

Abstract
SAA has been shown to have potential proinflammatory properties in inflammatory diseases such as atherosclerosis. These include induction of tumor necrosis factor α, interleukin-6, and monocyte chemoattractant protein 1 in vitro. However, concern has been raised that these effects might be due to use of recombinant SAA with low level of endotoxin contaminants or its non-native forms. Therefore, physiological relevance has not been fully elucidated. In this study, we investigated the role of SAA in the production of inflammatory cytokines. Stimulation of mouse monocyte J774 cells with lipid-poor recombinant human SAA and purified SAA derived from cardiac surgery patients, but not ApoA-I and ApoA-II, elicited pro-inflammatory cytokines like granulocyte colony stimulating factor (G-CSF). However, HDL-associated SAA failed to stimulate production of these cytokines. Using neutralizing antibodies against toll like receptor (TLR) 2 and 4, we could evaluate that TLR 2 is responsible for G-CSF production by lipid-poor SAA. To confirm these data in vivo, we expressed mouse SAA in SAA deficient C57BL/6 mice using an adenoviral vector. G-CSF was identically expressed in SAA-Adenoviral infected mice as well as in control null-Adenoviral mice at the early time points (4–8 h) and could not be detected in plasma 24 h after infection when plasma SAA levels were maximally elevated, indicating that adenoviral vector rather than SAA affected G-CSF levels. Taken together, our findings suggest that lipid-poor SAA, but not HDL-associated SAA, stimulates G-CSF production and this stimulation is mediated through TLR 2 in J774 cells. However, its physiological role in vivo remains ambiguous.
Keywords: SAA, Cytokine, G-CSF, Acute phase, HDL
1. Introduction
Notable amongst the metabolic changes that affect lipid and lipoproteins during inflammation are the structural alterations in HDL [1]. Serum amyloid A protein (SAA) is induced by cytokines with the plasma concentration increasing up to a 1000-fold [2]. SAA is mostly associated with HDL and can even become the major apolipoprotein of this particle [2]. Teleologically these dramatic changes in HDL composition during inflammation likely serve a short term purpose to allow the organism to survive a noxious assault. However, the precise role that SAA plays during inflammation remains unclear. Induced by inflammatory cytokines, SAA itself has been reported to have a number of pro-inflammatory effects, particularly its ability to induce cytokines, possibly setting up an “autocrine” amplification mechanism [3,4]. It is thus important to establish definitively whether SAA can initiate, maintain or even amplify inflammation. Human and mouse SAAs are expressed as a number of distinct, yet highly homologous isoforms and it is the major acute phase isoforms that have been linked to cytokine induction [5]. The two major acute phase SAAs of humans and mice, SAA1/SAA2 and SAA1.1/SAA2.1 respectively, are produced mainly in the liver and are encoded by two homologous genes. Mouse SAA3 is produced extrahepatically while in humans SAA3 is a pseudogene [5]. However, the vast majority of studies have been conducted with a recombinant human SAA (rSAA) that constitutes a hybrid molecule that is not identical to any of the known human SAA isoforms. This recombinant chimera has been reported to have a number of pro-inflammatory effects. Recently it was reported that the pro-inflammatory activity of rSAA is not shared by endogenous human SAA [6].
Here we set out to validate the ability of rSAA, as well as isolated human and mouse acute phase SAAs, to induce cytokines, both in lipid-poor and HDL-associated forms. Results from these studies confirmed the ability of native lipid-poor human and mouse SAA to induce cytokines in vitro. However HDL-associated SAA lacked this ability and adenoviral expression of SAA failed to induce cytokines in vivo. The physiological relevance of cytokine induction by lipid-poor SAAs needs to be further examined. Given the lipophilic nature of this apolipoprotein it will only occur at sites when SAA is in a “free” lipid-poor form. Such putative sites are likely restricted to where high concentrations of remodeling factors are present.
2. Materials and methods
2.1. Reagents
Recombinant human SAA (rSAA), corresponding to human SAA1α except for an amino terminal methionine and the substitution of asparagine for aspartic acid at position 60 and arginine for histidine at position 71, was purchased from Peprotech (Rocky Hill, NJ) and contained less than 0.1 ng endotoxin/μg protein as stated by vendor or determined by limulus assay. Apolipoprotein AII (apoA-II) was obtained from Biodesign International (Saco, Maine). Lipopolysaccharides (LPS) from Escherichia coli 0111:B4 was purchased from Sigma–Aldrich (St. Louis, MO). The blocking antibodies anti-Toll Like Receptor 2 (clone T2.5), anti-Toll Like Receptor 4 (clone MTS510) and their isotype control IgG (mouse IgG1 and rat IgG2a) were from eBioscience (San Diego, CA). G-CSF enzyme-linked immunosorbent assay (ELISA) kit was purchased from R&D System (Minneapolis, MN). Other ELISA Assay Kits were obtained from BD Biosciences (Franklin Lakes, NJ). The Limulus Amebocyte Lysate Kit (QCL-1000) was from Cambrex (East Rutherford, NJ).
2.2. Human subjects
Blood was collected from healthy volunteers for isolation of normal (N) HDL, and from patients undergoing cardiac surgery using a membrane oxygenator (coronary artery bypass, valve replacement), 24 h post-operatively for isolation of acute phase (AP) HDL [7]. Blood was only collected from patients who underwent successful uncomplicated surgery and who gave informed consent. The study was approved by the University of Kentucky Medical Institutional Review Board.
2.3. Animals
C57BL/6 mice were obtained from Jackson Laboratories. Mice lacking SAA1.1 and SAA 2.1 (SAAKO mice) were generated by targeted deletion of both mouse acute phase SAA genes SAA1 and SAA2 (InGenious Targeting Laboratory Inc. Stony Brook, NY) using embryonic stem cells derived from C57BL/6×129 SVEV mice [8]. The targeting vector contained a neo cassette that replaced ∼10.1 kb of SAA1 and SAA2, including exon 2 of both oppositely orientated genes. Mice were maintained in a pathogen-free facility under equal light–dark cycles with free access to water and food. To elicit an AP response, 12–16 week-old mice were injected intra-peritoneally with 1 μg LPS per gram of body weight or, alternatively, subcutaneously with 0.5 ml 2% (w/v) AgNO3 for a sterile AP response. After 24 h the mice were humanely killed and plasma was collected for preparation of HDL. All procedures were carried out in accordance with PHS policy and approved by the Veterans Administration Medical Center Institutional Animal Care and Use Committee (Assurance number A3506-01).
2.4. HDL isolation
Mouse and human HDL (m HDL, hu HDL) (d = 1.063–1.21 g/mL) were isolated from C57BL/6 mouse plasma or human plasma by density gradient ultracentrifugation, dialyzed against 150mmol/L NaCl, 0.01% (w/v) EDTA, sterile filtered, and stored under argon gas at 4 °C [2]. Protein concentrations were determined by the method of Lowry et al. [9]. HDL apoproteins were quantitated by densitometric scanning of SDS gels.
2.5. Electrofocusing
Aliquots of mouse plasma (7 μL) were subjected to electrofocusing as previously described [10], using an ampholine gradient consisting of 20% (v/v) ampholines pH 3–10, 40% (v/v) ampholines pH 4–6.5, and 40% (v/v) ampholines pH 7–9 (Pharmacia LKB Biotechnology Inc.). Electrofocused samples were subjected to immunochemical analysis as described using rabbit anti mouse SAA antisera [10].
2.6. Purification of SAA and apoA-I
Human SAA (huSAA), human apoA-I and mouse SAA (mSAA) were purified, essentially as described [11] from AP HDL isolated from the plasma of patients undergoing cardiac surgery or mice injected with LPS or AgNO3. Briefly, ∼20 mg AP HDL was delipidated and the delipidated proteins separated by gel filtration on a Seph-acryl S-200 column in a buffer containing 7 mol/L urea, 20 mmol/L Tris, 150 mmol/L NaCl, 1 mmol/L EDTA pH 8.4. SAA-containing fractions were identified by SDS-PAGE, pooled and dialyzed against 2 mmol/L Tris, 15 mmol/L NaCl, and 0.1 mmol/L EDTA pH 8.4 prior to 10-fold concentration.
2.7. AdSAA1.1. and AdSAA2.1 construction
AdSAA1.1 and AdSAA2.1 were constructed using a published method [12]. Briefly, SAA coding sequences were amplified from cDNA prepared from the liver of mice injected with LPS using Platinum® Taq DNA polymerase high fidelity (Invitrogen, Carlsbad, CA) and the following oligonucleotide primers: 5′-GCCGCAGGTACCAT-GAAGCTACTCACCAGCCTG (forward primer for SAA1.1 and SAA2.1); 5′-GCGCTCGAGAGGACCCCAACACAGCCTTCTGAA (reverse primer for SAA1.1); 5′-GCGCTCGAGAGGCCCCCAGCACAACCTACT-GAG (reverse primer for SAA2.1). The amplified fragment was inserted into the pAdTrackCMV adenovirus expression vector containing the cytomegalovirus (CMV) immediate early enhancer-promoter element [12] and introduced into BJ5183-AD-1 cells (Invitrogen). The adenoviral vector DNAs were analyzed to confirm their sequence, and then transfected into HEK-293 cells to produce replication defective recombinant viruses. Adnull consists of pAd-Track CMV, which does not contain a transgene.
2.8. SAA expression in SAAKO mice
For baseline measurements, blood was obtained by retro-orbital bleed 24 h prior to adenoviral administration. The isoform-specific adenoviruses were titrated so that combined expression of both would mimic total SAA expression under inflammatory conditions. SAA expression was elicited by tail vein injection of 100 μL aliquots PBS containing 1.5 × 1011 particles AdSAA1.1, 3.0 × 1011 particles AdSAA2.1, or a combination of AdSAA1.1 and AdSAA2.1 at 1.5 and 3.0 × 1011 particles respectively, into SAAKO mice [8]. Adnull (4.5 × 1011 particles) was used as a control. Blood was taken from mice at the indicated time points for plasma measurement of cytokines and quantitative immunoblot for SAA. Western analyses were performed on plasma obtained 24 h after viral injection.
2.9. Cell culture
The mouse macrophage-like cell line J774 was obtained from American Type Culture Collection and maintained in Dulbecco Modified Eagle Medium supplemented with 10%(v/v) heat inactivated fetal bovine serum, 2 mM glutamine, 50 units/mL penicillin G, and 50 μg/mL streptomycin. Cells were plated one day prior to treatment in 24 well plates at 5 × 105 cells per well. Culture media were replaced with fresh media containing stimulants as indicated in figure legends. The concentration of selected cytokines in cell-free supernatants was determined by ELISA. Protein concentration of cell lysates was determined by BCA. Cell culture was performed with endotoxin free-reagents. Polymyxin was avoided due to its known interactions with lipoproteins [13].
2.10. Statistical analysis
Data was analyzed for statistical significance using Prism software (version 4.0; GraphPad Software, San Diego, CA) and is expressed as mean ± SEM. Statistical significance and p values are indicated on the figures where appropriate. The p values < 0.05 were considered statistically significant.
3. Results
3.1. Lipid-poor SAA, but not apoA-I or apoA-II, induces G-CSF in vitro
We investigated whether purified human or mouse acute phase SAAs elicit pro-inflammatory responses in macrophages as has been reported for rSAA, a hybrid molecule comprised of sequences derived from both human SAA1 and SAA2. The ability of mouse SAA1.1/2.1 and human SAA1/2 (10 μg/mL) to induce G-CSF secretion by J774 cells was assessed. Purified human SAA induced G-CSF secretion in J774 cells after a 24 h incubation period. A level of 35 ng/mL was achieved, which represents approximately 50% of that induced by 10 ng/mL LPS. Mouse SAA was somewhat less effective than human SAA to induce G-CSF in vitro (Fig. 1A). As SAA is an apolipoprotein of HDL, sharing structural features with other HDL apolipoproteins, the specificity of G-CSF induction by SAA was explored by determining whether human apoA-I or apoA-II produced a similar pro-inflammatory effect. Human apoA-I was co-purified with human SAA from acute phase patients post cardiac surgery; human apoA-II was obtained commercially. It is notable that unlike SAA, neither human apoA-I nor apoA-II was capable of inducing G-CSF expression in J774 cells (Fig. 1A). Human SAA induced G-CSF in a concentration dependent manner (Fig. 1B). The time course of induction was comparable to LPS, reaching a maximum at 12 h (Fig. 1C).
Fig. 1.
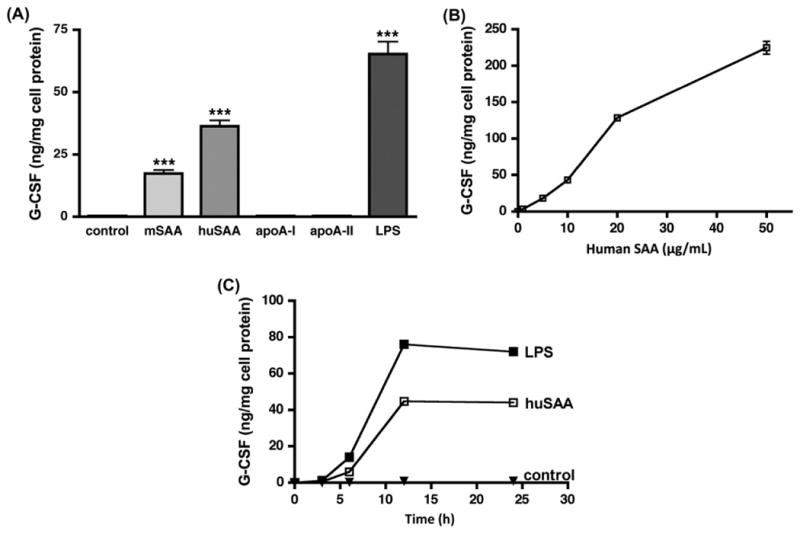
Lipid-poor SAA induces G-CSF secretion in macrophages. G-CSF was measured in the cell-free supernatant of J774 murine macrophage cells incubated with: (A) cell culture media only (control), or media containing the purified apoproteins mSAA, huSAA, apoA-I, apoA-II (all 10 μg/mL) or LPS (10 ng/mL) for 24 h. Values are the mean ± SEM of triplicate determinations from a representative experiment, performed twice. Significance between cells treated with media only (control) and cells treated with SAA or LPS was determined by Student t-test. *** indicates p < 0.001; (B) the indicated concentration of purified huSAA for 24 h. Values are the mean ± SEM of triplicate determinations; (C) culture media only (control), LPS (10 ng/mL) or purified huSAA (10μg/mL) for the indicated times. Values are the mean ± SEM of triplicate determinations from a representative experiment, performed twice.
3.2. G-CSF secretion by lipid-poor SAA requires TLR2
It was reported that recombinant SAA (rSAA) induces cytokine secretion through a TLR2-dependent mechanism [14,15]. We used purified huSAA (10 μg/mL for 16 h) to confirm that, analogous to rSAA, induction of G-CSF in J774 cells is inhibited when TLR2 signaling is blocked by neutralizing antibodies. Pre-incubation with anti-TLR2 antibody (5 μg/mL for 1 h) reduced G-CSF production by 80% compared to an isotype-matched control antibody. After pre-incubation with anti-TLR4 antibody G-CSF production was only 14% lower than with an isotype-matched control antibody (Fig. 2).
Fig. 2.
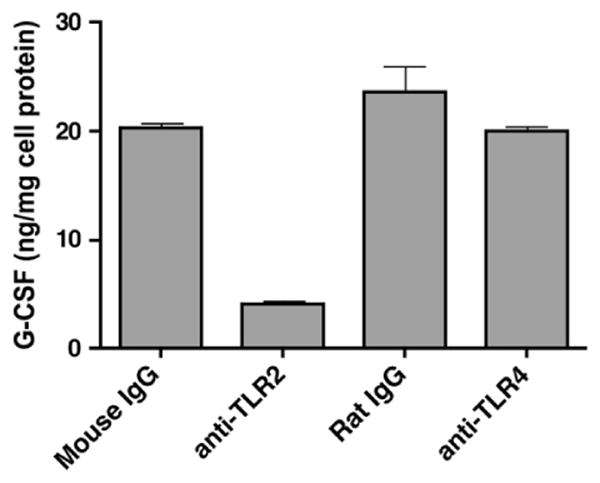
SAA induces G-CSF secretion through TLR2. G-CSF content was determined in the cell-free supernatant of J774 murine macrophage cells pre-treated with anti-TLR2 antibody, anti-TLR4 antibody or isotype-specific IgG controls (all 5 μg/mL) for 1 h prior to incubation with 10 μg/mL huSAA for 16 h. Bars represent the average of duplicate determinations.
3.3. Lipid-poor SAA, but not HDL-associated SAA induces cytokine production in vitro
The vast majority of SAA, a lipophilic apolipoprotein, is associated with HDL in plasma [2]. We therefore determined whether HDL-associated SAA (its natural state), as in AP HDL, induces G-CSF expression and compared it to G-CSF produced by purified huSAA. N HDL was purified from normal human subjects or control mice, and AP HDL was purified from cardiac surgery patients, or mice injected with LPS or AgNO3 as described in Section 2. HDL apoprotein composition was analyzed by SDS-PAGE (Fig. 3A). SAA comprised approximately 30% of the protein component of AP human HDL and AP mouse HDL from AgNO3-injected animals, and approximately 40% of the protein of HDL from LPS-injected mice. AP m HDL (100 μg/mL) from LPS-injected animals induced detectable G-CSF production, albeit 7-fold less than purified huSAA at 10 μg/mL. Neither m HDL (100 μg/mL) from control or AgNO3-injected mice nor hu HDL (100 μg/mL) from normal controls and cardiac surgery patients stimulated G-CSF production (Fig. 3B). Purified mSAA and huSAA also elicited the production of TNF-α while purified apoA-I and apoA-II failed to do so; neither did N or AP HDL isolated from mice or humans (Fig. 3C). Similar data was obtained for the induction of IL-6 and MCP-1 (Supplementary data). Taken together, these data indicate that only lipid-poor SAA, but not HDL-associated SAA is an inflammatory mediator.
Fig. 3.
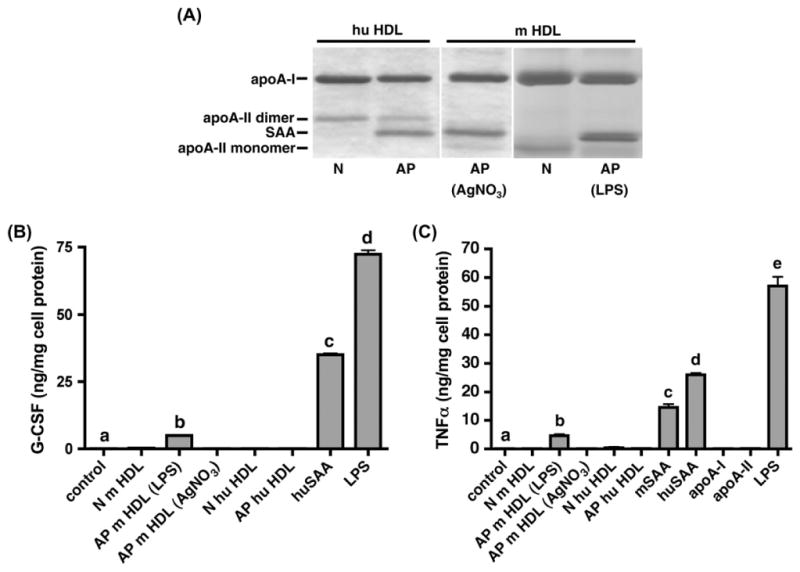
HDL-associated SAA does not induce G-CSF secretion in macrophages. HDL (d = 1.063–1.21 g/mL) was isolated from the plasma of normal human subjects (N) and from patients 24 h after cardiac surgery (AP), and from control C57BL/6 mice (N) or C57BL/6 mice 24 h after injection of LPS or AgNO3 (AP), as set out in Section 2. (A) HDL (5 μg protein) were subjected to SDS–PAGE utilizing a 4–20% (v/v) acrylamide gel and visualized by Coomassie staining. (B) G-CSF content in the cell-free supernatant of J774 murine macrophage cells incubated overnight (16 h) with the indicated HDLs (100 μg/mL), huSAA (10 μg/mL) or LPS (10 ng/mL). Values are the mean ± SEM of triplicate determinations from a representative experiment, performed twice. (C) TNFα in the cell-free supernatant of J774 murine macrophage cells incubated overnight (16 h) with the indicated HDLs (100 μg/mL), apoproteins (10 μg/mL) or LPS (10 ng/mL). Values are the mean ± SEM of triplicate determinations from a representative experiment, performed twice. Significance for GCSF and TNFα measurements between cells treated with media only (control) and SAA, HDL or LPS was determined by one way ANOVA; different letters correspond to different means (p < 0.001).
3.4. Overexpression of SAA in SAA-deficient mice does not induce G-CSF
To evaluate the ability of SAA to generate an inflammatory response in vivo, we generated recombinant adenoviruses expressing the individual mouse SAA isoforms i.e. SAA1.1 and SAA2.1. The expression of these isoforms was confirmed in SAAKO mice [8] by isoelectric focusing of plasma aliquots obtained 24 h after adenovirus administration followed by quantitative immunoblotting (Fig. 4A). Combined administration of both SAA adenoviruses as set out in Section 2 achieved plasma SAA concentrations of 327 μg/mL, similar to the levels produced in mice injected with 2.5 μg LPS (360 μg/mL) (Fig. 4A). Increased plasma SAA levels were maintained 72 h after viral vector administration (Fig. 4B). SAA was not detected in plasma of mice administered a control adenoviral vector, Adnull at any time during the course of our experiments (Fig. 4A, B).
Fig. 4.

In vivo SAA over expression does not induce G-CSF secretion in SAAKO mice. SAAKO mice were injected with AdSAA1.1 and/or AdSAA2.1; Adnull, an adenovirus that does not express a protein product; or LPS (2.5 μg/mouse) as set out in Section 2. (A) Plasma was obtained 24 h after injection and 7 μL aliquots were subjected to IEF followed by immunoblotting for SAA. Each lane represents plasma from a single mouse. (B) Plasma aliquots (1.5 μL), collected at the indicated times after injection of Adnull or AdSAA1.1/2.1, were subjected to SDS–PAGE followed by immunoblotting for SAA. Each time point represents plasma collected from three individual animals. (C) G-CSF was measured in plasma collected from mice at the indicated times after administration of Adnull or AdSAA1.1/2.1. Values represent mean ± SEM, n = 4.
G-CSF production was determined by assessing plasma aliquots obtained before and at selected intervals after injection of equal particle numbers of Adnull or AdSAA1.1/2.1 (Fig. 4C). While G-CSF was not detected in any of the mice at baseline, administration of either Adnull or AdSAA resulted in a similar transient peak of G-CSF, suggesting that the adenoviral vectors and not SAA were responsible for the induction of G-CSF. G-CSF could be detected as early as 2 h after viral injection, reached maximal levels at 8 h post virus injection and was back to baseline levels by 24 h, when plasma SAA concentrations in AdSAA1.1/2.1 injected mice were maximal. From these data, we concluded that SAA does not induce G-CSF in this system.
3.5. SAA-containing AP HDL can protect against LPS-induced inflammation
HDL can bind (neutralize) LPS and mitigate against the pro-inflammatory responses of endotoxin [16]. It has also been reported that rSAA can bind to and neutralize LPS in a dose dependent manner and negate the actions of LPS [17]. Therefore, it was of interest to determine whether the presence of SAA alters the ability of HDL to neutralize LPS. To this end we pretreated J774 cells without or with 100 μg/mL m HDL (N or AP) for 1 h prior to adding LPS (10 ng/mL) to the incubation mixes and incubating for an additional 16 h (Fig. 5).
Fig. 5.
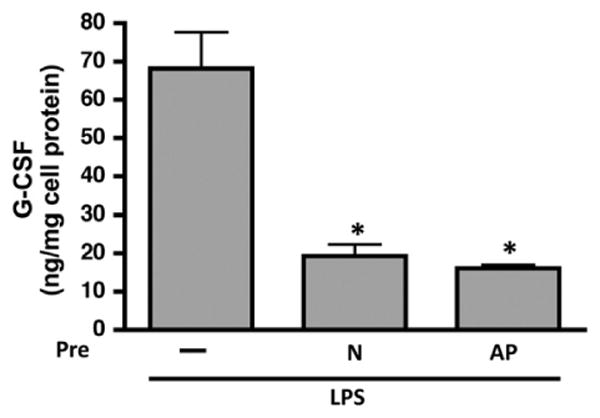
N and AP HDL reduces LPS-induced G-CSF secretion by macrophages. G-CSF content in the cell-free supernatant of J774 murine macrophage cells pretreated for 1 h with 100 μg/mL HDL (N – normal mouse HDL; AP – acute phase mouse HDL from AgNO3 injected mice) prior to 16 h incubation with LPS (10 ng/mL). J774 murine macrophage cells that received no pretreatment prior to LPS incubation were included as a control. Values are the mean ± SEM of triplicate determinations from a representative experiment, performed twice. Significance between groups was determined by one way ANOVA (p < 0.01).
As before, LPS treatment resulted in robust G-CSF secretion by J774 cells to ∼68 ng/mg cell protein, but pretreatment of cells with N m HDL dramatically reduced G-CSF production by LPS to ∼19 ng/mg. Similar results were obtained when cells were pretreated with AgNO3-generated mouse AP HDL. This indicates that the capacity of HDL to protect monocytes/macrophages against LPS-induced inflammation is unaltered by the presence of SAA.
4. Discussion
Numerous pro-inflammatory actions have been ascribed to lipid-poor SAA. These include increased chemotaxis of inflammatory cells [18], metalloproteinase induction [19] and inflammatory cytokine production [20]. These studies were preponderantly performed with rSAA, a hybrid molecule for which no natural counterpart exists. Mammalian proteins synthesized in a prokaryotic system contain a formylated amino terminal methionine. Incomplete removal of this amino acid during purification could result in a protein that will induce an inflammatory response. Furthermore, limited amino acid differences between SAA isoforms have profound functional effects [21]. The amino terminus of SAA is the lipid binding domain and the addition of a methionine could impact lipid association. This is supported by data that either deletion or mutation of a single amino acid impacts HDL association and amyloid formation [22]. There is a paucity of studies conducted with naturally occurring SAAs in the appropriate biological context as an apolipoprotein of HDL. One such study used isolated SAA from acute phase patients and showed the induction of cytokines [20]. A more recent study, however, suggested that the pro-inflammatory activity of the recombinant hybrid is not shared by the SAA protein in the circulation [6]. In our studies we investigated the ability of isolated human and mouse SAA, both lipid-poor and in HDL-associated context, to induce cytokines. These studies are important because, given the extensive literature on rSAA acting in a pro-inflammatory manner, naturally occurring SAAs could be major contributors in amplifying inflammatory tissue destruction. It is critically important to conduct these studies in a physiological context with naturally occurring SAAs. Further it is relevant when using purified SAA to appropriately control experiments with other apolipoproteins of HDL such as apoA-I and apoA-II. The lipophilic nature of these molecules could conceivably produce artifacts when interacting with lipid in cell membranes when used outside of their naturally occurring lipid context.
For our studies we focused on G-CSF as a representative cytokine, which is well established to be induced by rSAA [15]. G-CSF plays a central role in the regulation of neutrophilia and is induced by exogenous stimuli such as LPS, lipoteichoic acid (LTA) and phorbol-12-myristate-13-acetate (PMA) as well as endogenous cytokines and other hematopoietic growth factors [23].
Given the plethora of pro-inflammatory actions ascribed to SAA, a number of distinct and overlapping mechanisms have been suggested. SAA was shown to interact with the N-formyl peptide receptor 2 (FPR2), a membrane G protein coupled receptor [24]. This interaction resulted in diverse inflammatory sequelae including leukocyte migration, metalloproteinase induction and IL8 induction [24,25]. Evidence was also produced that SAA binds to and signals through the human HDL receptor scavenger receptor class BI (SR-BI) resulting in cytokine production [26]. Further, SAA was reported to induce G-CSF by interacting with the Toll-like receptor 2 (TLR2) [14,15]. Others, however, have indicated that SAA can activate TLR4 to stimulate NO production in macrophages [27]. It is difficult to comprehend how such a wide spectrum of mechanisms and actions can all be operative in a pathophysiological context. When one considers that SAA is very lipophilic and able to bind to proteoglycans present on the cell surface [28], it must be considered that the suggested mechanisms could be influenced by an amphophilic molecule taken out of its lipid context and interacting with proteoglycans and membrane lipids indirectly impacting these receptors in a complex integral.
We have generated adenoviral vectors that express the classic major acute phase mouse SAA's, SAA1.1 and SAA2.1. By using these vectors in combination we can mimic the SAA profile produced in mice during an acute phase response without significant concomitant inflammatory response (Fig 4A). This is important as different physical and functional properties, particularly related to amyloi-dogenesis, have been ascribed to these respective isoforms [21,29]. The use of SAA knockout mice precludes any influence of endogenous acute phase SAAs. Our results show that the induction of G-CSF is identical when SAA-expressing and control adenoviral vectors were compared. Peak G-CSF induction was at approximately 5 h after viral vector injection. SAA can be detected by immunoblot at that time. However, peak SAA levels occurred much later (24–72 h) and at that time G-CSF levels have returned to normal. This suggests that the transient induction of G-CSF was an adenoviral effect and not due to elevated SAA levels. In contrast to our results, in vivo over expression of SAA by lentivirus in apoE−/− mice resulted in low levels of plasma SAA (35 mg/mL) and elevated levels of IL-6 and TNFα 14 weeks after virus administration. These effects were not shared by the null virus [30]. This variation in experimental outcomes can likely be explained on the basis of differences in the viral vectors employed, the type of SAA expressed and particularly differences in the time of measurement.
We isolated SAA from the plasma of mice that were injected with AgNO3 to initiate a “sterile” inflammatory response. We also isolated human SAA from the plasma of patients undergoing cardiac surgery. Both human and mouse SAA thus purified were able to induce G-CSF production in macrophages, but purified human apoA-I and apoA-II did not. There is thus some specificity in the ability of SAA to induce G-CSF when compared to other apolipoproteins. G-CSF induction by SAA is both time and concentration dependent. However the concentration curve did not saturate and the possibility that this is a high capacity, low affinity interaction must be considered. The likely mechanism for G-CSF induction by these naturally occurring SAAs is interaction with TLR2 as reported by others for rSAA [14,15]. However, when SAA is associated with HDL the induction of G-CSF and other cytokines is totally abrogated. Only when the acute phase mouse HDL was induced with LPS was a minor elevation in G-CSF detected. Acute phase induction with AgNO3 failed to show any G-CSF induction. We speculate that the modest induction of G-CSF by LPS-induced AP mHDL was due to endotoxin contamination of the HDL preparation [31,32].
The drastic lipid remodeling that the HDL particle undergoes during inflammation mediated by group IIA secretory phospholipase A II (sPLA2-IIA) and cholesteryl ester transfer protein (CETP) impacts both the surface and the core of the particle resulting in the liberation of substantial amounts of lipid poor apoA-I, which has the potential to remove cholesterol and phospholipid from cells in an ABCA1-dependent manner [33]. Although lipid-poor SAA can be an effective acceptor of cellular cholesterol in lipid efflux [34], SAA tends to remain more lipid-associated during remodeling of acute phase HDL by sPLA2-IIA and CETP, liberating less SAA than apoA-I [7]. This is in keeping with SAA being more lipophilic than apoA-I. Microenvironments could exist where sufficient SAA is liberated to promote inflammation. This is possible given that numerous acute phase proteins impact HDL remodeling during inflammation [7]. Factors that impact the phospholipid surface, such as the acute phase protein sPLA2-IIA, and factors that concomitantly alter the neutral lipid core, such as CETP, are particularly potent to alter the equilibrium between lipid bound and lipid free apolipoproteins [7]. During chronic inflammation SAA can indeed be removed from HDL to be deposited as fibrils resulting in amyloidosis. Proteoglycan binding by SAA is important in this process [21]. The degree to which this could impact cytokine production on merits future consideration. Caution should therefore be applied to the interpretation of experiments performed in vitro with purified SAA, particularly rSAA that is structurally different from native SAA.
Care should be taken when using purified SAA in aqueous solutions as aggregates may form. Best results can be obtained with fresh preparations (<2 weeks old), avoiding repeated freeze–thaw cycles and using buffer conditions described in Section 2. The amyloidogenic mouse SAA1.1 even has the ability to form micro fibrils in solution [29]. The artefactual potential of these aggregates when interacting with lipid and proteoglycan in membranes is likely substantial.
We conclude that sites could exist where HDL remodeling factors are present in sufficient concentrations to achieve SAA liberation. If pro-inflammatory activities are ascribed to SAA, proof should be provided that SAA exist in such a physical form that it could induce cytokines.
Supplementary Material
Acknowledgments
This work was supported by the National Institutes of Health (Grant P01 HL-086670).
Abbreviations
- SAA
serum amyloid A protein
- rSAA
recombinant human SAA
- mSAA
mouse SAA
- huSAA
human SAA
- apo-AI
apolipoprotein AI
- apoA-II
apolipoprotein AII
- LPSs
lipopolysaccharides
- G-CSF
granulocyte colony-stimulating factor
- AP
acute phase
- N
normal
- N m HDL
normal mouse HDL
- N hu HDL
normal human HDL
- AP m HDL
acute phase mouse HDL
- AP hu HDL
acute phase human HDL
- IEF
isoelectrofocusing
- GGE
gradient gel electrophoresis
- SAAKO
mice with targeted deletion of SAA1.1 and SAA2.1
- WT
wild-type
Footnotes
Appendix A. Supplementary material: Supplementary data associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.cyto.2012.10.019.
References
- 1.Van Lenten BJ, Reddy ST, Navab M, Fogelman AM. Understanding changes in high density lipoproteins during the acute phase response. Arterioscler Thromb Vasc Biol. 2006;26:1687–8. doi: 10.1161/01.ATV.0000232522.47018.a6. [DOI] [PubMed] [Google Scholar]
- 2.Coetzee GA, Strachan AF, van der Westhuyzen DR, Hoppe HC, Jeenah MS, de Beer FC. Serum amyloid A-containing human high density lipoprotein 3. Density, size, and apolipoprotein composition. J Biol Chem. 1986;261:9644–51. [PubMed] [Google Scholar]
- 3.Koga T, Torigoshi T, Motokawa S, Miyashita T, Maeda Y, Nakamura M, et al. Serum amyloid A-induced IL-6 production by rheumatoid synoviocytes. FEBS Lett. 2008;582:579–85. doi: 10.1016/j.febslet.2008.01.022. [DOI] [PubMed] [Google Scholar]
- 4.Song C, Hsu K, Yamen E, Yan W, Fock J, Witting PK, et al. Serum amyloid A induction of cytokines in monocytes/macrophages and lymphocytes. Atherosclerosis. 2009;207:374–83. doi: 10.1016/j.atherosclerosis.2009.05.007. [DOI] [PubMed] [Google Scholar]
- 5.Uhlar CM, Whitehead AS. Serum amyloid A, the major vertebrate acute-phase reactant. Eur J Biochem. 1999;265:501–23. doi: 10.1046/j.1432-1327.1999.00657.x. [DOI] [PubMed] [Google Scholar]
- 6.Bjorkman L, Raynes JG, Shah C, Karlsson A, Dahlgren C, Bylund J. The proinflammatory activity of recombinant serum amyloid A is not shared by the endogenous protein in the circulation. Arthritis Rheum. 2010;62:1660–5. doi: 10.1002/art.27440. [DOI] [PubMed] [Google Scholar]
- 7.Jahangiri A, de Beer MC, Noffsinger V, Tannock LR, Ramaiah C, Webb NR, et al. HDL remodeling during the acute phase response. Arterioscler Thromb Vasc Biol. 2009;29:261–7. doi: 10.1161/ATVBAHA.108.178681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.de Beer MC, Webb NR, Wroblewski JM, Noffsinger VP, Rateri DL, Ji A, et al. Impact of serum amyloid A on high density lipoprotein composition and levels. J Lipid Res. 2010;51:3117–25. doi: 10.1194/jlr.M005413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. Protein measurement with the Folin phenol reagent. J Biol Chem. 1951;193:265–75. [PubMed] [Google Scholar]
- 10.Strachan AF, Brandt WF, Woo P, van der Westhuyzen DR, Coetzee GA, de Beer MC, et al. Human serum amyloid A protein. The assignment of the six major isoforms to three published gene sequences and evidence for two genetic loci. J Biol Chem. 1989;264:18368–73. [PubMed] [Google Scholar]
- 11.Strachan AF, Shephard EG, Bellstedt DU, Coetzee GA, van der Westhuyzen DR, de Beer FC. Human serum amyloid A protein. Behaviour in aqueous and urea-containing solutions and antibody production. Biochem J. 1989;263:365–70. doi: 10.1042/bj2630365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.He TC, Zhou S, da Costa LT, Yu J, Kinzler KW, Vogelstein B. A simplified system for generating recombinant adenoviruses. Proc Natl Acad Sci USA. 1998;95:2509–14. doi: 10.1073/pnas.95.5.2509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liao W, Floren CH. Complexes with and cationizes low density lipoproteins. The cause of polymyxin B-induced enhancement of endocytotic catabolism of low density lipoproteins. Biochem Pharmacol. 1993;45:1835–43. doi: 10.1016/0006-2952(93)90441-x. [DOI] [PubMed] [Google Scholar]
- 14.Cheng N, He R, Tian J, Ye PP, Ye RD. Cutting edge: TLR2 is a functional receptor for acute-phase serum amyloid A. J Immunol. 2008;181:22–6. doi: 10.4049/jimmunol.181.1.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.He RL, Zhou J, Hanson CZ, Chen J, Cheng N, Ye RD. Serum amyloid A induces G-CSF expression and neutrophilia via Toll-like receptor 2. Blood. 2009;113:429–37. doi: 10.1182/blood-2008-03-139923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sprong T, Netea MG, van der Ley P, Verver-Jansen TJ, Jacobs LE, Stalenhoef A, et al. Human lipoproteins have divergent neutralizing effects on E. coli LPS, N. meningitidis LPS, and complete Gram-negative bacteria. J Lipid Res. 2004;45:742–9. doi: 10.1194/jlr.M300453-JLR200. [DOI] [PubMed] [Google Scholar]
- 17.Van Leeuwen H, van Isterdael M, Van Strijp J, Van Kessel K, Verhoef J. The role of serum amyloid A (Saa) in sepsis and LPS neutralization. Crit Care Med. 1998;26:130A. [Google Scholar]
- 18.Badolato R, Wang JM, Murphy WJ, Lloyd AR, Michiel DF, Bausserman LL, et al. Serum amyloid A is a chemoattractant: induction of migration, adhesion, and tissue infiltration of monocytes and polymorphonuclear leukocytes. J Exp Med. 1994;180:203–9. doi: 10.1084/jem.180.1.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.O'Hara R, Murphy EP, Whitehead AS, FitzGerald O, Bresnihan B. Local expression of the serum amyloid A and formyl peptide receptor-like 1 genes in synovial tissue is associated with matrix metalloproteinase production in patients with inflammatory arthritis. Arthritis Rheum. 2004;50:1788–99. doi: 10.1002/art.20301. [DOI] [PubMed] [Google Scholar]
- 20.Patel H, Fellowes R, Coade S, Woo P. Human serum amyloid A has cytokine-like properties. Scand J Immunol. 1998;48:410–8. doi: 10.1046/j.1365-3083.1998.00394.x. [DOI] [PubMed] [Google Scholar]
- 21.de Beer MC, de Beer FC, McCubbin WD, Kay CM, Kindy MS. Structural prerequisites for serum amyloid A fibril formation. J Biol Chem. 1993;268:20606–12. [PubMed] [Google Scholar]
- 22.Patel H, Bramall J, Waters H, De Beer MC, Woo P. Expression of recombinant human serum amyloid A in mammalian cells and demonstration of the region necessary for high-density lipoprotein binding and amyloid fibril formation by site-directed mutagenesis. Biochem J. 1996;318(Pt 3):1041–9. doi: 10.1042/bj3181041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hareng L, Hartung T. Induction and regulation of endogenous granulocyte colony-stimulating factor formation. Biol Chem. 2002;383:1501–17. doi: 10.1515/BC.2002.172. [DOI] [PubMed] [Google Scholar]
- 24.He R, Sang H, Ye RD. Serum amyloid A induces IL-8 secretion through a G protein-coupled receptor, FPRL1/LXA4R. Blood. 2003;101:1572–81. doi: 10.1182/blood-2002-05-1431. [DOI] [PubMed] [Google Scholar]
- 25.Bjorkman L, Karlsson J, Karlsson A, Rabiet MJ, Boulay F, Fu H, et al. Serum amyloid A mediates human neutrophil production of reactive oxygen species through a receptor independent of formyl peptide receptor like-1. J Leukoc Biol. 2008;83:245–53. doi: 10.1189/jlb.0607-408. [DOI] [PubMed] [Google Scholar]
- 26.Mullan RH, McCormick J, Connolly M, Bresnihan B, Veale DJ, Fearon U. A role for the high-density lipoprotein receptor SR-B1 in synovial inflammation via serum amyloid-A. Am J Pathol. 2010;176:1999–2008. doi: 10.2353/ajpath.2010.090014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sandri S, Rodriguez D, Gomes E, Monteiro HP, Russo M, Campa A. Is serum amyloid A an endogenous TLR4 agonist? J Leukoc Biol. 2008;83:1174–80. doi: 10.1189/jlb.0407203. [DOI] [PubMed] [Google Scholar]
- 28.Chiba T, Chang MY, Wang S, Wight TN, McMillen TS, Oram JF, et al. Serum amyloid A facilitates the binding of high-density lipoprotein from mice injected with lipopolysaccharide to vascular proteoglycans. Arterioscler Thromb Vasc Biol. 2011;31:1326–32. doi: 10.1161/ATVBAHA.111.226159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yu J, Zhu H, Guo JT, de Beer FC, Kindy MS. Expression of mouse apolipoprotein SAA1.1 in CE/J mice. isoform-specific effects on amyloidogenesis. Lab Invest. 2000;80:1797–806. doi: 10.1038/labinvest.3780191. [DOI] [PubMed] [Google Scholar]
- 30.Dong Z, Wu T, Qin W, An C, Wang Z, Zhang M, et al. Serum amyloid A directly accelerates the progression of atherosclerosis in apolipoprotein E-deficient mice. Mol Med. 2011;17:1357–64. doi: 10.2119/molmed.2011.00186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tobias PS, Ulevitch RJ. Control of lipopolysaccharide-high density lipoprotein binding by acute phase protein(s) J Immunol. 1983;131:1913–6. [PubMed] [Google Scholar]
- 32.Vishnyakova TG, Bocharov AV, Baranova IN, Chen Z, Remaley AT, Csako G, et al. Binding and internalization of lipopolysaccharide by Cla-1, a human orthologue of rodent scavenger receptor B1. J Biol Chem. 2003;278:22771–80. doi: 10.1074/jbc.M211032200. [DOI] [PubMed] [Google Scholar]
- 33.Lawn RM, Wade DP, Garvin MR, Wang X, Schwartz K, Porter JG, et al. The Tangier disease gene product ABC1 controls the cellular apolipoprotein-mediated lipid removal pathway. J Clin Invest. 1999;104:R25–31. doi: 10.1172/JCI8119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.van der Westhuyzen DR, Cai L, de Beer MC, de Beer FC. Serum amyloid A promotes cholesterol efflux mediated by scavenger receptor B-I. J Biol Chem. 2005;280:35890–5. doi: 10.1074/jbc.M505685200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.