

Background: The role of DNA methylation in musculoskeletal diseases is poorly understood.
Results: Methylation of specific CpG sites in the MMP13 and IL1B promoters correlates strongly with gene activity in human cartilage.
Conclusion: Specific CpG site methylation inhibits MMP13 transactivation by HIF-2α.
Significance: Our findings offer new insight on the impact of epigenetic control of a joint disease that can affect adults throughout life.
Keywords: Cartilage, DNA Methylation, Hypoxia-inducible Factor (HIF), Interleukin, Matrix Metalloproteinase (MMP), Transcription Promoter
Abstract
The role of DNA methylation in the regulation of catabolic genes such as MMP13 and IL1B, which have sparse CpG islands, is poorly understood in the context of musculoskeletal diseases. We report that demethylation of specific CpG sites at −110 bp and −299 bp of the proximal MMP13 and IL1B promoters, respectively, detected by in situ methylation analysis of chondrocytes obtained directly from human cartilage, strongly correlated with higher levels of gene expression. The methylation status of these sites had a significant impact on promoter activities in chondrocytes, as revealed in transfection experiments with site-directed CpG mutants in a CpG-free luciferase reporter. Methylation of the −110 and −299 CpG sites, which reside within a hypoxia-inducible factor (HIF) consensus motif in the respective MMP13 and IL1B promoters, produced the most marked suppression of their transcriptional activities. Methylation of the −110 bp CpG site in the MMP13 promoter inhibited its HIF-2α-driven transactivation and decreased HIF-2α binding to the MMP13 proximal promoter in chromatin immunoprecipitation assays. In contrast to HIF-2α, MMP13 transcriptional regulation by other positive (RUNX2, AP-1, ELF3) and negative (Sp1, GATA1, and USF1) factors was not affected by methylation status. However, unlike the MMP13 promoter, IL1B was not susceptible to HIF-2α transactivation, indicating that the −299 CpG site in the IL1B promoter must interact with other transcription factors to modulate IL1B transcriptional activity. Taken together, our data reveal that the methylation of different CpG sites in the proximal promoters of the human MMP13 and IL1B genes modulates their transcription by distinct mechanisms.
Introduction
Somatic cell phenotypes can be stably regulated by epigenetic mechanisms, including DNA methylation (1, 2), which may regulate gene transcription by recruiting chromatin remodeling proteins and by modulating the binding affinities of specific transcription factors (3–7). In contrast to the more dynamic and reversible histone modifications (1), CpG methylation is generally stable in somatic cells throughout adult life (8, 9). Investigations have mainly focused on the so-called CpG islands but have ignored the contributions of sparse CpG sites. Moreover, only a few reports have addressed how methylation of individual CpG sites influences the promoter activity of a specific gene (6, 10, 11), and the relative contributions of the methylation status of each individual CpG site to the transcriptional regulation of a given gene in vivo remain largely unexplored. In addition, although epigenetic gene regulation has been broadly investigated in other contexts, little is known about the specific impact of this mechanism of gene control in musculoskeletal diseases (12–29).
Osteoarthritis (OA)3 is characterized by the progressive loss of cartilage matrix, accompanied by deleterious changes in the synovium and subchondral bone (30, 31). In OA disease, chondrocytes undergo phenotypic changes with altered patterns of gene expression that are stably transmitted to subsequent cell generations (25, 32), suggesting the involvement of epigenetic mechanisms in disease onset and progression. We previously described a novel relationship between CpG demethylation within the CpG-sparse promoters of the MMP3, -9, -13, and ADAMTS4 genes and their aberrant, enhanced expression in OA chondrocytes, although a direct relationship between the methylation status of a specific CpG site and the respective promoter activity was not investigated (25). We also found that long term, repetitive stimulation of primary human chondrocytes with inflammatory cytokines induced demethylation of specific CpG sites in the IL1B proximal promoter, leading to increased and sustained IL1B expression (22). More recently, Reynard et al. (24) reported CpG methylation-dependent regulation of growth differentiation factor 5 expression in OA, along with direct evidence of suppressed gene activity in response to methylation of its CpG islands in vitro.
Here, we report that the methylation of specific CpG sites in the proximal promoters of both the human MMP13 and IL1B genes correlates with their expression in cartilage and alters their transcriptional activities in chondrocytes. In addition, we provide the first evidence that binding to and transactivation of the MMP13 promoter by hypoxia-inducible factor-2α (HIF-2α), also known as EPAS1 (endothelial PAS domain protein 1), are controlled by these epigenetic events. Importantly, our in vitro results correlate strongly with the in vivo methylation status of the same individual CpG sites in the MMP13 and IL1B promoters and with their relative levels of gene activity in chondrocytes isolated directly from healthy and OA human cartilage.
EXPERIMENTAL PROCEDURES
Human Primary Chondrocyte Isolation
Human articular cartilage was obtained after hemiarthroplasty following femoral neck fracture or total hip arthroplasty for OA with patient consent and prior approval of the local Institutional Review Board. Cartilage was dissected within 6 h of surgery, and non-OA fractured neck of femur and OA human primary chondrocytes were isolated as described previously (22). Briefly, samples were obtained from the deep zone of cartilage from patients with femoral neck fracture for isolation of non-OA/healthy chondrocytes, whereas cartilage pieces adjacent to weight-bearing areas of OA femoral heads (lacking surface zones) were harvested to obtain OA chondrocytes. Cartilage samples were then cut in small fragments and digested with 10% trypsin (Lonza) in PBS for 30 min, followed by sequential digestions in 1 mg/ml of hyaluronidase (Sigma-Aldrich) in PBS for 15 min, and in 10 mg/ml of collagenase B (Roche Applied Science) in α-modified Eagle's medium (Sigma-Aldrich) for 12–15 h at 37 °C. Following isolation, the chondrocytes were either subjected to bisulfite pyrosequencing or placed in primary culture.
Pyrosequencing
Genomic DNA extracted directly from neck of femur and OA chondrocytes or from cultured chondrocytes was treated with sodium bisulfite to convert unmethylated cytosine in CpG sites to uracil using the EZ DNA methylation Gold kit (Zymo Research). PCR and sequencing primers for pyrosequencing were designed using the Biotage Assay Design Software (version 2.0; Table 1). PCR was performed with Premium® PCR Supermix High Fidelity (Invitrogen). Pyrosequencing reactions were run in the PyroMark Q96 MD system (Qiagen), using 20 μl and 5 μl of the PCR products for CpG methylation analysis of MMP13 and IL1B promoters, respectively. Data are represented as percentage of methylation of each CpG site in OA or 5-aza-deoxycytidine (5-aza-dC)-treated samples set against 100% in the non-OA or untreated controls.
TABLE 1.
Primer sequences for pyrosequencing
| Name (range)/direction/purpose (covering CpG sites) | Sequence (5′ to 3′) |
|---|---|
| MMP13-Pyro-1 (−402/−188) | |
| Forward | AATTAGTATTAAGTTTTTTTTTATGGAAGT |
| Reverse | TTCAACAAAATCTCAAAACCCATCTAA |
| Sequencing 1 | AAATTTTTTTTTTTTTACCTTCTAT |
| Sequencing 2 | CTCAAAACCCATCTAAC |
| MMP13-Pyro-2 (−210/+36) | |
| Forward | ATGGGTTTTGAGATTTTG |
| Reverse | ACCCCTAAATACATCTTAAATA |
| Sequencing 1 | CAATCACTTAAAAATAAACATACTT |
| Sequencing 2 | AATAATACCTAAAAACTATTATC |
| Forward | ATGGAAGGGTAAGGAGTAGTAA |
| IL1B-Pyro-1 (−331/−175) | |
| Reverse | CCCACATATACTAAATTTAAACATTCTT |
| Sequencing | ATACTAAATTTAAACATTCTTCTA |
| IL1B-Pyro-2 (−64/+65) | |
| Forward | ATGAAGATTGGTTGAAGAGAATTTTAGA |
| Reverse | ATTTCTCAACCTCCTACTTCTACTTTTAA |
| Sequencing | ATTTTAGAGTAGTTTGTTGTG |
Human Primary Chondrocyte Culture and 5-Aza-dC Treatment
For culture and in vitro analysis, only deep-zone primary non-OA chondrocytes were utilized, as described (21). Before treatment, chondrocytes were cultured for 48 h at an average density of 16 × 103 cells/cm2 in a 25-cm2 flask in 5 ml of α-modified Eagle's medium supplemented with 10% FBS (Invitrogen), 1% insulin-transferrin-selenium (Sigma-Aldrich), 100 units/ml of penicillin, and 100 μg/ml of streptomycin (Lonza) and 100 μg/ml of ascorbic acid (Sigma-Aldrich). The chondrocytes were then left untreated (control) or incubated with 2 μm 5-aza-dC and added freshly at twice weekly medium changes for 4 to 5 weeks until confluence.
DNA and RNA Extraction
Genomic DNA and total RNA were extracted simultaneously from the harvested chondrocytes using AllPrep DNA/RNA mini kit (Qiagen). RNA was reverse-transcribed with avian myeloblastosis virus reverse transcriptase and combination of oligo(dT)15 and random primers (Promega).
Quantitative RT-PCR
Gene expression was analyzed with the ABI Prism 7500 gene detection system (Applied Biosystems) using the following primers, designed using Primer Express software (version 3.0; Applied Biosystems) to bracket exon-exon boundaries: GAPDH, 5′-CCAGGTGGTCTCCTCTGACTTC-3′ (forward) and 5′-TCATACCAGGAAATGAGCTTGACA-3′ (reverse); MMP13, 5′-TTAAGGAGCATGGCGACTTCT-3′ (forward) and 5′-CCCAGGAGGAAAAGCATGAG-3′ (reverse). The IL1B primers were designed by and purchased from PrimerDesign, Ltd. (Southampton, UK): 5′-TGGCAATGAGGATGACTTGTTC-3′ (forward) and 5′-CTGTAGTGGTGGTCGGAGATT-3′ (reverse). Reactions were performed in triplicate, with GAPDH as the normalizing control gene. The 2−ΔΔCt method was employed for relative quantification.
Plasmid Constructions
A CpG-free reporter vector, pCpGfree-Luc vector, was generated according to the literature (33). The MMP13 promoter constructs spanning −372/+14, −214/+14, and −86/+14 bp of the human MMP13 promoter sequence and containing seven, four, and one CpG sites, respectively, were generated by PCR amplification, utilizing genomic DNA from the human chondrocyte cell line C28/I2 (34) as a template and the following PCR primers: MMP13(−372/+14), 5′-CCGACTAGTGGAAGTAAACATGCCATCTTGATA-3′(forward); MMP13(−214/+14), 5′-CCGACTAGTATTTTGCCAGATGGGTTTTG-3′ (forward); MMP13(−86/+14), forward: 5′-CCGACTAGTCAAGTGACTAGGAAGTGGAAACC-3′; and reverse (common to all three MMP13 promoter constructs): 5′-CCGAAGCTTCCTGGGGACTGTTGTCTTT-3′. Underlined letters indicate SpeI and HindIII recognition sequences in the forward and reverse primers, respectively. The resultant PCR products were digested with SpeI and HindIII and transferred into the multiple cloning site of a pCpGfree-Luc vector treated with the same enzymes using TaKaRa DNA ligation kit (version 2.1; TaKaRa).
The human IL1B promoter constructs spanning −521/+56, −276/+56, and −183/+56 bp and containing four, three, and two CpG sites, respectively, were generated by PCR amplification utilizing the following PCR primers: IL1B(−521/+56), 5′-CCGACTAGTAGAGAGCTCCTGAGGCAGAG-3′ (forward); IL1B(−276/+56), 5′-CCGACTAGTTGTGGACATCAACTGCACAA-3′ (forward); IL1B(−183/+56), 5′-CCGACTAGTCCCTTCCATGAACCAGAGA-3′ (forward); and common to all three IL1B promoter constructs, 5′-CCGAGATCTGGCTGAAGAGAATCCCAGAG-3′ (reverse). Underlined letters indicate SpeI and BglII recognition sequences on the forward and reverse primers, respectively. The resultant PCR products were digested with SpeI and BglII and transferred into the multiple cloning site of the pCpGfree-Luc vector.
Point mutations of CpG sites were generated by converting CG to TG by a two-step PCR mutagenesis approach as described previously (35) using the primers shown in Table 2 and the wild type (WT) −214/+14 bp MMP13 (MMP13–214) or −521/+56 bp IL1B (IL1B-521) promoter constructs, linearized by double enzyme digestion by SacI and KpnI as templates. After two-step PCR, the resulting products were digested with SpeI/HindIII and SpeI/BglII for MMP13–214 and IL1B-521, respectively, and cloned into the pCpGfree-Luc vector backbone treated with the same enzymes using TaKaRa DNA ligation kit (version 2.1, TaKaRa). Promoter constructs with multiple CpG mutations were generated by a repeated two-step PCR mutagenesis, utilizing different PCR mutagenesis primers and linearized CpG-mutated MMP13–214 or IL1B-521 construct as template. Expression vectors encoding ELF3, HIF1α, HIF-2α, Sp1, RUNX2, NF-κB, and AP-1 (cFos/cJun) were purchased from Addgene and/or described elsewhere (35–37). The sequences of all constructs were confirmed by DNA sequencing at the Cornell University Life Sciences Core Laboratories Center.
TABLE 2.
Primer sequences for two-step promoter mutagenesis
| Name (location of mutated CpG)/part of amplicon direction | Sequence (5′ to 3′)a |
|---|---|
| MMP13–214-Mut1 (-14) | |
| 1st half (forward) | TATGTGAGCAAACAGCAGATTAAAA |
| 1st half (reverse) | TTGTCTTTCCaCAGAGATTACCTTT |
| 2nd half (forward) | AAAGGTAATCTCTGtGGAAAGACAA |
| 2nd half (reverse) | AGACATCTCAAAGTATTCAGCATAGGT |
| MMP13–214-Mut2 (−110) | |
| 1st half (forward)b | AATAAATCTCTTTGTTCAGCTCTCTGTTTC |
| 1st half (reverse) | CTTACaTGGCGACTTTTTCTTTTCCCT |
| 2nd half (forward) | AGGGAAAAGAAAAAGTCGCCAtGTAAG |
| 2nd half (reverse)c | AGACATCTCAAAGTATTCAGCATAGGTGAT |
| MMP13–214-Mut3 (−115) | |
| 1st half (forward)b | AATAAATCTCTTTGTTCAGCTCTCTGTTTC |
| 1st half (reverse) | CTTACGTGGCaACTTTTTCTTTTCCCT |
| 2nd half (forward) | AGGGAAAAGAAAAAGTtGCCACGTAAG |
| 2nd half (reverse)c | AGACATCTCAAAGTATTCAGCATAGGTGAT |
| MMP13–214-Mut4 (−135) | |
| 1st half (forward) | ACCCAATAAATAATAAATCTCTTTGTTCAGCTCT |
| 1st half (reverse) | TTCTTTTCCCTCCCaAGTGTGGTTT |
| 2nd half (forward) | AAACCACACTtGGGAGGGAAAAGAA |
| 2nd half (reverse) | GACATCTCAAAGTATTCAGCATAGGTGATG |
| IL1B-521-Mut1 (+13) | |
| 1st half (forward)d | TTGTTTATGTGAGCAAACAGCA |
| 1st half (reverse) | CCTTGTGCCTCaAAGAGGTT |
| 2nd half (forward) | AACCTCTTtGAGGCACAAGG |
| 2nd half (reverse)e | CCAGCCTCACAGACATCTCA |
| IL1B-521-Mut2 (−20) | |
| 1st half (forward)d | TTGTTTATGTGAGCAAACAGCA |
| 1st half (reverse) | CCCTCaCTGTTTTTATGGCTTT |
| 2nd half (forward) | AAAGCCATAAAAACAGtGAGGG |
| 2nd half (reverse)e | CCAGCCTCACAGACATCTCA |
| IL1B-521-Mut3 (−256) | |
| 1st half (forward) | TTTGTTTATGTGAGCAAACAGCAG |
| 1st half (reverse) | TTTTCCTGACAATCaTTGTGCAGT |
| 2nd half (forward) | ACTGCACAAtGATTGTCAGGAAAA |
| 2nd half (reverse) | TGCCATCTTCCAGAGGGTAG |
| IL1B-521-Mut4 (−299) | |
| 1st half (forward) | TATGTGAGCAAACAGCAGATTAAAAG |
| 1st half (reverse) | AATACTGGATTTTCCCACaTTAGAAG |
| 2nd half (forward) | CTTCTAAtGTGGGAAAATCCAGTATT |
| 2nd half (reverse) | ACTAAGCTTAAGATCTGGCTGAAGAG |
a Lowercase letters indicate a mutated base.
b Common primers.
c Common primers.
d Common primers.
e Common primers.
In Vitro Methylation, Transfections, and Luciferase Assay
The methylated plasmids were generated by incubating 1 μg of plasmid DNA with 4 units/μl of the CpG methyltransferase, M.SssI (Zymo Research) in reaction buffer supplemented with 1600 μm S-adenosylmethionine according to the manufacturer's instructions. Complete methylation was verified by plasmid DNA bisulfite modification and pyrosequencing using specific primers. The methylated plasmid DNA was purified by the PureLink® Quick Plasmid Miniprep kit (Invitrogen). The methylated and non-methylated vectors were then transfected in immortalized human chondrocytes (C28/I2) seeded 24 h prior to transfection in 24-well tissue culture plates at 3.0 × 104 cells/well, using Lipofectamine PLUSTM reagents as described (35). Briefly, transfections were carried out in triplicate in serum-free antibiotic-free conditions, with a total of no more than 475 ng of plasmid DNA per well, including 300 ng of non-methylated or methylated MMP13 or IL1B luciferase reporter construct and the expression vectors. The ratios of luciferase reporter construct to expression vector were established by titrations to ensure that concentrations of expressed transcription factors were not limiting to the promoter transactivations under the conditions of the co-transfections. Reporter constructs were treated with DNA methyltransferase to methylate all available CpG sites in the promoters. Luciferase assays were performed using the luciferase assay system (Promega) and detected using a LMaxII384 Luminometer system (Molecular Devices). Unless otherwise specified, the firefly luciferase activities were normalized to the Renilla activity of pGL4.74-hRluc-TK (Promega).
ChIP Assay
The ChIP-IT Express Enzymatic Kit (ActiveMotif) was used to perform ChIP assays according to the manufacturer's instructions with minor modifications, as described (35), using C28/I2 chondrocytic cells co-transfected with non-methylated or methylated pCpGfree-Luc-MMP13 vector (−214/+14bp) and expression vectors encoding HA-tagged HIF-2α or FLAG-tagged ELF3. At 24 h after transfection, precleared chromatin was stored as assay input or incubated at 4 °C overnight with 1.25 μg of rabbit anti-HA antibody (Invitrogen), rabbit anti-FLAG antibody (Sigma), or normal rabbit IgG (Cell Signaling). After reverse cross-linking and purification on DNA mini-columns (Qiagen), the final DNA preparations were subjected to quantitative PCR analysis using 5 μl of the eluted DNA and the following primers specific for the transiently transfected MMP13 reporter construct: 5′-TCAAATTCTACCACAAACCACA-3′ (forward) and 5′-TAATATTCTTGGCATCCTCCAT-3′ (reverse). Primer efficiency was calculated utilizing serial dilutions of the pooled input DNA samples. For real time PCR analysis, the CT of each sample was normalized to the CT of the input sample (10%).
Statistical Analysis
Each data point is reported as the means ± S.E. of at least three independent experiments. Statistical significance was evaluated using Student's t test, with p values of < 0.05 considered significant.
RESULTS
Specific MMP13 and IL1B Proximal Promoter CpG Sites Are Demethylated in OA Chondrocytes and Correlate with Higher Levels of Gene Expression in OA Disease
We initially quantified the CpG methylation status of the −343/−14 bp MMP13 and −299/+13 bp IL1B proximal promoter sequences in human primary chondrocytes isolated from articular cartilage obtained from non-OA donors (n = 12) and patients with OA (n = 17). Pyrosequencing analysis of the MMP13 promoter revealed that five of the seven CpG sites contained in the proximal promoter region were significantly demethylated in OA compared with non-OA chondrocytes. The highest degree of demethylation was observed at the −110-bp CpG site (Fig. 1A). The IL1B promoter was also significantly demethylated at the −299-bp CpG and −256-bp CpG sites in OA compared with non-OA chondrocytes, whereas there was no significant difference in methylation status at the −20-bp CpG and +13-bp CpG sites (Fig. 1B). Finally, the decreased methylation of the MMP13 (Fig. 1C) and IL1B (Fig. 1D) proximal promoters correlated with higher levels of their mRNAs in OA chondrocytes (Fig. 1C).
FIGURE 1.

Demethylation of specific MMP13 and IL1B proximal promoter CpG sites in OA chondrocytes correlates with higher levels of gene expression in OA disease. Bisulfite pyrosequencing analysis was performed to determine the relative methylation status of the indicated CpG sites on the (A) MMP13 or (B) IL1B proximal promoter sequences in healthy (fractured neck of femur (NOF; n = 12) and OA (n = 17) human chondrocytes isolated directly from cartilage. Data are represented as percentage of methylation in OA samples relative to that in non-OA specimens set at 100% for each CpG site. MMP13 (C) or IL1B (D) mRNA was analyzed by quantitative RT-PCR and normalized against GAPDH. All of the data are shown as means ± S.E. (*, p < 0.05; **, p < 0.01; ***, p < 0.001).
CpG Demethylation in Vitro Correlates with Enhanced Levels of MMP13 and IL1B Gene Expression in Cultured Human Primary Chondrocytes
To determine the functional effects of demethylating the CpG sites in the MMP13 and IL1B promoters, we cultured human primary chondrocytes in the presence or absence of 5-aza-dC, a CpG demethylating agent, as described (22). Treatment with 5-aza-dC resulted in significant demethylation of the CpG sites in the MMP13 promoter at −343 bp, −115 bp, −110 bp, and −14 bp (Fig. 2A). In the IL1B promoter, significant loss of methylation occurred at the −299 bp CpG after 5-aza-dC treatment (Fig. 2B). In accordance with these results, we had reported previously that MMP13 and IL1B mRNA levels increased by 2- and 5-fold, respectively, in response to treatment with 5-aza-dC (22, 38).
FIGURE 2.

CpG demethylation in vitro correlates with enhanced levels of MMP13 and IL1B gene expression in cultured human primary chondrocytes. Primary human chondrocytes were cultured without or with 5-aza-dC and the CpG methylation status of the MMP13 proximal promoter (A) or the IL1B proximal promoter (B) was analyzed. All data are shown as means ± S.E. and represented as percentage of methylation in the 5-aza-dC-treated cells relative to that in the non-treated control cells set at 100% for each CpG site (*, p < 0.05).
Methylation of Unique CpG Sites in the Proximal Human MMP13 and IL1B Gene Promoters Directly Affects Transcriptional Activities
We next investigated the contribution of each of the methylation-sensitive CpG sites to MMP13 and IL1B promoter activities using a CpG-free reporter system. The activities of deletion constructs of the MMP13 promoter, spanning −372/+14, −214/+14, and −86/+14 bp, were significantly reduced in their methylated state (Fig. 3A). Next, we generated mutant constructs of the −214/+14-bp MMP13 promoter harboring site-specific CpG mutation(s), replacing unique cytosine (C) with thymine (T) residues, as indicated in Fig. 3B. A single CpG mutation at −110 bp (−110 mt) significantly decreased promoter activity compared with the WT promoter; in contrast, no other individual CpG mutant caused a significant change in MMP13 promoter activity. In addition, methylation significantly suppressed the activities of all single CpG mutants except for the −110 mt and −14 mt (Fig. 3B). Moreover, a triple CpG mutant construct (−135/−115/−14 mt), which preserves the −110 bp CpG site, retained promoter activity comparable with WT; all other triple mutants with a mutated −110 bp CpG site showed significantly decreased promoter activity (Fig. 3C). Importantly, CpG methylation treatment of the −135/−115/−14-mt construct significantly decreased reporter activity. These results indicate that the methylation status of the −110-bp CpG is crucial for MMP13 promoter activity. In contrast, activity of the methylated −135/−110/−14-mt construct, which only preserves the −115-bp CpG site, increased by 3.7-fold (Fig. 3C).
FIGURE 3.

Methylation of unique CpG sites in the proximal human MMP13 and IL1B gene promoters directly affects transcriptional activity. Basal activities of MMP13 and IL1B promoter constructs with deletions from 5′ ends or specific CpG mutation(s) were analyzed with (Meth+, closed bars) or without (Meth−, open bars) CpG methylation treatment in luciferase assays performed in C28/I2 chondrocytes. MMP13 promoter activity was analyzed in cells transfected with WT −372/+14, −214/+14, and −86/+14-bp constructs (A), −214/+14-bp constructs containing single CpG mutations (B), and −214/+14-bp promoter constructs containing triple CpG mutations (C), leaving only one intact CpG site. IL1B promoter activity was analyzed in cells transfected with −521/+56, −276/+56, and −183/+56-bp constructs (D), −521/+56-bp constructs with single CpG mutations (E), and −521/+56-bp constructs with triple-CpG mutations (F), leaving only one intact CpG site. All data are shown as means ± S.E. (*, p < 0.05; **, p < 0.01; ***, p < 0.001). Luc, luciferase. N.S., not significant.
We also generated three IL1B promoter constructs. Methylation treatment significantly decreased the activities of the WT −521/+56 and −276/+56 reporter constructs, with no significant effect on the −183/+56-bp promoter (Fig. 3D). Single and triple CpG mutant constructs of the −521/+56 WT IL1B promoter revealed that a single CpG mutation at −299 bp (−299 mt) significantly decreased promoter activity, whereas the −256-bp CpG mutant was significantly more active than WT; mutations of the other two CpG sites had no effect on promoter activity (Fig. 3E). Among the triple CpG mutants with only one intact CpG site, the basal activity of the −256/−20/+13-mt promoter did not change, whereas the basal activities of all constructs containing a mutated −299-bp CpG decreased significantly (Fig. 3F). Furthermore, methylation treatment significantly decreased the activity of the −256/−20/+13 mutant (Fig. 3F). These results indicate that, similar to the −110-bp CpG site in the MMP13 promoter, the methylation status of one CpG at −299 bp in the IL1B promoter uniquely impacts on its transcriptional activity.
CpG Methylation Impairs HIF-2α-driven MMP13 Promoter Transactivation but Not IL1B Promoter Activation
CpG methylation may regulate gene transcriptional activities by differentially and directly modulating the binding affinities of trans-acting factors (6). Hence, we next investigated the effects of CpG methylation status on the actions of MMP13 trans-acting factors selected based on previous reports (35, 39–44) and on bioinformatic analyses using the Transcription Element Search System (Fig. 4A). In agreement with prior work (43, 44), HIF-2α overexpression increased the activity of the nonmethylated WT −214/+14-bp promoter construct; interestingly, CpG methylation significantly attenuated this HIF-2α-mediated promoter activation (Fig. 4B). In addition, HIF-2α overexpression did not significantly activate the nonmethylated −110-mt construct (Fig. 4B), whereas HIF-2α transactivated the non methylated triple mutant with an intact −110-bp CpG to an extent comparable with the WT construct. All other triple mutant constructs showed decreased HIF-2α-driven transactivation, compared with the WT reporter construct (Fig. 4C). Methylation treatment of the triple mutant with an intact −110-bp CpG significantly decreased MMP13 promoter activity in response to HIF-2α overexpression (Fig. 4C). Together, these results indicate that targeted −110-bp CpG methylation impairs HIF-2α transactivation of the MMP13 promoter in vitro.
FIGURE 4.

CpG methylation impairs the HIF-2α-driven MMP13 promoter transactivation. Shown is the schematic representation of the proximal human MMP13 (42) (A) and IL1B (D) promoter sequences. Putative binding sites for trans-acting factors are underlined. Methylated (Meth+; closed bars) or non-methylated (Meth−; open bars) MMP13 promoter constructs with single CpG mutations (B) or triple CpG mutations (C) were co-transfected with the empty control vector (pcDNA3) or with the HIF-2α expression vector. Methylated (Meth+; closed bars) or non-methylated (Meth−; open bars) IL1B promoter constructs with single CpG mutations (E) or triple CpG mutations (F) were co-transfected with the empty control vector (pcDNA3) or with the HIF-2α expression vector. All data are shown as means ± S.E. Luc, luciferase.
To assess the specificity of the above effects of HIF-2α, we interrogated the dependence of activities of multiple transcription factors on CpG site-specific methylation of the MMP13 promoter. Overexpression of RUNX2, ELF3, or AP-1 (c-Fos/c-Jun) activated the MMP13 promoter constructs as shown previously (35, 42); but in contrast to our results with HIF-2α, these transactivations were unaffected by any CpG site mutation or by alteration of methylation status (data not shown). Moreover, in contrast to HIF-2α, HIF-1α had no significant effect when co-transfected with any MMP13 promoter construct in the absence or presence of methylation (data not shown). Similarly, overexpressing Sp1, USF-1, or GATA-1 had no effect on MMP13 promoter activity and did not correlate with mutation or methylation of any specific CpG site (data not shown).
We also investigated the effects of CpG methylation on the actions of candidate IL1B trans-acting factors (Fig. 4D). HIF-2α overexpression had no significant effect on IL1B promoter activity (Fig. 4, E and F). In contrast, HIF-1α or NF-κB (p65/p50) overexpression significantly increased IL1B promoter activity, but this enhancement was independent of mutation of any CpG site or alteration of CpG methylation status (data not shown). Furthermore, overexpression of USF-1 or the combination of Single-minded (Sim) 2 and aryl hydrocarbon receptor nuclear translocator (ARNT) significantly repressed IL1B promoter activity, but the latter responses were not significantly changed by the methylation status of the promoter or by targeted mutation of any CpG site (data not shown).
CpG Methylation Attenuates HIF-2α Binding to the MMP13 Promoter
Given the results above, we investigated whether CpG methylation status directly affected HIF-2α binding to the proximal MMP13 promoter in ChIP assays. In the context of endogenous chromatin, methylating/demethylating agents would impact transcription affecting not only in the proximal promoter region analyzed in our study but also distal sequences, therefore introducing further complexity to the analysis. Hence, to avoid confounding effects of CpG sites in distal MMP13 promoter regions and their contributions to overall endogenous chromatin structure, ChIP assays were performed in chondrocytes transfected with methylated and nonmethylated WT −214/+14-bp MMP13 constructs in combination with expression vectors encoding HA-tagged HIF-2α or FLAG-tagged ELF3; and MMP13 promoter binding was analyzed with specific PCR primers that only recognize the transiently transfected promoter constructs, as described in the “Experimental Procedures.” ChIP assays revealed that methylation treatment significantly reduced HIF-2α binding to the MMP13 proximal promoter (Fig. 5A), in agreement with our results showing decreased HIF-2α-driven MMP13 transactivation after methylation treatment of the WT and/or mutant MMP13 reporter constructs (Fig. 4, B and C). In contrast, as a control, ELF3 binding to the proximal MMP13 promoter was not affected by promoter methylation status (Fig. 5B), in accordance with our results showing that ELF3-driven MMP13 transactivation is methylation-insensitive, as indicated above. Our results show that CpG methylation of the MMP13 proximal promoter specifically impairs HIF-2α-driven promoter activation by altering its binding to DNA.
FIGURE 5.
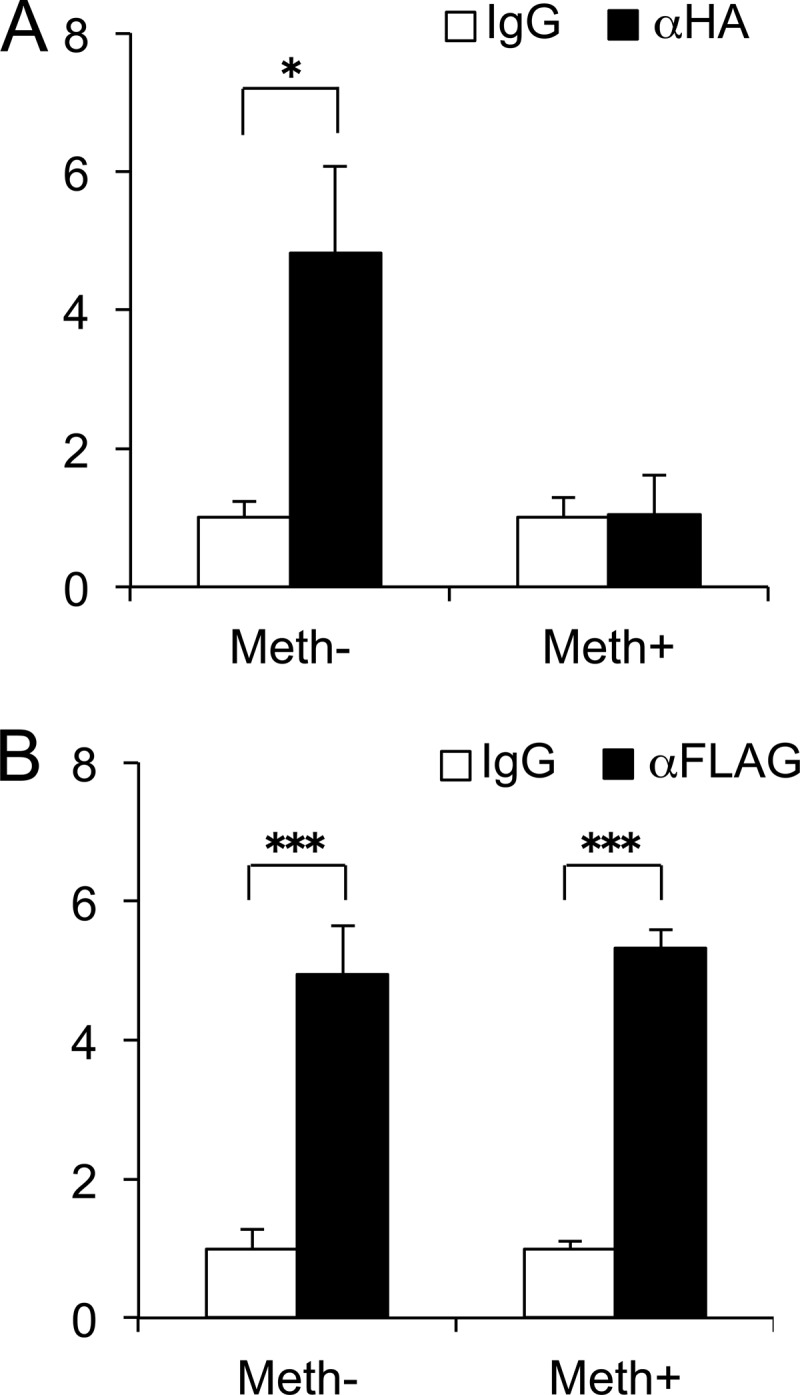
CpG methylation attenuates HIF-2α binding to the MMP13 promoter. ChIP assays were performed using cell lysates of C28/I2 cells transfected with methylated (Meth−) or non-methylated (Meth+) WT −214/+14-bp MMP13 promoter constructs and expression vectors encoding HA-tagged HIF-2α (A) or FLAG-tagged ELF3 (B). At 24 h after transfection, chromatin was cross-linked and enzymatically sheared, and after reverse cross-linking of the DNA-protein complexes, the precleared lysates were incubated with antibodies against HA, FLAG, or normal IgG overnight at 4 °C. The HA-HIF-2α and FLAG-ELF3 binding to the human MMP13 promoter region was analyzed by real time PCR, utilizing PCR primers bracketing the transfected reporter vector boundaries. All data are shown as means ± S.E. and represented as fold-change versus IgG (*, p < 0.05; ***, p < 0.001).
DISCUSSION
MMP-13, or collagenase 3, is the major type II collagen-degrading enzyme that drives erosion of the cartilage collagen network in OA disease (45–48). This enzyme plays essential roles in extracellular matrix remodeling in the late hypertrophic zone of the growth plate prior to endochondral ossification, but articular chondrocytes express low basal MMP-13 levels under normal, physiological conditions in vivo (49). High levels and activity of MMP-13 and other matrix-degrading enzymes in OA chondrocytes are due in part to their activated state in response to enhanced stress and inflammatory signaling (reviewed in Ref. 39). MMP13 transcription is subject to regulation by a number of transcription factors, including ELF3, RUNX2, AP-1 (cFos/cJun), p130cas nuclear matrix transcription factor 4 (NMP-4), the canonical NF-κB subunits p65/p50, and HIF-2α (35, 40–44, 50). NF-κB also plays a major role in increased IL1B expression in various cell types under stress/proinflammatory conditions by directly activating the IL1B promoter (51, 52). Moreover, as shown previously, NF-κB modulates gene expression in some contexts via epigenetic mechanisms, affecting the level of CpG demethylation (53).
Our previous studies showed epigenetic changes associated with abnormal MMP13 and IL1B gene expression in chondrocytes of OA cartilage (25) and in long term cytokine-treated human primary chondrocyte cultures (22). Although other studies have also described associations between CpG methylation status and deregulation of OA-associated genes (23, 24, 54), this does not appear to be the case for all genes in human chondrocytes (26, 55). However, most previous studies of OA-associated genes have investigated their CpG islands, ignoring the fact that 5′-UTR with non-island CpG sites may also impact functionally on gene regulation by alterations in CpG methylation status (56). Indeed, there is a paucity of studies that have functionally analyzed alterations in promoter activity induced by the methylation of a single CpG site (6, 10, 11). In our study, the use of a CpG-free luciferase reporter gene system (33) facilitated the investigation of the direct cause-and-effect relationships between CpG methylation and altered promoter activities of specific genes, and the dissection of the relative contribution of individual CpG sites to the transcriptional control of a given gene.
Here, we have shown that methylation of unique CpG sites in the proximal MMP13 and IL1B promoters regulates their transcriptional activities in human chondrocytes via different mechanisms (Fig. 6). We initially quantified the methylation status of the promoters of the human genes in chondrocytes isolated from normal and OA articular cartilage and found that the −110-bp CpG and −299-bp CpG in the proximal MMP13 and IL1B promoters, respectively, were most subject to demethylation. Interestingly, 5-aza-dC treatment of human primary chondrocyte cultures induced demethylation of the same CpG sites in MMP13 and IL1B promoters, accompanied with increased expression of both genes. Although the possible involvement of other pathways in promoter-activating effects of 5-aza-dC cannot be excluded, these findings indicate that each CpG site has a different sensitivity to demethylation and that demethylation of specific CpG sites has the potential to activate or derepress MMP13 and IL1B gene expression.
FIGURE 6.

Scheme of CpG sites on the proximal MMP13 and IL1B promoters. The CpG sites are indicated by open ovals. The positive (+) and negative (−) transcription factors that bind to these promoters are indicated with arrows above and below, respectively. In co-transfection experiments, only HIF-2α-driven MMP13 promoter activity was modulated by CpG methylation status and methylation attenuated HIF-2α binding to the −110 CpG in ChIP assays. On the IL1B promoter, HIF-1α and NF-κB, but not HIF-2α, enhanced transcriptional activity, but there was no relationship with CpG methylation status for activities of the transcription factors studied.
We directly investigated the cause-and-effect relationship between the methylation of these specific CpG sites and the transcriptional activities of MMP13 and IL1B promoters by comparative analysis of their non-methylated and methylated forms. The activities of WT MMP13 and IL1B promoters with methylated CpG sites were reduced compared with their non-methylated counterparts; this was true even for promoter constructs containing longer 5′-flanking promoter sequences, indicating that the methylation of sparse CpG sites in the proximal promoters of these two genes can have a direct suppressive effect on transcriptional activity. In addition, analyses of single and triple CpG mutants within the proximal MMP13 and IL1B promoters further clarified that their respective −110-bp CpG and −299-bp CpG sites have key roles in the methylation-dependent regulation of these two genes in human chondrocytes. Importantly, the latter specific CpG sites correspond to the most demethylated CpG sites in OA chondrocytes ex vivo and in 5-aza-dC-treated primary chondrocytes in vitro.
The −110-bp CpG and −299-bp CpG sites in the proximal promoters of the human MMP13 and IL1B genes contain putative ACGT hypoxia-responsive element consensus core sequences, which in some contexts respond to both HIF-1α and HIF-2α (57–61). HIF-2α, but not HIF-1α, controls MMP-13 expression in chondrocytes in vitro and in vivo and regulates both endochondral ossification processes and cartilage destruction in OA disease (43, 44). HIF-2α, the gene product of EPAS1, is an IL-1β-inducible member of the basic helix-loop-helix Per-ARNT-Sim (bHLH-PAS) family of transcription factors that directly targets the hypoxia-responsive elements within the COL10A1, MMP13, and VEGFA promoters in chondrocytic ATDC5 cells and in HeLa cells (43). HIF-2α binding to the endogenous MMP13 promoter containing the proximal hypoxia-responsive element has been demonstrated previously by ChIP in vivo and by EMSA in vitro (43). In addition, HIF-2α-mediated MMP13 transcriptional control has been established both in vitro and in vivo (43, 44). Moreover, enforced expression of HIF-2α in vivo has been shown to result in enhanced cartilage damage in association with enhanced MMP-13 expression. Furthermore, Epas1+/− mice, which are haploinsufficient in HIF-2α, have reduced levels of MMP13 mRNA and are resistant to surgically or chemically induced knee OA disease (44).
Here, we have shown that enforced HIF-2α expression transactivates a nonmethylated WT MMP13 promoter construct and that its transactivating effect is reduced by CpG methylation. Moreover, HIF-2α-driven MMP13 activation is solely dependent on the presence of the −110-bp CpG site in the proximal promoter of the gene and targeted methylation of the −110-bp CpG also diminishes MMP13 promoter transactivation by HIF-2α, suggesting that demethylation of this specific CpG site is essential for HIF-2α-mediated MMP13 promoter activation and contributes to increased MMP13 expression in OA chondrocytes. Consistent with our finding that CpG methylation treatment suppresses HIF-2α-driven activation of the MMP13 promoter, ChIP assays performed with cells co-transfected with HIF-2α and a MMP13 promoter construct with a methylation-resistant reporter gene revealed that targeted methylation of the MMP13 proximal promoter significantly decreased HIF-2α binding affinity.
In addition to our observations, Bui et al. (10) recently reported that the same −110-bp CpG site (positioned at −104 in their report, based on the selected TSS) affects MMP13 transactivation via basal and IL1-β-induced CREB recruitment in vitro. The latter observations, taken together with our results demonstrating that HIF-2α-driven MMP13 control depends upon the methylation status of the −110-bp CpG site, highlight the multifaceted importance of the methylation status of this specific CpG site in the control of MMP13 transcription by different trans-acting factors responding to diverse disease-associated environmental cues and thereby revealing its potentially important role in the dysregulation of MMP13 expression observed in OA cartilage.
In contrast to the MMP13 promoter, HIF-2α had no activating effect on the IL1B promoter, even though the critical −299-bp CpG for IL1B promoter activity is located within a putative hypoxia-responsive element core sequence (Fig. 4D), a non-canonical “E-box-like” sequence (CANNTG), to which several bHLH-PAS family transcription factors can bind. A heterodimer of two bHLH-PAS family members, Sim2 and ARNT, binds to non-canonical E-boxes, repressing (62, 63) or activating (64) transcription depending upon the promoter and cellular contexts. However, overexpression of Sim2 and ARNT repressed IL1B promoter activity independent of CpG methylation status. Although we found that the −299-bp CpG is essential for IL1B promoter activity and that its demethylation correlates with enhanced IL1B gene expression in human primary chondrocytes ex vivo and in vitro, mutation of this site does not attenuate IL1B promoter transactivation by NF-κB. Early studies in myeloid cell lines demonstrated that two other functional NF-κB sites, which exist upstream in the IL1B promoter, were influenced by CREB, whereas mutation of the −299-bp NF-κB site attenuated transactivation by C/EBPβ but not by CREB (51, 52). Whether methylation at −299-bp CpG directly affects the binding and activities of other bHLH family members or it modulates IL1B transcription via other indirect mechanisms merits future investigation.
Our finding that methylation of the −115-bp CpG site enhances MMP13 promoter activity (Fig. 3C), as opposed to the inhibitory effect of −110-bp CpG methylation, suggests alternative mechanisms, as reported for half-Cre sequences, where CpG methylation creates C/EBPα binding sites and activates gene expression (6). The −115-bp CpG overlaps a GATA-1 binding sequence, although the inhibitory effect of GATA-1 overexpression on MMP13 promoter activity was not altered by methylation status (data not shown). Further analysis will be required to elucidate the role of the −115-bp CpG site in methylation-dependent MMP13 promoter activation.
Interestingly, treatment with 5-aza-dC in vitro produced a more marked decrease in the methylation of CpG sites in the proximal MMP13 and IL1B promoters than that observed ex vivo comparing OA and non-OA articular chondrocytes. However, whereas OA chondrocytes showed 25-fold and 800-fold MMP13 and IL1B gene expression compared with non-OA chondrocytes, the MMP13 and IL-1β expression levels were only modestly elevated by the more pronounced 5-aza-dC-induced demethylation in vitro. Moreover, in reporter assays, differences in promoter activities with or without methylation were also modest even when the methylation status of a specific CpG was all or none in the reporter constructs. Taken together, our observations suggest that even a small change in CpG methylation status may have a significant impact on gene expression in vivo, where CpG methylation is likely to be a contributing factor not only by altering the binding of trans-acting factors, but also by acting together with histone-modifying, chromatin-remodeling activities and other transcriptional regulatory elements. Indeed, 5-aza-dC treatment of primary cultures does not necessarily reflect the more complex in vivo physiological or pathological condition, where more prolonged exposure to different stimuli may induce not only differential methylation patterns but also other epigenetic changes that may be absent in our chondrocyte culture systems and reporter assays. Furthermore, analysis of the cell populations in vivo/ex vivo is limited in that cell-to-cell variability is not taken into account, whereas in vitro studies are performed with synchronized cells. Hence, limitations in current methylation and epigenomic mechanistic studies stem from the lack of technology and models that more precisely reflect the in vivo scenario. Techniques that allow for specific modulation of CpG methylation patterns in vivo are required, therefore, for a better understanding of the precise contributions of this means of transcriptional control (65, 66).
In summary, we have investigated the relative contributions of the methylation status of individual CpG sites in the MMP13 and IL1B proximal promoters to modulation of their transcriptional activities and found that methylation of unique CpG sites has promoter-specific effects. Moreover, we show that the DNA binding and transactivation activity of HIF-2α, a key transcription factor in MMP13 gene regulation, is prevented by methylating only one unique CpG site in the proximal MMP13 promoter. Although the use of epigenetic inhibitors as cancer therapies is closer to reality than their use in musculoskeletal diseases (67), specific targeting of DNA methylation may eventually represent a potential therapeutic strategy to prevent or delay OA disease progression by suppressing the activities of catabolic genes causing cartilage matrix destruction. Our findings may also suggest the utility of methylation marks as advantageous early diagnostic markers of OA disease.
Acknowledgment
We are grateful to Prof. Nicholas Clarke (University of Southampton, Southampton, UK) for providing human cartilage specimens.
This work was supported, in whole or part, by National Institutes of Health Grants R21-AR054887, R01-AG022021, and RC4-AR060546 (to M. B. G.), an Arthritis Foundation Postdoctoral Fellowship (to M. O.), and Biotechnology and Biological Sciences Research Council (BBSRC) Grant sLOLA BB/GO10579/1 (to R.O.C.O.).
- OA
- osteoarthritis
- MMP
- matrix metalloproteinase
- HIF-2α
- hypoxia-inducible factor-2α
- EPAS1
- endothelial PAS domain protein 1
- 5-aza-dC
- 5-aza-deoxycytidine
- bHLH-PAS
- basic helix-loop-helix Per-ARNT-Sim
- mt
- mutant.
REFERENCES
- 1. Berger S. L. (2007) The complex language of chromatin regulation during transcription. Nature 447, 407–412 [DOI] [PubMed] [Google Scholar]
- 2. Razin A. (1998) CpG methylation, chromatin structure and gene silencing-a three-way connection. EMBO J. 17, 4905–4908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Irvine R. A., Lin I. G., Hsieh C. L. (2002) DNA methylation has a local effect on transcription and histone acetylation. Mol. Cell Biol. 22, 6689–6696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Nan X., Ng H. H., Johnson C. A., Laherty C. D., Turner B. M., Eisenman R. N., Bird A. (1998) Transcriptional repression by the methyl-CpG-binding protein MeCP2 involves a histone deacetylase complex. Nature 393, 386–389 [DOI] [PubMed] [Google Scholar]
- 5. Okitsu C. Y., Hsieh C. L. (2007) DNA methylation dictates histone H3K4 methylation. Mol. Cell Biol. 27, 2746–2757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Rishi V., Bhattacharya P., Chatterjee R., Rozenberg J., Zhao J., Glass K., Fitzgerald P., Vinson C. (2010) CpG methylation of half-CRE sequences creates C/EBPalpha binding sites that activate some tissue-specific genes. Proc. Natl. Acad. Sci. U.S.A. 107, 20311–20316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wade P. A., Gegonne A., Jones P. L., Ballestar E., Aubry F., Wolffe A. P. (1999) Mi-2 complex couples DNA methylation to chromatin remodelling and histone deacetylation. Nat. Genet. 23, 62–66 [DOI] [PubMed] [Google Scholar]
- 8. Bird A. (2002) DNA methylation patterns and epigenetic memory. Genes Dev. 16, 6–21 [DOI] [PubMed] [Google Scholar]
- 9. Reik W. (2007) Stability and flexibility of epigenetic gene regulation in mammalian development. Nature 447, 425–432 [DOI] [PubMed] [Google Scholar]
- 10. Bui C., Barter M. J., Scott J. L., Xu Y., Galler M., Reynard L. N., Rowan A. D., Young D. A. (2012) cAMP response element-binding (CREB) recruitment following a specific CpG demethylation leads to the elevated expression of the matrix metalloproteinase 13 in human articular chondrocytes and osteoarthritis. FASEB J. 26, 3000–3011 [DOI] [PubMed] [Google Scholar]
- 11. Murayama A., Sakura K., Nakama M., Yasuzawa-Tanaka K., Fujita E., Tateishi Y., Wang Y., Ushijima T., Baba T., Shibuya K., Shibuya A., Kawabe Y., Yanagisawa J. (2006) A specific CpG site demethylation in the human interleukin 2 gene promoter is an epigenetic memory. EMBO J. 25, 1081–1092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ballestar E. (2011) Epigenetic alterations in autoimmune rheumatic diseases. Nat. Rev. Rheumatol. 7, 263–271 [DOI] [PubMed] [Google Scholar]
- 13. Grabiec A. M., Krausz S., de Jager W., Burakowski T., Groot D., Sanders M. E., Prakken B. J., Maslinski W., Eldering E., Tak P. P., Reedquist K. A. (2010) Histone deacetylase inhibitors suppress inflammatory activation of rheumatoid arthritis patient synovial macrophages and tissue. J. Immunol. 184, 2718–2728 [DOI] [PubMed] [Google Scholar]
- 14. Horiuchi M., Morinobu A., Chin T., Sakai Y., Kurosaka M., Kumagai S. (2009) Expression and function of histone deacetylases in rheumatoid arthritis synovial fibroblasts. J. Rheumatol. 36, 1580–1589 [DOI] [PubMed] [Google Scholar]
- 15. Huber L. C., Brock M., Hemmatazad H., Giger O. T., Moritz F., Trenkmann M., Distler J. H., Gay R. E., Kolling C., Moch H., Michel B. A., Gay S., Distler O., Jüngel A. (2007) Histone deacetylase/acetylase activity in total synovial tissue derived from rheumatoid arthritis and osteoarthritis patients. Arthritis. Rheum. 56, 1087–1093 [DOI] [PubMed] [Google Scholar]
- 16. Kawabata T., Nishida K., Takasugi K., Ogawa H., Sada K., Kadota Y., Inagaki J., Hirohata S., Ninomiya Y., Makino H. (2010) Increased activity and expression of histone deacetylase 1 in relation to tumor necrosis factor-α in synovial tissue of rheumatoid arthritis. Arthritis. Res. Ther. 12, R133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Maciejewska-Rodrigues H., Karouzakis E., Strietholt S., Hemmatazad H., Neidhart M., Ospelt C., Gay R. E., Michel B. A., Pap T., Gay S., Jüngel A. (2010) Epigenetics and rheumatoid arthritis: the role of SENP1 in the regulation of MMP-1 expression. J. Autoimmun 35, 15–22 [DOI] [PubMed] [Google Scholar]
- 18. Neidhart M., Rethage J., Kuchen S., Künzler P., Crowl R. M., Billingham M. E., Gay R. E., Gay S. (2000) Retrotransposable L1 elements expressed in rheumatoid arthritis synovial tissue: association with genomic DNA hypomethylation and influence on gene expression. Arthritis. Rheum. 43, 2634–2647 [DOI] [PubMed] [Google Scholar]
- 19. Strietholt S., Maurer B., Peters M. A., Pap T., Gay S. (2008) Epigenetic modifications in rheumatoid arthritis. Arthritis. Res. Ther. 10, 219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Cheung K. S., Hashimoto K., Yamada N., Roach H. I. (2009) Expression of ADAMTS-4 by chondrocytes in the surface zone of human osteoarthritic cartilage is regulated by epigenetic DNA de-methylation. Rheumatol. Int. 29, 525–534 [DOI] [PubMed] [Google Scholar]
- 21. da Silva M. A., Yamada N., Clarke N. M., Roach H. I. (2009) Cellular and epigenetic features of a young healthy and a young osteoarthritic cartilage compared with aged control and OA cartilage. J. Orthop. Res. 27, 593–601 [DOI] [PubMed] [Google Scholar]
- 22. Hashimoto K., Oreffo R. O., Gibson M. B., Goldring M. B., Roach H. I. (2009) DNA demethylation at specific CpG sites in the IL1B promoter in response to inflammatory cytokines in human articular chondrocytes. Arthritis. Rheum. 60, 3303–3313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Loeser R. F., Im H. J., Richardson B., Lu Q., Chubinskaya S. (2009) Methylation of the OP-1 promoter: potential role in the age-related decline in OP-1 expression in cartilage. Osteoarthritis Cartilage 17, 513–517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Reynard L. N., Bui C., Canty-Laird E. G., Young D. A., Loughlin J. (2011) Expression of the osteoarthritis-associated gene GDF5 is modulated epigenetically by DNA methylation. Hum. Mol. Genet. 20, 3450–3460 [DOI] [PubMed] [Google Scholar]
- 25. Roach H. I., Yamada N., Cheung K. S., Tilley S., Clarke N. M., Oreffo R. O., Kokubun S., Bronner F. (2005) Association between the abnormal expression of matrix-degrading enzymes by human osteoarthritic chondrocytes and demethylation of specific CpG sites in the promoter regions. Arthritis. Rheum. 52, 3110–3124 [DOI] [PubMed] [Google Scholar]
- 26. Sesselmann S., Söder S., Voigt R., Haag J., Grogan S. P., Aigner T. (2009) DNA methylation is not responsible for p21WAF1/CIP1 down-regulation in osteoarthritic chondrocytes. Osteoarthritis Cartilage 17, 507–512 [DOI] [PubMed] [Google Scholar]
- 27. Zimmermann P., Boeuf S., Dickhut A., Boehmer S., Olek S., Richter W. (2008) Correlation of COL10A1 induction during chondrogenesis of mesenchymal stem cells with demethylation of two CpG sites in the COL10A1 promoter. Arthritis. Rheum. 58, 2743–2753 [DOI] [PubMed] [Google Scholar]
- 28. Goldring M. B., Marcu K. B. (2012) Epigenomic and microRNA-mediated regulation in cartilage development, homeostasis, and osteoarthritis. Trends Mol. Med. 18, 109–118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Barter M. J., Bui C., Young D. A. (2012) Epigenetic mechanisms in cartilage and osteoarthritis: DNA methylation, histone modifications and microRNAs. Osteoarthritis Cartilage 20, 339–349 [DOI] [PubMed] [Google Scholar]
- 30. Aigner T., Sachse A., Gebhard P. M., Roach H. I. (2006) Osteoarthritis: pathobiology-targets and ways for therapeutic intervention. Adv. Drug. Deliv. Rev. 58, 128–149 [DOI] [PubMed] [Google Scholar]
- 31. Goldring M. B., Goldring S. R. (2007) Osteoarthritis. J. Cell Physiol. 213, 626–634 [DOI] [PubMed] [Google Scholar]
- 32. Roach H. I., Aigner T. (2007) DNA methylation in osteoarthritic chondrocytes: a new molecular target. Osteoarthritis Cartilage 15, 128–137 [DOI] [PubMed] [Google Scholar]
- 33. Klug M., Rehli M. (2006) Functional analysis of promoter CpG methylation using a CpG-free luciferase reporter vector. Epigenetics 1, 127–130 [DOI] [PubMed] [Google Scholar]
- 34. Goldring M. B., Birkhead J. R., Suen L. F., Yamin R., Mizuno S., Glowacki J., Arbiser J. L., Apperley J. F. (1994) Interleukin-1 β-modulated gene expression in immortalized human chondrocytes. J. Clin. Invest. 94, 2307–2316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Otero M., Plumb D. A., Tsuchimochi K., Dragomir C. L., Hashimoto K., Peng H., Olivotto E., Bevilacqua M., Tan L., Yang Z., Zhan Y., Oettgen P., Li Y., Marcu K. B., Goldring M. B. (2012) E74-like factor 3 (ELF3) impacts on matrix metalloproteinase 13 (MMP13) transcriptional control in articular chondrocytes under pro-inflammatory stress. J. Biol. Chem. 287, 3559–3572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ijiri K., Zerbini L. F., Peng H., Correa R. G., Lu B., Walsh N., Zhao Y., Taniguchi N., Huang X. L., Otu H., Wang H., Wang J. F., Komiya S., Ducy P., Rahman M. U., Flavell R. A., Gravallese E. M., Oettgen P., Libermann T. A., Goldring M. B. (2005) A novel role for GADD45β as a mediator of MMP-13 gene expression during chondrocyte terminal differentiation. J. Biol. Chem. 280, 38544–38555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Peng H., Tan L., Osaki M., Zhan Y., Ijiri K., Tsuchimochi K., Otero M., Wang H., Choy B. K., Grall F. T., Gu X., Libermann T. A., Oettgen P., Goldring M. B. (2008) ESE-1 is a potent repressor of type II collagen gene (COL2A1) transcription in human chondrocytes. J. Cell Physiol. 215, 562–573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hashimoto K., Kokubun S., Itoi E., Roach H. I. (2007) Improved quantification of DNA methylation using methylation-sensitive restriction enzymes and real-time PCR. Epigenetics 2, 86–91 [DOI] [PubMed] [Google Scholar]
- 39. Goldring M. B., Otero M., Plumb D. A., Dragomir C., Favero M., El Hachem K., Hashimoto K., Roach H. I., Olivotto E., Borzì R. M., Marcu K. B. (2011) Roles of inflammatory and anabolic cytokines in cartilage metabolism: signals and multiple effectors converge upon MMP-13 regulation in osteoarthritis. Eur. Cell Mater. 21, 202–220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Jiménez M. J., Balbín M., López J. M., Alvarez J., Komori T., López-Otín C. (1999) Collagenase 3 is a target of Cbfa1, a transcription factor of the runt gene family involved in bone formation. Mol. Cell Biol. 19, 4431–4442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Liacini A., Sylvester J., Li W. Q., Zafarullah M. (2002) Inhibition of interleukin-1-stimulated MAP kinases, activating protein-1 (AP-1) and nuclear factor κ B (NF-κB) transcription factors down-regulates matrix metalloproteinase gene expression in articular chondrocytes. Matrix Biol. 21, 251–262 [DOI] [PubMed] [Google Scholar]
- 42. Mengshol J. A., Vincenti M. P., Brinckerhoff C. E. (2001) IL-1 induces collagenase-3 (MMP-13) promoter activity in stably transfected chondrocytic cells: requirement for Runx-2 and activation by p38 MAPK and JNK pathways. Nucleic Acids Res. 29, 4361–4372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Saito T., Fukai A., Mabuchi A., Ikeda T., Yano F., Ohba S., Nishida N., Akune T., Yoshimura N., Nakagawa T., Nakamura K., Tokunaga K., Chung U. I., Kawaguchi H. (2010) Transcriptional regulation of endochondral ossification by HIF-2α during skeletal growth and osteoarthritis development. Nat. Med. 16, 678–686 [DOI] [PubMed] [Google Scholar]
- 44. Yang S., Kim J., Ryu J. H., Oh H., Chun C. H., Kim B. J., Min B. H., Chun J. S. (2010) Hypoxia-inducible factor-2α is a catabolic regulator of osteoarthritic cartilage destruction. Nat. Med. 16, 687–693 [DOI] [PubMed] [Google Scholar]
- 45. Mitchell P. G., Magna H. A., Reeves L. M., Lopresti-Morrow L. L., Yocum S. A., Rosner P. J., Geoghegan K. F., Hambor J. E. (1996) Cloning, expression, and type II collagenolytic activity of matrix metalloproteinase-13 from human osteoarthritic cartilage. J. Clin. Invest. 97, 761–768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Reboul P., Pelletier J. P., Tardif G., Cloutier J. M., Martel-Pelletier J. (1996) The new collagenase, collagenase-3, is expressed and synthesized by human chondrocytes but not by synoviocytes. A role in osteoarthritis. J. Clin. Invest. 97, 2011–2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Neuhold L. A., Killar L., Zhao W., Sung M. L., Warner L., Kulik J., Turner J., Wu W., Billinghurst C., Meijers T., Poole A. R., Babij P., DeGennaro L. J. (2001) Postnatal expression in hyaline cartilage of constitutively active human collagenase-3 (MMP-13) induces osteoarthritis in mice. J. Clin. Invest. 107, 35–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Little C. B., Barai A., Burkhardt D., Smith S. M., Fosang A. J., Werb Z., Shah M., Thompson E. W. (2009) Matrix metalloproteinase 13-deficient mice are resistant to osteoarthritic cartilage erosion but not chondrocyte hypertrophy or osteophyte development. Arthritis Rheum. 60, 3723–3733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Inada M., Wang Y., Byrne M. H., Rahman M. U., Miyaura C., López-Otín C., Krane S. M. (2004) Critical roles for collagenase-3 (Mmp13) in development of growth plate cartilage and in endochondral ossification. Proc. Natl. Acad. Sci. U.S.A. 101, 17192–17197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Fan Z., Tardif G., Boileau C., Bidwell J. P., Geng C., Hum D., Watson A., Pelletier J. P., Lavigne M., Martel-Pelletier J. (2006) Identification in human osteoarthritic chondrocytes of proteins binding to the novel regulatory site AGRE in the human matrix metalloprotease 13 proximal promoter. Arthritis Rheum. 54, 2471–2480 [DOI] [PubMed] [Google Scholar]
- 51. Cogswell J. P., Godlevski M. M., Wisely G. B., Clay W. C., Leesnitzer L. M., Ways J. P., Gray J. G. (1994) NF-κB regulates IL-1 β transcription through a consensus NF-κB binding site and a nonconsensus CRE-like site. J. Immunol. 153, 712–723 [PubMed] [Google Scholar]
- 52. Hiscott J., Marois J., Garoufalis J., D'Addario M., Roulston A., Kwan I., Pepin N., Lacoste J., Nguyen H., Bensi G. (1993) Characterization of a functional NF-κB site in the human interleukin 1 β promoter: evidence for a positive autoregulatory loop. Mol. Cell Biol. 13, 6231–6240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Kirillov A., Kistler B., Mostoslavsky R., Cedar H., Wirth T., Bergman Y. (1996) A role for nuclear NF-κB in B-cell-specific demethylation of the Igκ locus. Nat. Genet. 13, 435–441 [DOI] [PubMed] [Google Scholar]
- 54. Iliopoulos D., Malizos K. N., Tsezou A. (2007) Epigenetic regulation of leptin affects MMP-13 expression in osteoarthritic chondrocytes: possible molecular target for osteoarthritis therapeutic intervention. Ann. Rheum. Dis. 66, 1616–1621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Pöschl E., Fidler A., Schmidt B., Kallipolitou A., Schmid E., Aigner T. (2005) DNA methylation is not likely to be responsible for aggrecan down regulation in aged or osteoarthritic cartilage. Ann. Rheum. Dis. 64, 477–480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Eckhardt F., Lewin J., Cortese R., Rakyan V. K., Attwood J., Burger M., Burton J., Cox T. V., Davies R., Down T. A., Haefliger C., Horton R., Howe K., Jackson D. K., Kunde J., Koenig C., Liddle J., Niblett D., Otto T., Pettett R., Seemann S., Thompson C., West T., Rogers J., Olek A., Berlin K., Beck S. (2006) DNA methylation profiling of human chromosomes 6, 20 and 22. Nat. Genet. 38, 1378–1385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Harris A. L. (2002) Hypoxia–a key regulatory factor in tumour growth. Nat. Rev. Cancer 2, 38–47 [DOI] [PubMed] [Google Scholar]
- 58. Huang L. E., Bunn H. F. (2003) Hypoxia-inducible factor and its biomedical relevance. J. Biol. Chem. 278, 19575–19578 [DOI] [PubMed] [Google Scholar]
- 59. Pescador N., Cuevas Y., Naranjo S., Alcaide M., Villar D., Landázuri M. O., Del Peso L. (2005) Identification of a functional hypoxia-responsive element that regulates the expression of the egl nine homologue 3 (egln3/phd3) gene. Biochem. J. 390, 189–197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Saito T., Kawaguchi H. (2010) HIF-2α as a possible therapeutic target of osteoarthritis. Osteoarthritis Cartilage 18, 1552–1556 [DOI] [PubMed] [Google Scholar]
- 61. Semenza G. L. (2000) HIF-1 and human disease: one highly involved factor. Genes Dev. 14, 1983–1991 [PubMed] [Google Scholar]
- 62. Ema M., Morita M., Ikawa S., Tanaka M., Matsuda Y., Gotoh O., Saijoh Y., Fujii H., Hamada H., Kikuchi Y., Fujii-Kuriyama Y. (1996) Two new members of the murine Sim gene family are transcriptional repressors and show different expression patterns during mouse embryogenesis. Mol. Cell Biol. 16, 5865–5875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Moffett P., Reece M., Pelletier J. (1997) The murine Sim-2 gene product inhibits transcription by active repression and functional interference. Mol. Cell Biol. 17, 4933–4947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Woods S., Farrall A., Procko C., Whitelaw M. L. (2008) The bHLH/Per-Arnt-Sim transcription factor SIM2 regulates muscle transcript myomesin2 via a novel, non-canonical E-box sequence. Nucleic Acids Res. 36, 3716–3727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Carvin C. D., Parr R. D., Kladde M. P. (2003) Site-selective in vivo targeting of cytosine-5 DNA methylation by zinc-finger proteins. Nucleic Acids Res. 31, 6493–6501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Smith A. E., Ford K. G. (2007) Specific targeting of cytosine methylation to DNA sequences in vivo. Nucleic Acids Res. 35, 740–754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Dawson M. A., Kouzarides T. (2012) Cancer epigenetics: from mechanism to therapy. Cell 150, 12–27 [DOI] [PubMed] [Google Scholar]