

Abstract
N-methyl-D-aspartate (NMDA) receptors are ion channels activated by the neurotransmitter glutamate and are highly expressed by neurons. These receptors are critical for excitatory synaptic signaling and inhibition of NMDA receptors leads to impaired cognition and learning. Ethanol inhibits NMDA currents at concentrations associated with intoxication and this action may underlie some of the behavioral effects of ethanol. Although numerous sites and mechanisms of action have been tested, the manner in which ethanol inhibits NMDA receptors remains unclear. Recent findings in the literature suggest that ethanol, via facilitation of tyrosine phosphatase activity, may dephosphorylate key tyrosine residues in the C-terminus of GluN2B subunits resulting in diminished channel function. To directly test this hypothesis, we engineered GluN2B mutants that contained phenylalanine in place of tyrosine at three different sites and transiently expressed them with the GluN1 subunit in human embryonic kidney (HEK) cells. Whole-cell patch clamp electrophysiology was used to record glutamate-activated currents in the absence and presence of ethanol (10–600 mM). All mutants were functional and did not differ from one another with respect to current amplitude, steady-state to peak ratio, or magnesium block. Analysis of ethanol dose-response curves showed no significant difference in IC50 values between wild-type receptors and Y1252F, Y1336F, Y1472F or triple Y-F mutants. These findings suggest that dephosphorylation of C-terminal tyrosine residues does not account for ethanol inhibition of GluN2B receptors.
Keywords: Ethanol, phosphorylation, GluN2B, Electrophysiology
Introduction
The N-methyl-D-aspartate receptor (NMDAR) is a ligand-gated ionotropic receptor activated by the neurotransmitter glutamate. NMDARs are highly calcium permeable and show enriched expression in the post-synaptic density where they interact with a host of scaffolding proteins and kinases (Gladding and Raymond, 2011). NMDA receptors are critical to fast excitatory neurotransmission and numerous studies have shown that they are essential for many forms of neuronal plasticity including long-term potentiation. Aberrant function or altered expression of NMDARs is associated with a number of neuropathological states including excitotoxic cell death, schizophrenia, and drug addiction (Tzschentke and Schmidt, 2003). Functional receptors are tetrameric and composed of two obligate GluN1 subunits as well as two GluN2 subunits that bind glycine and glutamate respectively (Traynelis et al, 2010). A third subunit, dubbed GluN3, has been discovered and imparts unique functional characteristics to NMDARs (Cavara and Hollmann, 2008; Smothers and Woodward, 2007). GluN2-containing NMDARs, however, are the most widely studied receptor composition, and a significant body of literature has shown that NMDAR complexes are inhibited by physiologically relevant doses of alcohol across many brain areas (Lovinger et al, 1989; Roberto et al, 2004; Yaka et al, 2003).
Despite much effort, it remains unclear what discrete mechanism(s) underlie ethanol inhibition of NMDARs. Data from most studies suggest that ethanol inhibition is not due to competitive antagonism at either the glutamate or glycine binding sites, nor due to direct channel block (Masood et al, 1994; Mirshahi and Woodward, 1995; Peoples and Weight, 1992). Studies of single channel behavior have shown that ethanol affects channel gating (Wright et al, 1996) and results from this laboratory and others have identified possible sites of action for ethanol within transmembrane domains of GluN1 and GluN2 subunits (Ren et al, 2003; Ren et al, 2012; Ronald et al, 2001; Smothers and Woodward, 2006).
In addition to possible direct sites of action for ethanol, ethanol may impact post-translational modifications of NMDARs such as phosphorylation that may then influence receptor activity. For example, the GluN2B C-terminal region is the longest of the GluN2 family and is known to interact with a variety of kinases and phosphatases including those that target tyrosine residues (Salter and Kalia, 2004). Indeed, it has been shown that physiologically relevant doses of ethanol result in significant alterations in the phosphorylation state of tyrosine 1472 (Y1472) of the GluN2B subunit (Wu et al, 2010). The extent of this modification arises by the opposing actions of the neuronally enriched tyrosine kinase Fyn and striatal-enriched protein phosphatase (STEP) (Wang et al, 2007; Hicklin et al, 2011). As the phosphorylation status of Y1472 influences the surface expression of GluN2B containing receptors, it has been suggested that ethanol-induced dephosphorylation of this residue may underlie ethanol’s inhibition of NMDAR currents in brain slices (Alvestad et al, 2003; Hicklin et al, 2011; Prybylowski et al, 2005; Wu et al, 2010). To directly determine whether dephosphorylation of key C-terminal tyrosine residues accounts for the inhibitory actions of ethanol, a series of tyrosine to phenylalanine phospho-site mutants were generated in the GluN2B subunit. If dephosphorylation of these tyrosines represents a key step in the inhibition of channel currents by alcohol, then rendering these sites permanently dephosphorylated should mimic ethanol inhibition and occlude any further action of ethanol.
Materials and Methods
Cell Culture and Transfection
Human embryonic kidney (HEK) 293 cells were obtained from ATCC (Manassas, VA) and maintained in 10 cm culture dishes containing serum-supplemented DMEM in a humidified incubator with 5% CO2. Cells were split every 48 hours. For recordings, cells were split on poly-ornithine coated 35 mm dishes and 24 hours later transfected using Lipofectamine 2000 reagent (Invitrogen Inc, Carlsbad, CA). Plasmids containing rat GluN1, wild-type or mutant rat GluN2B, and an enhanced green fluorescent protein for cell selection were transfected at a 2:2:1 ratio unless otherwise noted. Mutant receptors were generated using the Quik Change II site-directed mutagenesis kit (Agilent Technologies, Santa Clara, CA) and nucleotide substitutions were verified via sequencing (Genewiz, South Plainfield, NJ). Prior to transfection, media was exchanged with fresh serum-supplemented DMEM containing 0.5 mM AP5 to prevent glutamate-mediated excitotoxicity (Cik et al, 1994). AP5 was removed by extensive washing prior to recording.
Electrophysiology
Dishes containing transfected cells were mounted on an Olympus IX50 inverted microscope and perfused with an extracellular recording solution at a rate of 1–2mL/min. Extracellular recording solution contained the following (in mM); NaCl (135), KCl (5.4), CaCl2 (1.8), HEPES (5), glucose (10), (pH adjusted to 7.4 with 1M NaOH, and osmolarity adjusted to 315–325 mOsm with sucrose). Patch pipettes (2–4 MOhms) were pulled from standard wall borosilicate glass (1.5 × 0.85 mm) and filled with internal solution containing the following (in mM); CsCl (140), MgCl2 (2), EGTA (5), HEPES (10), NaATP (2), NaGTP (0.3), (pH adjusted to 7.2 with 2M CsOH, and osmolarity adjusted to 290–295 mOsm with sucrose). Transfected cells were identified by eGFP fluorescence and whole-cell voltage clamp recordings were performed at room temperature using an Axon Instruments 200B microamplifier (Molecular Devices, Union City, CA). Cells were held at −70 mV to monitor breakthrough and maintained at this potential unless otherwise noted. Whole-cell capacitance and series resistance were compensated for and access resistance was monitored throughout the experiment. Cells demonstrating unstable holding currents or significant changes in series resistance were excluded from analysis. NMDA currents were evoked using a Warner FastStep multi-barrel perfusion system programmed to switch between extracellular recording solution and solution containing agonist (10 μM glutamate and glycine) or agonist plus ethanol (10–600 mM). Glutamate dose-response curves were established using a similar method by increasing the concentration of glutamate from 0.03–10 μM. The order of solutions was interleaved to monitor current rundown and changes >15% were excluded from analysis. For current-voltage relationship experiments, stable whole-cell configurations were first established at −70 mV, after which cells were held at 0 mV. Extracellular solution containing agonist and MgCl2 was then washed on and the cell was subjected to a ramp protocol consisting of a jump to −80 mV, a 1.3s ramp to +80 mV, and a return to 0 mV holding potential. This protocol was repeated three times and I–V curves were obtained from the average of the three traces. All data were filtered at 1–2 kHz and acquired at 5 kHz using an Instrutech ITC-16 digital interface (HEKA Instruments, Bellmore, NY) and analyzed offline by Axograph X software (Axograph Scientific, Sydney, NSW, Australia).
Data Analysis
Data are expressed as mean ± SEM and were analyzed by analysis of variance using Prism 5.0 software (Graphpad Software, San Diego, CA) where indicated. Concentration-response curves for ethanol and glutamate were analyzed using non-linear regression. Not all ethanol concentrations could be tested on each cell thus preventing using Prism to calculate individual IC50s for each cell. Instead for these data, a two-parameter (slope and EC50) logistic function was estimated for all curves using SPSS (IBM Corporation, Armonk, NY) and individual curve IC50s were then analyzed for group differences via ANOVA. Preliminary analysis indicated that slopes were not different and all curves for each group were estimated simultaneously with shared logistic slopes. For all analyses, statistical significance was set at p<0.05.
Results
Figure 1 shows a schematic of the GluN2B subunit, highlighting the N-terminal domain, the glutamate binding site, transmembrane domains, and the intracellular C-terminal region of the GluN2B subunit. Residues that were mutated from tyrosine to phenylalanine were at positions 1252, 1336 and 1472 of the full length rat GluN2B subunit. Wild-type and mutant GluN2B constructs were co-expressed with GluN1-1a subunits in HEK293 cells and whole-cell patch clamp electrophysiology was used to determine the sensitivity of ethanol.
Figure 1.
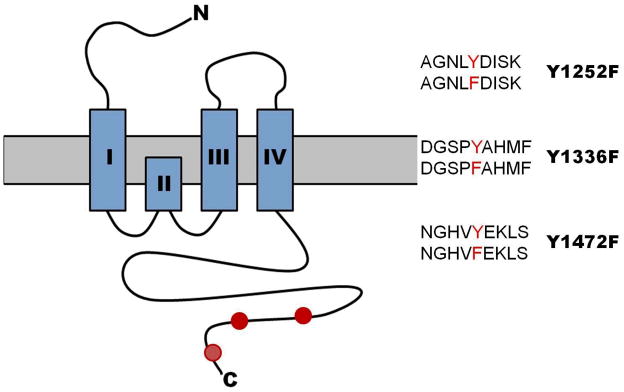
Schematic cartoon of the NMDA GluN2B subunit. The extracellular N-terminus, four transmembrane domains denoted by the four blue boxes, and the intracellular C-terminus are depicted in the diagram. Red circles on the intracellular C-terminus indicate the approximate location of tyrosine residues 1252, 1336, and 1472. Also depicted are comparisons of wild-type (top) and mutant (bottom) amino acid sequences for each mutation studied.
Table 1 summarizes the functional characteristics of wild-type and mutant receptors in terms of peak and steady-state amplitude and the steady-state to peak ratio, a marker of macroscopic receptor desensitization. While no significant differences were observed between wild-type and mutant receptors for peak current amplitude, one-way ANOVA revealed an overall significant effect between groups for steady-state current amplitude and steady-state to peak ratio (f(4,119)=2.2694, p=0.034; f(4,119)=2.729, p=0.032 respectively). Post-hoc analysis using Bonferroni and Dunnett’s tests, however, demonstrated no significant differences between wild-type and mutant receptors for either parameter.
Table 1.
Functional characteristics of mutant and wild-type receptors. Values for peak current, steady-state current, and steady-state to peak ratio are presented as mean ± SEM (N). While no significant effect in peak current amplitude was observed, one-way ANOVA did reveal a significant overall effect in steady-state current amplitude and steady-state to peak ratio (p = 0.032, p = 0.034 respectively). Dunnett’s post-hoc testing, however, demonstrated no significant effect between pair-wise comparisons of wild-type and mutant receptors.
| NR1-1a | |||
|---|---|---|---|
| Mutant Expressed | Peak Current, PA | Steady-state Current, pA | SS: Peak Ratio |
| NR2B-WT | −261.6 ± 19.6 (41) | −182.4 ± 14.0 (41) | 0.70 ± 0.02 (41) |
| Y1472F | −226.1 ± 26.3 (22) | −152.4 ± 18.9 (22) | 0.67 ± 0.02 (22) |
| Y1336F | −205.6 ± 23.6 (25) | −134.7 ± 15.8 (25) | 0.65 ± 0.02 (25) |
| Y1252F | −280.6 ± 17.6 (17) | −215.1 ± 14.2 (17) | 0.78 ± 0.04 (17) |
| Triple Mutant | −305.8 ± 59.4 (20) | −190.4 ± 27.9 (20) | 0.68 ± 0.04 (20) |
To determine whether manipulation of the three tyrosine residues altered the receptor’s affinity for agonist, concentration-response curves for glutamate were generated. As shown in Figure 2, increasing concentrations of glutamate evoked increases in whole-cell current in both wild-type and mutant receptors. Nonlinear regression analysis revealed no significant difference in glutamate affinity for any of the mutants tested. Calculated EC50 values for glutamate were 0.92 μM (wild-type), 0.88 μM (Y1472F), 0.95 μM (Y1336F), 0.78 μM (Y1252F), and 0.69 μM (triple mutant). ANOVA revealed no significant differences between groups for glutamate EC50 values (f(4,27)=0.822, p=0.523).
Figure 2.
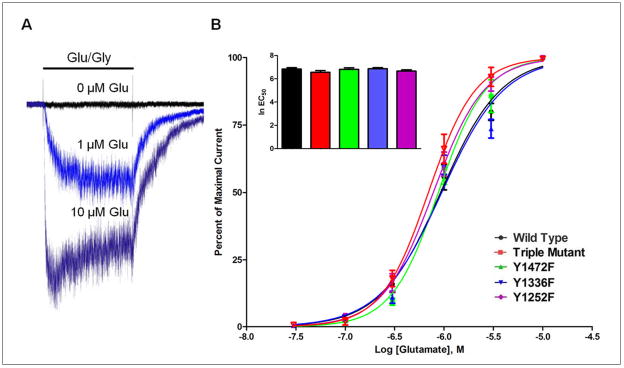
Concentration-response relationship for glutamate activation of mutant and wild-type GluN2B-containing NMDARs. (A) Example traces showing currents from wild-type NMDAR transfected cell during exposure to to 0.03 μM (black), 1 μM (light blue), and 10 μM glutamate (dark blue). (N = 6 – 8 cells). All recordings performed in the presence of 10 μM glycine. (B) Currents are expressed as percent of maximal response to various concentrations of glutamate (0.03–10 μM). Non-linear regression yielded EC50 values of 0.92 μM (wild-type), 0.88 μM (Y1472F), 0.95 μM (Y1336F), 0.78 μM (Y1252F), and 0.69 μM (triple mutant). Inset bar graph shows a comparison of ln EC50 values expressed as mean ± SEM.
As a follow-up to the agonist dose-response experiments, the effect of tyrosine mutations on the voltage-dependent magnesium block of NMDA receptors was also determined. Figure 3A shows glutamate-evoked currents in the presence of 2 mM Mg++ in cells held at different membrane potentials. In these studies, the concentrations of glutamate and glycine were held at 10 μM and current amplitude was expressed as a function of cell capacitance. Analysis of the current-voltage relationship revealed no significant differences between wild-type and mutants receptors although overall amplitudes did tend to vary. To account for this variability, I/V curves generated in each cell were normalized to the amplitude measured at +80 mV where Mg block is minimal. Figure 3B shows these normalized data and confirms there were no differences in voltage-dependent block between wild-type and mutant receptors.
Figure 3.

Current-voltage relationship of wild-type and phospho-mutant NMDARs. (A) Cells were voltage clamped at 0 mV and then stepped to −80 mV for 200 ms followed by a 1.3s ramp to +80 mV for another 200 ms before returning to 0 mV. All recordings were performed in the presence of 2 mM Mg++. (N = 7 – 9 cells) (B) Current-voltage relationship of wild-type and mutant receptors after normalizing current to that obtained at +80 mV.
Finally, the effect of a series of ethanol concentrations on glutamate-activated currents was determined for wild-type and mutant receptors. In these studies, currents were evoked with 10 μM glutamate/glycine in the absence or presence of ethanol (10–600 mM). As shown in Figure 4, ethanol caused a concentration-dependent inhibition of wild-type and mutant receptors. At the lowest concentration tested (10 mM), currents were inhibited by approximately 5–15% while 600 mM ethanol completely eliminated NMDA receptor currents. In fact, in all receptors tested, 600 mM ethanol resulted in slightly greater than 100% inhibition due to the presence of outward currents at this concentration. These currents may have resulted from ethanol’s inhibition of leak current or from the slight change in ionic strength (~4%) produced when preparing the 600 mM ethanol solution. To examine these possibilities, non-transfected cells were voltage-clamped at -70 mV and holding currents were monitored before and during administration of ethanol. At a concentration of 600 mM, ethanol produced outward currents in non-transfected cells with an average amplitude of 11.63 pA ± 5.25 pA (mean ± SEM). No such effect was observed when ethanol was replaced with water suggesting that ethanol’s effect on holding current was due to inhibition of a leak current and not a change in ionic strength. To account for this effect, the mean amplitude of the outward current measured in non-transfected cells was subtracted from the currents obtained in transfected cells during exposure to 600 mM ethanol. Regression analysis of the ethanol concentration-response curves yielded estimated IC50 values of 136 mM for wild-type, 135 mM for Y1472F, 118 mM for Y1336F, 104 mM for Y1252F, and 156 mM for the triple mutant. There were no significant differences between groups for ethanol IC50 (f(4,86)=1.48, p=0.215).
Figure 4.
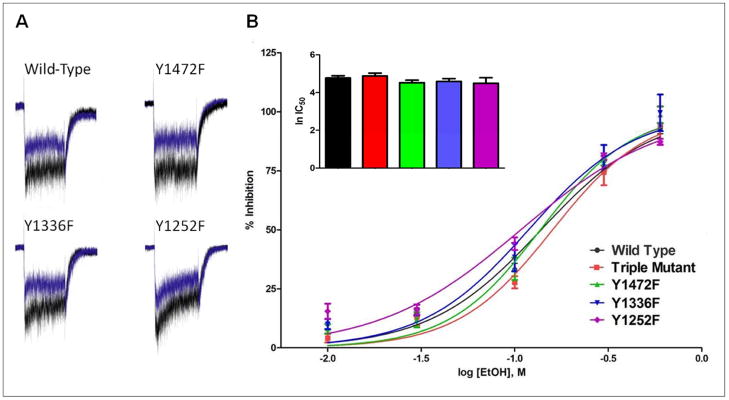
Concentration-response effect of ethanol on wild-type and mutant GluN2B-containing NMDARs. (A) Example traces from a cell expressing wild-type and mutant GluN2B receptors. Agonist-only (blue) current; agonist + 100 mM EtOH (black). (B) Data are expressed as percent inhibition by ethanol (10–600 mM) of glutamate-evoked currents. (N = 6–8 cells per concentration). Nonlinear regression yielded estimated IC50 values for ethanol of 136 mM (wild-type), 135 mM (Y1472F), 118 mM (Y1336F), 104 mM (Y1252F), and 156 mM (triple mutant). Inset bar graph shows a comparison of ln IC50 values expressed as mean ± SEM.
Discussion
In the present study, site-directed mutagenesis and whole-cell patch-clamp electrophysiology were used to examine the ethanol sensitivity of recombinant NMDARs with mutations that mimic permanently dephosphorylated tyrosine residues implicated in the inhibitory actions of ethanol. All mutant receptors tested in HEK293 cells showed robust glutamate-activated currents and all mutants retained wild-type like sensitivity to ethanol. The results of this study strongly suggest that dephosphorylation of these C-terminal tyrosine residues does not mediate the acute inhibitory actions of ethanol on recombinant NMDARs. They also suggest that ethanol inhibition of NMDA receptors expressed in native neurons is not solely due to dephosphorylation at these sites as has been suggested from studies in brain slices (Wu et al, 2010; Hicklin et al, 2011). Indeed, it is more likely that the phosphorylation state of these residues may represent intermediate sites of regulation in a larger cascade of events that are induced during long exposures to ethanol.
NMDA receptors are subject to tyrosine phosphorylation and the Src-family kinase Fyn is highly enriched in neurons where it associates with GluN2B-containing NMDARs. Over-expression of a constitutively active form of Fyn kinase in cerebellar granule cells resulted in robust increases in NMDA mini-EPSCs with little change in overall kinetics, suggesting increased membrane insertion of NMDARs (Prybylowski et al, 2005). Co-expression of Fyn kinase with recombinant NMDA receptors to enhance tyrosine phosphorylation had no effect on the ethanol sensitivity of GluN1/GluN2B currents, supporting the hypothesis that changes in NMDAR currents observed in neuronal tissue by Fyn is due to altered trafficking (Kohr and Seeburg, 1996; Anders et al, 1999). Work conducted by Nakazawa et al (2001) further showed that Fyn preferentially phosphorylates residues Y1252, Y1336, and Y1472 on GluN2B.
The interaction of Fyn kinase with GluN2B receptors is highly brain region dependent. Work by Yaka et al (2003) showed that compartmentalization of Fyn with NMDARs occurs prominently in hippocampus and dorsal striatum but not in cerebral cortex. This interaction requires the scaffolding protein RACK1 and enhanced scaffolding of Fyn and GluN2B via RACK1 during ethanol exposure allows for increased membrane insertion of GluN2B-containing NMDARs, resulting in enhanced currents and the development of a functional form of acute ethanol tolerance (Wang et al, 2007; Wang et al, 2010). This mechanism is consistent with the lack of acute ethanol tolerance at the electrophysiological and behavioral level observed in Fyn-deficient mice (Miyakawa et al, 1997).
While activation of Fyn and enhanced NMDAR membrane trafficking may compensate for reduced NMDA signaling during acute ethanol inhibition, recent data from other laboratories suggests that dephosphorylation of key GluN2B tyrosine residues by striatal enriched protein phosphatase (STEP) may directly underlie the inhibitory actions of alcohol. STEP is abundantly expressed in the striatum and hippocampus, and associates with GluN2B-containing NMDARs (Pelkey et al, 2002). It has been hypothesized that STEP mediates the inhibitory actions of ethanol on NMDARs by dephosphorylating Y1472 on the C-terminus of GluN2B. Using a phospho-Y1472 GluN2B antibody, Wu et al. (2010) demonstrated a significant reduction in immunoreactivity in hippocampal slices following acute administration of 80 mM ethanol. Further work showed that mice lacking STEP showed no change in the phosphorylation of Y1472 following ethanol administration (Hicklin et al, 2011). Inhibiting STEP with dominant-negative peptide eliminated ethanol inhibition of NMDAR-mediated EPSCs in hippocampal slices. Interestingly, however, a compensatory rebound in NMDAR current was observed after washout of ethanol in these experiments, likely due to the activity of Fyn (Yaka et al., 2003).
In the present study, mutating Y1472 to phenylalanine in order to mimic a permanently dephosphorylated state had no effect on ethanol sensitivity or channel function of recombinant GluN2B receptors. These results are consistent with prior work by our laboratory that found co-expression of various kinases including c-Src and Fyn also had no effect on the ethanol sensitivity of recombinant GluN2B-containing NMDARs, despite significant enhancement of tyrosine phosphorylation of these subunits (Anders et al, 1998; Anders et al, 1999). Results from recombinant and brain slice studies instead suggest that acute administration of ethanol likely induces multiple effects. First, ethanol directly inhibits channel function on a sub-second time scale, perhaps by interacting with regions of GluN1 and GluN2 subunits that are involved in receptor gating (Ren et al, 2012; Ronald et al, 2001; Smothers and Woodward, 2006; Xu et al., 2011). Next, via an unknown mechanism, STEP is activated and preferentially recruited to GluN2B subunits where it dephosphorylates Y1472, promoting their removal from the post-synaptic density and internalization (Hicklin et al, 2011; Prybylowksi et al, 2005). Finally, ethanol-induced activity of Fyn via Rack1 scaffolding compensates for STEP-induced internalization, producing enhanced surface expression of NMDARs that is revealed following ethanol washout (Wang et al; 2007). Such a scenario may exist in brain regions such as hippocampus and striatum where STEP and Fyn prominently associate with GluN2B-containing NMDARs. Other brain areas such as cortex that do not show robust interactions with STEP and Fyn show significant inhibition of NMDA-mediated EPSCs without evidence of a rebound in NMDA EPSCs following cessation of ethanol exposure.
In summary, the results of this study show that the phospho-state of key C-terminal tyrosine residues of the GluN2B subunit do not directly regulate ethanol sensitivity. Rather, these residues likely influence apparent ethanol sensitivity via alterations in the trafficking and surface expression of GluN2B-containing NMDARs. Such data demonstrate that the actions of ethanol are myriad and complex and may modulate NMDAR activity through a number of different mechanisms.
Acknowledgments
The authors thank Dr. Patrick Russell for his assistance with the statistical analysis of the data. This work was supported by a grant to JJW from the U.S. National Institutes of Health (R37 AA009986) and a traineeship to BAH from NIH (T32 AA007474).
Footnotes
The authors declare no competing financial interests
References
- Alvestad RM, Grosshans DR, Coultrap SJ, Nakazawa T, Yamamoto T, Browning MD. Tyrosine dephosphorylation and ethanol inhibition of N-methyl-D-aspartate receptor function. J Biol Chem. 2003;278(13):11020–11025. doi: 10.1074/jbc.M210167200. [DOI] [PubMed] [Google Scholar]
- Anders DL, Blevins T, Sutton G, Chandler LJ, Woodward JJ. Effects of c-Src tyrosine kinase on ethanol sensitivity of recombinant NMDA receptors expressed in HEK 293 cells. Alcohol Clin Exp Res. 1999;23(2):357–362. [PubMed] [Google Scholar]
- Anders DL, Blevins T, Sutton G, Swope S, Chandler LJ, Woodward JJ. Fyn tyrosine kinase reduces the ethanol inhibition of recombinant NR1/NR2A but not NR1/NR2B NMDA receptors expressed in HEK 293 cells. J Neurochem. 1999;72(4):1389–1393. doi: 10.1046/j.1471-4159.1999.721389.x. [DOI] [PubMed] [Google Scholar]
- Cavara NA, Hollmann M. Shuffling the deck anew: how NR3 tweaks NMDA receptor function. Mol Neurobiol. 2008;38(1):16–26. doi: 10.1007/s12035-008-8029-9. [DOI] [PubMed] [Google Scholar]
- Cik M, Chazot PL, Stephenson FA. Expression of NMDAR1–1a (N598Q)/NMDAR2A receptors results in decreased cell mortality. Eur J Pharmacol. 1994;266:R1–R3. doi: 10.1016/0922-4106(94)90146-5. [DOI] [PubMed] [Google Scholar]
- Gladding CM, Raymond LA. Mechanisms underlying NMDA receptor synaptic/extrasynaptic distribution and function. Mol Cell Neurosci. 2011;48(4):308–320. doi: 10.1016/j.mcn.2011.05.001. [DOI] [PubMed] [Google Scholar]
- Hicklin TR, Wu PH, Radcliffe RA, Freund RK, Goebel-Goody SM, Correa PR, Proctor WR, Lombroso PJ, Browning MD. Alcohol inhibition of the NMDA receptor function, long-term potentiation, and fear learning requires striatal-enriched protein tyrosine phosphatase. Proc Natl Acad Sci U S A. 2011;108(16):6650–6655. doi: 10.1073/pnas.1017856108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohr G, Seeburg PH. Subtype-specific regulation of recombinant NMDA receptor-channels by protein tyrosine kinases of the src family. J Physiol. 1996;492(2):445–452. doi: 10.1113/jphysiol.1996.sp021320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovinger DM, Whilte G, Weight FF. Ethanol inhibits NMDA-activated ion current in hippocampal neurons. Science. 1989;243(4899):1721–1724. doi: 10.1126/science.2467382. [DOI] [PubMed] [Google Scholar]
- Masood K, Wu C, Brauneis U, Weight FF. Differential ethanol sensitivity of recombinant N-methyl-D-aspartate receptor subunits. Mol Pharmacol. 1994;45(2):324–329. [PubMed] [Google Scholar]
- Mirshahi T, Woodward JJ. Ethanol sensitivity of heteromeric NMDA receptors: effects of subunit assembly, glycine and NMDAR1 Mg(2+)-insensitive mutants. Neuropharmacology. 1995;34(3):347–355. doi: 10.1016/0028-3908(94)00155-l. [DOI] [PubMed] [Google Scholar]
- Miyakawa T, Yagi T, Kitazawa H, Yasuda M, Kawai N, Tsuboi K, Niki H. Fyn-kinase as a determinant of ethanol sensitivity: relation to NMDA-receptor function. Science. 1997;278:698–701. doi: 10.1126/science.278.5338.698. [DOI] [PubMed] [Google Scholar]
- Nakazawa T, Komai S, Tezuka T, Hisatsune C, Umemori H, Semba K, Mishina M, Manabe M, Yamamoto T. Characterization of Fyn-mediated tyrosine phosphorylation sites on GluR epsilon 2 (NR2B) subunit of the N-methyl-D-aspartate receptor. J Biol Chem. 2001;276(1):693–699. doi: 10.1074/jbc.M008085200. [DOI] [PubMed] [Google Scholar]
- Pelkey KA, Askalan R, Paul S, Kalia LV, Nguyen TH, Pitcher GM, Salter MW, Lombroso PJ. Tyrosine phosphatase STEP is a tonic brake on induction of long-term potentiation. Neuron. 2002;34(1):127–138. doi: 10.1016/s0896-6273(02)00633-5. [DOI] [PubMed] [Google Scholar]
- Peoples RW, Weight FF. Ethanol inhibition of N-methyl-D-aspartate-activated ion current in rat hippocampal neurons is not competitive with glycine. Brain Res. 1992;571(2):342–344. doi: 10.1016/0006-8993(92)90674-x. [DOI] [PubMed] [Google Scholar]
- Prybylowski K, Chang K, Sans N, Kan L, Vicini S, Wenthold RJ. The synaptic localization of NR2B-containing NMDA receptors is controlled by interactions with PDZ proteins and AP-2. Neuron. 2005;47(6):845–857. doi: 10.1016/j.neuron.2005.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren H, Honse Y, Karp BJ, Lipsky RH, Peoples RW. A site in the fourth membrane-associated domain of the N-methyl-D-aspartate receptor regulates desensitization and ion channel gating. J Biol Chem. 2003;278(1):276–283. doi: 10.1074/jbc.M209486200. [DOI] [PubMed] [Google Scholar]
- Ren H, Zhao Y, Dwyer DS, Peoples RW. Interactions among positions in the third and fourth membrane-associated domains at the intersubunit interface of the N-methyl-D-aspartate receptor forming sites of alcohol action. J Biol Chem. 2012;287:27302–27312. doi: 10.1074/jbc.M111.338921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberto M, Schweitzer P, Madamba SG, Stouffer DG, Parsons LH, Siggins GR. Acute and chronic ethanol alter glutamatergic transmission in rat central amygdale: and in vitro and in vivo analysis. J Neurosci. 2004;24(7):1594–1603. doi: 10.1523/JNEUROSCI.5077-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronald KM, Mirshahi T, Woodward JJ. Ethanol inhibition of N-methyl-D-aspartate receptors is reduced by site-directed mutagenesis of a transmembrane domain phenylalanine residue. J Biol Chem. 2001;276(48):44729–44735. doi: 10.1074/jbc.M102800200. [DOI] [PubMed] [Google Scholar]
- Salter MW, Kalia LV. Src kinases: a hub for NMDA receptor regulation. Nat Rev Neurosci. 2004;5(4):317–328. doi: 10.1038/nrn1368. [DOI] [PubMed] [Google Scholar]
- Smothers CT, Woodward JJ. Effects of amino acid substitutions in transmembrane domains of the NR1 subunit on the ethanol inhibition of recombinant N-methyl-D-aspartate receptors. Alcohol Clin Exp Res. 2006;30(3):523–530. doi: 10.1111/j.1530-0277.2006.00058.x. [DOI] [PubMed] [Google Scholar]
- Smothers CT, Woodward JJ. Pharmacological characterization of glycine-activated currents in HEK 293 cells expressing N-methyl-D-aspartate NR1 and NR3 subunits. J Pharmacol Exp Ther. 2007;322:739–748. doi: 10.1124/jpet.107.123836. [DOI] [PubMed] [Google Scholar]
- Traynelis SF, Wollmuth LP, McBain CJ, Menniti FS, Vance KM, Ogden KK, Hansen KB, Yuan H, Myers SJ, Dingledine R. Glutamate receptor ion channels: structure, regulation, and function. Pharmacol Rev. 2010;62(3):405–496. doi: 10.1124/pr.109.002451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tzschentke TM, Schmidt WJ. Glutamatergic mechanisms in addiction. Mol Psychiatry. 2003;8(4):373–382. doi: 10.1038/sj.mp.4001269. [DOI] [PubMed] [Google Scholar]
- Wang J, Carnicella S, Phamluong K, Jeanblanc J, Ronesi JA, Chaudhri N, Janak PH, Lovinger DM, Ron D. Ethanol induces long-term facilitation of NR2B-NMDA receptor activity in the dorsal striatum: implications for alcohol drinking behavior. J Neurosci. 2007;27(13):3593–3602. doi: 10.1523/JNEUROSCI.4749-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Lanfranco MF, Gibb SL, Yowell QV, Carnicella S, Ron D. Long-lasting adaptations of the NR2B-containing NMDA receptors in the dorsomedial striatum play a crucial role in alcohol consumption and relapse. J Neurosci. 2010;30(30):10187–10198. doi: 10.1523/JNEUROSCI.2268-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wills TA, Klug JR, Silberman Y, Baucum AJ, Weitlauf C, Colbran RJ, Delpire E, Winder DG. GluN2B subunit deletion reveals key role in acute and chronic ethanol sensitivity of glutamate synapses in bed nucleus of the stria terminalis. Proc Natl Acad Sci U S A. 2012;109(5):E278–E287. doi: 10.1073/pnas.1113820109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright JM, Peoples RW, Weight FF. Single-channel and whole-cell analysis of ethanol inhibition of NMDA-activated currents in cultured mouse cortical and hippocampal neurons. Brain Res. 1996;738:249–256. doi: 10.1016/s0006-8993(96)00780-9. [DOI] [PubMed] [Google Scholar]
- Wu PH, Coultrap S, Browning MD, Proctor WR. Correlated changes in NMDA receptor phosphorylation, functional activity, and sedation by chronic ethanol consumption. J Neurochem. 2010;115(5):1112–1122. doi: 10.1111/j.1471-4159.2010.06991.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu M, Chandler LJ, Woodward JJ. Ethanol inhibition of recombinant NMDA receptors is not altered by coexpression of CaMKII-alpha or CaMKII-beta. Alcohol. 2008;42(5):425–432. doi: 10.1016/j.alcohol.2008.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu M, Smothers CT, Woodward JJ. Effects of ethanol on phosphorylation site mutants of recombinant N-methyl-D-aspartate receptors. Alcohol. 2011;45(4):373–380. doi: 10.1016/j.alcohol.2010.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yaka R, Phamluong K, Ron D. Scaffolding of Fyn kinase to the NMDA receptor determines brain region sensitivity to ethanol. J Neurosci. 2003;23(9):3623–3632. doi: 10.1523/JNEUROSCI.23-09-03623.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]