

Abstract
Nitric oxide (NO) released from NO donors can be cytotoxic in tumor cells and can enhance the transport of drugs into brain tumors by altering blood-tumor barrier permeability. The NO donor JS-K [O2-(2,4-dinitrophenyl) 1-[(4-ethoxycarbonyl)piperazin-1-yl]diazen-1-ium-1,2-diolate] releases NO upon enzymatic activation selectively in cells overexpressing glutathione-S-transferases (GSTs) such as gliomas. Thus, JS-K-dependent NO effects - especially on cell viability and vascular permeability - were investigated in U87 glioma cells in vitro and in an orthotopic U87 xenograft model in vivo by magnetic resonance imaging (MRI). In vitro experiments showed dose-dependent antiproliferative and cytotoxic effects in U87 cells. In addition, treatment of U87 cells with JS-K resulted in a dose-dependent activation of soluble guanylate cyclase and intracellular accumulation of cyclic guanosine monophosphate (cGMP) which was irreversibly inhibited by the selective inhibitor of soluble guanylate cyclase ODQ (1H-[1,2,4]oxadiazolo(4,3a)quinoxaline-1-one). Using dynamic contrast enhanced MRI (DCE-MRI) as a minimally invasive technique, we demonstrated for the first time a significant increase in the DCE-MRI read-out initial area under the concentration curve (iAUC60) indicating an acute increase in blood-tumor barrier permeability after i.v. treatment with JS-K. Repeated MR imaging of animals with intracranial U87 gliomas under treatment with JS-K (3.5 μmol/kg JS-K 3×/week) and of untreated controls on day 12 and 19 after tumor inoculation revealed no significant changes in tumor growth, edema formation or tumor perfusion. Immunohistochemical workup of the brains showed a significant antiproliferative effect of JS-K in the gliomas. Taken together, in vitro and in vivo data suggest that JS-K has antiproliferative effects in U87 gliomas and opens the blood-tumor barrier by activation of the NO/cGMP signaling pathway. This might be a novel approach to facilitate entry of therapeutic drugs into brain tumors. DCE-MRI is a non-invasive, repeatable imaging modality to monitor biological effects of NO donors and other experimental therapeutics in intracranial tumor models.
Keywords: Nitric oxide, JS-K, blood-brain barrier, blood-tumor barrier, DCE-MRI, U87 glioma, cGMP
1. Introduction
Glioblastoma multiforme is the most malignant primary brain tumor with a median survival of one year and a 5-year-survival of less than 3–5% [1]. Standard glioblastoma therapy includes resection followed by radiotherapy and chemotherapy [2]. However, the response of malignant gliomas to systemic chemotherapy is limited by the blood-tumor barrier which impairs drug delivery to brain tumors and by strong chemoresistance. Under physiological conditions, nitric oxide (NO) is involved in multiple cellular processes such as regulation of vasodilation, cerebral blood flow, vascular permeability and may promote tumor cell survival at low concentrations [3; 4; 5; 6]. Promising experimental strategies of NO-based therapy of gliomas focus on the delivery of high NO concentrations into the tumor to either increase the drug uptake across the blood-tumor barrier, to sensitize tumor cells to radiation therapy [7], to sensitize tumor cells to chemotherapeutic drugs and/or to induce apoptotic cell death selectively in tumor cells [8; 9; 10; 11; 12; 13; 14]. Exogenous NO can be administered using NO-releasing substances (NO donors) of the diazeniumdiolate group. These substances generate bioactive NO in physiological fluids spontaneously or after enzymatic activation in a controlled manner with reliable half-lives ranging from 2 s to 20 h [15].
JS-K [O2-(2,4-dinitrophenyl) 1-[(4-ethoxycarbonyl)piperazin-1-yl]diazen-1-ium-1,2-diolate] is a NO donor which releases NO upon enzymatic activation by glutathione-S-transferases (GST), a family of phase II detoxification enzymes [16; 17]. Prodrugs like JS-K which require activation by GST were specifically designed to exert their biological effect selectively in tumor cells overexpressing GST such as glioblastoma cells [18; 19]. Consequently, we studied JS-K as a NO-releasing prodrug in U87 glioma cells in vitro and in a glioma xenograft model in vivo.
Exposure of tumor cells to NO released from NO donors and NO derivates such as peroxynitrite results in accumulation of DNA double strand breaks, lipid and protein modifications (such as modification of potassium channels by S-nitrosylation), and impairment of the mitochondrial energy cycle with resultant breakdown of cellular energy generation [20; 21; 22]. Furthermore, they induce a variety of cellular pathways in glioma cells like the mitogen-activated protein (MAP) kinase cascades (PI3K/AKT pathway) [23]. All these effects lead to the amplified induction of proapoptotic mechanisms. Additionally, matrix metalloproteinases (MMPs) are targets of reactive nitrogen and oxygen species (ROS/RNS) at the transcriptional and post-translational level. Activation of MMP2/9 might contribute to loosening of the extracellular matrix [24; 25; 26]. Exposure of brain or brain tumor endothelial cells to NO leads to activation of the NO/cGMP pathway, cyclic guanosine monophosphate (cGMP) dependent phosphorylation of extracellular signal-regulated kinases (ERK), and modifications of tight junctions contributing to opening of the blood-brain or blood-tumor barrier [27; 28; 29; 30; 31; 32].
Previous experiments demonstrated a survival benefit in a C6 glioma model after combined treatment with the NO donor PROLI/NO and carboplatin chemotherapy in vivo. This effect was due to increased vascular permeability of the blood-tumor barrier resulting in enhanced uptake and efficacy of the chemotherapy [9]. Furthermore, our group could demonstrate with quantitative autoradiography (QAR) studies that the permeabilizing effect of PROLI/NO at the blood-tumor barrier is predominantly mediated by activation of the NO/cGMP pathway [33].
In addition to its effect on the permeability of brain tumor vasculature, NO has chemoand radio-sensitizing effects in glioma cells [10] and is cytotoxic in vitro. JS-K showed intrinsic anti-neoplastic activity in vitro and in vivo in a myeloid leukemia cell line [17]. Our group demonstrated a dose-dependent cytotoxic effect of JS-K in U87 gliomas in vitro and retardation of tumor growth in a subcutaneous xenograft model in vivo [34].
So far, therapeutic blood-brain barrier disruption was predominantly studied with invasive techniques such as fluorescence-based intracranial windows techniques or quantitative autoradiography [9; 33; 35; 36] or post-mortem methods like Evans blue staining. Our aim in this study was to investigate the JS-K-induced blood-brain barrier disruption in vivo by application of magnetic resonance imaging (MRI) in combination with MR contrast agents. Dynamic contrast-enhanced magnetic resonance imaging (DCE-MRI) is a noninvasive functional imaging technique that permits indirect measurement of hemodynamics in tissues and tumors. DCE-MRI utilizes a low molecular weight paramagnetic contrast agent (CA) such as gadolinium-DTPA, which readily diffuses from the blood to the extravascularextracellular space (EES). CA leads to a change in MR relaxation time T1 and consequently to a change in MR image contrast in perfused tissue. By acquiring a series of rapid MR images, the time course of the CA-induced change in T1 can be followed and the CA concentration can be calculated. This CA concentration time course can be characterized by the initial area under the concentration curve (iAUC). In general, iAUC is sensitive to changes in a number of hemodynamic parameters, including blood flow, blood volume, vessel permeability, and vessel surface area [37]. DCE-MRI has been used in several preclinical and clinical studies for monitoring of anti-angiogenic therapies of tumors, including vascular endothelial growth factor (VEGF) inhibition in animal tumor models and cancer patients [38; 39; 40; 41].
In diseased brain (e.g. multiple sclerosis) and in brain tumors, DCE-MRI is the in vivo method of choice for investigating and quantifying blood-brain barrier impairment [42; 43]. In rodent models, DCE-MRI was used to investigate the opening of the blood-brain barrier after treatment with ultrasound [44] or pharmacological agents such as bradykinin receptor antagonists [45; 46].
In this study we investigated the biological effects of JS-K on cell viability, proliferation, and the NO/cGMP signaling pathway in U87 cells in vitro and in an orthotopic glioma xenograft model in vivo. Serial morphological MRI and DCE-MRI were used to assess the acute and prolonged effects of NO donor treatment on vascular permeability and perfusion, edema formation, and tumor growth.
2. Material and methods
The NO donor JS-K [O2-(2,4-dinitrophenyl)1 [(4-ethoxycarbonyl)piperazin-1-yl]diazen-1-ium-1,2-diolate] was synthesized as described earlier and prepared as 10 mM stock solution in DMSO [47] For the in vivo experiments, JS-K was formulated in Pluronic Micelles P123 (BASF, Florham, NJ, USA).
Data are presented as the mean / SD. Statistical significance was tested with an unpaired two-tailed Student's t-test vs. control group. Differences were considered statistically significant at * p 0.05, ** p 0.01 and *** p 0.001.
2.1 Cell culture
Human U87 glioma cells were obtained by American Tissue Type Collection (ATCC HTB-14 Rockville, MD, USA) and cultivated in Dulbecco's modified Eagle Medium (DMEM) supplemented with 10% FCS, 100 U/ml penicillin, and 100 μg/ml streptomycin (Gibco, Invitrogen, Darmstadt, Germany). Cells were incubated in a 95% air/5% CO2 atmosphere at 37°C.
2.2 Cell viability and cytotoxicity assay
JS-K dependent effects on cell viability were determined using the multitox-fluor multiplex cytotoxicity assay (Promega, Madison, WI, USA). U87 cells were seeded at a density of 10.000 cells/200 μl on 96-well plates. JS-K was applied to the cells in concentrations of 5 μM, 10 μM and 15 μM JS-K for 4 h. The maximal dose of DMSO (0.3%) was added to the cells as solvent control and untreated cells were considered as control. Cytotoxicity assay was performed at 4 h and 24 h after treatment according to the manufacturer's instruction.
2.3 Immunocytochemistry
In order to determine the antiproliferative effect of JS-K on U87 cells in vitro, cells were seeded on glass cover slips (12 mm2) in 24-well plates at a density of 5.000 cells/750 μl medium. Cells were treated with either 5 μM, 10 μM or 15 μM JS-K. In addition, the maximal dose of DMSO (0.3%) was applied to exclude side effects caused by the solvent. After 4 h the compounds were removed and replaced by fresh medium. Untreated cells served as control. Fixation of cells was performed on ice in 4% paraformaldehyde for 30 min at 4 h and 24 h after treatment. Afterwards, cells were washed in PBS and permeabilized in acetone for 10 min at −20°C. Unspecific binding was minimized by blocking with 10% normal goat serum in PBS for 1 h at room temperature. Cells were incubated overnight at 4°C with primary antibodies (polyclonal rabbit anti-Ki-67, Abcam, Cambridge, UK, 1:1.000) diluted in 0.1% Triton X-100/PBS. The following day cells were incubated with FITC-conjugated anti-rabbit IgG secondary antibodies (Invitrogen, Karlsruhe, Germany, 1:600) diluted in 0.1% PBST for 1 h at room temperature. Cell nuclei were counterstained with 4',6'-diamidino-2-phenylindole dihydrochloride (DAPI) diluted 1:1.000 in PBS for 5 min. The cover slips were washed with PBS and mounted with Fluoromount G (DakoCytomation, Glostrup, Denmark). Ki-67-positive cells were counted in three visual fields on three cover fields using a Zeiss Axioskop fluorescence microscope (magnification 20, Zeiss, Jena, Germany).
2.4 Guanosine 3`,5`cyclic monophosphate (cGMP) analysis
Enzyme linked immunosorbent assay (ELISA) was used to determine the accumulation of intracellular cGMP (cGMP-EIA Kit, Cayman Chemical Company, MI, USA). U87 cells were seeded in uncoated multi-well cell culture dishes at a density of 0.5 106 cells and grown until confluency was reached. The next day, cells were treated with 50 μM ODQ (1H-[1,2,4]oxadiazolo(4,3a)quinoxaline-1-one, Sigma-Aldrich, St. Louis, MO, USA) for 30 min to block NO-specific soluble guanylate cyclase (sGC) activation. Cells treated with ODQ and untreated cells were exposed to increasing concentrations of JS-K (1 μM – 15 μM) for 5 – 120 min, cells exposed to DMSO (max. concentration of 0.3%) were used as solvent control. Cells were lysed in 0.1 M trichloroacetic acid for 20 min at room temperature and scraped off the cell culture flask. Cell culture extracts were homogenized thoroughly and centrifuged at 1,000 ×g for 10 min. Acetylation of intracellular cGMP (standards and probes) was performed according to the manufacturer's protocol. Total protein concentration of the supernatants was determined according to Bradford [48] to assure comparability of the samples. Lysates of treated cells were assayed for cGMP by a cGMP competitive enzyme immunoassay. Spectro-photometrical readings (=410 nm) were performed using the Tecan i-Control infinite 200 photometer and software (Tecan, Männedorf, Switzerland).
2.5 Animal model and preparation
Institutional guidelines for animal welfare and experimental conduct were followed for all animal experiments which were approved by the Institutional Animal Care and Use Committee and the Regional Administrative Authority under protocol G08/28. The animals were housed in individually ventilated cages (IVC) and received food and water ad libitum. Nude rats (rnu/nu, n=10) underwent stereotactic implantation of 50.000 U87 glioma cells into the right striatum under ketamine/xylazine anesthesia.
Rats were randomly assigned to two groups (n=5/group) and treatment was performed 3 times per week starting on day 5 after inoculation: 1. control: phosphate-buffered saline (PBS) i.p.; 2. JS-K (3.5 μmol/kg in 2.25% Pluronic P123 in PBS i.v.). MRI was performed on 10 rats on day 12 (1 week of treatment) and on the remaining 6 rats (2 controls, 4 treated rats) on day 19 (2 weeks of treatment). Four rats had to be sacrificed prior to the study endpoint due to tumor burden.
For the MRI experiments, animals were anaesthetized with isoflurane (1.0–1.5%) in O2 (1.2 l/min) applied with a face mask. A custom-built catheter was inserted into the tail vein (30 G needle) for CA administration. The animal body temperature was kept at 37°C with a circulating water pad, and respiration (spontaneous breathing) was continuously monitored.
2.6 Magnetic resonance imaging
12 and 19 days after tumor inoculation MRI measurements were performed on a 9.4 T animal scanner (BioSpec 94/20, Bruker Biospin, Ettlingen, Germany) using a transmit birdcage resonator and a receive four channel array surface coil. T1-weighted and T2-weighted multislice RARE scans were acquired to localize the tumor with following parameters: 20 axial slices, slice thickness 1.0 mm, field of view 30 mm × 30 mm, matrix size 256 × 256, in-plane resolution 0.12 mm × 0.12 mm; T1-weighted scan: TR/TEeffective = 1.5 s/8.0 ms, RARE factor 4; T2-weighted scan: TR/TEeffective = 2.7 s/33 ms, matrix size 256 × 256, in-plane resolution 0.12 mm × 0.12 mm, RARE factor 8.
A time series of DCE-MR images was acquired pre- and post-injection of Gd-DTPA with an inversion recovery (IR) balanced steady state free precision (TrueFISP) sequence (1 axial slice of 2 mm thickness positioned in the tumor center, field of view 30 mm × 30 mm, matrix size 128×96, in-plane resolution 0.23 mm × 0.31 mm, half Fourier factor 1.4, TE/TR 1.45/2.91 ms, flip angle 40 deg.). To obtain absolute T1-relaxation rates at each time point of the time series, images at ten inversion times (TIs: 110 ms, 311 ms, 412 ms, 1918 ms) after each inversion pulse were acquired [49]. The dynamic changes were monitored for a period of 12 min (120 scans) with a temporal resolution of 6 s. The CA gadolinium diethylenetriamine pentaacetic acid (Gd-DTPA, Magnevist, Bayer-Schering, Germany; dose 0.1 mmol Gd/kg) was manually injected as a bolus via the tail vein catheter. To obtain a baseline measurement without CA, the measurement started 1 min before CA administration resulting in 10 baseline scans to determine baseline T1.
To assess the acute effect of JS-K on the blood-tumor barrier DCE-MRI was repeated 18 min after the first DCE-MRI scan and 3 minutes after i.v. injection of JS-K (JS-K group: n = 4) or saline (control rats: n = 3). This experiment was performed on day 12 after inoculation.
2.7 MRI data processing
Regions of interest (ROIs) containing the whole tumor in the slice were manually drawn on DCE-MRI images using the T1 contrast between tumor and surrounding tissue in the image series. The ROI for the reference region was manually drawn in the contralateral brain. Quantitative T1 was calculated in the ROIs for each scan of the DCE-MRI series [50] and CA concentration curves were calculated from the change in 1/T1 after CA injection using a relaxivity of 3.9 s−1mM−1 [51] for Gd-DTPA at 9.4 T.
The initial area under the CA concentration curve iAUC60 [52] was calculated for the time range 0 to 60 s after CA injection and normalized against iAUC60 in the contralateral brain (reference region). Accordingly, iAUC120 and iAUC300 were calculated and normalized for the time ranges 0 to 120 s and 300 s after CA injection.
Tumor and peritumoral edema areas were traced manually on each slice of the T2 weighted multislice RARE dataset and total tumor and edema volumes were calculated.
2.8 Immunohistochemistry
After euthanizing the animals, the brains were quickly removed from the skull and embedded in Tissue Tek OCTVR compound (Sakura Finetek, Alphen, Netherlands). The specimen were quickly frozen in isopentane in liquid nitrogen and stored at −80°C. Immunofluorescence staining was performed for Ki-67, a marker of cell proliferation (monoclonal mouse, Dako, Glostrup, Denmark). Brains were sliced into 10 μm sections which were fixed with acetone for 10 min at −20°C, rehydrated in 0.1% PBST and treated with blocking solution for 1 h at room temperature (2% FCS in 0.1% PBST). Sections were exposed to the primary antibody anti-Ki-67 (1:150 in blocking solution) overnight at 4°C. After three wash cycles with PBS, sections were incubated in presence of the secondary antibody (FITC-conjugated anti-mouse IgG, Invitrogen, Karlsruhe, Germany, 1:600 in 0.1% PBST) at room temperature for 1 h. The specimen were washed and exposed to DAPI (1:1.000) for 5 min at room temperature. Sections were washed in PBS and mounted in Fluoromount G. Immunofluorescence was visualized using a Zeiss Axioskop microscope (magnification 20). Ki-67-positive cells were counted in three visual fields per slide of at least three different tumor sections per animal per treatment group (JS-K group: n=4, controls: n=4).
3. Results
3.1 In vitro experiments
3.1.1 Cytotoxic and antiproliferative effects of JS-K
JS-K had a dose-dependent cytotoxic effect on U87 cells in vitro. Cell viability was significantly reduced after 4 h (Fig. 1a) and to a greater extent after 24 h (Fig. 1b). The increased number of dead cells indicated induction of apoptotic and non-apoptotic cell death compared to the controls. Significant impairment of cell viability could be achieved at doses as low as 5 μM JS-K. Immunocytochemistry for Ki-67 revealed a time- and dose-dependent effect of JS-K on proliferation activity of U87 cells 4 and 24 h after treatment compared to untreated controls (Fig. 2a, p 0.05 *, p 0.01 **). A maximal decrease in proliferation was determined after 24 h at doses as low as 10 μM JS-K. Treatment groups were statistically compared to solvent control to exclude a DMSO-dependent reduction of cell proliferation. Ki-67 expression was decreased by 10 and 15 μM JS-K after 24 h (Fig. 2a, p 0.05 #). Representative photomicrographs of Ki-67 immunostaining to illustrate the time- and concentration-dependent decrease in proliferation after JS-K treatment are depicted in Fig. 2b.
Figure 1.
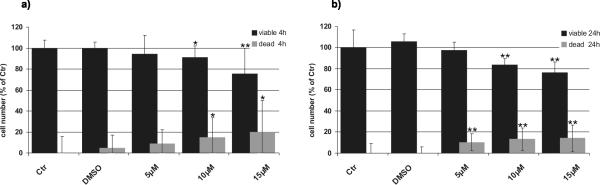
JS-K is cytotoxic in U87 cells. Cells were exposed to increasing concentrations of JS-K (5 μM, 10 μM and 15 μM) for 4 hours. Cells exposed to DMSO (max. concentration of 0.3%) were used as solvent control. The percentage of viable (black) and dead (grey) cells are displayed (untreated control = 100%) at 4 (a) and 24 (b) hours after treatment. *p 0.05, **p 0.01, ***p 0.001 t-test compared to control (Ctr).
Figure 2.

a) JS-K inhibits proliferation in U87 cells in vitro. The percentage of Ki-67-positive, proliferative cells is displayed at 4 (black) and 24 (grey) hours after treatment (untreated control = 100%). The cells were exposed for 4 hours to different concentrations of JS-K (5 μM, 10 μM and 15 μM). Cells exposed to DMSO (max. concentration of 0.3%) were used as solvent control. * p 0.05, ** p 0.01, treatment groups compared to untreated control; # p 0.05 treatment groups compared to solvent control. b) Immunocytochemistry for Ki-67 (green) in U87 cells at 4 h (upper row) and 24 h (lower row) after treatment (untreated control; DMSO control; 5 μM, 10 μM and 15 μM JS-K). Cell nuclei were counterstained with DAPI (magnification 20, scale bar = 100 m).
3.1.2 Effect of JS-K on cGMP synthesis
Exposure of U87 cells to JS-K (1–15 μM) led to a dose-dependent, intracellular accumulation of cGMP via NO-dependent activation of cytosolic guanylate cyclase reaching a maximum concentration of cGMP after 60–90 min (Fig. 3 and supplementary data 1). This effect was thoroughly abolished by the specific sGC inhibitor ODQ. 50 μM ODQ was sufficient to reduce the cGMP synthesis to the synthesis rate of untreated controls independently of the applied JS-K concentration (1–15 μM) in U87 cells (Fig. 3).
Figure 3.

JS-K induces cGMP formation in U87 cells which can be inhibited by ODQ. Cells were exposed to increasing concentrations of JS-K (1 μM, 5 μM, 10 μM and 15 μM) with and without pretreatment with ODQ. Cells exposed to DMSO (max. concentration of 0.3%) were used as solvent control. The maximum of intracellular cGMP release can be detected 60 min (black) and 90 min (grey) after JS-K exposure. **p 0.01, ***p 0.001 t-test compared to control (Ctr).
3.2 In vivo MRI experiments
3.2.1 Acute effect of JS-K
Injection of JS-K between two consecutive DCE-MRI experiments led to an obvious difference in the time courses of the CA concentration: CA concentration was considerably higher post injection of JS-K compared to pre-injection (Fig. 4a, solid grey and black curves, respectively), especially in the first minutes of the DCE-MRI experiments. In contrast, no changes in the CA concentrations curves were observed in the saline group, especially in the first minutes (Fig. 4b, solid curves). The CA concentration curves in contralateral brain were similar in both groups (Fig. 4a and b, dashed curves; positions of ROIs are shown in Fig. 4c). The increase in intratumoral CA uptake induced by JS-K can also be demonstrated in the time series of DCE-MRI scans showing a stronger and longer-lasting contrast uptake (Fig. 4d). Quantitative analysis of the CA curves in gliomas revealed a significant difference in the change in normalized iAUC60 after JS-K injection compared to saline controls (p=0.043; Fig. 4e) where the mean difference in iAUC60 between consecutive experiments was zero. However, this early NO effect on the tumor vasculature disappeared when longer normalized iAUC intervals (iAUC120, iAUC300) were analyzed (p=0.136, p=0.759). The difference in iAUC60 in the reference region contralateral brain was not significantly different between the JS-K and the saline group (p=0.559) justifying the calculation of normalized iAUC60 to compare the two groups. This was also the case for iAUC120 and iAUC300 in contralateral brain (p=0.928, p=0.812). These findings demonstrate that JS-K had a selective acute effect on CA uptake into gliomas but not in normal brain tissue.
Figure 4.

Acute vascular effect of JS-K: a) Contrast agent concentration curves of the first (black curves, pre-JSK, baseline) and second (grey curves, post-JS-K) DCE-MRI scan in tumor (solid upper curves) and contralateral brain (dashed lower curves). The second DCE-MRI scan was started 18 min after end of the first DCE-MRI scan. Drug injection was 3 min before the start of the second DCE-MRI scan. The grey area visualizes the iAUC60 for the concentration time course before JS-K injection in tumor. b) Contrast agent concentration curves in a control rat pre- and post saline injection. c) Regions of interest (ROIs) for DCE-MRI in tumor (shown right, solid) and contralateral brain (shown left, dashed) depicted on an anatomical T2-weighted image in the rat shown in a). d) Time series of DCE-MRI-scans showing the contrast agent-induced change in image contrast in the tumor pre- (upper row) and post- (lower row) JS-K injection (other rat than in a–c). CA was administered at time point 1 min. JS-K was injected 3 min before the second DCE-MRI time series. e) Change in normalized iAUC60 and iAUC120 in the control group (n=3) and the JS-K group (n=4) after injection of saline (Ctr, grey diamonds) or JS-K (white triangles). * p 0.05 t-test compared to control.
3.2.2 Long-term effect of JS-K
Repetitive i.v. administration of the NO donor prodrug JS-K was well tolerated by the animals with no major side effects.
Tumor and edema volumes were not significantly different between control and JS-K groups at both time points (Fig. 5a,b; tumor volume p=0.821/0.788; edema volume day 19 p=0.462). All tumors grew considerably within one week. JS-K did not lead to tumor growth attenuation between day 12 and 19. Mild peritumoral edema was detected in one control tumor and in two treated tumors on day 19. Necrotic cores were found in one control tumor and in three JS-K treated tumors on day 19. Normalized iAUC60 was not significantly different between control and JS-K groups at either time point (Fig. 5c, p=0.664 (12 d) and p=0.828 (19 d)). iAUC60 dropped in both groups between day 12 and 19, most likely due to the necrotic tissue without perfusion included in the ROI analysis leading to a lower mean CA accumulation in the whole tumor. As iAUC60 in normal contralateral brain tissue was not significantly different between control and treatment groups (p=0.324), normalization of iAUC in tumors did not influence the group differences. There was no difference in baseline T1 (before CA injection) between the groups (p=0.995, data not shown).
Figure 5.
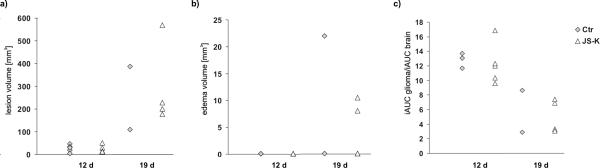
Effect of JS-K on day 12 and day 19 after tumor inoculation: a) tumor volume, b) edema volume, and c) normalized iAUC60 (normalized to contralateral brain) in control (Ctr, grey diamonds) and JS-K-treated (white triangles) rats. There was no significant difference between the two groups on either time point. Tumor volumes increased with time, whereas iAUC60 decreased. Mild peritumoral edema was detected in some animals on day 19.
3.3 Survival study and histology
NO donor therapy with JS-K did not lead to prolonged survival in this intracranial U87 glioma model (supplementary data 2). Four rats (3 controls, 1 treated rat) had to be euthanized before MRI on day 19 due to tumor burden. The remaining rats were sacrificed after the second MRI experiment (control: on day 19–22; JS-K: on day 19–20) because of the tumor burden. Histological workup of the brain specimen, however, showed a significant reduction in Ki-67-positive cells up to 44% in JS-K treated animals (Fig. 6a, p 0.001). Fig. 6b illustrates the reduction of proliferating Ki-67 expressing cells within the tumor in JS-K treated rats compared to controls.
Figure 6.
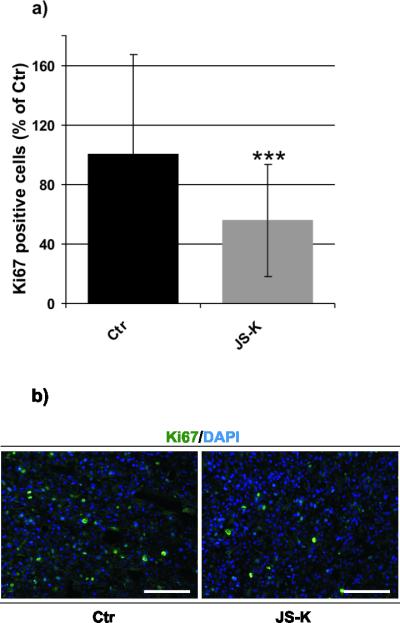
JS-K inhibits proliferation in U87 cells in vivo. a) The number of Ki-67-positive, proliferation active cells is shown for untreated control rats (Ctr, black, n=4) and JS-K treated rats (grey, n=4, *** p 0.001). b) Immunohistochemistry for Ki-67 illustrating the JS-K-dependent reduction of proliferating cells (green) within the tumor in a JS-K treated rat (right) compared to a control animal (left). Cell nuclei were counterstained with DAPI (magnification 20, scale bar = 50 m).
4. Discussion
Our experiments in U87 cells confirmed the ab ility of JS-K to induce cell death in U87 glioma cells in vitro after GST-mediated release of NO which was already shown in previous experiments of our group [34]. Cytotoxicity was dose-dependent but there was no difference between 4 h or 24 h exposure to JS-K. Dose-dependent inhibition of U87 cell proliferation was demonstrated by immunocytochemical detection of Ki-67 confirming the previous experiments using a 5`-bromodeoxyuridine incorporation assay [34].
There is strong experimental evidence that the biological effect of NO released by NO donors at the blood-tumor barrier is mediated by activation of the intracellular NO/cGMP pathway [27; 28; 29; 30; 31; 32]. The maximum of cGMP formation was reached within 60–90 min after JS-K exposure. In earlier autoradiography studies in orthotopic C6 gliomas, we could show that selective opening of the blood-tumor barrier using the NO donor PROLI/NO could be blocked by pretreatment with the sGC inhibitor LY83583, thus preventing cGMP formation [33]. Similar to those earlier in vivo findings [33], the process of cGMP generation by NO donor treatment can be blocked by sGC inhibition with ODQ in vitro (Fig. 3). Therefore, we hypothesize that activation of the NO/cGMP pathway by JS-K may be the mechanism by which NO donor treatment mediates its effect on blood-brain barrier integrity.
This is the first study using DCE-MRI to assess the impact of NO donor therapy with JS-K on blood-brain barrier integrity and tumor growth in an orthotopic U87 xenograft model in nude rats in vivo. Due to an impaired blood-tumor barrier CA diffuses from the blood stream into tumor tissue making gliomas visible on contrast enhanced MR images. NO released from JS-K led to increased CA uptake into the tumor, possibly as a result of opening tight junctions in the glioma blood vessels, thereby increasing blood-tumor barrier permeability. This acute effect of JS-K was demonstrated in vivo by DCE-MRI pre and post JS-K injection: iAUC60 was higher after JS-K injection compared to saline injection, i.e. more CA molecules diffused from the blood vessels into the tumor. We also showed that the permeability effect was tumor-selective as no changes were detected in contralateral normal brain confirming the glioma-specific release of NO. The calculation of normalized iAUC was therefore not influenced by an acute drug effect on the reference tissue. As the short time window directly after entry of the contrast agent bolus is crucial, it is standard to calculate iAUC60 for the time range 0 to 60 s. iAUC60 is influenced both by perfusion and permeability. In brain tumors, iAUC60 is dominated by permeability because a partially intact blood tumor barrier renders brain tumors less permeable than extra-cranial tumors [37]. Calculating iAUC values for time ranges up to 300 sec did not generate additional meaningful information on the permeability effect of JS-K. In our experiment the second DCE-MRI scan was performed 3 min after JS-K injection. Increased permeability of the blood-tumor barrier secondary to JS-K could therefore be confirmed for a period of at least 4 to 6 min after i.v. injection. In order to investigate the duration of the permeabilizing JS-K effect, future studies have to focus on performing DCE-MRI scans at different time points after NO donor treatment to determine the therapeutic window of blood-tumor barrier disruption.
The selective opening of the blood-tumor barrier after administration of NO donors has previously been demonstrated in gliomas in rodents post-mortem using quantitative autoradiography [9]. DCE-MRI is an ideal imaging modality for in vivo treatment monitoring of substances acting on tumor physiology, i.e. to obtain functional biomarkers. Coté et al. and Sarin et al. chose the increased contrast agent distribution volume and iAUC, respectively, as indicators of increased vascular permeability in their DCE-MRI studies with bradykinin receptor antagonists [45; 46].
In contrast to previous studies which revealed a strong cytotoxic effect in U87 gliomas in vitro and growth retardation in a flank tumor model in vivo [17], JS-K therapy did not result in tumor growth retardation or extended survival in this intracranial tumor model. Although, MRI and histology did not reveal increased intratumoral necrosis after NO donor treatment, a significant antiproliferative effect was demonstrated in vitro as well as in vivo by Ki-67-staining. In the previous study, JS-K was administered at a dose of 6 mg/kg/d (or 15 mol/kg/d) s.c. for 7 consecutive days, whereas in the current study, JS-K was administered i.v. at a lower dose (3.5 mol/kg) 3 times a week for 1 or 2 weeks. As biological effects of NO are highly dose- and time-dependent, we postulate that the NO released from JS-K after i.v. injection might have been sufficient to alter blood-tumor barrier integrity and to exert a local antiproliferative effect in tumor cells. However, the intratumoral NO levels were not high enough to induce a long-lasting antitumor effect in this experimental setup.
Upon vascular delivery, JS-K could react with glutathione in the blood, making it difficult to estimate the actual amount of JS-K reaching its target in the brain. Higher JS-K doses may also result in depletion of serum glutathione, which is involved in the maintenance of membrane integrity and in proper functioning of the blood-brain barrier. In addition to the NO effects at the blood-brain barrier, glutathione depletion might contribute to a further destabilization of the blood-brain barrier [53; 54].
Peritumoral edema is a typical feature of malignant gliomas in humans indicating impaired blood-brain barrier integrity around the tumor. It has been observed earlier that NO-induced opening of the blood-brain barrier also affects the peritumoral zone around the contrast-enhancing tumor margin in C6 gliomas [9]. As rodent gliomas differ considerably from human gliomas with respect to invasive growth pattern and edema formation, it was not surprising that peritumoral edema was detected in only half of the tumors in both groups and at a late time point. Treatment with JS-K did not lead to increased peritumoral edema formation. Other biological effects of long-term NO donor treatment may include changes in angiogenesis and an extended opening of the blood-tumor barrier since NO activates the VEGF pathway [55]. These effects may also be monitored by calculation of the iAUC and could induce detrimental side effects such as intratumoral hemorrhage or increased intracranial pressure in the animals. As no significant changes of iAUC was detected in tumors under repetitive JS-K treatment over two weeks, we conclude that JS-K treatment is safe and did not result in undesirable side effects.
Taken together, this study elucidates the biological effects of the NO-donating prodrug JS-K on cell viability, proliferation, cGMP signaling, tumor growth and vascular permeability in U87 gliomas in vitro and in vivo. A selective increase in blood-tumor barrier permeability upon i.v. JS-K delivery was detected in intracranial U87 gliomas using DCE-MRI. Using this non-invasive imaging technique to assess physiological and biological parameters in brain tumors offers the possibility for serial monitoring of the safety and efficacy of experimental treatment strategies in orthotopic xenograft models in rodents. Exploring NO-based antitumor treatment may be a promising strategy in gliomas due to their significant treatment resistance and privileged location in the brain.
Supplementary Material
Time course of cGMP release in U87 cells. Cells treated with ODQ and untreated cells were exposed to increasing concentrations of JS-K (1 μM, 5 μM, 10 μM and 15 μM) for 5 – 120 min, cells exposed to DMSO (max. concentration of 0.3%) were used as solvent control. *p 0.05, # p 0.01 and § p 0.001 t-test compared to control (Ctr)
Cumulative survival of rats with orthotopic U87 gliomas. Treatment groups (n=9/group) consist of five rats of the MRI study and four additional rats that were treated accordingly. There was no significant difference in the survival of rats in the JS-K treated group (light) compared to untreated controls (Ctr, dark) (20.1 vs. 19.7 days).
Highlights #NOX-12-106
NO donor JS-K selectively increases blood-tumor barrier permeability in gliomas.
JS-K has cytotoxic and antiproliferative effects in U87 gliomas.
JS-K increases cGMP formation in U87 cells.
Dynamic contrast enhanced MRI is a non-invasive tool for treatment monitoring.
DCE-MRI detects blood-tumor barrier opening in U87 gliomas in JS-K-treated rats.
Acknowledgments
Financial Support This research was supported in part by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research (JS) and by Federal funds from the National Cancer Institute, National Institutes of Health, under contract HHSN261200800001E. It was also supported in part by ENCITE which is co-funded by the European Commission under the 7th Framework Programme.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Ohgaki H. Epidemiology of brain tumors. Methods in Molecular Biology. 2009;472:323–342. doi: 10.1007/978-1-60327-492-0_14. [DOI] [PubMed] [Google Scholar]
- [2].Stupp R, Mason WP, Bent M.J.v.d., Weller M, Fisher B, Taphoorn MJB, Belanger K, Brandes AA, Marosi C, Bogdahn U, Curschmann J, Janzer RC, Ludwin SK, Gorlia T, Allgeier A, Lacombe D, Cairncross JG, Eisenhauer E, Mirimanoff RO. Radiotherapy plus Concomitant and Adjuvant Temozolomide for Glioblastoma. New England Journal of Medicine. 2005;352:987–996. doi: 10.1056/NEJMoa043330. [DOI] [PubMed] [Google Scholar]
- [3].Lam-Himlin D, Espey MG, Perry G, Smith MA, Castellani RJ. Malignant glioma progression and nitric oxide. Neurochem Int. 2006;49:764–8. doi: 10.1016/j.neuint.2006.07.001. [DOI] [PubMed] [Google Scholar]
- [4].Ridnour LA, Thomas DD, Donzelli S, Espey MG, Roberts DD, Wink DA, Isenberg JS. The biphasic nature of nitric oxide responses in tumor biology. Antioxid Redox Signal. 2006;8:1329–37. doi: 10.1089/ars.2006.8.1329. [DOI] [PubMed] [Google Scholar]
- [5].Wink DA, Mitchell JB. Nitric oxide and cancer: an introduction. Free Radic Biol Med. 2003;34:951–4. doi: 10.1016/s0891-5849(02)01362-x. [DOI] [PubMed] [Google Scholar]
- [6].Kroncke KD, Fehsel K, Kolb-Bachofen V. Nitric oxide: cytotoxicity versus cytoprotection--how, why, when, and where? Nitric Oxide. 1997;1:107–20. doi: 10.1006/niox.1997.0118. [DOI] [PubMed] [Google Scholar]
- [7].de Ridder M, Verellen D, Verovski V, Storme G. Hypoxic tumor cell radiosensitization through nitric oxide. Nitric Oxide. 2008;19:164–169. doi: 10.1016/j.niox.2008.04.015. [DOI] [PubMed] [Google Scholar]
- [8].Huerta S, Chilka S, Bonavida B. Nitric oxide donors: novel cancer therapeutics (review) Int J Oncol. 2008;33:909–27. [PubMed] [Google Scholar]
- [9].Weyerbrock A, Walbridge S, Pluta RM, Saavedra JE, Keefer LK, Oldfield EH. Selective opening of the blood-tumor barrier by a nitric oxide donor and long-term survival in rats with C6 gliomas. Journal of Neurosurgery. 2003;99:728–737. doi: 10.3171/jns.2003.99.4.0728. [DOI] [PubMed] [Google Scholar]
- [10].Weyerbrock A, Baumer B, Papazoglou A. Growth inhibition and chemosensitization of exogenous nitric oxide released from NONOates in glioma cells in vitro. Journal of Neurosurgery. 2009;110:128–136. doi: 10.3171/2008.6.17607. [DOI] [PubMed] [Google Scholar]
- [11].Kogias E, Osterberg N, Baumer B, Psarras N, Koentges C, Papazoglou A, Saavedra JE, Keefer LK, Weyerbrock A. Growth-inhibitory and chemosensitizing effects of the glutathione-S3 transferase-pi activated nitric oxide donor PABA/NO in malignant gliomas. International Journal of Cancer. 2011 doi: 10.1002/ijc.26106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Bonavida B, Baritaki S, Huerta-Yepez S, Vega MI, Chatterjee D, Yeung K. Novel therapeutic applications of nitric oxide donors in cancer: roles in chemo- and immunosensitization to apoptosis and inhibition of metastases. Nitric Oxide. 2008;19:152–7. doi: 10.1016/j.niox.2008.04.018. [DOI] [PubMed] [Google Scholar]
- [13].Singh S, Gupta AK. Nitric oxide: role in tumour biology and iNOS/NO-based anticancer therapies. Cancer Chemother Pharmacol. 67:1211–24. doi: 10.1007/s00280-011-1654-4. [DOI] [PubMed] [Google Scholar]
- [14].Griffin RJ, Makepeace CM, Hur WJ, Song CW. Radiosensitization of hypoxic tumor cells in vitro by nitric oxide. Int J Radiat Oncol Biol Phys. 1996;36:377–83. doi: 10.1016/s0360-3016(96)00329-x. [DOI] [PubMed] [Google Scholar]
- [15].Keefer LK. Progress toward clinical application of the nitric oxide-releasing diazeniumdiolates. Annual Review of Pharmacology and Toxicology. 2003;43:585–607. doi: 10.1146/annurev.pharmtox.43.100901.135831. [DOI] [PubMed] [Google Scholar]
- [16].Shami PJ, Saavedra JE, Bonifant CL, Chu J, Udupi V, Malaviya S, Carr BI, Kar S, Wang M, Jia L, Ji X, Keefer LK. Antitumor Activity of JS-K [O2-(2,4-Dinitrophenyl) 1-[(4-Ethoxycarbonyl)piperazin-1-yl]diazen-1-ium-1,2-diolate] and Related O2-Aryl Diazeniumdiolates in Vitro and in Vivo. Journal of Medicinal Chemistry. 2006;49:4356–4366. doi: 10.1021/jm060022h. [DOI] [PubMed] [Google Scholar]
- [17].Shami PJ, Saavedra JE, Wang LY, Bonifant CL, Diwan BA, Singh SV, Gu Yijun, Fox SD, Buzard GS, Citro ML, Waterhouse DJ, Davies KM, Ji X, Keefer LK. JS-K, a Glutathione/Glutathione S-Transferase-activated Nitric Oxide Donor of the Diazeniumdiolate Class with Potent Antineoplastic Activity. Molecular Cancer Therapeutics. 2003;2:409–417. [PubMed] [Google Scholar]
- [18].Chakrapani H, Kalathur RC, Maciag AE, Citro ML, Ji X, Keefer LK, Saavedra JE. Synthesis, mechanistic studies, and anti-proliferative activity of glutathione/glutathione S-transferase-activated nitric oxide prodrugs. Bioorg Med Chem. 2008;16:9764–71. doi: 10.1016/j.bmc.2008.09.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Kiziltepe T, Hideshima T, Ishitsuka K, Ocio EM, Raje N, Catley L, Li CQ, Trudel LJ, Yasui H, Vallet S, Kutok JL, Chauhan D, Mitsiades CS, Saavedra JE, Wogan GN, Keefer LK, Shami PJ, Anderson KC. JS-K, a GST-activated nitric oxide generator, induces DNA double-strand breaks, activates DNA damage response pathways, and induces apoptosis in vitro and in vivo in human multiple myeloma cells. Blood. 2007;110:709–18. doi: 10.1182/blood-2006-10-052845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Brookes PS, Bolanos JP, Heales SJ. The assumption that nitric oxide inhibits mitochondrial ATP synthesis is correct. FEBS Lett. 1999;446:261–3. doi: 10.1016/s0014-5793(99)00217-3. [DOI] [PubMed] [Google Scholar]
- [21].Yabuki M, Tsutsui K, Horton AA, Yoshioka T, Utsumi K. Caspase activation and cytochrome c release during HL-60 cell apoptosis induced by a nitric oxide donor. Free Radic Res. 2000;32:507–14. doi: 10.1080/10715760000300511. [DOI] [PubMed] [Google Scholar]
- [22].Leon L, Jeannin JF, Bettaieb A. Post-translational modifications induced by nitric oxide (NO): implication in cancer cells apoptosis. Nitric Oxide. 2008;19:77–83. doi: 10.1016/j.niox.2008.04.014. [DOI] [PubMed] [Google Scholar]
- [23].Liu J, Li C, Leslie C, Bonifant CL, Buzard GS, Saavedra JE, Keefer LK, Waalkes M. JS-K and CB-3-100 enhance arsenic and cisplatin cytolethality by increasing cellular accumulation. Molecular Cancer Therapeutics. 2004;3:709–714. [PubMed] [Google Scholar]
- [24].Ishii Y, Ogura T, Tatemichi M, Fujisawa H, Otsuka F, Esumi H. Induction of matrix metalloproteinase gene transcription by nitric oxide and mechanisms of MMP-1 gene induction in human melanoma cell lines. Int J Cancer. 2003;103:161–8. doi: 10.1002/ijc.10808. [DOI] [PubMed] [Google Scholar]
- [25].Wu J, Akaike T, Hayashida K, Okamoto T, Okuyama A, Maeda H. Enhanced vascular permeability in solid tumor involving peroxynitrite and matrix metalloproteinases. Jpn J Cancer Res. 2001;92:439–51. doi: 10.1111/j.1349-7006.2001.tb01114.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Haorah J, Ramirez SH, Schall K, Smith D, Pandya R, Persidsky Y. Oxidative stress activates protein tyrosine kinase and matrix metalloproteinases leading to blood-brain barrier dysfunction. J Neurochem. 2007;101:566–76. doi: 10.1111/j.1471-4159.2006.04393.x. [DOI] [PubMed] [Google Scholar]
- [27].Koesling D, Bohme E, Schultz G. Guanylyl cyclases, a growing family of signal-transducing enzymes. Faseb J. 1991;5:2785–91. doi: 10.1096/fasebj.5.13.1680765. [DOI] [PubMed] [Google Scholar]
- [28].Bohme E, Graf H, Schultz G. Effects of sodium nitroprusside and other smooth muscle relaxants on cyclic GMP formation in smooth muscle and platelets. Adv Cyclic Nucleotide Res. 1978;9:131–43. [PubMed] [Google Scholar]
- [29].Maulik D, Ashraf QM, Mishra OP, Delivoria-Papadopoulos M. Activation of p38 mitogen-activated protein kinase (p38 MAPK), extracellular signal-regulated kinase (ERK) and c-jun N-terminal kinase (JNK) during hypoxia in cerebral cortical nuclei of guinea pig fetus at term: role of nitric oxide. Neurosci Lett. 2008;439:94–9. doi: 10.1016/j.neulet.2008.02.037. [DOI] [PubMed] [Google Scholar]
- [30].Lander HM, Jacovina AT, Davis RJ, Tauras JM. Differential activation of mitogen-activated protein kinases by nitric oxide-related species. J Biol Chem. 1996;271:19705–9. doi: 10.1074/jbc.271.33.19705. [DOI] [PubMed] [Google Scholar]
- [31].Pun PB, Lu J, Moochhala S. Involvement of ROS in BBB dysfunction. Free Radic Res. 2009;43:348–64. doi: 10.1080/10715760902751902. [DOI] [PubMed] [Google Scholar]
- [32].Schreibelt G, Kooij G, Reijerkerk A, van Doorn R, Gringhuis SI, van der Pol S, Weksler BB, Romero IA, Couraud PO, Piontek J, Blasig IE, Dijkstra CD, Ronken E, de Vries HE. Reactive oxygen species alter brain endothelial tight junction dynamics via RhoA, PI3 kinase, and PKB signaling. Faseb J. 2007;21:3666–76. doi: 10.1096/fj.07-8329com. [DOI] [PubMed] [Google Scholar]
- [33].Weyerbrock A, Walbridge S, Saavedra JE, Keefer LK, Oldfield EH. Differential effects of nitric oxide on blood–brain barrier integrity and cerebral blood flow in intracerebral C6 gliomas. Neuro-Oncology. 2011;13:203–211. doi: 10.1093/neuonc/noq161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Weyerbrock A, Osterberg N, Psarras N, Baumer B, Kogias E, Werres A, Bette S, Saavedra JE, Keefer LK, Papazoglou A. JS-K, a glutathione S-transferase-activated nitric oxide donor with antineoplastic activity in malignant gliomas. Neurosurgery. 2011 doi: 10.1227/NEU.0b013e31823209cf. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Blasberg R, Kobayashi T, Horowitz M, Rice J, Groothuis D, Molnar P, Fenstermacher J. Regional blood flow in ethylnitrosourea-induced brain tumors. Annals of Neurology. 1983;14:189–201. doi: 10.1002/ana.410140206. [DOI] [PubMed] [Google Scholar]
- [36].Blasberg R, Molnar P, Groothuis D, Patlak C, Owens E, Fenstermacher J. Concurrent measurements of blood flow and transcapillary transport in avian sarcoma virus-induced experimental brain tumors: implications for chemotherapy. Journal of Pharmacology and Experimental Therapeutics. 1984;231:724–735. [PubMed] [Google Scholar]
- [37].Padhani AR, Khan AA. Diffusion-weighted (DW) and dynamic contrast-enhanced (DCE) magnetic resonance imaging (MRI) for monitoring anticancer therapy. Targeted Oncology. 2010;5:39–52. doi: 10.1007/s11523-010-0135-8. [DOI] [PubMed] [Google Scholar]
- [38].Morgan B, Thomas AL, Drevs J, Hennig J, Buchert M, Jivan A, Horsfield MA, Mross K, Ball HA, Lee L, Mietlowski W, Fuxius S, Unger C, O'Byrne K, Henry A, Cherryman GR, Laurent D, Dugan M, Marmé D, Steward WP. Dynamic Contrast-Enhanced Magnetic Resonance Imaging As a Biomarker for the Pharmacological Response of PTK787/ZK222584, an Inhibitor of the Vascular Endothelial Growth Factor Receptor Tyrosine Kinases, in Patients With Advanced Colorectal Cancer and Liver Metastases: Results From TwoPhase I Studies. Journal of Clinical Oncology. 2003;21:3955–3964. doi: 10.1200/JCO.2003.08.092. [DOI] [PubMed] [Google Scholar]
- [39].Checkley D, Tessier J, Wedge S, Dukes M, Kendrew J, Curry B, Middleton B, Waterton J. Dynamic contrast-enhanced MRI of vascular changes induced by the VEGF-signalling inhibitor ZD4190 in human tumour xenografts. Magnetic Resonance Imaging. 2003;21:475–482. doi: 10.1016/s0730-725x(02)00644-6. [DOI] [PubMed] [Google Scholar]
- [40].Rudin M, McSheehy PMJ, Martin PRA, Baumann RD, Becquet M, Brecht K, Brueggen J, Ferretti S, Schaeffer F, Schnell C, Wood J. PTK787/ZK222584, a tyrosine kinase inhibitor of vascular endothelial growth factor receptor, reduces uptake of the contrast agent GdDOTA by murine orthotopic B16/BL6 melanoma tumours and inhibits their growth in vivo. NMR in Biomedicine. 2005;18:308–321. doi: 10.1002/nbm.961. [DOI] [PubMed] [Google Scholar]
- [41].Galbraith S, Maxwell R, Lodge M, Tozer G, Wilson J, Taylor N, Stirling J, Sena L, Padhani A, Rustin G. Combretastatin A4 phosphate has tumor antivascular activity in rat and man as demonstrated by dynamic magnetic resonance imaging. Journal of Clinical Oncology. 2003;21:2831–2834. doi: 10.1200/JCO.2003.05.187. [DOI] [PubMed] [Google Scholar]
- [42].Larsson H, Stubgaard M, Frederiksen J, Jensen M, Henriksen O, Paulson O. Quantitation of blood-brain barrier defect by magnetic resonance imaging and gadolinium-DTPA in patients with multiple sclerosis and brain tumors. Magnetic Resonance in Medicine. 1990;16:117–131. doi: 10.1002/mrm.1910160111. [DOI] [PubMed] [Google Scholar]
- [43].Sourbron S, Ingrisch M, Siefert A, Reiser M, Herrmann K. Quantification of cerebral blood flow, cerebral blood volume, and blood-brain-barrier leakage with DCE-MRI. Magnetic Resonance in Medicine. 2009;62:205–217. doi: 10.1002/mrm.22005. [DOI] [PubMed] [Google Scholar]
- [44].Vlachos F, Tung Y, Konofagou E. Permeability dependence study of the focused ultrasound-induced blood-brain barrier opening at distinct pressures and microbubble diameters using DCE-MRI. Magnetic Resonance in Medicine. 2011;66:821–830. doi: 10.1002/mrm.22848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Côté J, Savard M, Bovenzi V, Dubuc C, Tremblay L, Tsanaclis A, Fortin D, Lepage M, Gobeil F., Jr Selective tumor blood-brain barrier opening with the kinin B2 receptor agonist [Phe(8)psi(CH(2)NH)Arg(9)]-BK in a F98 glioma rat model: an MRI study. Neuropeptides. 2010;44:177–185. doi: 10.1016/j.npep.2009.12.009. [DOI] [PubMed] [Google Scholar]
- [46].Sarin H, Kanevsky AS, Fung SH, Butman JA, Cox RW, Glen D, Reynolds R, Auh S. Metabolically stable bradykinin B2 receptor agonists enhance transvascular drug delivery into malignant brain tumors by increasing drug half-life. Journal of Translational Medicine. 2009;7 doi: 10.1186/1479-5876-7-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Saavedra JE, Srinivasan A, Bonifant CL, Chu J, Shanklin AP, Flippen-Anderson JL, Rice W, Turpin JA, Davies KM, Keefer LK. The secondary amine/nitric oxide complex ion R(2)N[N(O)NO](−) as nucleophile and leaving group in S9N)Ar reactions. Journal of Organic Chemistry. 2001;66:3090–3098. doi: 10.1021/jo0016529. [DOI] [PubMed] [Google Scholar]
- [48].Bradford MM. Rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Analytical Biochemistry. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- [49].Scheffler K, Hennig J. T1 quantification with inversion recovery TrueFISP. Magnetic Resonance in Medicine. 2001;45:720–723. doi: 10.1002/mrm.1097. [DOI] [PubMed] [Google Scholar]
- [50].Scheffler K. On the transient phase of balanced SSFP sequences. Magnetic Resonance in Medicine. 2003;49:781–783. doi: 10.1002/mrm.10421. [DOI] [PubMed] [Google Scholar]
- [51].Thelwall PE, Neves AA, Brindle KM. Measurement of bioreactor perfusion using dynamic contrast agent-enhanced magnetic resonance imaging. Biotechnology and Bioengineering. 2001;75:682–690. doi: 10.1002/bit.10039. [DOI] [PubMed] [Google Scholar]
- [52].Evelhoch JL. Key factors in the acquisition of contrast kinetic data for oncology. Journal of Magnetic Resonance Imaging. 1999;10:254–259. doi: 10.1002/(sici)1522-2586(199909)10:3<254::aid-jmri5>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- [53].Agarwal R, Shukla GS. Potential role of cerebral glutathione in the maintenance of blood-brain barrier integrity in rat. Neurochem Res. 1999;24:1507–14. doi: 10.1023/a:1021191729865. [DOI] [PubMed] [Google Scholar]
- [54].Ballabh P, Braun A, Nedergaard M. The blood-brain barrier: an overview: structure, regulation, and clinical implications. Neurobiol Dis. 2004;16:1–13. doi: 10.1016/j.nbd.2003.12.016. [DOI] [PubMed] [Google Scholar]
- [55].Mocellin S, Bronte V, Nitti D. Nitric oxide, a double edged sword in cancer biology: searching for therapeutic opportunities. Med Res Rev. 2007;27:317–52. doi: 10.1002/med.20092. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Time course of cGMP release in U87 cells. Cells treated with ODQ and untreated cells were exposed to increasing concentrations of JS-K (1 μM, 5 μM, 10 μM and 15 μM) for 5 – 120 min, cells exposed to DMSO (max. concentration of 0.3%) were used as solvent control. *p 0.05, # p 0.01 and § p 0.001 t-test compared to control (Ctr)
Cumulative survival of rats with orthotopic U87 gliomas. Treatment groups (n=9/group) consist of five rats of the MRI study and four additional rats that were treated accordingly. There was no significant difference in the survival of rats in the JS-K treated group (light) compared to untreated controls (Ctr, dark) (20.1 vs. 19.7 days).