

Abstract
Deformities of cranial sutures such as craniosynostosis and enlarged parietal foramina greatly impact human development and quality of life. Here we have examined the role of the extracellular matrix protein ameloblastin (Ambn), a recent addition to the family of non-collagenous extracellular bone matrix proteins, in craniofacial bone development and suture formation. Using RT-PCR, western blot and immunohistochemistry, Ambn was localized in mouse calvarial bone and adjacent condensed mesenchyme. Five-fold Ambn overexpression in a K14-driven transgenic mouse model resulted in delayed posterior frontal suture fusion and incomplete suture closure. Moreover, Ambn overexpressor skulls weighed 13.2% less, their interfrontal bones were 35.3% thinner, and the width between frontal bones plus interfrontal suture was 14.3% wider. Ambn overexpressing mice also featured reduced cell proliferation in suture blastemas and in mesenchymal cells from posterior frontal sutures. There was a more than 2-fold reduction of Msx2 in Ambn overexpressing calvariae and suture mesenchymal cells, and this effect was inversely proportionate to the level of Ambn overexpression in different cell lines. The reduction of Msx2 expression as a result of Ambn overexpression was further enhanced in the presence of the MEK/ERK pathway inhibitor O126. Finally, Ambn overexpression significantly reduced Msx2 down-stream target gene expression levels, including osteogenic transcription factors Runx2 and Osx, the bone matrix proteins Ibsp, ColI, Ocn and Opn, and the cell cycle-related gene CcnD1. Together, these data suggest that Ambn plays a crucial role in the regulation of cranial bone growth and suture closure via Msx 2 suppression and proliferation inhibition.
Introduction
During skull development, calvarial bones expand from initial ossification centers toward the suture interface between adjacent bones [1]. Developmentally, this gradual intramembranous ossification is accomplished by the differentiation of ectomesenchymal cells into calvarial osteoblasts [2]. On a molecular level, membranous bone ossification is regulated by a number of growth and transcription factors as well as extracellular matrix proteins, including fibroblast growth factor receptors FGFR1 and FGFR2 [3], transcription factors CBFA1/RUNX2 [4]–[6], MSX2 [7], TWIST [8],[9], and matrix proteins osteopontin and tenascin [10],[11]. Abnormal changes in these factors not only affect bone thickness and mineralization, but also the expansion of bone along the ossification center/suture axis, resulting in either synostosis or open foramina.
From a clinical perspective, disturbances in normal skull ossification result in a number of pathologies with often dramatic consequences for children's health. Premature ossification of one or several sutures of the skull leads to an abnormal skull shape or retarded skull growth (Craniosynostosis, CS). The most common type of CS involves disturbances in the formation of a single suture (simple or non syndromic CS), and its etiology remains unexplained. Multiple suture synostoses (syndromic CS) are often associated with syndromes of genetic origin i.e. Apert syndrome, Crouzon syndrome, Pfeiffer syndrome and Saethre-Chotzen syndrome [12]. The incidence of non-syndromic CS in United States is about 34.3/100,000 live births while syndromic CS is rare; their incidence is about 1.5/100,000 [13]. The treatment of CS is composed of multiple surgical interventions to correct the abnormal shape of the head, give space for the brain to grow in a normal fashion and prevent the development of intracranial pressure leading to neurological and brain damage complications or death [14]. Complications from surgical treatment can lead to infection, re-ossification, and death from hemorrhage [15].
Cranial osteogenesis and the differentiated state of the bone/suture interface are regulated by extracellular matrix molecules that transmit signals across the cell membrane into the cytoplasm, activating a number of signaling pathways, which subsequently affect gene expression [16],[17]. One of the recently discovered members of the bone extracellular matrix is ameloblastin (AMBN), a glycoprotein previously only associated with the extracellular enamel matrix [18],[19]. Recent studies have noted Ambn expression also in dentin, cementum, pulp, cranial bones, and primary osteoblasts [20]–[25]. Moreover, Ambn has been shown to regulate Msx2 expression in ameloblasts [18] [26], and Msx2 mRNA expression was dysregulated in Ambn deficient mice [26]. In the present study, we hypothesized that Ambn might affect the growth of craniofacial bones and the patency of cranial sutures through its effect on Msx2 and possibly other mechanisms. Here we have tested this hypothesis using Ambn transgenic mice and in vitro models. Our studies reveal a distinct effect of Ambn on craniofacial bone growth and suture closure, and suggests possible mechanisms of action.
Table 1. Primer sequences for RT-PCR analysis.
| AMBN | 5′ aaaacccaccaacacctgag | 3′ cattggtccccgagatattg |
| β-ACTIN | 5′ gatcattgctcctcctgagc | 3′ acatctgctggaaggtggac |
| IBSP | 5′ gaagcaggtgcagaaggaac | 3′ gaaacccgttcagaaggaca |
| CCND1 | 5′ cacacggactacaggggagt | 3′ cgcggagtctgtagctctct |
| ColI A | 5′ caccctcaagagcctgagtc | 3′ tccgctcttccagtcagagt |
| MSX2 | 5′ agacatatgagccccaccac | 3′ caaggctagaagctgggatg |
| OCN | 5′ tcgtgtgtcttctccacagc | 3′ tggccacttacccaaggtag |
| OPN | 5′ ttccaaagagagccaggaga | 3′ ttgtggctctgatgttccag |
| OSX | 5′ cccctgttcttcacagcttc | 3′ gggaaaacggcaaataggat |
| RunX2 | 5′ ccaccactcactaccacacg | 3′ tatggagtgctgctggtctg |
Materials and Methods
Transgene constructs and transgenic mice
Two transgenic constructs were generated using a modified pSKII-trans vector in which the K14 promoter, the polyA signal (a generous gift from Dr. Elaine Fuchs, Rockefeller University), the β-globulin intron, the mouse Ambn coding region, or the LacZ gene was inserted. The β-globulin intron was used to ensure that the transgenes were transcripted properly. The transgenic fragments were freed from pSKII-K14-AMBN or pSKII-K14-LacZ by digesting the constructs with Sac I and Hind III, gel purified and microinjected into mouse zygotes [19]. Mice used in the present study included human keratin 14 (K14) promoter-driven Ambn transgenic mice (AmbnTg, E18.5, P1, 20, 35 and 60 days of age, n = 14) and K14 promoter-driven LacZ transgenic mice (P1, n = 3). This study was carried out in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. The protocol was approved by the Committee on the Ethics of Animal Experiments of the University of Illinois at Chicago (Permit Number: 11–077). For further analysis, mice were euthanized in a CO2 chamber followed by cervical dislocation, and calvarial bone and suture tissues were collected for fixation or RNA/protein extraction.
Genotyping
Genotyping was carried out using tails collected from AmbnTg or LacZ transgenic heterozygous litters [19]. The tails were lysed in DirectPCR (Tail) buffer (Viagen, Los Angeles, CA) at 50°C overnight and then 85°C for 40 min. PCR was performed using a set of specific primers for K14 promoters: a forward primer 5′GCTTAGCCAGGGTGACAGAG 3′ and a reverse primer 5′CACAGAGGCGTAAATGCAGA3′. After 35 reaction cycles, aliquots of the PCR products were separated on a 2.0% TAE (Tris acetate EDTA) agarose gel, stained with ethidium bromide, and photographed under UV light [19].
Dry skull preparation, whole mount X-gal staining and alcian blue/alizarin red staining
Dry skulls of three adult mice from wild-type and Ambn transgenic mice at postnatal day 60(n = 3) were prepared using Dermestes beetles. Briefly, the crude muscle and soft tissues were removed from the animal heads and left cool-dry for several days. The dry skulls were gently placed in a container with Dermestes beetles for several days at 30°C under close observation. Once the soft tissues of skulls were completely removed, the dry skulls were collected and cleaned meticulously with a soft brush. The dry skull, and especially the cranial suture of each animal was examined and photographed under a stereo microscope. Width of the posterior frontal suture of each animal was measured with a digital caliper and recorded in mm units. For whole mount X-gal staining, de-skinned animal heads were fixed with 4% paraformaldehyde in PBS at 4°C overnight. The samples were rinsed with PBS at room temperature 3 times for 15 minutes each and then incubated in the dark with a staining buffer containing 0.05 mM K3Fe(CN)6, 0.05 mM K4Fe(CN)6, 1 mM MgCl2 and 1 mg/ml X-gal at 37°C for 7 hours. For whole mount alcian blue/alizarin red staining, three embryos from the wild type and Ambn transgenic groups at embryonic day 18.5(n = 3) were fixed and dehydrated with 70%, 100% ethanol and acetone. The samples were stained with saturated alcian blue (Sigma, St Louis, MO) in 95% ethanol for 2 days, destained with 95% ethanol, and rehydrated. Subsequently, samples were then stained with saturated alizarin red S(Sigma) in 0.5% potassium hydroxide (KOH) solution for 2 days, destained in 0.5% KOH solution until all soft connective tissue turned clear and then stored in 80% glycerol.
Tissue processing
Calvaria and cranial sutures from wildtype, Ambn or LacZ transgenic mice at age of embryonic 18 and postnatal 35 days (n = 3) were dissected and fixed with 10% formalin at 4°C and demineralized in a solution containing 45% EDTA, 4.5% NaOH and 1% formalin at 4°C until the demineralization was completed. The demineralized samples were embedded in paraffin, cut in 5 µm sagittal sections and mounted on glass slides. Thereafter, the samples were deparaffinized, rehydrated, and stained with Hematoxylin and Eosin or subjected to immunohistochemistry.
Immunohistochemistry
Sections were deparaffinized, rehydrated, and treated with 6% peroxide and methanol followed by a brief incubation in 10 mM sodium citrate buffer with 0.05% Tween 20 at pH 6.0 for antigen retrieval. Sections were then incubated in 2% bovine serum albumin (BSA) for 30 minutes at room temperature to block nonspecific binding of the antibody. After blocking, sections were first incubated with affinity purified anti-Ambn antibody [19],[27] at dilution of 1∶200, and then with anti-rabbit secondary antibody (Abcam, Cambridge, MA) at dilution of 1∶2000. The expression of Ambn protein was visualized with a Histomouse Broad Spectrum AEC kit (Invitrogen, Carlsbad, CA) under a light microscope. As a negative control, non-immune rabbit serum was used instead of the primary antibody.
BrdU and TUNEL staining
For BrdU staining, pregnant mice at E18 were injected with BrdU (100 mg/kg) intraperitoneally prior to euthanization. Calvaria and cranial sutures were dissected and processed for paraffin sections. The sections were deparaffinated, rehydrated, and stained with biotinylated anti-BrdU antibody. The BrdU positive cells were detected with the streptavidine-biotin system (Invitrogen). For TUNEL staining, sections were first exposed to proteinase K(20 µg/ml) at ambient temperature for 10 min, and then incubated with TdT staining solution at 37°C for 1 hour according to the manufacturer's instruction (Promega, Madison, WI). The fluorescence-dUTP-labeled DNA was visualized using a fluorescence microscope.
Cell Culture
The mouse suture mesenchymal cells were isolated from the underlying dura mater and overlying pericranium of posterior frontal and sagittal sutures of wildtype and Ambn transgenic mice at postnatal 3 days, and maintained in α-minimum essential medium supplemented with 10% fetal bovine serum, 100 U/ml penicillin, 100 µg/ml streptomycin and 25 ng/ml Amphotericin B in a 5% CO2 atmosphere at 37°C. The medium was changed twice a week. To study the effects of Ambn on cell proliferation and differentiation, suture mesenchymal cells from wildtype and Ambn transgenic mice were subjected to regular culture medium or a mineralization induction medium containing 50 µg/ml ascorbic acid and 2 mM β-glycerophosphate, and cultured for 2–21 days. Upon terminating the culture, cells were used for MTT assay, alkaline phosphatase activity test, alizarin red staining, or RNA and protein preparation.
Alkaline phosphatase and in vitro mineralization assay
After 7 days of culture in osteogenic media, cells were washed and stained with alkaline phosphatase substrate (Roche Diagnostic, Indianapolis, IN) to verify early osteogenic activity. After 21 days of culture in osteogenic media, the cells were fixed with methanol, stained with 10% alizarin red solution, and mineralized nodules were identified as red spots on the culture dish.
Cell proliferation assay
Prior to termination of culture, cells were incubated in MTT solution (2 mg/ml of MTT in DMEM with 2% FBS) for 4 hours. To quantify proliferative activity, the MTT stained cells were lysed in HCL/Isopropanol, and the absorbance was detected at 570 nm with background subtraction at 630 nm [28].
RNA extraction and RT-real time PCR
Total RNAs were isolated from mouse suture tissues or cultured cells using the TRIZOL LS Reagent (Invitrogen) according to the manufacturer's instructions. Two micrograms of total extracted RNA was applied toward cDNA generation with the Sprint RT Complete kit® (Clontech, Mountain View, CA). To quantify the mRNA expression levels of transcription factors and bone marker genes, real-time PCR primers were designed based on EMBL/GenBank searches (shown in Table I). Real-time PCR was performed using sequence specific sybergreen primers and the ABI Prism 7000 Sequence detection system (Applied Biosystems, Foster City, CA). Reaction conditions were as follows: 2 min at 50°C (one cycle), 10 min at 95°C (one cycle), and 15 sec at 95°C, and 1 min at 60°C (40 cycles). Samples were normalized using GAPDH. The analyses were performed in triplicate for three independent experiments to confirm reproducibility of the results. Relative expression levels were calculated using the 2–ΔΔCt method [29], and values were graphed as the mean expression level ± standard deviation.
Protein extraction and western blot analysis
Calvaria from 3 transgenic and 3 control animals at age P20 were collected and homogenized under liquid N2. Equal amounts of protein extracts in a lysis buffer containing 100 mM Tris HCl pH 9.0, 200 mM KCl, 25 mM EGTA, 36 mM MgCl2, 2% deoxycholic acid and 10% DTT v/v were subjected to SDS–polyacrylamide gel electrophoresis (Biorad, Hercules, CA), and the separated proteins were transferred to a PVDF (Polyvinylidene Difluoride) membrane (Immobilon P®, Millipore, Billerica, MA). The membrane was incubated with rabbit anti-mouse Ambn [19],[27], Msx2, or CcnD1 antibodies (Abcam). Immune complexes were detected with goat anti-rabbit IgG horseradish peroxidase-conjugated secondary antibody (Molecular Probes®, Carlsbad, CA) and enhanced chemiluminescence reagents (Pierce Biotechnology, Rockford, IL). The amount of protein expression was compared after normalization with the amount of β-actin as an internal calibrator in each lane.
Statistical analysis
Quantitative data were presented as means ± SD from three independent experiments and compared using Kruskall-Wallis one-way analysis of variance. The difference between groups was considered statistically significant at P<0.05.
Results
AMBN is expressed in calvarial bone
Previous studies have demonstrated that the extracellular matrix protein Ambn is expressed in cranial tissues including enamel [30], cementoblasts [22], and cranial bones [24]. To examine the function of Ambn in cranial bone and suture formation, Ambn expression in mouse posterior frontal suture structures was characterized. RT-PCR analysis of samples ranging between postnatal days 1–20 revealed Ambn mRNA expression in postnatal cranial sutures (Fig. 1A). Western blotting recognized 50 and 55 kDa bands positive for Ambn in calvarial bone extracts (Fig. 1B). Using immunohistochemistry, Ambn protein was localized in calvarial bone, dura mater, and adjacent condensed mesenchyme (Fig. 1C).
Figure 1. Ambn expression in cranial bone and dura mater.
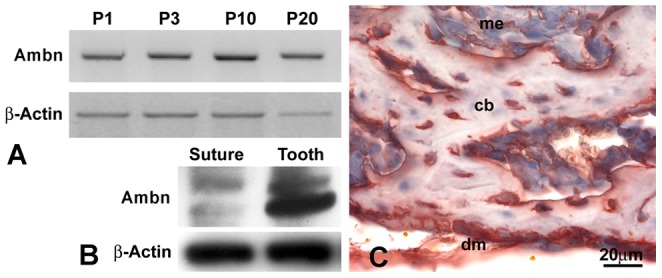
(A) RT-PCR analysis of Ambn mRNA expression in the posterior frontal suture region from 1, 3, 10, and 20 days postnatal WT mice. The β-Actin gene was used as internal control. (B) Western blot analysis of AMBN protein expression in postnatal day 3 posterior-frontal (PF) suture tissues and teeth. Two bands at 55 and 50 kDa were recognized. β-Actin was used as loading reference. (C) Immunostained sections of cranial bone and dura mater from 3 day postnatal wildtype (WT) mice demonstrated Ambn protein localization in calvarial bone (cb), dura mater (dm), and condensed mesenchymal cells (me). Note the high levels of Ambn protein in the dura mater and in calvarial osteoblasts. Bar = 20 µm.
Five-fold AMBN overexpression in a K14-driven transgenic mouse model
Gain of function mice are useful models to mimic syndromes of genetic origin such as suture synostoses (syndromic CS) [31]. We therefore generated Ambn transgenic mice using the human K14 promoter to drive Ambn gene expression [19]. In addition, K14- LacZ transgenic mice were created to verify the location of the enforced Ambn gene expression [19]. Whole mount β-gal staining of the K14-driven LacZ transgenic skull revealed dark blue staining in parietal, frontal, maxillary, ethmoid and occipital bones and in calvarial sutures (Fig. 2B), when compared to wildtype mice (Fig. 2A). Histological analysis demonstrated β-gal blue staining in calvarial osteoblasts in transgenic mouse sections (Fig. 2D), but not in control mouse sections (Fig. 2C). When compared on western blots and after normalization with β-actin, the amount of Ambn protein from calvarial bones and posterior frontal (PF) sutures was 5 times higher in the Ambn transgenic mice than in wildtype controls (Figs. 2E and F).
Figure 2. LacZ and Ambn transgene expression in cranial bone and sutures.
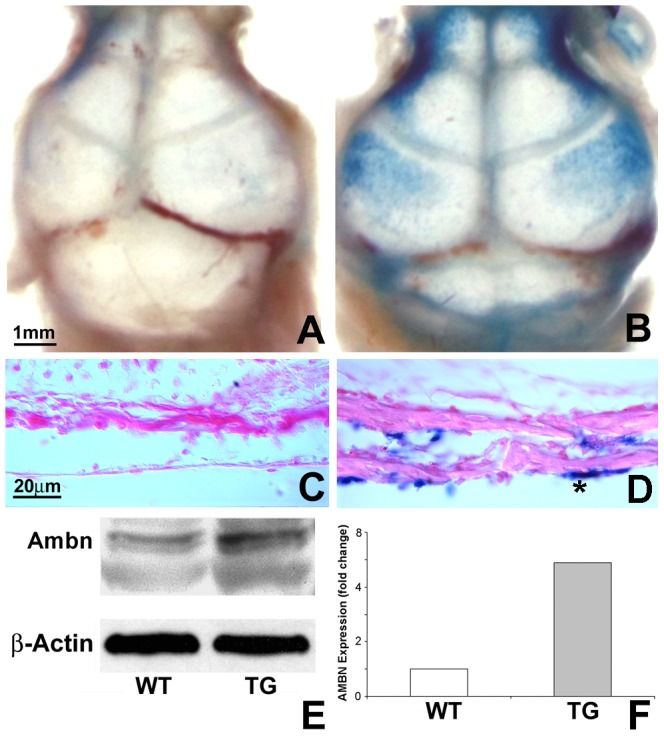
(A and B) Whole mount β-gal staining of wildtype (WT) and K14-LacZ overexpressing mouse skulls. The β-gal blue signal was localized in cranial bones and sutures of the Ambn overexpressor. (C and D) Histological sections of β-gal stained and Eosin counterstained skulls were prepared from the coronal plane of the calvaria in the posterior-frontal region. Note the β-gal positive calvarial osteoblasts in the Ambn transgenic mouse sections. No blue staining of β-gal was detected in the WT skull section. Bar for A and B = 1 mm, C and D = 20 µm. (E) Western blot analysis of protein extracts from WT and Ambn transgenic calvarial bone and sutures at age postnatal day 3. The Ambn protein level in the transgenic mouse (right lane, upper panel) was 5-fold higher than that in the control mouse (left lane, upper panel) after normalization with β-actin (lower panel).
Posterior frontal suture fusion was delayed in Ambn transgenic mice
To determine the effect of Ambn on cranial suture closure, skulls from Ambn transgenic and wildtype mice were compared using alcian blue/alizarin red staining for cartilage and bone mineralized tissue. Ambn transgenic mice at embryonic day 18.5 exhibited wider gaps of interfrontal and interparietal spaces (Figs. 3B and D) compared to those of their controls (Figs. 3A and C) (n = 3). There was less bone density and ossification along the suture (Fig. 3D), and the osteogenic margins of frontal bones were disorganized and not well defined (Fig. 3D), when compared to the wildtype control (Fig. 3C). This phenotype was indicative of a delayed development of the frontal bones. Skull samples of adult Ambn transgenic animals (age 60 days postnatal) revealed the patency of the posterior frontal sutures, revealing spanned gaps between the frontal bones (Figs. 3F and H), while the controls exhibited complete suture closure (Figs. 3E and G). The posterior frontal suture gap width in the Ambn transgenic mice was 0.12–0.42 µm (Fig. 3J), compared to 0.02–0.04 µm in controls (Fig. 3I) (n = 3).
Figure 3. Delayed posterior frontal suture closure in Ambn overexpressing mice.

(A and B) Whole mount alcian blue/alizarin red staining of wildtype (WT) (A) and Ambn transgenic (TG) (B) animals at embryonic stage day 18. The interfrontal gap was wider in the transgenic skulls compared to controls. (C and D) Higher magnification of the skulls shown in A and B. Note that the margins of the frontal bone osteogenic front were not well defined, and the interfrontal and parietal spaces were wider in transgenic mice compared to controls. (E and F) Dried skulls of WT and TG animals at 35 days postnatal. The PF suture was closed in WT mice (E), but remained patent in transgenic mice (F). (G and H) Higher magnification of the skulls shown in A and B. In TG mice, sutures remained open and adjacent bones overlapped (H). Cross-sections of the PF suture revealed the ossified areas of WT mice (I) and TG mice (J) at age 35 day postnatal. The PF suture was sealed by ossification in WT mice, while the left and right parietal bones were linked by soft tissue in TG mice.
Ambn overexpressor skulls weighed less, and their interfrontal bones were thinner and wider
To further characterize the phenotypes of Ambn transgenic mice, dried skull samples from wildtype and Ambn transgenic mice at age of postnatal day 60 were compared, resulting in at least three significant differences. First, there was a significant 13.2% reduction in weight of Ambn transgenic skulls compared to WT controls (Fig. 4A). Second, skull width of Ambn transgenic animals was 14.3% increased when measured at the interfrontal bone/coronal suture interface (Figs. 4B). Third, there was a significant 35.3% reduction in interfrontal bone thickness in transgenic mice (Fig. 4D). Interestingly, there was no significant difference in the length of skulls (data not shown), and in the length of the posterior frontal suture, when comparing wildtype and transgenic groups (Fig. 4C).
Figure 4. Weight and size comparison between wildtype and Ambn transgenic mice dried skulls.
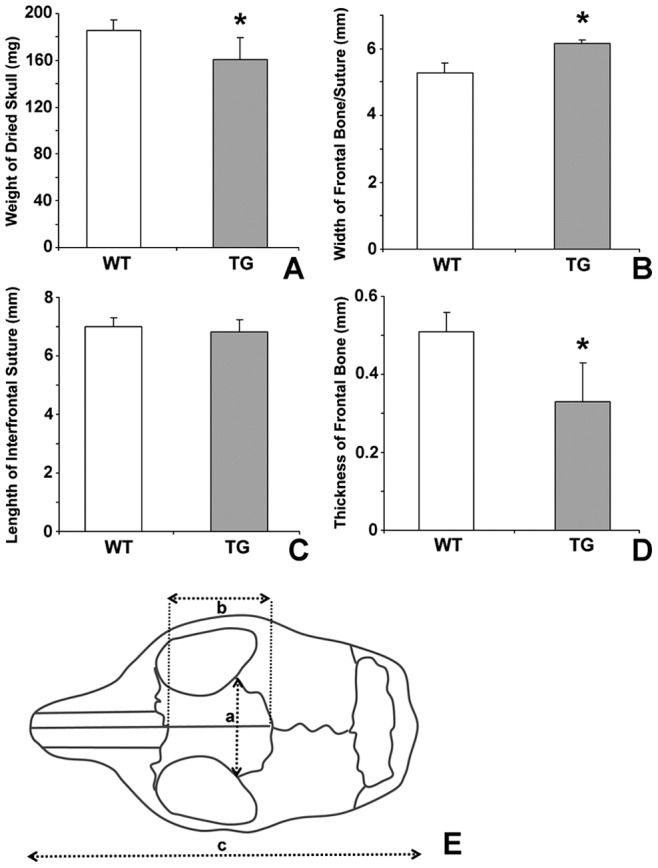
Dried skulls were prepared from 60 days postnatal wildtype (WT) and Ambn transgenic (TG) mice. Weight of dried skulls (A), width of frontal bone and suture (B), length of interfrontal suture (C) and thickness of frontal bone (D) were measured, and values were graphed as mean+/− standard deviation (SD). * represents significant difference, p<0.05 (Kruskall-Wallis one-way analysis). (E) Anatomical reference points for skull morphological measurements, including width of frontal bone and suture (a), length of interfrontal suture (b), and overall skull length (c).
Incomplete posterior frontal suture closure and reduced proliferation rates in Ambn transgenic mice
Histomorphological analysis demonstrated complete closure of the posterior frontal suture in control mice (Fig. 5A) in contrast to incomplete closure in transgenic mice (Fig. 5B). The micrograph was a fused bony bridge formation on the telencephalic aspect of the calvaria in control mice (Fig. 5A), while the frontal bone arch was interrupted by a soft tissue interface in the Ambn transgenic mice (Fig. 5B). The interrupting soft tissue extended from the periosteum to the dura mater in vertical direction (Fig. 5B). BrdU staining of the developing posterior frontal suture (E18) revealed 29% less proliferating cells in Ambn transgenic mice. The number of BrdU positive cells per 0.01 mm2 in the control group was 124+/−5.6 while the number in Ambn TG was 88+/−2.8. In contrast, the number of TUNEL positive cells was 1.3-fold higher in Ambn overexpressors (Figs. 5D and F) when compared to wildtype mice (Figs. 5C and E).
Figure 5. Role of Ambn in suture development.
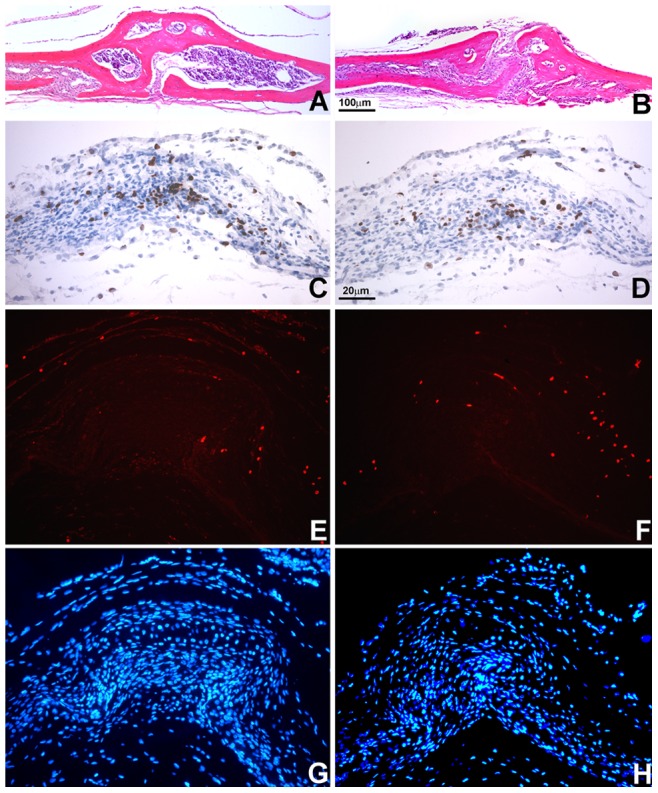
(A and B) Cross-sections of the PF suture revealed the ossified areas of wildtype (WT) and Ambn transgenic (TG) mice at age of 35 days postnatal. The PF suture was fused by ossification in WT mice (A), while the left and right parietal bones were interrupted by soft tissue in TG mice (B). (C and D) BrdU labeling of proliferative cells in developing sutures. Note the accumulation of BrdU-labeled cells in the suture center of WT mice (C) compared to TG mice (D). (E and F) TUNEL staining in developing sutures. Note the red labeling for TUNEL-positive cells in WT and TG mice. (G and H) DAPI counterstaining of (E and F).
Ambn inhibited mouse suture mesenchymal cell proliferation and differentiation in vitro
To determine how Ambn affects suture development, suture mesenchymal cells were isolated from underlying dura mater and overlying pericranium of posterior-frontal sutures of wildtype and transgenic mice. Suture mesenchymal cells from wildtype mice expressed basal Ambn protein levels (Fig. 6A), while the cells from two different transgenic mice over-expressed Ambn protein at about 4.05(TG1) and 2.5(TG2) times higher levels (Figs. 6A and 7B). To further examine whether Ambn expression levels affect cell function, proliferation of suture mesenchymal cells was analyzed using the MTT assay. There was an inverse relationship between Ambn expression levels and cell proliferation capacity, with highest MTT OD value in wildtype cells, followed by TG2 and then TG1 cells (Fig. 6B). In addition, there was a substantial reduction in alkaline phosphatase activity and Alizarin red staining in the two cell lines from Ambn overexpressor mice, TG1 and TG2 (Figs. 6C and D). Remarkably, the high Ambn overexpressing cell line TG1 had a stronger effect on Alizarin red-detected mineralization inhibition than the less Ambn overexpressing cell line TG2, suggesting that Ambn over-expression inhibited suture mesenchyme osteogenic cell differentiation in a dose-dependent manner (Figs. 6D).
Figure 6. Effect of Ambn on proliferation and differentiation of suture mesenchymal cells.

In this study, mesenchymal cells from wildtype (WT) mice and Ambn overexpressor (TG) sutures were cultured in osteogenic induction medium. (A) Ambn protein expression in suture-derived cells. β-Actin was used as loading control. (B) MTT assay to compare cell proliferation in WT and TG mesenchymal suture cells. The measured absorbance (mean+/− SD) is proportionate to the number of living cells. (C) Alkaline phosphotase activity staining at day 6 of culture identified differentiated cells positive for alkaline phosphatase with blue staining. (D) Alizarin red staining for mineral nodule formation. Cells were cultured for 21 days in osteogenic induction medium. Mineral nodules were stained in red. All experiments were carried out in triplicate and repeated three times. * represents significant difference, p<0.05 (Kruskall-Wallis one-way analysis).
Figure 7. Expression of Msx2 and downstream genes in suture and suture-derived mesenchymal cells of wildtype and Ambn transgenic mice.

(A) Western blot analysis of Msx2 and CcnD1 expression in sutures. Suture extracts from wildtype (WT) and Ambn transgenic (TG) mice were processed for immunoblotting with antibodies against Msx2 and CcnD1. β-Actin was detected as a loading control. (B and C) Quantitative RT real time PCR analysis of Ambn (B) and Msx2 (C) gene expression in suture-derived mesenchymal cells. (D and E) Quantitative RT real time PCR for Msx2 downstream target genes in cultured mesenchymal suture cells from WT and TG mice; (E) Runx2, Osterix (Osx), and Cyclin D1 (CcnD1) transcription factors; and (F) Bone sialoprotein (Ibsp), Collagen I (Col1A1), Osteocalcin (Ocn), and Osteopontin (Opn) matrix proteins. Expression levels were calculated relative to GAPDH gene expression levels using the 2–ΔΔCt method, and values were graphed as the mean expression level ± standard deviation. *,**, and *** represent significant difference, p<0.05, p<0.01, and p<0.001 respectively (Kruskall-Wallis one-way analysis).
Ambn regulated Msx2 and downstream gene expression
Ambn overexpressor mice displayed posterior frontal suture patency, a phenotype previously reported in Msx2 deficient mice [32]. In addition, Ambn over-expressing suture mesenchymal cells displayed lower proliferative capacity and delayed differentiation similar to the effects of Msx2 deficiency on mesenchymal cell function [32]. Therefore, Msx2 expression in suture tissues as well as suture mesenchymal cells from Ambn transgenic and wildtype mice was examined. Western blot analysis revealed that Msx2 and CcnD1 expression in calvarial bones and PF suture tissues of Ambn transgenic mice were downregulated (Fig. 7A). Msx2 mRNA expression in suture mesenchymal cells from wildtype and transgenic mice was reverse proportional to Ambn mRNA expression, when comparing cells from two different transgenic lines (Fig. 7B and C). To determine whether Ambn affects expression of Msx2 down-stream target genes, expression levels of osteogenic transcription factors Runx2 and Osx, bone matrix proteins Ibsp, ColI, Ocn and Opn, as well as the cell cycle-related gene CcnD1 were detected using quantitative RT real time PCR. Our data demonstrated that Ambn overexpression resulted in significant decreases in bone transcription factor and bone mineralization marker gene expression in a dose-dependent manner (Figs. 7D and E). Similarly, CcnD1 expression was downregulated in TG1 and TG2 cells. Together, these studies indicate that Ambn inhibits cell proliferation and reduces Msx2 and downstream gene expression in vivo and in vitro.
Discussion
Transgenic animal models are ideal biological systems to test the effect of forced gene expression on the development of a phenotype. In the present study we have used the human Keratin 14 promoter to overexpress the extracellular matrix protein Ambn in calvarial osteoblasts and suture mesenchymal cells. Keratin 14(K14, Cytokeratin 14 = CK14) is commonly known as an epithelial protein encoded by the KRT14 gene. However, K14 expression in calvarial osteoblasts has been previously reported and confirmed by Western blot [33]. In addition, reports of keratins in non-epithelial cell lineages have become more frequent in the literature, including mouse studies related to keratins involved in endodermal and mesoderm adhesion in early embryogenesis [34] (Vijayaraj et al. 2010, reports on K19 in dental papilla and dental pulp [35] (Webb et al. 1995), microarray and western blot detection of K18 in cementoblasts [36] (Dangaria et al. 2011), findings of K19 and K8 in mature striated muscle [37] (Shah et al. 2012), and K8 and K18 expression in mesenchymal progenitors of regenerating limbs [38] (Cocoran and Ferretti 1997). Here, K14 expression in calvarial osteoblasts was further confirmed by β-gal staining for the K14 LacZ transgene in K14 LacZ transgenic mice using both histology and whole mount staining. The K14 promoter system has become a promoter of choice in our and other laboratories because of its efficient expression of many transgenes [39],[40]. K14-driven overexpression of Ambn in mouse calvaria resulted in a robust 5-fold enhancement of Ambn levels in calvarial tissue extracts.
Our study indicates that Ambn was expressed in developing calvarial bone and sutures. This finding matches other reports of Ambn expression during bone formation [25],[41] and expands its original concept as an ameloblast-specific gene product [42]. However, when compared to the developing enamel matrix, expression levels were less, suggesting that Ambn may not act directly on bone development by controlling crystal growth as it does in the enamel matrix [29],[43], but rather indirectly as a signaling molecule affecting the expression of transcription factors and extracellular matrix signaling pathways [23],[44],[45]. This concept is supported by findings reported in the present study that already small differences in Ambn concentration have significant effects on cell behavior, including cell proliferation and mineralization.
Our human K14-driven Ambn overexpressor mice suffered from delayed and/or incomplete cranial suture closure and displayed patency of the posterior frontal suture. Moreover, frontal bones of Ambn overexpressors weighed less and were thinner, suggestive that Ambn was involved in tissue growth or mineralization or both. We interpret these findings to indicate that Ambn plays an important role in the growth and development of cranial bones and subsequent suture closure. Our concept that Ambn affects mineralized tissues outside of teeth is supported by earlier studies related to the effect of Ambn on bone [41]. In our study, the phenotype of incomplete suture closure in Ambn overexpressing mice is explained by two related in vitro findings, namely (i) a significant reduction in suture mesenchymal cell proliferation and (ii) a downregulation of the cell proliferation marker cyclin D1 (CcnD1). In addition, studies in our and other laboratories have also provided evidence for the inhibitory effect of Ambn on cell proliferation, e.g. an increase of the cell proliferation inhibitors p21 and p27 in Ambn overexpressing ameloblasts [26],[46]. In contrast, there have also been reports suggesting that Ambn promotes bone healing by enhancing progenitor cell proliferation [41] (Tamburstuen et al. 2011) and regulates osteogenic differentiation [47] (Iizuku et al.). We propose that such findings may be due to different concentrations of Ambn acting either as a signaling molecule or a structural matrix protein [48] (Diekwisch 2011).
Another factor responsible for the inhibition of suture closure by Ambn overexpression is the transcription factor Msx2, a causative gene involved in human cranial suture pathologies such as CS and EPF [49],[50]. Our finding that Msx2 is downregulated by Ambn is supported by earlier studies reporting upregulation of Msx2 in ameloblasts from Ambn deficient mice [19] and downregulation of MSX2 in human ameloblastoma cells overexpressing AMBN [26]. Notably, Msx2 deficient mice featured a patency of the posterior frontal suture [32], a phenotype resembling the Ambn overexpressor phenotype described in the present study. Underscoring the severe clinical impact of defects caused by changes in Msx levels and suture development, Msx1/2 double mutant embryos were lacking frontal bones and died at E17 to E18 [51]. Together, these studies indicate that changes in Ambn affect craniofacial growth and suture closure via Msx2.
Evidence from previous studies suggests that the effects of Ambn on Msx2 suppression and calvarial osteoblast proliferation inhibition may be related, as it has been shown that Msx2 increases the expression of CcnD1, inhibits cell differentiation [52], and influences the number of proliferative osteogenic cells in growth centers of the developing mouse skull [53]. Others have reported that calvarial osteoblasts derived from Msx2 deficient mice had a lower rate of proliferation and demonstrated accelerated osteoblastic differentiation at a later stage when compared to osteoblasts derived from wildtype mice [54]. Our finding that suture mesenchymal cells from transgenic mice reduced CcnD1 and osteogenic gene expression under sub-confluent condition supported a potential connection between the role of Ambn in osteogenic differentiation and its role in cell proliferation inhibition. Together, our data indicate that Ambn affects calvarial osteoblast proliferation and differentiation through suppression of the transcription factor Msx2.
Conclusions
These data suggested that the extracellular matrix protein Ambn plays a crucial role in cranial suture closure. The effect of Ambn on suture closure may be explained by the inhibitory effect of Ambn on the transcription factor Msx2 and its effect on cell proliferation, resulting in reduced growth and differentiation of calvarial osteoblasts as well as delayed suture closure in mice.
Funding Statement
This study has been generously supported by grant DE19155 from the National Institute of Dental and Craniofacial Ressearch (http://www.nidcr.nih.gov/). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Bruce JA (1941) Time and order of appearance of ossification centers and their development in the skull of the rabbit. Am J Anat 68: 41–67. [Google Scholar]
- 2.Hall BK (2005) Bones and Cartilage: development skeleton biology. San Diego: Academic press. 760p.
- 3. Ornitz DM, Marie PJ (2002) FGF signaling pathways in endochondral and intramembranous bone development and human genetic disease. Genes Dev 16: 1446–1465. [DOI] [PubMed] [Google Scholar]
- 4. Komori T, Yagi H, Nomura S, Yamaguchi A, Sasaki K, et al. (1997) Targeted disruption of Cbfa1 results in a complete lack of bone formation owing to maturational arrest of osteoblasts. Cell 89: 755–764. [DOI] [PubMed] [Google Scholar]
- 5. Lee B, Thirunavukkarasu K, Zhou L, Pastore L, Baldini A, et al. (1997) Missense mutations abolishing DNA binding of the osteoblast-specific transcription factor OSF2/CBFA1 in cleidocranial dysplasia. Nat Genet 16: 307–310. [DOI] [PubMed] [Google Scholar]
- 6. Mundlos S, Otto F, Mundlos C, Mulliken JB, Aylsworth AS, et al. (1997) Mutations involving the transcription factor CBFA1 cause cleidocranial dysplasia. Cell 89: 773–779. [DOI] [PubMed] [Google Scholar]
- 7. Dodig M, Tadic T, Kronenberg MS, Dacic S, Liu YH, et al. (1999) Ectopic Msx2 overexpression inhibits and Msx2 antisense stimulates calvarial osteoblast differentiation. Dev Biol 209: 298–307. [DOI] [PubMed] [Google Scholar]
- 8. el Ghouzzi V, Le Merrer M, Perrin-Schmitt F, Lajeunie E, Benit P, et al. (1997) Mutations of the TWIST gene in the Saethre-Chotzen syndrome. Nat Genet 15: 42–46. [DOI] [PubMed] [Google Scholar]
- 9. Howard TD, Paznekas WA, Green ED, Chiang LC, Ma N, et al. (1997) Mutations in TWIST, a basic helix-loop-helix transcription factor, in Saethre-Chotzen syndrome. Nat Genet 15: 36–41. [DOI] [PubMed] [Google Scholar]
- 10. Kopher RA, Mao JJ (2003) Suture growth modulated by the oscillatory component of micromechanical strain. J Bone Miner Res 18: 521–528. [DOI] [PubMed] [Google Scholar]
- 11. Opperman LA, Rawlins JT (2005) The extracellular matrix environment in suture morphogenesis and growth. Cells Tissues Organs 181: 127–135. [DOI] [PubMed] [Google Scholar]
- 12. Rice DP (2008) Clinical features of syndromic craniosynostosis. Front Oral Biol 12: 91–106. [DOI] [PubMed] [Google Scholar]
- 13.Cohen MJ, Maclean R (2000) Craniosynostosis, diagnosis, evaluation, and management. Oxford: Oxford University Press. 727p.
- 14. Hukki J, Saarinen P, Kangasniemi M (2008) Single suture craniosynostosis: Diagnosis and imaging. In: Front Oral Biol Rice DP, editor. Craniofacial sutures. Development, disease and treatment. 12: 79–90. [DOI] [PubMed] [Google Scholar]
- 15. Whitaker LA, Munro IR, Salyer KE, Jackson IT, Ortiz-Monasterio F, et al. (1979) Combined report of problems and complications in 793 craniofacial operations. Plast Reconstr Surg 64: 198–203. [DOI] [PubMed] [Google Scholar]
- 16. Adams JC, Watt FM (1993) Regulation of development and differentiation by the extracellular matrix. Development 117: 1183–1198. [DOI] [PubMed] [Google Scholar]
- 17. Carinci P, Becchetti E, Bodo M (2000) Role of the extracellular matrix and growth factors in skull morphogenesis and in the pathogenesis of craniosynostosis. Int J Dev Biol 44: 715–723. [PubMed] [Google Scholar]
- 18. Fukumoto S, Kiba T, Hall B, Iehara N, Nakamura T, et al. (2004) Ameloblastin is a cell adhesion molecule required for maintaining the differentiation state of ameloblasts. J Cell Biol 167: 973–983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lu X, Ito Y, Kulkarni A, Gibson C, Luan X, et al. (2011) Ameloblastin-rich enamel matrix favors short and randomly oriented apatite crystals. Eur J Oral Sci 119 Suppl: 254–260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Fong CD, Cerny R, Hammarstrom L, Slaby I (1998) Sequential expression of an amelin gene in mesenchymal and epithelial cells during odontogenesis in rats. Eur J Oral Sci 106 Suppl: 324–330 [DOI] [PubMed] [Google Scholar]
- 21. Hao J, He G, Narayanan K, Zou B, Lin L, et al. (2005) Identification of differentially expressed cDNA transcripts from a rat odontoblast cell line. Bone 37: 578–588. [DOI] [PubMed] [Google Scholar]
- 22. Nunez J, Sanz M, Hoz-Rodriguez L, Zeichner-David M, Arzate H (2010) Human cementoblasts express enamel-associated molecules in vitro and in vivo. J Periodontal Res 45: 809–814. [DOI] [PubMed] [Google Scholar]
- 23. Spahr A, Lyngstadaas SP, Slaby I, Haller B, Boeckh C, et al. (2002) Expression of amelin and trauma-induced dentin formation. Clin Oral Investig 6: 51–57. [DOI] [PubMed] [Google Scholar]
- 24. Spahr A, Lyngstadaas SP, Slaby I, Pezeshki G (2006) Ameloblastin expression during craniofacial bone formation in rats. Eur J Oral Sci 114: 504–511. [DOI] [PubMed] [Google Scholar]
- 25. Tamburstuen MV, Reseland JE, Spahr A, Brookes SJ, Kvalheim G, et al. (2010) Ameloblastin expression and putative autoregulation in mesenchymal cells suggest a role in early bone formation and repair. Bone 48: 406–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sonoda A, Iwamoto T, Nakamura T, Fukumoto E, Yoshizaki K, et al. (2009) Critical role of heparin binding domains of ameloblastin for dental epithelium cell adhesion and ameloblastoma proliferation. J Biol Chem 284: 27176–27184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zhang X, Diekwisch TGH, Luan X (2011) structure and function of ameloblastin as an extracellular matrix protein: adhesion, calcium binding, and CD63 interaction in human and mouse. Eur J Oral Sci 119 Suppl: 270–279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Loveland BE, Johns TG, Mackay IR, Vaillant F, Wang ZX, et al. (1992) Validation of the MTT dye assay for enumeration of cells in proliferative and antiproliferative assays. Biochem Int 27: 501–510. [PubMed] [Google Scholar]
- 29. Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 25: 402–408. [DOI] [PubMed] [Google Scholar]
- 30. Lee SK, Krebsbach PH, Matsuki Y, Nanci A, Yamada KM, et al. (1996) Ameloblastin expression in rat incisors and human tooth germs. Int J Dev Biol 40: 1141–1150. [PubMed] [Google Scholar]
- 31. Shen K, Krakora SM, Cunningham M, Singh M, Wang X, et al. (2009) Medical treatment of craniosynostosis: recombination Noggin inhibits coronal sture closure in the rat craniosynostosis model. Orthod Craniofac Res 12: 254–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Satokata I, Ma L, Ohshima H, Bei M, Woo I, et al. (2000) Msx2 deficiency in mice causes pleiotropic defects in bone growth and ectodermal organ formation. Nat Genet 24: 391–395. [DOI] [PubMed] [Google Scholar]
- 33. Kumar A, Singh AK, Gautam AK, Chandra D, Singh D, et al. (2010) Identification of kaempferol-regulated proteins in rat calvarial osteoblasts during mineralization by proteomics. Proteomics 10: 1730–1739. [DOI] [PubMed] [Google Scholar]
- 34. Vijayaraj P, Kroeger C, Reuter U, Hartmann D, Magin TM (2010) Keratins regulate yolk sac hematopoiesis and vasculogenesis through reduced BMP-4 signaling. Eru J Cell Biol 89: 299–306. [DOI] [PubMed] [Google Scholar]
- 35. Webb PP, Moxham BJ, Ralphs JR, Benjamin M (1995) Cytoskeleton of the mesenchymal cells of the rat dental papilla and dental pulp. Connect Tissue Res 32: 71–76. [DOI] [PubMed] [Google Scholar]
- 36. Dangaria S, Ito Y, Luan X, Diekwisch TG (2011) Differentiation of neural-crest-derived intermediate pluripotent progenitors into committed periodontal populations involves unique molecular signature changes, cohort shifts, and epigenetic modifications. Stem Cell Dev 20: 39–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shah SB, Love JM, O'Neill A, Lovering RM, Bloch RJ (2012) Influences of desmin and keratin 19 on passive biomechanical properties of mouse skeletal muscle J Biomed Biotech doi:10.1155/2012/704061. [DOI] [PMC free article] [PubMed]
- 38. Corcoran JP, Ferretti P (1997) Keratin 8 and 18 expression in mesenchymal progenitor cells of regenerating limbs is associated with cell proliferation and differentiation Dev Dyn. 210: 355–370. [DOI] [PubMed] [Google Scholar]
- 39. Wang X, Zinkel S, Polonsky K, Fuchs E (1997) Transgenic studies with a keratin promoter-driven growth hormone transgene: prospects for gene therapy. Proc Natl Acad Sci U S A 94: 219–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Luan X, Ito Y, Diekwisch TG (2006) Evolution and development of Hertwig's epithelial root sheath. Dev Dyn 235: 1167–1180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Tamburstuen MV, Reppe S, Spahr A, Sabetrasekh R, Kvalheim G, et al. (2010) Ameloblastin promotes bone growth by enhancing proliferation of progenitor cells and by stimulating immunoregulators. Eur J Oral Sci 118: 451–459. [DOI] [PubMed] [Google Scholar]
- 42. Krebsbach PH, Lee SK, Matsuki Y, Kozak CA, Yamada KM, et al. (1996) Full-length sequence, localization, and chromosomal mapping of ameloblastin. A novel tooth-specific gene. J Biol Chem 271: 4431–4435. [DOI] [PubMed] [Google Scholar]
- 43. Lee SK, Kim SM, Lee YJ, Yamada KM, Yamada Y, et al. (2003) The structure of the rat ameloblastin gene and its expression in amelogenesis. Mol Cells 15: 216–225. [PubMed] [Google Scholar]
- 44. Fukumoto S, Yamada A, Nonaka K, Yamada Y (2005) Essential roles of ameloblastin in maintaining ameloblast differentiation and enamel formation. Cells Tissues Organs 181: 189–195. [DOI] [PubMed] [Google Scholar]
- 45. Wazen RM, Moffatt P, Zalzal SF, Yamada Y, Nanci A (2009) A mouse model expressing a truncated form of ameloblastin exhibits dental and junctional epithelium defects. Matrix Biol 28: 292–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Zhang Y, Zhang X, Lu X, Atsawasuwan P, Luan X (2011) Ameloblastin regulate cel attachment and proliferation through RhoA and p27. Eur J Oral Sci 119 Suppl: 280–285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Iizuka S, Kudo Y, Yoshida M, Tsunematsu T, Yoshiko Y, et al. (2011) Ameloblastin regulates osteogenic differentiation by inhibiting Src kinase via cross talk between integrin β1 and CD63. Mol Cell Biol 31: 783–792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Diekwisch T (2011) Evolution and ameloblastin. J Eur Oral Sci 119 Suppl: 293–297 [DOI] [PubMed] [Google Scholar]
- 49. Jabs EW, Muller U, Li X, Ma L, Luo W, et al. (1993) A mutation in the homeodomain of the human MSX2 gene in a family affected with autosomal dominant craniosynostosis. Cell 75: 443–450. [DOI] [PubMed] [Google Scholar]
- 50. Wilkie AO, Tang Z, Elanko N, Walsh S, Twigg SR, et al. (2000) Functional haploinsufficiency of the human homeobox gene MSX2 causes defects in skull ossification. Nat Genet 24: 387–390. [DOI] [PubMed] [Google Scholar]
- 51. Han J, Ishii M, Bringas P Jr, Maas RL, Maxson RE Jr, et al. (2007) Concerted action of Msx1 and Msx2 in regulating cranial neural crest cell differentiation during frontal bone development. Mech Dev 124: 729–745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Hu G, Lee H, Price SM, Shen MM, Abate-Shen C (2001) Msx homeobox genes inhibit differentiation through upregulation of cyclin D1. Development 128: 2373–2384. [DOI] [PubMed] [Google Scholar]
- 53. Liu YH, Tang Z, Kundu RK, Wu L, Luo W, et al. (1999) Msx2 gene dosage influences the number of proliferative osteogenic cells in growth centers of the developing murine skull: a possible mechanism for MSX2-mediated craniosynostosis in humans. Dev Biol 205: 260–274. [DOI] [PubMed] [Google Scholar]
- 54. Marijanovic I, Kronenberg MS, Erceg I, Lichtler AC (2009) Comparison of proliferation and differentiation of calvarial osteoblast cultures derived from Msx2 deficient and wild type mice. Coll Antropol 33: 919–924. [PubMed] [Google Scholar]