2) Figure S1 showing spin density calculations of different protonation states of one-electron oxidized 5'-TMP; (3) Figure S2 showing the simulation of T(6OH)• formation in 5'-TMP and its comparison to the experimentally recorded T(6OH)• spectrum; (4) Figure S3 showing T(5OH)• formation and its conversion to T(6OH)• in 5'-TMP at high concentration (10 mg/ml); (5) Figure S4 showing T(5OH)• formation and its conversion to T(6OH)• in [5',5”-D,D]-dThd (3 mg/ml); (6) Figure S5 showing T(5OH)• formation and its conversion to T(6OH)• in [CD3,6D]-dThd (2 mg/ml); (7) Figure S6 showing T(5OH)• formation and its conversion to T(6OH)• in [5',5”-D,D]-5'-TMP (0.5 mg/ml); (8) Figure S7 showing T(5OH)• formation and its conversion to T(6OH)• in dThd (3 mg/ml); (9) Figure S8 showing T(5OH)• formation and its conversion to T(6OH)• in dThd at high concentration (10 mg/ml); (10) Figure S9 showing T(5OH)• formation and its conversion to T(6OH)• in 5'-TDP (3 mg/ml) and in 5'-TTP (3 mg/ml); (11) Figure S10 showing T(5OH)• formation and its conversion to T(6OH)• in [CD3]-dThd (3 mg/ml); (12) Figure S11 showing T(5OH)• formation and its conversion to T(6OH)• in dThd in H2O; (13) Figure S12 showing T(5OH)• formation and its conversion to T(6OH)• in L-dThd; (14) Figure S13 showing T(5OH)• formation and its conversion to T(6OH)• in α-dThd (3 mg/ml); (15) Figure S14 showing T(5OH)• formation and its conversion to T(6OH)• in α-dThd (10 mg/ml); (16) Figure S15 showing the optimized structure with spin densities and distribution of charge in N3-Me-dThd•+ of N3-Me-dThd; (17) Figure S16 showing dependence of the number of phosphate groups at 5'-site on the conformation of T(6OH)• formed on annealing at 170 to 175 K; (18) Figure S17 showing formation of N3-Me-dThd•+ in N3-Me-dThd via one-electron oxidation; (19) Figure S18 showing the lack of a reaction of OH− with T(−H)• formed in 5'-TMP at pH ca. 10; (20) Figure S19 showing the T(6OH)• formation in N3-Me-dThd at pH ca. 9 and at pH ca. 12 via annealing at 180 K; (21) Figure S20 showing the T(6OH)• formation (experimentally reorded spectrum and its simulation in N3-Me-dThd at pH ca. 12 via annealing upto 175 K) (22) Table T1 showing B3LYP/6-31G** calculated Mulliken charge and spin density distribution (in brackets) in T(−H)• + −OH and Thymine + •OH in C5- and C6- adduct radicals and at different dihedral angles. This information is available free of charge via the internet at http://pubs.acs.org/.
References
- 1.von Sonntag C. Free-radical-induced DNA Damage and Its Repair. Springer-Verlag; Berlin, Heidelberg: 2006. pp. 222–227. [Google Scholar]
- 2.von Sonntag C. Adv. Quantum Chem. 2007;52:5–20. [Google Scholar]
- 3.Myers LS, Jr., Hollis LM, Theard LM, Peterson FC, Warnick A. J. Am. Chem. Soc. 1970;92:2875–2882. doi: 10.1021/ja00712a046. [DOI] [PubMed] [Google Scholar]
- 4.Fujita S, Steenken S. J. Am. Chem. Soc. 1981;103:2540–2545. [Google Scholar]
- 5.Al-Sheikhly M, von Sonntag C. Z. Naturforsch. 1983;38b:1622–1629. [Google Scholar]
- 6.Schchmann MN, Steenken S, Wroblewski J, von Sonntag C. Int. J. Radiat. Biol. 1984;49:225–232. doi: 10.1080/09553008414551341. [DOI] [PubMed] [Google Scholar]
- 7.von Sonntag C, Schuchmann H-P. Int. J. Radiat. Biol. 1986;49:1–34. doi: 10.1080/09553008514552201. [DOI] [PubMed] [Google Scholar]
- 8.Schuchmann H-P, Deeble DJ, Olbrich G, von Sonntag C. Int. J. Radiat. Biol. 1987;51:441–453. doi: 10.1080/09553008714550931. [DOI] [PubMed] [Google Scholar]
- 9.von Sonntag C. Free Rad. Res. Commun. 1987;2:217–224. doi: 10.3109/10715768709065286. [DOI] [PubMed] [Google Scholar]
- 10.Rashid R, Mark F, Schuchmann H-P, von Sonntag C. Int. J. Radiat. Biol. 1991;59:1081–1100. doi: 10.1080/09553009114551001. [DOI] [PubMed] [Google Scholar]
- 11.Fujita S, Horii H, Taniguchi R, Lakshmi S, Renganathan R. Radiat. Phys. Chem. 1996;48:643–649. [Google Scholar]
- 12.Naumov S, von Sonntag C. Radiat. Res. 2008;169:355–363. doi: 10.1667/RR1081.1. [DOI] [PubMed] [Google Scholar]
- 13.Wardman P. J. Phys. Chem. Ref. Data. 1989;18:1637–1755. [Google Scholar]
- 14.Steenken S, Jovanovic SV. J. Am. Chem. Soc. 1997;119:617–618. [Google Scholar]; The reduction potential at pH 7 (i.e., midpoint potential (E7)) values of the DNA components at room temperature in aqueous solutions are: E7 (dG(−H)•/dG) = 1.29 V, E7 (dA(−H)•/dA) = 1.42 V, E7 (dT•/dT) = 1.7 V, E7 (dC•/dC) =1.6 V, and E7 (dR•/dR) >> 1.8 V (dR = 2'-deoxyribose). The pKa of the guanine cation radical (G•+) being 3.9, the reduction potential of G•+ is reported as (E° =1.58 V) –see
- 15.Khanduri D, Adhikary A, Sevilla MD. J. Am. Chem. Soc. 2011;133:4527–4537. doi: 10.1021/ja110499a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Adriaanse C, Sulpzi M, VandeVondele J, Sprik M. J. Am. Chem. Soc. 2009;131:6046–6047. doi: 10.1021/ja809155k. [DOI] [PubMed] [Google Scholar]
- 17.Pryor WA. Free Radical Biol. Med. 1988;4:219–223. doi: 10.1016/0891-5849(88)90043-3. [DOI] [PubMed] [Google Scholar]
- 18.Wu YD, Mundy CJ, Colvin ME, Car R. J. Phys Chem A. 2004;108:2922–2927. [Google Scholar]
- 19.Deeble DJ, Schuchmann MN, Steenken S, von Sonntag C. J. Phys. Chem. 1990;94:8186–8192. [Google Scholar]
- 20.Behrens G, Hildenbrand K, Schulte-Frohlinde D, Herak JN. J. Chem. Soc. Perkin Trans. II. 1988:305–317. [Google Scholar]
- 21.Hildenbrand K, Behrens G, Schulte-Frohlinde D, Herak JN. J. Chem. Soc. Perkin Trans. II. 1989:283–289. [Google Scholar]
- 22.Hildenbrand K. Z. Naturforsch. C. 1990;45:47–58. doi: 10.1515/znc-1990-1-210. [DOI] [PubMed] [Google Scholar]
- 23.Sevilla MD. J. Phys. Chem. 1971;75:626–631. doi: 10.1021/j100675a005. [DOI] [PubMed] [Google Scholar]
- 24.Sevilla MD, Van Paemel C, Nichols C. J. Phys. Chem. 1972;76:3571–3577. doi: 10.1021/j100668a012. [DOI] [PubMed] [Google Scholar]
- 25.Sevilla MD, Van Paemel C, Zorman G. J. Phys. Chem. 1972;76:3577–3582. doi: 10.1021/j100668a013. [DOI] [PubMed] [Google Scholar]
- 26.Sevilla MD. J. Phys. Chem. 1976;80:1898–1901. [Google Scholar]
- 27.Sevilla MD, Engelhardt ML. Faraday Discussions Chem. Soc. 1978;63:255–263. [Google Scholar]
- 28.Malone ME, Symons MCR, Parker AW. J. Chem. Soc. Perkin Trans 2. 1993:2067–2075. [Google Scholar]
- 29.Lange M, Weiland B, Hüttermann J. Int. J. Radiat. Biol. 1995;68:475–486. doi: 10.1080/09553009514551441. [DOI] [PubMed] [Google Scholar]
- 30.Adhikary A, Malkhasian AYS, Collins S, Koppen J, Becker D, Sevilla MD. Nucleic Acids Res. 2005;33:5553–5564. doi: 10.1093/nar/gki857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Adhikary A, Becker D, Collins S, Koppen J, Sevilla MD. Nucleic Acids Res. 2006;34:1501–1511. doi: 10.1093/nar/gkl026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Adhikary A, Kumar A, Becker D, Sevilla MD. J. Phys Chem. B. 2006;110:24170–24180. doi: 10.1021/jp064361y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Khanduri D, Collins S, Kumar A, Adhikary A, Sevilla MD. J. Phys. Chem. B. 2008;112:2168–2178. doi: 10.1021/jp077429y. [DOI] [PubMed] [Google Scholar]
- 34.Adhikary A, Khanduri D, Kumar A, Sevilla MD. J. Phys. Chem. B. 2008;112:15844–15855. doi: 10.1021/jp808139e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Verheyden JPH, Moffatt JG. In: Synthetic Procedures in Nucleic Acid Chemistry. Zorbach WW, Tipson RS, editors. Wiley-Interscience; New York: 1968. pp. 383–387. [Google Scholar]
- 36.Yoshikawa M, Kato T, Takenishi T. Tetrahedron Lett. 1967;8:5065–5068. doi: 10.1016/s0040-4039(01)89915-9. [DOI] [PubMed] [Google Scholar]
- 37.Adhikary A, Becker D, Palmer BJ, Heizer AN, Sevilla MD. J. Phys. Chem. B. 2012;116:5900–5906. doi: 10.1021/jp3023919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shukla LI, Adhikary A, Pazdro R, Becker D, Sevilla MD. Nucleic Acids Res. 2004;32:6565–6574. doi: 10.1093/nar/gkh989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Becke AD. J. Chem. Phys. 1993;98:1372–1377. [Google Scholar]
- 40.Stephens PJ, Devlin FJ, Frisch MJ, Chabalowski CF. J. Phys. Chem. 1994;98:11623–11627. [Google Scholar]
- 41.Lee C, Yang W, Parr RG. Phys. Rev. B. 1988;37:785–789. doi: 10.1103/physrevb.37.785. [DOI] [PubMed] [Google Scholar]
- 42.Hermosilla L, Calle P, García de la Vega J. M., Sieiro C. J. Phys. Chem. A. 2005;109:1114–1124. doi: 10.1021/jp0466901. [DOI] [PubMed] [Google Scholar]
- 43.Hermosilla L, Calle P, García de la Vega J. M., Sieiro C. J. Phys. Chem. A. 2006;110:13600–13608. doi: 10.1021/jp064900z. [DOI] [PubMed] [Google Scholar]
- 44.Barone V, Cimino P. Chem. Phys. Lett. 2008;454:139–143. [Google Scholar]
- 45.Close DM. J. Phys. Chem. A. 2010;114:1860–1187. doi: 10.1021/jp906963f. [DOI] [PubMed] [Google Scholar]
- 46.Cance's MT, Mennucci B, Tomasi J. J. Chem. Phys. 1997;107:3032–3041. [Google Scholar]
- 47.Mennucci B, Tomasi J. J. Chem. Phys. 1997;106:5151–5158. [Google Scholar]
- 48.Mennucci B, Cance's E, Tomasi J. J. Phys. Chem. B. 1997;101:10506–10517. [Google Scholar]
- 49.Tomasi J, Mennucci B, Cance's E. J. Mol. Struct. (THEOCHEM) 1999;464:211–226. [Google Scholar]
- 50.Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA, Nakatsuji H, Caricato M, Li X, Hratchian HP, Izmaylov AF, Bloino J, Zheng G, Sonnenberg JL, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Montgomery JA, Jr., Peralta JE, Ogliaro F, Bearpark M, Heyd JJ, Brothers E, Kudin KN, Staroverov VN, Kobayashi R, Normand J, Raghavachari K, Rendell A, Burant JC, Iyengar SS, Tomasi J, Cossi M, Rega N, Millam JM, Klene M, Knox JE, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Martin RL, Morokuma K, Zakrzewski VG, Voth GA, Salvador P, Dannenberg JJ, Dapprich S, Daniels AD, Farkas O, Foresman JB, Ortiz JV, Cioslowski J, Fox DJ. Gaussian 09. Gaussian, Inc.; Wallingford CT: 2009. [Google Scholar]
- 51.GaussView. Gaussian, Inc.; Pittsburgh, PA: 2003. [Google Scholar]
- 52.Jmol: An open-source Java viewer for chemical structures in 3D; Jmol Development Team, An Open-Science Project. 2004 available at http://jmol.sourceforge.net.
- 53.SPARTAN. version 10 Wavefunction, Inc.; Irvine, CA: 2010. [Google Scholar]
- 54.Adhikary A, Collins S, Khanduri D, Sevilla MD. J. Phys. Chem. B. 2007;111:7415–7421. doi: 10.1021/jp071107c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Adhikary A, Khanduri D, Pottiboyina V, Rice CT, Sevilla MD. J. Phys. Chem. B. 2010;114:9289–9299. doi: 10.1021/jp103403p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Heller C, McConnell HM. J. Chem. Phys. 1960;32:1535–1539. [Google Scholar]
- 57.Schweizer MP, Broom AD, Ts'o POP, Hollis DP. J. Am. Chem. Soc. 1968;68:1042–1055. doi: 10.1021/ja01006a035. [DOI] [PubMed] [Google Scholar]
- 58.Sprecher CA, Johnson WC., Jr. Biopolymers. 1977;16:2243–2264. doi: 10.1002/bip.1977.360161012. [DOI] [PubMed] [Google Scholar]
- 59.Bernhard WA. Adv. Radiat. Biol. 1981;9:249–251. [Google Scholar]
- 60.Kumar A, Pottiboyina V, Sevilla MD. J. Phys. Chem. B. 2012;116:9409–9416. doi: 10.1021/jp3059068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Rao PS, Hayon E. J. Phys. Chem. 1975;79:397–402. [Google Scholar]
- 62.Crespo-Herńandez CE, Close DM, Gorb L, Leszczynski J. J. Phys. Chem. B. 2007;111:5386–5395. doi: 10.1021/jp0684224. [DOI] [PubMed] [Google Scholar]
- 63.Tureček F. Adv. Quantum Chem. 2007;52:89–120. [Google Scholar]
- 64.Saidel CAM, Schulz A, Sauer M. J. Phys. Chem. 1996;100:5541–5553. [Google Scholar]
- 65.Colson A-O, Sevilla MD. J. Phys. Chem. 1995;99:13033–13037. [Google Scholar]
- 66.Riederer H, Hüttermann J. J. Phys. Chem. 1982;86:3454–3463. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.