

Background: Chemokine receptors are post-translationally sulfated on tyrosine residues.
Results: A tyrosine-sulfated fragment of CCR2 binds more tightly to the monomeric form than the dimeric form of the chemokine MCP-1.
Conclusion: Binding to sulfated CCR2 promotes conversion of MCP-1 from inactive dimer to active monomer.
Significance: Tyrosine sulfation may regulate the ability of chemokine receptors to be activated by chemokines.
Keywords: Chemokines, Nuclear Magnetic Resonance, Receptors, Structural Biology, Tyrosine Sulfation, CCR2, Dimerization, MCP-1
Abstract
Chemokine receptors are commonly post-translationally sulfated on tyrosine residues in their N-terminal regions, the initial site of binding to chemokine ligands. We have investigated the effect of tyrosine sulfation of the chemokine receptor CCR2 on its interactions with the chemokine monocyte chemoattractant protein-1 (MCP-1/CCL2). Inhibition of CCR2 sulfation, by growth of expressing cells in the presence of sodium chlorate, significantly reduced the potency for MCP-1 activation of CCR2. MCP-1 exists in equilibrium between monomeric and dimeric forms. The obligate monomeric mutant MCP-1(P8A) was similar to wild type MCP-1 in its ability to induce leukocyte recruitment in vivo, whereas the obligate dimeric mutant MCP-1(T10C) was less effective at inducing leukocyte recruitment in vivo. In two-dimensional NMR experiments, sulfated peptides derived from the N-terminal region of CCR2 bound to both the monomeric and dimeric forms of wild type MCP-1 and shifted the equilibrium to favor the monomeric form. Similarly, MCP-1(P8A) bound more tightly than MCP-1(T10C) to the CCR2-derived sulfopeptides. NMR chemical shift mapping using the MCP-1 mutants showed that the sulfated N-terminal region of CCR2 binds to the same region (N-loop and β3-strand) of both monomeric and dimeric MCP-1 but that binding to the dimeric form also influences the environment of chemokine N-terminal residues, which are involved in dimer formation. We conclude that interaction with the sulfated N terminus of CCR2 destabilizes the dimerization interface of inactive dimeric MCP-1, thus inducing dissociation to the active monomeric state.
Introduction
Chemokines, or chemoattractant cytokines, are a family of small (8–10 kDa) globular proteins that function to direct leukocyte migration (1). The ability of chemokines to recruit leukocytes is mediated by high affinity interactions with G protein-coupled receptors expressed in leukocyte membranes (1). According to the prevailing two-site model of these interactions (2), chemokines first use residues in the N-loop region (following the CC or CXC motif) and the second and third β-strands to bind to the receptor N terminus. Subsequently, the chemokine N terminus activates the receptor by binding to its extracellular loops and/or transmembrane helices.
A key factor that regulates chemokine-receptor interactions is the sulfation of tyrosine residues on the receptor N terminus. Tyrosine sulfation is a post-translational modification prevalent among membrane-bound proteins such as chemokine receptors, as well as among secreted proteins such as peptide hormones, enzymes, blood coagulants, and complement proteins (3). This modification is catalyzed in the Golgi apparatus by the enzymes tyrosylprotein sulfotransferases 1 and 2, which preferentially sulfate tyrosine residues located near acidic residues (4), a motif found in the N-terminal regions of most chemokine receptors (3). The chemokine receptors CCR2, CCR5, CCR8, CXCR3, CXCR4, CX3CR1, and Duffy antigen and receptor for chemokines (DARC) have been demonstrated to contain sulfated tyrosine residues that modulate chemokine binding (5–11). Moreover, studies using differentially sulfated N-terminal peptides from various chemokine receptors have shown that tyrosine sulfation increases the binding affinity of the receptor peptides to their cognate chemokines (12–16).
Among the chemokine receptors demonstrated to possess a tyrosine sulfation motif, CCR2 has received considerable attention because of its role in inflammatory diseases such as atherosclerosis and multiple sclerosis (17, 18). CCR2 is a major chemokine receptor expressed on monocytes, and activation by its chemokine ligands monocyte chemoattractant proteins 1 to 4 (MCP-1 to -4; systematic names CCL2, CCL8, CCL7, and CCL13, respectively)6 stimulates the migration of monocytes across the vascular wall into tissues where they mediate chronic inflammation (19). The tyrosine sulfation motif of CCR2 has the sequence 25DYDY28. In a HEK293 cell line transfected to express CCR2, mutation of Tyr26 resulted in a dramatic decrease in sulfation, a 10-fold decrease in binding affinity for MCP-1 and almost complete loss of activation in response to MCP-1 (5). Because the Tyr26 mutant had no detectable sulfation, the authors concluded that Tyr28 on CCR2 is not sulfated. However, it is possible that Tyr28 is at least partially sulfated in the wild type receptor. An independent study reported that a double Asp → Ala mutation in the DYDY motif, which is likely to decrease the extent of tyrosine sulfation, resulted in a large (>50-fold) decrease in MCP-1 binding affinity (20).
Although the above mutational experiments strongly suggest that sulfation of Tyr26 in CCR2 can modulate the strength of the interaction with MCP-1, they do not directly compare sulfated with nonsulfated forms of the wild type receptor. Thus, it is instructive to complement the mutational approach with experiments that examine the influence of post-translational sulfation without introducing receptor mutations. In the current study, we have used two independent approaches to achieve this. First, we measured CCR2 activation by MCP-1 using cells in which tyrosine sulfation was inhibited by sodium chlorate. Second, we used NMR spectroscopy to monitor the binding of MCP-1 to CCR2 N-terminal peptides possessing defined patterns of sulfation on Tyr26 and/or Tyr28. Because MCP-1 exists in a monomer-dimer equilibrium in solution, we distinguished the binding abilities of the monomer and dimer by using a previously characterized obligate monomer, MCP-1(P8A) (21), and obligate dimer, MCP-1(T10C) (22). Our results indicate that tyrosine sulfation of the CCR2 N terminus increases the binding affinity for both the MCP-1 monomer and dimer and, moreover, induces dissociation of the dimer into its functional monomeric units. These findings suggest that the sulfation state of CCR2 influences its ability to be activated by MCP-1 and is therefore likely to play a role in the physiological regulation of MCP-1/CCR2 interactions.
EXPERIMENTAL PROCEDURES
Expression and Purification of Wild Type and Mutant MCP-1
MCP-1 was expressed and purified as described previously (22). Briefly, an N-terminally His6-tagged form of MCP-1 was expressed in Escherichia coli using minimal media to allow 15N enrichment. Inclusion bodies containing the fusion protein were isolated, dissolved in denaturing buffer, and purified by Ni2+ affinity chromatography. After refolding of the fusion protein by dropwise dilution into native buffer, the His6 tag was removed using thrombin, and the mature MCP-1 protein was purified by cation exchange chromatography. Mass spectral analysis indicated that a small proportion (15–25%) of each protein had spontaneously undergone pyroglutamate formation from the N-terminal Gln residue, a natural modification of MCP-1 (23).
Cell-based Assays and Inhibition of Sulfation
Cell-based assays were performed in inducible FlpIn TRex HEK293 cells expressing FLAG-CCR2, as described previously (22). Inhibition of sulfation was performed by growing the cells in media containing 30 mm sodium chlorate for 48 h prior to the experiment.
ERK1/2 Phosphorylation Assays
FlpIn TRex HEK293 cells expressing FLAG-CCR2 (22) were seeded at 50,000 cells/well into a poly-d-Lys-coated 96-well plate and grown overnight in the presence of 1 μg/μl tetracycline. Initial time course experiments were used to determine the time required to stimulate maximum ERK1/2 phosphorylation by each ligand, and subsequent concentration response experiments were conducted by stimulating cells with wild type or mutant MCP-1 for 2.5 min, which represented the time at which maximal ERK1/2 phosphorylation was achieved. The reaction was terminated by the removal of media and the addition of lysis buffer. Samples were processed following the AlphaScreen SureFire pERK1/2 kit manufacturer's instructions (PerkinElmer Life Sciences). The data were normalized to the response of 10% (v/v) FBS and analyzed in GraphPad Prism 5.02. All of the experiments were performed at least three times in duplicate.
Intracellular Ca2+ Mobilization Assays
FlpIn TRex HEK293 cells expressing FLAG-CCR2 (22) were seeded at 30,000 cells/well into a poly-d-Lys-coated 96-well plate and grown overnight in the presence of 1 μg/μl tetracycline. The cells were washed twice in Ca2+ assay buffer (150 mm NaCl, 2.6 mm KCl, 1.2 mm MgCl2, 10 mm d-glucose, 10 mm HEPES, 2.2 mm CaCl2, 0.5% (w/v) BSA, and 4 mm probenecid), replaced with assay buffer containing 1 μm Fluo-4-AM (Invitrogen), and incubated for 1 h at 37 °C in 5% CO2. The cells were washed twice more and replaced with warm assay buffer. The addition of the drugs and fluorescence measurements were performed in a FlexstationTM (Molecular Devices) using 485-nm excitation and 520-nm emission wavelengths. Peak fluorescence was measured as a marker for Ca2+ mobilization and used in further analyses. The data were normalized to the response of 1 μm ionomycin and analyzed using GraphPad Prism 5.02. All of the experiments were performed at least three times in duplicate.
Leukocyte Adhesion and Emigration by Intravital Imaging
MCP-1-induced leukocyte adhesion and emigration were assessed as described previously (24, 25). In brief, mice were injected subcutaneously, adjacent to the cremaster muscle, with 345 ng of either wild type or mutant MCP-1, in 150 μl of saline. Four hours later, the cremaster muscle was exteriorized for intravital microscopy as described previously (25). Three or four postcapillary venules (25–40 μm in diameter) were examined in each animal, and leukocyte adhesion (cells/100 μm venule length) and emigration (cells/field of view) were quantitated using standard techniques (25). In some experiments, we examined recombinant MCP-1 that had been denatured by boiling to exclude the possibility of endotoxin contamination.
NMR Titration of Wild Type and Mutant MCP-1 with CCR2 Peptides
NMR experiments were conducted at 25 °C on a Bruker Avance 600 MHz NMR spectrometer equipped with a triple resonance cryoprobe. Chemical shifts were referenced to internal or external 4,4-dimethyl-4-silapentane-1-sulfonic acid. Peptides R2A-R2D, containing residues 18–31 of CCR2, were prepared by solid phase synthesis and purified as described previously (26). NMR samples for peptide titrations initially contained wild type or mutant MCP-1 at a concentration of 50 μm in NMR buffer (20 mm sodium acetate-d4, 5% D2O, 0.02% NaN3, pH 7.0). The peptide was added from a 1 mm stock solution (in NMR buffer) in aliquots such that the final peptide/protein molar ratios were 0.25, 0.50, 0.75, 1.00, 1.25, 1.50, 2.00, 3.50, and 5.00, respectively. The initial volume of MCP-1 was 500 μl, and the volume after the final peptide addition was 625 μl. For the initial sample and after each addition of peptide, a 1H-15N heteronuclear single quantum correlation (HSQC) spectrum was recorded using 96 and 1024 complex points and spectral widths of 24.0 and 12.0 ppm in the 15N and 1H dimensions, respectively.
The NMR data were processed using Bruker TopSpin and analyzed using Sparky (T. D. Goddard and D. G. Kneller, University of California, San Francisco, CA). Chemical shift assignments for wild type MCP-1 and for both P8A and T10C mutants have been reported previously (22, 27). For each MCP-1 mutant, the weighted changes in chemical shift (ΔδNH = |ΔδH| + 0.2|ΔδN|) were fit simultaneously for all residues exhibiting final ΔδNH values above a threshold of 0.02 ppm (for peptide R2A) or 0.04 ppm (for peptides R2B-R2D). Fitting was performed using GraphPad Prism to a 1:1 binding equation (Equation 1) to give a single dissociation constant (Kd) value for the binding between the peptide and MCP-1 variant of interest,
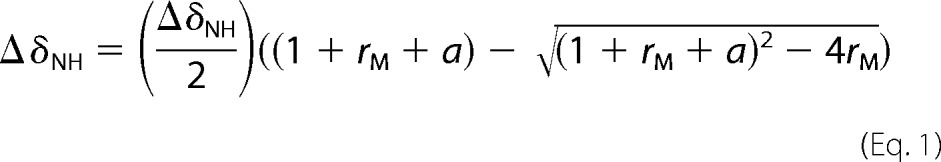 |
where a is Kd (L0 + P0rM)/(P0L0), P0 is the initial concentration of protein, L0 is the stock concentration of ligand, and rM is the molar ratio ([peptide]/[protein]).
RESULTS
Activation of Sulfated and Nonsulfated CCR2
To determine the influence of CCR2 tyrosine sulfation on activation by MCP-1, we cultured FlpIn TRex HEK293 cells that express FLAG-tagged CCR2 in an inducible fashion in the presence and absence of sodium chlorate. Sodium chlorate inhibits the biosynthesis of 3′-phosphoadenosine 5′-phosphosulfate, the source of sulfate in tyrosylprotein sulfotransferase-catalyzed tyrosine sulfation reactions. Chlorate did not influence the growth rate of CCR2-expressing HEK293 cells. An ELISA directed to the FLAG tag of the receptor indicated that the FLAG-CCR2 expression level increased slightly in the chlorate-treated cells (data not shown). However, it should be noted that the FLAG sequence (DYKDDDDK) has the potential to be sulfated, thereby reducing the ELISA signal in the absence of chlorate (28). Thus, the increased FLAG signal in the ELISA could simply indicate a reduction in FLAG tag sulfation rather than an increase in FLAG tag expression in the chlorate-treated cells. We monitored MCP-1 activation of CCR2 using both Ca2+ mobilization and ERK1/2 phosphorylation assays. The concentration-response curves from these experiments are shown in Fig. 1, with the corresponding pEC50 values listed in Table 1. In each assay, growth of cells in the presence of sodium chlorate significantly reduced CCR2 activation by wild type MCP-1 and by the obligate monomer MCP-1(P8A), with potencies decreasing by 3.5–10-fold (Table 1); the obligate dimer does not activate the receptor (22). These results support the previous observation (5) that CCR2 is tyrosine-sulfated and indicate that sulfation enhances the ability of CCR2 to be activated in response to MCP-1.
FIGURE 1.

Concentration-response curves for activation of CCR2 by MCP-1. Shown are the results for Ca2+ mobilization (A) and ERK1/2 phosphorylation (B) assays for wild type MCP-1 (circles) and obligate monomer MCP-1(P8A) (squares) using FlpIn TRex HEK293 cells expressing FLAG-CCR2 cells grown in the absence of sodium chlorate (filled symbols and solid lines) or the presence of chlorate (open symbols and dotted lines). The data points represent the means ± S.E. of at least three independent experiments performed in triplicate; in most cases the error bars are smaller than the data symbols.
TABLE 1.
Summary of cell-based activation data testing the effect of CCR2 tyrosine sulfation on chemokine potency
pEC50 data are shown as the mean values ± S.E. from three independent experiments. EC50 values (nm) are shown in parentheses.
| Chemokine | pEC50 (EC50) |
|||
|---|---|---|---|---|
| Ca2+ mobilization |
ERK1/2 phosphorylation |
|||
| Chlorate (−) | Chlorate (+) | Chlorate (−) | Chlorate (+) | |
| Wild type MCP-1 | 7.78 ± 0.09 (16) | 6.82 ± 0.16 (151)a | 8.66 ± 0.07 (2.2) | 7.58 ± 0.12c (26) |
| MCP-1(P8A) | 8.02 ± 0.10 (9) | 7.47 ± 0.22 (34)b | 8.32 ± 0.12 (4.8) | 7.48 ± 0.42 (33)b |
a p < 0.01 compared with pEC50 values from chlorate-free assays (Student's t test).
b p < 0.05 compared with pEC50 values from chlorate-free assays (Student's t test).
c p < 0.0001 compared with pEC50 values from chlorate-free assays (Student's t test).
Tyrosine-sulfated CCR2 Peptides
To investigate the specific effects of Tyr26 and Tyr28 sulfation in the CCR2 N terminus, we used NMR spectroscopy to monitor MCP-1 binding by a series of peptides spanning residues 18–31 of the CCR2 N terminus with different patterns of tyrosine sulfation (Fig. 2). Peptide R2A is nonsulfated, peptides R2B and R2C are sulfated on Tyr26 and Tyr28, respectively, and peptide R2D is sulfated on both Tyr residues. As described previously, we have also prepared a corresponding set of peptides spanning residues 1–31 of CCR2 (26). However, we found that the longer peptides were poorly soluble in aqueous buffers. Only the doubly sulfated variant was sufficiently soluble to obtain qualitative NMR binding data. The observed changes of MCP-1 chemical shifts upon binding to this peptide correlated closely with those observed upon binding to the corresponding shorter peptide R2D (data not shown), suggesting that the major binding determinants were located in the residue 18–31 region. Therefore, herein we present data for only the shorter set of peptides.
FIGURE 2.
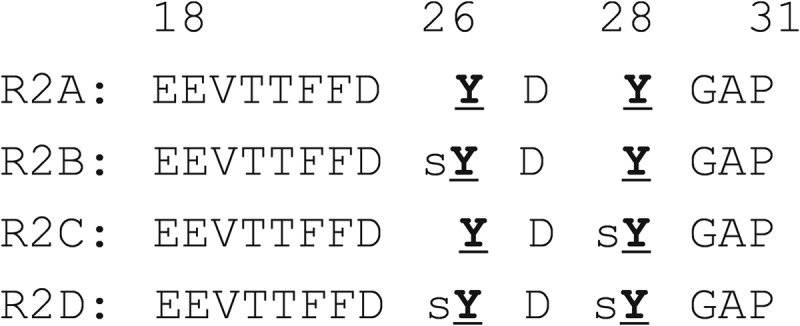
Primary sequences of peptides R2A-R2D. Standard amino acids are shown using single-letter codes. sY signifies sulfated tyrosine residues.
Binding of Wild Type MCP-1 to CCR2 Peptides
To enable characterization of the binding properties of the wild type MCP-1 monomer and dimer, we used an MCP-1 concentration of 50 μm, at which resonances of both species are observable in two-dimensional NMR (15N-1H HSQC) spectra (Fig. 3A). These resonances were observed as two separate sets of signals indicating that the monomer and dimer exchange slowly on the NMR chemical shift time scale (approximately milliseconds). Upon addition of the nonsulfated CCR2 peptide R2A, there were no significant changes in the NMR signals of wild type MCP-1. In contrast, upon addition of the sulfated CCR2 peptides R2B-R2D, peaks assigned to both monomer and dimer species shifted in a concentration-dependent manner, indicating that both monomeric and dimeric wild type MCP-1 could bind to the sulfated CCR2 peptides and that the bound and unbound species were in fast exchange with each other on the NMR chemical shift time scale (Fig. 3A). Moreover, binding of the sulfated peptides led to a reduction in the intensities of dimer peaks and an increase in the intensities of monomer peaks (Fig. 3B). These observations indicate that: 1) sulfopeptide binding is thermodynamically coupled to MCP-1 dimerization; 2) sulfopeptides bind more strongly to monomeric than to dimeric MCP-1; and 3) sulfopeptide binding shifts the monomer-dimer equilibrium toward the monomeric form of MCP-1, which is the state capable of activating the receptor. Fig. 3 (C and D) shows the weighted changes in chemical shift (ΔδNH) for several MCP-1 monomer and dimer signals upon binding to each CCR2 peptide. These data have been fitted to a simple 1:1 binding model to yield the equilibrium dissociation constants (Kd) shown in Table 2. These data, together with qualitative comparison of the binding curves, indicate that sulfation of Tyr26 and/or Tyr28 substantially increases binding affinity for MCP-1. However, it should be noted that the binding model used to fit these data does not account for the dependence of the relative concentrations of monomeric and dimeric species on peptide concentration. Moreover, only five sets of monomer and dimer peaks could be monitored over the full peptide titration because of substantial resonance overlap resulting from the presence of both species. To overcome the difficulties of coupled equilibria and peak overlap, we repeated the peptide binding experiments using mutants of MCP-1 that are trapped in the monomeric or dimeric states, MCP(P8A) and MCP-1(T10C), respectively (21, 22).
FIGURE 3.

Binding of wild type MCP-1 to CCR2 peptides monitored by two-dimensional NMR. A, an expanded region of the 1H-15N HSQC spectrum of wild type MCP-1, showing the movements of monomer (M) and dimer (D) peaks of residue Lys19 upon addition of each peptide (R2A-R2D). Within each panel, spectra are shown for the unbound sample and three molar ratios of peptide:protein (1:1, 2:1, and 5:1), with the direction of peak movement indicated by arrows. B, concentration dependence of the peak intensities of monomer (filled symbols) and dimer (open symbols) peaks; each point (and error bar) represents the average (and standard deviation) of the normalized peak height for the five residues for which both monomer and dimer peaks were resolved across the full range of peptide concentrations used (Lys19, Leu25, Ile42, Phe43, and Cys52). C and D, concentration dependence of the peak positions for monomer (C) and dimer (D) peaks are shown for each of the five residues for which both monomer and dimer peaks were resolved across the full range of peptide concentrations used: Lys19 (circles), Leu25 (squares), Ile42 (triangles), Phe43 (inverted triangles), and Cys52 (diamonds). The solid lines represent the best fits of the data to Equation 1; in these fits, the initial concentration of the protein monomer or dimer (P0) is assumed to be halfway between the two extreme concentrations deduced from the normalized peak heights in B.
TABLE 2.
Kd values for binding of wild type MCP-1 to R2A-R2D
The values in the table are Kd ± standard error as obtained from simultaneous fitting of ΔδNH values for the five residues for which both monomer and dimer peaks were resolved across the full range of peptide concentrations used (Lys19, Leu25, Ile42, Phe43, and Cys52). Fits were performed assuming the concentration of the monomer or dimer was halfway between the two extreme concentrations deduced from the normalized peak heights in Fig. 3B. Changing the monomer or dimer concentration to the extreme values caused only small (less than ∼10%) changes in the fitted Kd values. ND, not determinable because of insignificant changes in chemical shift values.
| Peptide | Monomer | Dimer |
|---|---|---|
| μm | μm | |
| R2A | ND | ND |
| R2B | 11.3 ± 1.1 | 105.3 ± 14.0 |
| R2C | 13.3 ± 3.4 | 39.1 ± 7.9 |
| R2D | 13.9 ± 1.2 | 37.8 ± 4.3 |
Obligate Monomeric and Dimeric Mutants of MCP-1
The MCP-1 mutant P8A, which is unable to dimerize, has previously been shown to activate CCR2 in vitro but to be incapable of inducing leukocyte recruitment in vivo (29) and to inhibit leukocyte recruitment by wild type MCP-1 (30). We recently reported that the MCP-1 mutant T10C is an obligate dimer and does not activate CCR2 in vitro at concentrations as high as ∼1 μm (22). We have now compared the capacity of these mutants to induce leukocyte recruitment in vivo with that of wild type MCP-1. These experiments made use of a mouse model of MCP-1-induced leukocyte recruitment in which leukocyte adhesion in postcapillary venules of the cremaster muscle and subsequent emigration are monitored by intravital microscopy (25) (Fig. 4). In these experiments, wild type MCP-1 induced robust leukocyte adhesion and emigration within 4 h of injection. This response did not occur for boiled MCP-1, indicating that it was solely due to the bioactive chemokine. In contrast to the previous finding that MCP-1(P8A) was unable to induce leukocyte recruitment into the peritoneal cavity of mice (29, 30), we observed that MCP-1(P8A) did not significantly differ from wild type MCP-1 in its ability to induce leukocyte adhesion or emigration in the murine skeletal muscle microvasculature. However, MCP-1(T10C) was significantly impaired relative to wild type MCP-1 in its ability to induce both leukocyte adhesion and emigration. This is consistent with observation that MCP-1(T10C) is inactive in vitro; the small residual activity could be due to a minor (∼5%) impurity of monomeric chemokine (22).
FIGURE 4.

In vivo activity of monomeric and dimeric MCP-1. Shown are the results for mean leukocyte adhesion (A) and leukocyte emigration (B) in skeletal muscle postcapillary venules 4 h after local injection of either wild type MCP-1 (wtMCP-1), obligate monomeric mutant P8A, obligate dimeric mutant T10C, or wild type MCP-1 denatured by boiling. The data represent analysis of three or four venules in seven (wild type), nine (P8A and T10C), or two (denatured MCP-1) mice per group. The data are shown as the means ± S.E. *, p < 0.05; **, p < 0.01 versus wtMCP-1.
Binding of MCP-1(P8A) and MCP-1(T10C) to Receptor Peptides
Signals in the HSQC spectra of MCP-1(P8A) and MCP-1(T10C) shifted with increasing peptide concentration (Fig. 5, A–F), confirming that both monomeric and dimeric forms of MCP-1 could bind to the peptides. Global fitting of the resultant ΔδNH values to a 1:1 binding model was conducted separately for each protein/peptide pair. For all protein/peptide pairs, the titration curves (Fig. 5, A–F) were consistent with simple 1:1 binding, allowing determination of the equilibrium dissociation constants (Kd) shown in Table 3 and Fig. 5G. For MCP-1(T10C), a binding stoichiometry of 1:1 indicates that two peptide molecules bind to each MCP-1 dimer, but the binding curves show no evidence of cooperativity between the binding sites.
FIGURE 5.

Binding of obligate monomeric and dimeric MCP-1 to CCR2 peptides monitored by two-dimensional NMR. A–F, left panels, expanded regions of the HSQC spectra of MCP-1(P8A) (A–C) and MCP-1(T10C) (D–F) showing movements of the indicated peaks during peptide titrations. Each set of four panels shows the same spectral region for titrations of MCP-1(P8A) or MCP-1(T10C) with each of the four peptides (R2A, top left; R2B, top right; R2C, bottom left; and R2D, bottom right). Within each panel, spectra are shown for the unbound sample and three concentrations of bound sample, including the concentration that induced the greatest changes in chemical shift, with the direction of peak movement indicated by arrows. A–F, right panels, titration curves showing changes in backbone amide chemical shift for the indicated protein and residue plotted against peptide/protein molar ratio for each peptide (R2A, open triangles; R2B, filled diamonds; R2C, open squares; and R2D, filled circles). The solid lines were obtained from global fitting of ΔδNH values conducted separately for each peptide/protein pair. G, globally fitted Kd values (and standard errors) for binding of MCP-1(P8A) and MCP-1(T10C) to R2A-R2D.
TABLE 3.
Kd values for binding of MCP-1(P8A) and MCP-1(T10C) to R2A-R2D
The values in the table are Kd values ± standard error as obtained from simultaneous fitting of ΔδNH values for all resolved residues from each protein/peptide titration.
| Peptide | MCP-1(P8A) | MCP-1(T10C) |
|---|---|---|
| μm | μm | |
| R2A | 396 ± 66 | 198 ± 44 |
| R2B | 12.3 ± 0.2 | 57.1 ± 3.1 |
| R2C | 16.2 ± 0.5 | 29.8 ± 1.3 |
| R2D | 0.21 ± 0.11 | 1.24 ± 0.24 |
The affinity data (Table 3) indicate that tyrosine sulfation of the CCR2 N-terminal peptides increased their binding affinities for both MCP-1(P8A) and MCP-1(T10C). Whereas the obligate monomer MCP-1(P8A) bound to the unsulfated peptide R2A with low affinity (Kd = 492 ± 67 μm), sulfation of either Tyr26 (R2B) or Tyr28 (R2C) increased the affinity by ∼30-fold, and sulfation of both residues (R2D) enhanced the affinity by more than 3 orders of magnitude. Similarly, the obligate dimer MCP-1(T10C) bound to R2A with low affinity (Kd = 188 ± 30 μm), but sulfation of Tyr26 and Tyr28 increased the binding affinity by only 3- and 6-fold, respectively, whereas double sulfation enhanced the affinity by approximately 2 orders of magnitude. As a consequence of these changes, the sulfated peptides bound 2- to 5-fold more tightly to MCP-1(P8A) than to MCP-1(T10C), consistent with the observation that sulfated peptides shift the monomer-dimer equilibrium of wild type MCP-1 to favor the monomeric form.
Regions of MCP-1(P8A) and MCP-1(T10C) that Interact with R2A-R2D
The chemical shift changes of MCP-1 variants observed upon peptide binding were used to tentatively identify the peptide binding sites on monomeric and dimeric MCP-1. Fig. 6 (A and B) shows the maximum ΔδNH values for all assigned residues of MCP-1(P8A) and MCP-1(T10C) after binding to R2A-R2D. Several trends can be observed. First, for each form of MCP-1, the same set of residues exhibited chemical shift changes for all four peptides. In most cases, the directions of peak shifts were also similar for all peptides, although there were a few exceptions to this trend (e.g., Lys19 in Fig. 5) Second, the residues of MCP-1(P8A) generally had higher ΔδNH values than the residues of MCP-1(T10C), particularly for binding to R2B and R2D. As we have noted in previous sulfopeptide binding studies (13, 15), larger chemical shift changes generally occur for tighter binding protein-peptide pairs. Third, in many cases the same regions of the two MCP-1 mutants displayed chemical shift changes upon binding to the peptides; these regions included the N-loop (residues 13–21), the 310 turn (residues 22–24), the 40s loop (residues 46–49), and the third β-sheet (residues 50–54). However, several residues were used exclusively by one mutant or the other, e.g., residues 9, 11, and 12 by MCP-1(P8A) and residues 4 and 5 by MCP-1(T10C).
FIGURE 6.

Structural mapping of the chemical shift changes upon binding of peptide R2D to MCP-1(P8A) and MCP-1(T10C). A and B, maximum changes in weighted amide chemical shift (ΔδNH) of MCP-1(P8A) (A) and MCP-1(T10C) (B) residues upon binding to R2A (black), R2B (cyan), R2C (red), and R2D (green). The data for each protein are shown in two panels. The secondary structure of MCP-1 is indicated at the top of each graph. Black dots indicate unassigned residues or prolines. C, surface representation of the MCP-1 monomer (one monomer from Protein Data Bank file 1DOM) color-coded according to maximum ΔδNH values upon binding of obligate monomer MCP-1(P8A) to peptide R2D. The two diagrams show the two different sides of MCP-1(P8A) linked by a 180° rotation around the vertical axis. D, surface and ribbon representations of the MCP-1 dimer (Protein Data Bank file 1DOM) colored to show the changes in weighted amide chemical shift (ΔδNH) upon binding of obligate dimer MCP-1(T10C) to peptide R2D. The right half of the protein is related the left half by a 180° rotation around the vertical 2-fold axis. The bottom two structures are related to the top two structures by a 90° rotation around the horizontal axis. In the two ribbon representations (right), the side chains of N-terminal residues Ala4, Ile5, Ala7, and Tyr12 are shown as sticks. Residues in the one of the monomer units of the dimer are denoted with a prime symbol. E, structure of a cross-linked dimer of CXC chemokine CXCL12/SDF-1 bound to two sulfated peptides corresponding to residues 1–38 of receptor CXCR4, with Tyr21 sulfated (Protein Data Bank file 2K03). SDF-1 monomers are shown as red and blue ribbons, and the CXCR4 peptides are shown as cyan and yellow backbone stick structures with the sulfotyrosine residues shown as spheres.
DISCUSSION
CCR2 Sulfation Enhances MCP-1 Binding
Previous studies investigating the 25DYDY28 tyrosine sulfation motif of CCR2 have shown that Tyr26 is sulfated and that this motif facilitates binding by MCP-1 (5, 20). The experiments described herein build upon the previous mutational studies by demonstrating that inhibition of CCR2 sulfation causes a reduction in the potency of MCP-1 at this receptor and that sulfation of both Tyr26 and Tyr28 in N-terminal peptides of CCR2 enhances the affinity of these receptors for MCP-1. Taken together, these results clearly indicate that post-translational sulfation of residues in the N-terminal region of CCR2 enhances the ability of this receptor to be activated by MCP-1.
Previous studies of chemokine binding by sulfated peptides derived from chemokine receptor CCR3 have shown that affinity enhancements are dependent on the position of sulfation within the CCR3 16YYDD19 motif and that the effect of sulfating both tyrosine residues may be additive or positively cooperative depending on the chemokine binding partner. In the current study, the affinity of the CCR2 peptide for the MCP-1 obligate monomer was enhanced by approximately the same amount (∼30-fold) upon sulfation of either Tyr26 or Tyr28, and these affinity enhancements were approximately additive (in terms of free energy) for binding of the doubly sulfated peptide (Fig. 5G), suggesting that the interactions of the two sulfated tyrosine groups with monomeric MCP-1 are independent of each other. Binding enhancements for the MCP-1 obligate dimer were much smaller than for the obligate monomer and were weakly dependent on the position of sulfation (∼3- and ∼6-fold enhancements for sulfation of Tyr26 and Tyr28, respectively). Notably, the sulfation of both tyrosine residues resulted in a ∼160-fold enhancement of binding to obligate dimeric MCP-1, substantially greater than the ∼18-fold enhancement expected if the effects of Tyr26 and Tyr28 sulfation were energetically additive. Thus, affinity enhancements of this form of MCP-1 exhibit positive cooperativity, suggesting that the interactions of one sulfotyrosine group enhance those of the second sulfotyrosine with dimeric MCP-1.
The current in vitro data suggest that tyrosine sulfation is likely to alter the ability of CCR2 to interact with chemokine ligands. However, the in vivo consequences of CCR2 sulfation remain largely unexplored. Tyrosine sulfation of chemokine receptors is likely to be incomplete and heterogeneous (10) depending on the activity of the two tyrosylprotein sulfotransferase isoforms in the cells expressing these receptors (3). It remains very difficult to determine the sulfation states of naturally expressed receptors in primary cells or even heterologously expressed in cell lines. Currently we have no information on the populations of the differently sulfated forms of CCR2 on monocytes or macrophages. With respect to cell lines, the previous mutational study suggests that Tyr26 is more highly sulfated than Tyr28 in HEK293 cells (5), but it remains possible that Tyr28 alone or both Tyr residues are sulfated in different cell types or under different biological circumstances. In light of the known sulfation of Tyr26, it is noteworthy that the CCR2 peptide sulfated at this position (R2B) was the most effective peptide at shifting the wild type MCP-1 equilibrium from dimer to monomer (Table 2 and Fig. 3B) and that this form induced the largest chemical shift changes of MCP-1(P8A) (Fig. 6A). Thus, it is tempting to speculate that the chemokine may have evolved to be optimally responsive to the biologically dominant form of the receptor.
Our data suggest two ways in which the tissue-specific regulation of CCR2 tyrosine sulfation could affect the cellular response to MCP-1. First, cells expressing CCR2 with greater levels of sulfation would be more responsive to MCP-1 simply because of stronger interactions with the chemokine. Second, close inspection of the MCP-1 NMR spectra reveals a small group of peaks (e.g., Lys19; Fig. 5B) that respond differently to binding by two peptides with identical binding affinities (e.g., R2B and R2C). This observation suggests that the precise position or orientation of the chemokine on the N terminus of the receptor may be dependent on the sulfation state of the receptor, potentially leading to sulfation-dependent activation of different signaling pathways and cellular responses. Whereas the influence of receptor sulfation on such signaling is currently speculative, the ability of CCR2 to signal through alternative pathways has been observed previously for MCP-1 mutants (31). Future studies are required to investigate the effect of sulfation in differential receptor signaling.
Interactions of the CCR2 N Terminus with Monomeric MCP-1
Although the structural basis of chemokine function has been of long-standing interest, there are still no reported structures of CC chemokines bound to receptors or receptor fragments. Nevertheless, extensive mutational analysis has identified the residues on MCP-1 that are involved in receptor binding and activation (20, 31). Critical residues for binding to CCR2 were found to be: Tyr13, Arg18, Lys19 (in the N-loop), Arg24 (in the 310-turn), Lys35, Lys38 (in the 30s loop), and Lys49 (in the β3-strand) (20). The chemical shift changes observed in the current study allow us to identify a putative binding site on MCP-1 for the N-terminal region of CCR2. Fig. 6C shows a surface representation of the MCP-1 monomer color-coded according to maximum ΔδNH values upon binding to R2D. The residues undergoing the largest chemical shift changes are all located on the face of the chemokine defined by the N-loop and β3-strand. Thus, the current NMR data are consistent with the previous mutational data and indicate that the likely binding site for the receptor N terminus is a shallow cleft between the N-loop and β3-strand on the chemokine. Previous NMR studies of other chemokines have also implicated the N-loop and β3-strand regions in recognition of the N-terminal regions of chemokine receptors (13–15, 32–37). In contrast, the 30s loop residues of MCP-1 only showed minor changes of chemical shift, but mutations in this region decreased CCR2 binding (20). It is therefore likely that the 30s loop of MCP-1 either interacts with non-N-terminal residues of the receptor or indirectly affects the interactions of the N-loop/β3-strand regions, perhaps via the Cys11 to Cys36 disulfide bond.
In Vivo Effects of Obligate Monomer MCP-1(P8A)
Here we found that the obligate monomer MCP-1(P8A) induced leukocyte adhesion and transmigration with efficiency similar to that of wild type MCP-1, whereas the obligate dimer MCP-1(T10C) was significantly less effective. The findings with T10C are in agreement with our recent description of the reduced ability of this mutant to activate CCR2-dependent signaling (22). However, the findings with P8A differ from previous studies reporting that the monomeric mutant was unable to induce leukocyte recruitment into the peritoneal cavity (29). The different modes of administration of MCP-1 and the different doses used provide some potential explanations for this discrepancy. Proudfoot et al. (29) administered 10 μg of P8A intraperitoneally and examined the effect after 18 h. This route allows absorption of the injected protein into the bloodstream, with the downstream effect of reduced CCR2 expression on circulating leukocytes. These systemic effects have the potential to modulate the chemotactic response of leukocytes in the bloodstream to locally expressed chemokine gradients. In contrast, in the present study, we injected 0.34 μg subcutaneously, adjacent to the cremaster muscle, and examined the response only 4 h later. Under the latter conditions, it is likely that the systemic effect of the injected MCP-1 on leukocyte CCR2 expression was substantially less than in the previous study, allowing circulating leukocytes to respond effectively to the local effects of MCP-1 in the muscle microvasculature. Alternatively, it is conceivable that in the subcutaneous injection model, MCP-1 injected locally induced leukocyte chemotaxis indirectly via interaction with CCR2-expressing tissue-resident cells such as mast cells or macrophages, with the responding cells releasing additional chemoattractants to induce leukocyte arrest and transmigration. This would avoid any potential differences in the ability of wild type versus obligate monomer MCP-1 to undergo shear-resistant interactions with glycosaminoglycans (GAGs) on the endothelial surface, a possibility raised in previous studies (29).
Finally, it is possible that the MCP-1 samples produced in the current and previous studies had different levels of pyroglutamate formation from the N-terminal Gln residue. Although this modification does not appear to influence receptor binding or activation or chemotactic activity in vitro (38), we cannot exclude the possibility that it affects the in vivo activity of MCP-1.
Interactions of the Sulfated CCR2 N Terminus with Dimeric MCP-1
Although the CCR2-derived sulfopeptides bind to both monomeric and dimeric MCP-1, the NMR data clearly indicate that dimer binding is weaker than monomer binding, both for the wild type and mutant forms of MCP-1 studied here. The dimerization and receptor sulfopeptide binding of MCP-1 can be described by the thermodynamic model shown in Fig. 7. According to this model, weaker peptide binding by the dimer compared with the monomer (KDP > KMP) gives rise to a greater tendency of the peptide-bound dimer (DP2) to dissociate than for the free dimer (D) to dissociate (KMDP2 > KMD). Thus, binding of receptor peptides to the dimer weakens the dimer interface and induces dissociation to the monomeric form of the chemokine. This model is supported not only by the Kd values (Tables 2 and 3) but also by the observation that the fraction of wild type MCP-1 in the monomeric form increases at the expense of dimer as the receptor peptide concentration is increased (Fig. 3B).
FIGURE 7.
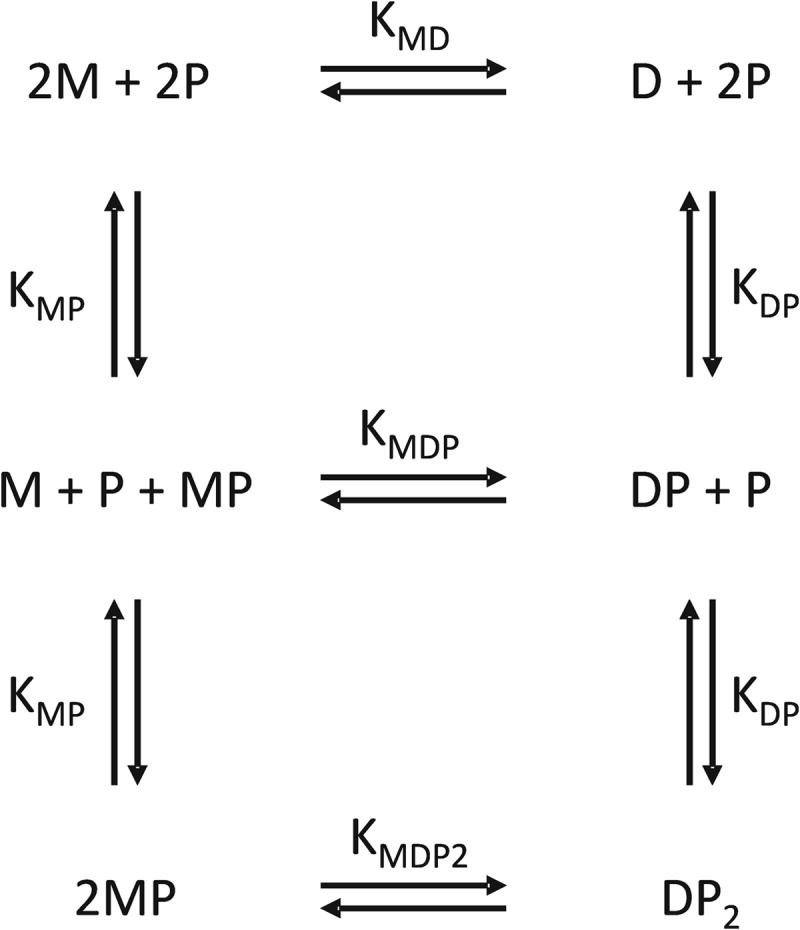
Coupled thermodynamic model to account for binding of a receptor peptide (P) to both monomeric (M) and dimeric (D) forms of MCP-1. Stepwise binding of two peptide molecules to the dimer is assumed to be noncooperative. Each binding or dimerization equilibrium is labeled with the corresponding bimolecular equilibrium dissociation constant.
The observation that sulfated CCR2 peptides stabilize the MCP-1 monomer relative to the dimer contrasts with the previous finding that sulfated peptides derived from the chemokine receptor CXCR4 enhance dimerization of the CXC chemokine CXCL12/SDF-1 (12). The stabilization of dimeric SDF-1 is consistent with structures showing that two CXCR4 peptides bind to one SDF-1 dimer and that each peptide forms interactions with residues on both monomeric units of the chemokine dimer (Fig. 6E) (35). However, the structural basis of receptor peptide binding is expected to be quite different for dimeric MCP-1 compared with dimeric SDF-1 because CC chemokines have substantially different dimer structures from CXC chemokines, although the monomer structures are very similar (Fig. 6, D and E). Structural mapping of our chemical shift data for dimeric MCP-1 (Fig. 6D) shows that the majority of residues undergoing substantial chemical shift changes are located in the N-loop and β3-strand regions, as observed for monomeric MCP-1, so this is expected to be the primary receptor sulfopeptide binding site. Because of the 2-fold symmetry of the dimer structure, the N-loop/β3-strand regions of the two monomers are located on opposite faces of the dimer structure. Thus, the chemical shift mapping data indicate that two sulfopeptide molecules bind to opposite faces of the dimer, consistent with the independent binding deduced from fitted binding curves. In addition to the primary binding regions, residues Ala4, Ile5, and Ala7 within the N-terminal region of MCP-1 undergo substantial chemical shift changes upon binding of CCR2 peptides (Fig. 6B). These residues from one monomer are close to the N-loop region of the other monomer in the dimer (Fig. 6D). Moreover, the side chain of residue Tyr13, in the N-loop of one monomer extends across the dimer interface to make contact with the equivalent side chain in the other monomer (Fig. 6D). Thus, it appears that sulfopeptide binding to the N-loop leads to modified interactions among these residues and hence weakening of the dimerization interface.
Unlike CXCL12, the related dimeric chemokine CXCL8/interleukin-8 is induced to dissociate to its monomeric form upon binding to (nonsulfated) peptides derived from the N terminus of the receptor CXCR1 (39). Both monomeric and dimeric CXCL8 species are able to bind and activate the receptors CXCR1 and CXCR2, although the relative activity of the two species varies depending on the cellular activity monitored (40). NMR data have suggested that binding of CXCR1 peptides weakens the dimer interface by inducing subtle structural and dynamical changes (34), similar to the mechanism suggested here for MCP-1. Considering that the structure of the CXCL8 dimer resembles that of dimeric CXCL12, it is also possible that binding of two CXCR1 peptides to the dimer gives rise to direct repulsion between the two bound peptides, thus contributing to dimer dissociation.
The conserved tendency of many chemokines to oligomerize and the observation that obligate monomers such as MCP-1(P8A) are able to activate chemokine receptors has led to an ongoing debate on whether dimeric CC chemokines are able to bind and activate chemokine receptors. As discussed in our recent paper describing the development of obligate dimeric mutant MCP-1(T10C), the consensus of these previous studies is that CC chemokine dimers are unable to bind or activate their receptors at concentrations up to ∼1 μm (22, 41), at least under the conditions of cell-based assays. The current in vivo data showing that dimeric MCP-1 does not induce leukocyte adhesion or emigration support the view that dimeric CC chemokines are unable to activate chemokine receptors. However, our observation that sulfation of a CCR2 peptide substantially increases affinity for dimeric MCP-1 raises the possibility that highly sulfated forms of CCR2 may be able to bind dimeric MCP-1 at biologically relevant concentrations. Such binding could be enhanced by receptor dimerization or by the relatively high local concentrations of chemokines formed by their interactions with GAGs on endothelial surfaces. Indeed it is even possible that dimeric or oligomeric chemokines could bind simultaneously to GAGs on endothelial cells and chemokine receptors on rolling leukocytes, thus preventing the loss of chemokine signals caused by blood flow.
Concluding Remarks
The data presented herein show that sulfation of Tyr26 and/or Tyr28 of CCR2 enhances the ability of the N-terminal region of the receptor to bind both monomeric and dimeric forms of MCP-1. Moreover, binding of CCR2 peptides weakens the dimerization of MCP-1, thereby inducing dissociation to the monomeric state. Considering that the MCP-1 dimer binds more avidly than the monomer to GAGs but the monomer is required for receptor activation, the current results suggest a model in which a substantial proportion of MCP-1 is transferred as a dimer from GAGs on the endothelial surface to CCR2 on the surface of a rolling leukocyte, and then the receptor-bound dimer subsequently dissociates to allow receptor activation.
This work was supported by Australian Research Council Grants DP0881570 and LE0989504 (to M. J. S.) and DP1094884 (to R. J. P. and M. J. S.) and by Australian National Health and Medical Research Council Grant 519461 (to A. C.).
- MCP
- monocyte chemoattractant protein
- GAG
- glycosaminoglycan
- HSQC
- heteronuclear single quantum coherence spectrum
- SDF-1
- stromal cell-derived factor-1.
REFERENCES
- 1. Thelen M., Stein J. V. (2008) How chemokines invite leukocytes to dance. Nat. Immunol. 9, 953–959 [DOI] [PubMed] [Google Scholar]
- 2. Crump M. P., Gong J. H., Loetscher P., Rajarathnam K., Amara A., Arenzana-Seisdedos F., Virelizier J. L., Baggiolini M., Sykes B. D., Clark-Lewis I. (1997) Solution structure and basis for functional activity of stromal cell-derived factor-1. Dissociation of CXCR4 activation from binding and inhibition of HIV-1. EMBO J. 16, 6996–7007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Stone M. J., Chuang S., Hou X., Shoham M., Zhu J. Z. (2009) Tyrosine sulfation. An increasingly recognised post-translational modification of secreted proteins. N. Biotechnol. 25, 299–317 [DOI] [PubMed] [Google Scholar]
- 4. Liu J., Louie S., Hsu W., Yu K. M., Nicholas H. B., Jr., Rosenquist G. L. (2008) Tyrosine sulfation is prevalent in human chemokine receptors important in lung disease. Am. J. Respir. Cell Mol. Biol. 38, 738–743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Preobrazhensky A. A., Dragan S., Kawano T., Gavrilin M. A., Gulina I. V., Chakravarty L., Kolattukudy P. E. (2000) Monocyte chemotactic protein-1 receptor CCR2B is a glycoprotein that has tyrosine sulfation in a conserved extracellular N-terminal region. J. Immunol. 165, 5295–5303 [DOI] [PubMed] [Google Scholar]
- 6. Fong A. M., Alam S. M., Imai T., Haribabu B., Patel D. D. (2002) CX3CR1 tyrosine sulfation enhances fractalkine-induced cell adhesion. J. Biol. Chem. 277, 19418–19423 [DOI] [PubMed] [Google Scholar]
- 7. Farzan M., Mirzabekov T., Kolchinsky P., Wyatt R., Cayabyab M., Gerard N. P., Gerard C., Sodroski J., Choe H. (1999) Tyrosine sulfation of the amino terminus of CCR5 facilitates HIV-1 entry. Cell 96, 667–676 [DOI] [PubMed] [Google Scholar]
- 8. Gutiérrez J., Kremer L., Zaballos A., Goya I., Martínez-A. C., Márquez G. (2004) Analysis of post-translational CCR8 modifications and their influence on receptor activity. J. Biol. Chem. 279, 14726–14733 [DOI] [PubMed] [Google Scholar]
- 9. Colvin R. A., Campanella G. S., Manice L. A., Luster A. D. (2006) CXCR3 requires tyrosine sulfation for ligand binding and a second extracellular loop arginine residue for ligand-induced chemotaxis. Mol. Cell Biol. 26, 5838–5849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Farzan M., Babcock G. J., Vasilieva N., Wright P. L., Kiprilov E., Mirzabekov T., Choe H. (2002) The role of post-translational modifications of the CXCR4 amino terminus in stromal-derived factor 1α association and HIV-1 entry. J. Biol. Chem. 277, 29484–29489 [DOI] [PubMed] [Google Scholar]
- 11. Choe H., Moore M. J., Owens C. M., Wright P. L., Vasilieva N., Li W., Singh A. P., Shakri R., Chitnis C. E., Farzan M. (2005) Sulphated tyrosines mediate association of chemokines and Plasmodium vivax Duffy binding protein with the Duffy antigen/receptor for chemokines (DARC). Mol. Microbiol. 55, 1413–1422 [DOI] [PubMed] [Google Scholar]
- 12. Veldkamp C. T., Seibert C., Peterson F. C., Sakmar T. P., Volkman B. F. (2006) Recognition of a CXCR4 sulfotyrosine by the chemokine stromal cell-derived factor-1α (SDF-1α/CXCL12). J. Mol. Biol. 359, 1400–1409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zhu J. Z., Millard C. J., Ludeman J. P., Simpson L. S., Clayton D. J., Payne R. J., Widlanski T. S., Stone M. J. (2011) Tyrosine sulfation influences the chemokine binding selectivity of peptides derived from chemokine receptor CCR3. Biochemistry 50, 1524–1534 [DOI] [PubMed] [Google Scholar]
- 14. Duma L., Häussinger D., Rogowski M., Lusso P., Grzesiek S. (2007) Recognition of RANTES by extracellular parts of the CCR5 receptor. J. Mol. Biol. 365, 1063–1075 [DOI] [PubMed] [Google Scholar]
- 15. Simpson L. S., Zhu J. Z., Widlanski T. S., Stone M. J. (2009) Regulation of chemokine recognition by site-specific tyrosine sulfation of receptor peptides. Chem. Biol. 16, 153–161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Seibert C., Veldkamp C. T., Peterson F. C., Chait B. T., Volkman B. F., Sakmar T. P. (2008) Sequential tyrosine sulfation of CXCR4 by tyrosylprotein sulfotransferases. Biochemistry 47, 11251–11262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Charo I. F., Peters W. (2003) Chemokine receptor 2 (CCR2) in atherosclerosis, infectious diseases, and regulation of T-cell polarization. Microcirculation 10, 259–264 [DOI] [PubMed] [Google Scholar]
- 18. Izikson L., Klein R. S., Charo I. F., Weiner H. L., Luster A. D. (2000) Resistance to experimental autoimmune encephalomyelitis in mice lacking the CC chemokine receptor (CCR)2. J. Exp. Med. 192, 1075–1080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Mestas J., Ley K. (2008) Monocyte-endothelial cell interactions in the development of atherosclerosis. Trends Cardiovasc. Med. 18, 228–232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hemmerich S., Paavola C., Bloom A., Bhakta S., Freedman R., Grunberger D., Krstenansky J., Lee S., McCarley D., Mulkins M., Wong B., Pease J., Mizoue L., Mirzadegan T., Polsky I., Thompson K., Handel T. M., Jarnagin K. (1999) Identification of residues in the monocyte chemotactic protein-1 that contact the MCP-1 receptor, CCR2. Biochemistry 38, 13013–13025 [DOI] [PubMed] [Google Scholar]
- 21. Paavola C. D., Hemmerich S., Grunberger D., Polsky I., Bloom A., Freedman R., Mulkins M., Bhakta S., McCarley D., Wiesent L., Wong B., Jarnagin K., Handel T. M. (1998) Monomeric monocyte chemoattractant protein-1 (MCP-1) binds and activates the MCP-1 receptor CCR2B. J. Biol. Chem. 273, 33157–33165 [DOI] [PubMed] [Google Scholar]
- 22. Tan J. H., Canals M., Ludeman J. P., Wedderburn J., Boston C., Butler S. J., Carrick A. M., Parody T. R., Taleski D., Christopoulos A., Payne R. J., Stone M. J. (2012) Design and receptor interactions of obligate dimeric mutant of chemokine monocyte chemoattractant protein-1 (MCP-1). J. Biol. Chem. 287, 14692–14702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Robinson E. A., Yoshimura T., Leonard E. J., Tanaka S., Griffin P. R., Shabanowitz J., Hunt D. F., Appella E. (1989) Complete amino acid sequence of a human monocyte chemoattractant, a putative mediator of cellular immune reactions. Proc. Natl. Acad. Sci. U.S.A. 86, 1850–1854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Apostolopoulos J., Hickey M. J., Sharma L., Davenport P., Moussa L., James W. G., Gregory J. L., Kitching A. R., Li M., Tipping P. G. (2008) The cytoplasmic domain of tissue factor in macrophages augments cutaneous delayed-type hypersensitivity. J. Leukocyte Biol. 83, 902–911 [DOI] [PubMed] [Google Scholar]
- 25. Fan H., Hall P., Santos L. L., Gregory J. L., Fingerle-Rowson G., Bucala R., Morand E. F., Hickey M. J. (2011) Macrophage migration inhibitory factor and CD74 regulate macrophage chemotactic responses via MAPK and Rho GTPase. J. Immunol. 186, 4915–4924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Taleski D., Butler S. J., Stone M. J., Payne R. J. (2011) Divergent and site-selective solid-phase synthesis of sulfopeptides. Chem. Asian J. 6, 1316–1320 [DOI] [PubMed] [Google Scholar]
- 27. Handel T. M., Domaille P. J. (1996) Heteronuclear (1H, 13C, 15N) NMR assignments and solution structure of the monocyte chemoattractant protein-1 (MCP-1) dimer. Biochemistry 35, 6569–6584 [DOI] [PubMed] [Google Scholar]
- 28. Schmidt P. M., Sparrow L. G., Attwood R. M., Xiao X., Adams T. E., McKimm-Breschkin J. L. (2012) Taking down the FLAG! How insect cell expression challenges an established tag-system. PLoS One 7, e37779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Proudfoot A. E., Handel T. M., Johnson Z., Lau E. K., LiWang P., Clark-Lewis I., Borlat F., Wells T. N., Kosco-Vilbois M. H. (2003) Glycosaminoglycan binding and oligomerization are essential for the in vivo activity of certain chemokines. Proc. Natl. Acad. Sci. U.S.A. 100, 1885–1890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Handel T. M., Johnson Z., Rodrigues D. H., Dos Santos A. C., Cirillo R., Muzio V., Riva S., Mack M., Déruaz M., Borlat F., Vitte P. A., Wells T. N., Teixeira M. M., Proudfoot A. E. (2008) An engineered monomer of CCL2 has anti-inflammatory properties emphasizing the importance of oligomerization for chemokine activity in vivo. J. Leukocyte Biol. 84, 1101–1108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Jarnagin K., Grunberger D., Mulkins M., Wong B., Hemmerich S., Paavola C., Bloom A., Bhakta S., Diehl F., Freedman R., McCarley D., Polsky I., Ping-Tsou A., Kosaka A., Handel T. M. (1999) Identification of surface residues of the monocyte chemotactic protein 1 that affect signaling through the receptor CCR2. Biochemistry 38, 16167–16177 [DOI] [PubMed] [Google Scholar]
- 32. Skelton N. J., Quan C., Reilly D., Lowman H. (1999) Structure of a CXC chemokine-receptor fragment in complex with interleukin-8. Structure 7, 157–168 [DOI] [PubMed] [Google Scholar]
- 33. Clubb R. T., Omichinski J. G., Clore G. M., Gronenborn A. M. (1994) Mapping the binding surface of interleukin-8 complexed with an N-terminal fragment of the type 1 human interleukin-8 receptor. FEBS Lett. 338, 93–97 [DOI] [PubMed] [Google Scholar]
- 34. Ravindran A., Joseph P. R., Rajarathnam K. (2009) Structural basis for differential binding of the interleukin-8 monomer and dimer to the CXCR1 N-domain. Role of coupled interactions and dynamics. Biochemistry 48, 8795–8805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Veldkamp C. T., Seibert C., Peterson F. C., De la Cruz N. B., Haugner J. C., 3rd, Basnet H., Sakmar T. P., Volkman B. F. (2008) Structural basis of CXCR4 sulfotyrosine recognition by the chemokine SDF-1/CXCL12. Sci. Signal. 1, ra4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Mayer K. L., Stone M. J. (2000) NMR solution structure and receptor peptide binding of the CC chemokine eotaxin-2. Biochemistry 39, 8382–8395 [DOI] [PubMed] [Google Scholar]
- 37. Ye J., Kohli L. L., Stone M. J. (2000) Characterization of binding between the chemokine eotaxin and peptides derived from the chemokine receptor CCR3. J. Biol. Chem. 275, 27250–27257 [DOI] [PubMed] [Google Scholar]
- 38. Gong J. H., Clark-Lewis I. (1995) Antagonists of monocyte chemoattractant protein 1 identified by modification of functionally critical NH2-terminal residues. J. Exp. Med. 181, 631–640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Fernando H., Chin C., Rösgen J., Rajarathnam K. (2004) Dimer dissociation is essential for interleukin-8 (IL-8) binding to CXCR1 receptor. J. Biol. Chem. 279, 36175–36178 [DOI] [PubMed] [Google Scholar]
- 40. Nasser M. W., Raghuwanshi S. K., Grant D. J., Jala V. R., Rajarathnam K., Richardson R. M. (2009) Differential activation and regulation of CXCR1 and CXCR2 by CXCL8 monomer and dimer. J. Immunol. 183, 3425–3432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Jin H., Shen X., Baggett B. R., Kong X., LiWang P. J. (2007) The human CC chemokine MIP-1β dimer is not competent to bind to the CCR5 receptor. J. Biol. Chem. 282, 27976–27983 [DOI] [PubMed] [Google Scholar]