

Background: Rheb (Ras homologue enriched in brain) regulates mammalian target of rapamycin complex 1 (mTORC1).
Results: mTORC1 activity and cardiac hypertrophy are attenuated in Rheb-deficient hearts after the early postnatal period.
Conclusion: Rheb-dependent mTORC1 activation becomes essential for cardiomyocyte hypertrophic growth after the early postnatal period.
Significance: The findings provide insight into the regulatory mechanism of mTORC1 in postnatal heart development.
Keywords: Autophagy, Cell Growth, Heart, Heart Development, mTOR Complex (mTORC), Protein Synthesis, Signal Transduction, Translation, 4E-BP1, Rheb
Abstract
Cardiomyocytes proliferate during fetal life but lose their ability to proliferate soon after birth and further increases in cardiac mass are achieved through an increase in cell size or hypertrophy. Mammalian target of rapamycin complex 1 (mTORC1) is critical for cell growth and proliferation. Rheb (Ras homologue enriched in brain) is one of the most important upstream regulators of mTORC1. Here, we attempted to clarify the role of Rheb in the heart using cardiac-specific Rheb-deficient mice (Rheb−/−). Rheb−/− mice died from postnatal day 8 to 10. The heart-to-body weight ratio, an index of cardiomyocyte hypertrophy, in Rheb−/− was lower than that in the control (Rheb+/+) at postnatal day 8. The cell surface area of cardiomyocytes isolated from the mouse hearts increased from postnatal days 5 to 8 in Rheb+/+ mice but not in Rheb−/− mice. Ultrastructural analysis indicated that sarcomere maturation was impaired in Rheb−/− hearts during the neonatal period. Rheb−/− hearts exhibited no difference in the phosphorylation level of S6 or 4E-BP1, downstream of mTORC1 at postnatal day 3 but showed attenuation at postnatal day 5 or 8 compared with the control. Polysome analysis revealed that the mRNA translation activity decreased in Rheb−/− hearts at postnatal day 8. Furthermore, ablation of eukaryotic initiation factor 4E-binding protein 1 in Rheb−/− mice improved mRNA translation, cardiac hypertrophic growth, sarcomere maturation, and survival. Thus, Rheb-dependent mTORC1 activation becomes essential for cardiomyocyte hypertrophic growth after early postnatal period.
Introduction
Growth of the heart during embryonic development occurs primarily through proliferation of cardiac myocytes. However, cardiac myocytes withdraw from the cell cycle soon after birth and further increases in cardiac mass are achieved predominantly through hypertrophic growth rather than proliferation of individual myocytes (1). Normal heart development in perinatal period requires cardiomyocyte maturation, proliferation, and hypertrophy. However, the precise molecular mechanisms underlying perinatal cardiac development remain to be elucidated. The serine/threonine protein kinase Akt is an important mediator of phosphatidylinositol-3 kinase signaling and involved in postnatal cardiac growth (2). The stimulation of Akt signaling pathway leads to the activation of a master regulator of cell growth and metabolism, mammalian target of rapamycin complex 1 (mTORC1).2 mTORC1 consists of five components, including mTOR, the Raptor (regulatory associated protein of mTOR), and PRAS40 (proline-rich Akt substrate 40 kDa) (3). One of the most important signaling molecules in the regulation of mTORC1 is the Ras homologue enriched in brain gene (Rheb). Rheb is ubiquitouly expressed in mammalian cells with the highest levels in skeletal and cardiac muscle (4). The Rheb gene encodes a small GTPase, closely related to Ras, that exists either in an active GTP-bound or an inactive GDP-bound state (5). Rheb is inactivated by the tuberous sclerosis complex 1 and 2 (TSC1 and TSC2, respectively) GTPase-activating protein complex that catalyzes the conversion of Rheb-GTP to Rheb-GDP (6). The role of Rheb toward mTORC1 activation in the postnatal hearts has not been elucidated.
Protein biosynthesis represents a major cellular process controled by mTORC1, which is coordinately regulated by the mTORC1–4E-BP1 and -4E-BP2 (eukaryotic initiation factor 4E-binding proteins 1 and 2) and mTORC1-p70 S6 kinase axis (3). In mammalian cells, translation is controlled at the initiation step, when the 40 S ribosomal subunit is recruited to the mRNA (7). The capping of the 5′ end of mRNA allows the recruitment of eIF4F, including eIF4E and the 40 S ribosomal subunit to the mRNA 5′-cap structure (8). The phosphorylation of 4E-BP1 by mTORC1 prevents its binding to eIF4E, enabling eIF4E to promote cap-dependent translation (8). The detailed role of the mTORC1-S6 kinase axis in translational control remains to be elucidated (9). S6 kinase phosphorylates several substrates that function in translation and drive protein production.
In addition to the role of mTORC1 in protein synthesis, it plays an important role in protein degradation. There are two protein degradation systems in mammalian cells, namely autophagy and ubiquitin/proteasome systems. mTORC1 suppresses autophagy in mammalian cells (10) by binding and phosphorylating the autophagy-initiating kinase ULK1 (11).
To investigate an in vivo role of Rheb, loss-of-function studies have been reported (12, 13). Rheb-deficient embryos died around midgestation with impaired development of the cardiovascular system (12). Rheb-deficient embryonic fibroblasts showed decreased mTORC1 activity, were smaller, and showed impaired proliferation compared with wild-type cells. Embryonic deletion of Rheb1 in neural progenitor cells abolished mTORC1 signaling in developing brain (13).
In adult hearts, Rheb has been reported to be involved in cardiac hypertrophy. Overexpression of Rheb in isolated adult rat cardiomyocytes activated mTORC1 and protein synthesis and induced enlargement of cell surface area (14). Although Rheb was inactivated during acute myocardial ischemia, overexpression of Rheb in the mouse heart reversed the down-regulation of mTORC1 activity during ischemia and increased infarct size accompanied by inhibition of autophagy (15). In the current study, we generated floxed Rheb mice to obtain cardiac-specific Rheb-deficient mice to elucidate the role of Rheb in the in vivo heart and examine the contribution of Rheb to mTORC1 signaling in postnatal cardiac development. We found that Rheb is not required for mTORC1 signaling during the early postnatal period but becomes essential for mTORC1 signaling and normal cardiac development after transition from proliferation to hypertrophy.
EXPERIMENTAL PROCEDURES
Generation of Cardiac-specific Rheb-deficient Mice
The Rheb gene-targeting vector was constructed using a mouse C57BL/6J BAC genomic library (BACPAC Resources Center) as reported previously (16). The targeting vector was electroporated into ES cells (F1; SVJ129 and C57BL/6J), and the transfected ES clones were selected for neomycin resistance according to standard protocols. The neomycin-resistant ES clones with targeted homologous recombination were screened by PCR and further confirmed by Southern blotting. Circular pCAG-FLpe plasmid and pPGK-Puro plasmid was electroporated into the selected ES clones, and the transfected ES clones were selected for puromycin resistance according to the standard protocols. The neomycin cassette excised ES clones were screened by PCR. Southern blotting and karyotyping analyses were performed to obtain ES clones exhibiting the desired homologous recombination and normal karyotype. These targeted ES clones were injected into blastocyst mouse embryos to generate chimeric mice. The chimeric mice were crossed with C57BL/6J mice to validate germ line transmission. The offspring with “floxed” Rheb mice were crossed with transgenic mice expressing Cre recombinase under the control of the α-myosin heavy chain promoter (αMHC-Cre mice) in the C57BL/6J background (17) to generate cardiac-specific deletion of Rheb. The genotype of the floxed mice was determined by PCR on tail genomic DNA using primers as follows: 5′-GCT GAC ACT CAC TAC AGA ATA ATG-3′ and 5′-CAA TCA TTA ACC TGA CTG GTC TCT-3′ to amplify the wild-type and floxed alleles (296 and ∼350 bp, respectively). The primers for the amplification of αMHC-Cre are 5′-GAA CAC ACC TGG AAG ATG CTC CT-3′ and 5′- CTG ATT CTG GCA ATT TCG GCA AT-3′ with the amplicon of 427 bp.
To generate double knock-out mice of Rheb and Eif4ebp1, Rhebflox/+;αMHC-Cre+ mice were crossed with Eif4ebp1−/− mice (18) in the C57BL/6J background. To generate double knock-out mice of Rheb and Atg5, Rhebflox/+;αMHC-Cre+ mice were crossed with Atg5flox/flox mice (19) in the C57BL/6J background.
Southern Hybridization
Southern blot analysis of embryonic stem cells was performed as reported previously (17). Genomic DNA was isolated from embryonic stem cells, digested with KpnI, and subjected to Southern blot analysis. The probe used was an 1174-bp PCR fragment amplified with 5′-ACT TCC CTT GTA GTT TAG CGT ATA GCA-3′ and 5′-ACC TAA CTA AAT GAA CAA ACA AAA ATG GCA-3′.
Echocardiographic Assessment
Vevo 770 with a 25-MHz imaging transducer or Vevo 2100 with a 40-MHz imaging transducer (Visual Sonics) was used for noninvasive transthoracic echocardiographic analysis on awake mice in water at 37 °C. Two-dimensional guided M-mode tracings were recorded. The internal diameter of the left ventricle in the short axis plane was measured at end diastole and end systole from M-mode recordings just below the tips of the mitral valve leaflets. The interventricular and left ventricular (LV) posterior wall thicknesses were measured at the end diastole.
Histological Analysis
Hematoxylin/eosin or Azan-Mallory staining was performed on paraffin-embedded sections. The cross-sectional areas of cardiomyocytes were determined as described previously (20). To determine the number of cells undergoing apoptosis, we performed a TUNEL assay on paraffin-embedded heart sections, using an in situ apoptosis detection kit (Takara Bio, Inc.). For electron microscopy, the hearts were perfused in retrograde and fixed with 2.5% glutaraldehyde (Wako) in 0.1 m phosphate buffer (pH 7.4). The LV tissues were processed for transmission electron microscopy H-7650 (Hitachi) as described previously (21). To evaluate the sarcomeric to cytosolic area ratio, the area of sarcomere or cytosol in the micrograph taken at 1000-fold magnification was evaluated using NIH ImageJ software (version 1.43u) for 20 fields per mouse.
Western Blot Analysis
Protein homogenates were subjected to Western blot analysis using antibodies against Rheb, eIF4E-BP1, proliferating cell nuclear antigen (PCNA; Abcam), cleaved caspase 3, phospho-eIF4E-BP1 (Ser-65), phospho-eIF4E-BP1 (Thr-70), phospho-eIF4E-BP1 (Thr-37/Thr-46), S6 ribosomal protein, phospho-S6 ribosomal protein, AKT, phospho-AKT (Thr-308), phospho-AKT (Ser-473), LC-3, PRAS40, phospho-PRAS40 and α-tubulin (Cell Signaling Technology), ubiquitin (Dako Cytomation), p62 (PROGEN Biotechnik) and GAPDH (Santa Cruz Biotechnology). NIH ImageJ software (version 1.43u) was used to perform densitometric analyses.
Quantitative Real-time RT-PCR
We isolated total RNA from the ventricle for analysis using the TRIzol reagent (Invitrogen). We determined mRNA levels for Nppa, Nppb, collagen type 1 α2 (Col1a2), and GAPDH by quantitative RT-PCR. For reverse transcription and amplification, we used the TaqMan Reverse Transcription Reagents (Applied Biosystems) and Platinum Quantitative PCR SuperMix-UDG (Invitrogen). The PCR primers and probes of Nppa (assay ID Mm01255747_g1), Nppb (assay ID Mm00435304_g1), Col1a2 (assay ID Mm01165187_m1), and GAPDH (4352339E) were obtained from Applied Biosystems. We constructed RT-PCR standard curves using the corresponding cDNA. All data were normalized to GAPDH content and are expressed as fold increase over the control group.
Myocyte Isolation and Cell Surface Area
Myocytes were enzymatically isolated from neonatal hearts. The cannulated heart was attached to Langendorff apparatus for coronary perfusion with calcium-free solution (120 mm NaCl, 5.5 mm KCl, 1.2 mm NaH2PO4, 20 mm NaHCO3, 1.6 mm MgCl2, 5.6 mm glucose, 5 mm taurine). After 2 min, the perfusion solution was switched to the calcium-free solution containing 350 units/ml collagenase (Worthington) and perfused for an additional 15 min. The heart was then minced in the calcium-free solution containing 1 mg/ml BSA (Sigma), and isolated cardiomyocytes were obtained. Cell images were taken under a phase contrast microscope DP21 (Olympus). NIH ImageJ software (version 1.43u) was used to analyze cell surface area. One hundred myocytes from each heart were used for the measurement of cell surface area.
Polysome Analysis
Polysome profile analysis was carried out as described previously (22). Hearts were lysed in a hypotonic lysis buffer (5 mm Tris-HCl (pH 7.5), 2.5 mm MgCl2, 1.5 mm KCl, 100 mg/ml cycloheximide, 2 mm DTT, 0.5% Triton X-100, and 0.5% sodium deoxycholate). Lysates were loaded onto 10–50% sucrose density gradients (20 mm HEPES-KOH (pH 7.6), 100 mm KCl, 5 mm MgCl2) and centrifuged at 40,000 rpm for 2.5 h at 4 °C using an ultracentrifuge Optima L-90K and SW 41 Ti (Beckman). Gradients were fractionated, and absorbance at 254 nm was continuously recorded using a gradient fractionator (BioComp Instruments) and a spectrophotometer UV-3100PC (Shimazu).
Immunoprecipitation
The hearts from 8-day-old mice were homogenized in TEN buffer (100 mm Tris-HCl, (pH 7.5), 100 mm NaCl, 10 mm EDTA, 0.5% Nonidet P-40) supplemented with 1× phosphatase inhibitor mixture (Roche Applied Science) and 1× Complete protease inhibitor mixture (Roche Applied Science) and clarified by centrifugation at 15,000 rpm for 20 min at 4 °C. Detergent-soluble fractions were incubated with protein A-Sepharose (GE Healthcare) preincubated with rabbit monoclonal anti-4E-BP1 antibody (Cell Signaling Technology), followed by overnight rotation at 4 °C. Protein A-Sepharose complexes were sequentially washed twice with TEN buffer supplemented with 500 mm NaCl and twice with TEN buffer alone. Immunoprecipitates were eluted by heating at 95 °C for 5 min in 2× Laemmli sample buffer containing 2% 2-mercaptoethanol. Immunoprecipitates and input lysates were resolved by SDS-PAGE and subjected to Western blot analysis using antibodies 4E-BP1 and eIF4E (Abcam).
Statistics
Results are expressed as the mean ± S.E. Comparisons between two groups were performed using Student's t test. One-way analysis of variance with Bonferroni post hoc test was used for multiple comparisons. The Kaplan-Meier method with a log-rank test was used for survival analysis. p < 0.05 was considered statistically significant.
Study Approval
Experiments using animals in this study were carried out under the supervision of the Animal Research Committee of Osaka University and in accordance with the Guidelines for Animal Experiments of Osaka University and the Japanese Act on Welfare and Management of Animals (no. 105).
RESULTS
Generation of Cardiac-specific Rheb-deficient Mice
To investigate the role of Rheb in the heart, we generated cardiac-specific Rheb-deficient mice. To obtain cardiac-specific Rheb-deficient mice, conditional inactivation of the Rheb gene was achieved by the inserting loxP sites cloned 5′ and 3′ of exon 3 (Fig. 1A). Selection cassettes comprising a neomycin resistance gene (neo), flanking two flippase recognition target sites, for positive selection and a diphtheria toxin gene for negative selection were positioned between exon 3 and the downstream loxP site and at the 3′ end of the targeting vector, respectively (Fig. 1A). Homologous recombinants were identified by PCR and Southern blotting. The ES cells with the Rheb-floxed allele and the selection marker gene, that is, the PGK-neo cassette, were transfected with plasmid encoding FLpe recombinase to obtain Rheb-floxed ES cells without the PGK-neo cassette, which were identified by PCR and Southern blotting (Fig. 1B). The homozygous Rheb-floxed mice (Rhebflox/flox) appeared normal and were externally indistinguishable from littermates of other genotypes. To investigate the in vivo role of Rheb, we generated cardiac-specific Rheb-deficient mice. We crossed Rhebflox/flox mice with α-myosin heavy chain promoter-driven Cre recombinase transgenic mice (17) (αMHC-Cre) to generate Rhebflox/flox;αMHC-Cre+ mice (Rheb−/−). We used Rhebflox/flox;αMHC-Cre− littermates as controls (Rheb+/+). Immunoblot analysis of heart extracts from mice indicated that there were no significant differences in Rheb protein level in Rheb+/+ hearts among at postnatal day 3, 5, and 8 (Fig. 1C). We observed a 70% reduction in Rheb protein level in Rheb−/− hearts relative to Rheb+/+ hearts at postnatal day 3 (Fig. 1D). It has been reported that the αMHC promoter becomes active in cardiomyocytes between embryonic days 7.5 and 8 in mice (23). In fact, there were no significant differences in relative Rheb protein level to Rheb+/+ in Rheb−/− hearts at postnatal days 3, 5, and 8 (70, 70.5, and 68.9% reduction at postnatal day 3, 5, and 8, respectively) (Fig. 1D). Expected Mendelian ratios of Rhebflox/flox;αMHC-Cre+, Rhebflox/flox;αMHC-Cre−, Rhebflox/+;αMHC-Cre+, Rhebflox/+;αMHC-Cre− mice (n = 115, 140, 114, and 118, respectively) were detected at postnatal day 3 among the offspring of Rhebflox/flox;αMHC-Cre− and Rhebflox/+;αMHC-Cre+ mice, indicating no significant embryonic lethality.
FIGURE 1.

Targeted modification of the Rheb gene. A, schematic structures of genomic Rheb sequences, the targeting construct, the targeted allele, the floxed allele, and the Rheb−/− allele (from top to bottom). The black and white arrowheads represent loxP and flippase recognition target sites, respectively. The targeting construct includes the PGK-neo cassette flanked by flippase recognition target sites and a diphtheria toxin gene (DTA). The arrows correspond to the primer sequences for PCR screening. The bar labeled probe corresponds to the sequence used for Southern blotting analysis in B. B, genomic analysis of ES cells (left panel). Genomic DNA was isolated from ES cells, digested with KpnI, and analyzed by Southern blotting with the probe. Shown is genomic analysis of mouse tails (right panel). Genomic DNA was isolated from mouse tail and subjected to PCR analysis. C and D, protein expression of Rheb at postnatal days 3, 5, and 8. Heart extracts from Rheb+/+ (C and D) and Rheb−/− (D) at postnatal day 3, 5, or 8 were subjected to Western blot analysis. The average value at postnatal day 3 was set equal to 1 (C). Right panels show densitometric analysis (n = 3). Values are expressed as the mean ± S.E. *, p < 0.05.
Cardiac-specific Deletion of Rheb Resulted in Premature Death and Cardiac Dysfunction after Early Postnatal Period
Rheb−/− mice started to die at postnatal day 8 (Fig. 2A). At postnatal day 10, all Rheb−/− mice had died, whereas no Rheb+/+ mice died at that time. Physiological parameters such as body weight, heart weight and lung weight were not significantly different between Rheb−/− and Rheb+/+ mice at postnatal days 3 and 5 (Fig. 2B). Body weight increased with age both in Rheb−/− and Rheb+/+ mice. The heart weight at postnatal day 8 was greater than that at postnatal day 3 or 5 in Rheb+/+ mice. In Rheb−/− mice, the heart weight at postnatal day 8 was greater than that at postnatal day 3 but was not significantly different from that at day 5. The heart weight in Rheb−/− mice was significantly lower than that in Rheb+/+ mice at postnatal day 8. The ratio of heart-to-body weight in Rheb−/− mice at postnatal day 8 was significantly lower than that in Rheb−/− at postnatal day 3 or 5 or that in Rheb+/+ mice at postnatal day 8. We performed echocardiographic analysis on Rheb−/− mice. The echocardiographic parameters were not significantly different at postnatal day 5 between Rheb−/− and Rheb+/+ mice (Fig. 2D). However, the diastolic interventricular septum thickness and diastolic LV posterior wall thickness in Rheb−/− mice were significantly smaller than those in Rheb+/+ mice at postnatal day 8. The end-diastolic and systolic LV dimensions were significantly larger and LV fractional shortening was significantly reduced in Rheb−/− mice at postnatal day 8 compared with Rheb+/+ mice at postnatal day 8 or Rheb−/− mice at postnatal day 5. We evaluated the level of atrial natriuretic factor and brain natriuretic peptide mRNA expression, which are biochemical markers for adverse cardiac remodeling, by means of quantitative RT-PCR at postnatal day 8 (Fig. 3A). The mRNA levels of atrial natriuretic factor and brain natriuretic peptide were significantly elevated in Rheb−/− mice compared with Rheb+/+ mice. The Rheb−/− hearts appeared to exhibit lower Hematoxylin/eosin stainability at postnatal day 8 (Fig. 3B). The mRNA level for collagen α-2(I) chain increased in Rheb−/− hearts (Fig. 3A), whereas Azan-Mallory staining indicated a slight increase in the extent of fibrosis at postnatal day 8 (Fig. 3C). These data indicate that Rheb−/− mice exhibited normal cardiac function until postnatal day 5 but developed cardiac dysfunction at postnatal day 8. Thus, Rheb is not required for embryonic heart development and maintenance of cardiac structure and function until postnatal day 5.
FIGURE 2.

Physiological and echocardiographic characterization of Rheb−/− mice. A, survival ratio of Rheb−/− and Rheb+/+ mice after birth. Open and closed circles represent Rheb+/+ (n = 11) and Rheb−/− mice (n = 9), respectively. *, p < 0.05. B, physiological parameters of Rheb−/− and Rheb+/+ mice at postnatal days 3 (n = 8 per group), 5 (n = 10–11 per group), and 8 (n = 16–20 per group). Open and closed bars represent Rheb+/+ and Rheb−/− mice, respectively. Values are expressed as the mean ± S.E. *, p < 0.05. BW and HW indicate body weight and heart weight, respectively. C, representative M-mode echocardiographic tracings at postnatal day 8. Scale bars, 0.1 s and 1 mm, respectively. D, echocardiographic parameters of Rheb−/− and Rheb+/+ mice at postnatal days 5 (n = 6–7 per group) and 8 (n = 8–9 per group). IVSd indicates diastolic interventricular septum thickness; PWThd, diastolic left ventricular posterior wall thickness; LVIDd, end diastolic left ventricle internal dimension; LVIDs, end-systolic left ventricle internal dimension; FS, left ventricle fractional shortening; HR, heart rate. Open and closed bars represent Rheb+/+ and Rheb−/− mice, respectively. Values are expressed as the mean ± S.E. *, p < 0.05.
FIGURE 3.
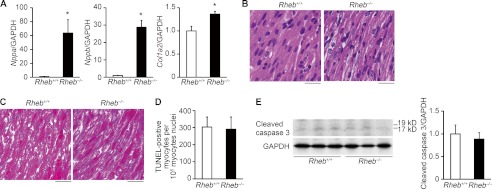
Biochemical and histological characterization of Rheb−/− mice at postnatal day 8. A, mRNA expressions of Nppa, Nppb, and Col1a2 (n = 3–5). GAPDH was used as the loading control. Open and closed bars represent Rheb+/+ and Rheb−/− mice, respectively. The average value for Rheb+/+ mice was set equal to 1. Values are expressed as the mean ± S.E. *, p < 0.05 versus Rheb+/+ controls. B, hematoxylin/eosin-stained heart sections. Scale bar, 20 μm. C, Azan-Mallory-stained heart sections. Scale bar, 20 μm. D. number of TUNEL-positive cardiomyocytes. Values are expressed as the mean ± S.E. (n = 3). E, Western blot analysis of heart extracts using cleaved caspase 3 antibody at postnatal day 8. Right panel shows densitometric analysis (n = 3). The average value for Rheb+/+ mice was set equal to 1. Values are expressed as the mean ± S.E.
TUNEL assay revealed that there was no significant difference in the number of TUNEL-positive cardiomyocytes between Rheb−/− and Rheb+/+ hearts at postnatal day 8 (Fig. 3D). Furthermore, there was no significant difference in the level of cleaved caspase 3 (Fig. 3E). These results suggest that apoptosis was not a major cause for the development of dilated cardiomyopathy in Rheb−/− mice.
Histological Analysis of Rheb-deficient Mice Revealed Small Cardiomyocyte Size
Because we observed a lower ratio of heart weight/body weight in Rheb−/− mice at postnatal day 8, we estimated cross-sectional area of cardiomyocytes on heart sections at postnatal day 3, 5, and 8 (Fig. 4, A and B). There were no significant differences in cross-sectional area of cardiomyocytes between 3 and 5 days after birth either in Rheb−/− or Rheb+/+ mice or between Rheb−/− and Rheb+/+ mice at postnatal day 3 or 5. In Rheb+/+ hearts, the cross-sectional area at postnatal day 8 was larger than that at postnatal day 5. However, there was no significant difference in the cross-sectional area between 5 and 8 days after birth in Rheb−/− mice. As a result, Rheb−/− hearts exhibited smaller cross-sectional area than Rheb+/+ hearts at postnatal day 8. To confirm this result, we isolated cardiomyocytes from the mice and estimated the cell surface area (Fig. 4, C and D). In Rheb+/+ mice, the cell surface area increased with age. Although the cell surface area increased by 35.5% between 3 and 5 days after birth, it increased by 88.9% for the next 3 days. In Rheb−/− mice, the cell surface area increased by 36.6% between 3 and 5 days after birth, but there was no further increase in the cell surface area between 5 and 8 days after birth. Consequently, Rheb−/− cardiomyocytes exhibited smaller cell surface area than Rheb+/+ cardiomyocytes at postnatal day 8. We examined the protein level of PCNA to evaluate the level of proliferation (Fig. 5). In Rheb+/+ hearts, the level of PCNA at postnatal day 8 was lower than that at postnatal day 3. There was no significant difference in the PCNA protein level between Rheb−/− and Rheb+/+ mice at postnatal day 3, 5, or 8. These results suggest that Rheb is not related to cardiomyocyte proliferation but is essential for postnatal hypertrophic growth of cardiomyocytes.
FIGURE 4.

Cardiomyocyte size of Rheb−/− mice. A, hematoxylin/eosin-stained heart sections at postnatal day 3, 5, and 8. Scale bar, 20 μm. Right panels show histograms of cross-sectional area of cardiomyocytes. B, cross-sectional area of cardiomyocytes. Values are expressed as the mean ± S.E. (n = 3–4 per group). *, p < 0.05. C, isolated cardiomyocytes from Rheb−/− mice. Scale bar, 20 μm. Right panels show histograms of cell surface area of cardiomyocytes. D, cell surface area of isolated cardiomyocytes. Values are expressed as the mean ± S.E. (n = 3–4 per group). *, p < 0.05.
FIGURE 5.

Proliferation in Rheb−/− hearts. Western blot analysis of heart extracts using anti-PCNA. Heart extracts from Rheb−/− and Rheb+/+ mice at postnatal days 3, 5, and 8. Lower panel shows densitometric analysis (n = 3). Open and closed bars represent Rheb+/+ and Rheb−/− mice, respectively. The average value for Rheb+/+ mice at postnatal day 3 was set equal to 1. Values are expressed as the mean ± S.E. *, p < 0.05 versus corresponding mice at postnatal day 3. †, p < 0.05 versus corresponding mice at postnatal day 5.
mTORC1 Signaling Was Impaired by Deletion of Rheb after Early Postnatal Period
The extent of reduction in Rheb protein level in Rheb−/− hearts compared with Rheb+/+ hearts was similar at postnatal days 3, 5, and 8 (Fig. 1D). Nevertheless, there was no significant difference in the phosphorylation level of S6 or 4E-BP1 between Rheb−/− and Rheb+/+ mice at postnatal day 3, indicating preservation of mTORC1 signaling (Fig. 6, A–D). The phosphorylation level of S6 or 4E-BP1 was significantly attenuated in Rheb−/− hearts compared with the control at postnatal day 5 or 8. The phosphorylation of 4E-BP1 was further analyzed using its phosphorylation site-specific antibodies. The phosphorylation levels of Thr-37/Thr-46 and Ser-65 in 4E-BP1 were attenuated in Rheb−/− hearts at postnatal day 8. It has been reported that deletion of Rheb resulted in an increase in Akt phosphorylation, suggesting a negative feedback in the mTORC1 signaling pathway (12). Consistent with the existence of a negative feedback loop, the phosphorylation level of Akt in Rheb−/− hearts was increased at postnatal day 5 or 8, but not at day 3, compared with Rheb+/+ hearts. The interaction of 4E-BP1 with eIF4E was enhanced in Rheb−/− hearts (Fig. 6E). The phosphorylation level of PRAS40 was increased in Rheb−/− hearts at postnatal day 3 (Fig. 6F).
FIGURE 6.

Signal transduction in Rheb−/− hearts. Open and closed bars represent Rheb+/+ and Rheb−/− mice, respectively. A, heart extracts from 3-, 5-, and 8-day-old mice were immunoblotted with the indicated antibodies. B, average of phospho-S6 to total S6 ratios from heart extracts (n = 3). The average value for Rheb+/+ mice at postnatal day 3 was set equal to 1. *, p < 0.05 versus corresponding Rheb+/+ controls. †, p < 0.05 versus corresponding mice at postnatal day 3. C, Western blot analysis on 4E-BP1. Heart extracts were immunoblotted (IB) with the indicated antibodies. D, average of γ-form 4E-BP1 to total 4E-BP1 ratios from heart extracts (n = 3). The average value for Rheb+/+ mice at postnatal day 3 was set equal to 1. *, p < 0.05 versus corresponding Rheb+/+ controls. †, p < 0.05 versus corresponding mice at postnatal day 3. ‡, p < 0.05 versus corresponding mice at postnatal day 5. E, coimmunoprecipitation of 4E-BP1 and elF4E in Rheb+/+ and Rheb−/− heart lysates at postnatal day 8 followed by Western blot analysis. F, Rheb-independent pathway at postnatal day 3. Western blot analysis of heart extracts using anti-phospho-PRAS40 or PRAS40 antibody. Heart extracts from Rheb−/− and Rheb+/+ mice at postnatal day 3. The lower panel shows densitometric analysis (n = 3). The average value for Rheb+/+ mice was set equal to 1. Values are expressed as the mean ± S.E. *, p < 0.05.
Rapamycin, an inhibitor of mTORC1, attenuated the PCNA protein level, the phosphorylation level of S6 or 4E-BP1, body weight, heart weight, and the ratio of heart-to-body weight in Rheb−/− and Rheb+/+ mice at postnatal day 3 (Fig. 7, A and B). There was no significant difference in body weight, heart weight, or the ratio of heart-to-body weight between rapamycin-treated Rheb−/− and Rheb+/+ mice at postnatal day 3. At postnatal day 8, rapamycin attenuated the PCNA protein level and the phosphorylation level of S6 or 4E-BP1 in Rheb−/− and Rheb+/+ mice. Heart weight or the ratio of heart-to-body weight in Rheb+/+ mice was greater than that in Rheb−/− mice. Rapamycin decreased the ratio of heart-to-body weight in Rheb+/+ but not in Rheb−/− mice.
FIGURE 7.

Effect of rapamycin on the proliferation and the cardiac growth. Rapamycin (2.5 mg/kg) or dimethyl sulfoxide was administered intraperitoneally at postnatal day 1 and 2 and analyzed at postnatal day 3. When mice were analyzed at postnatal day 8, rapamycin was administered at postnatal day 7. D and R indicate dimethyl sulfoxide and rapamycin-treated groups, respectively. A, heart extracts were immunoblotted with the indicated antibodies. B. physiological parameters. BW and HW indicate body weight and heart weight, respectively. Values are expressed as the mean ± S.E. (n = 3 per group). *, p < 0.05 versus corresponding dimethyl sulfoxide-treated mice. †, p < 0.05 versus dimethyl sulfoxide-treated Rheb+/+ control mice. ‡, p < 0.05 versus rapamycin-treated Rheb+/+ control mice.
Sarcomere Maturation and mRNA Translation Were Attenuated in Rheb−/− Hearts
To identify a molecular mechanism underlying the cardiac phenotypes observed in Rheb−/− mice, we performed ultrastructural analysis on the hearts (Fig. 8, A and B). At postnatal day 3 or 5, the sarcomere structures in both Rheb−/− and Rheb+/+ were immature, indicated by a narrow sarcomere width. There was no significant difference in sarcomeric to cytosolic area ratio between Rheb−/− and Rheb+/+ mice at postnatal day 3 or 5. At postnatal day 8, the width of the sarcomere became greater in Rheb+/+ mice, but it remained narrow in Rheb−/− mice. The sarcomeric to cytosolic area ratio was increased by 35% in Rheb+/+ hearts between 5 and 8 days after birth, whereas there was no significant difference during this period in Rheb−/− mice. The ratio was larger in Rheb+/+ hearts than that in Rheb−/− hearts at postnatal day 8. These results indicate that sarcomere maturation was impaired in Rheb−/− hearts. There was no significant difference in the number of mitochondria per cell area between 5 and 8 days after birth in Rheb−/− or Rheb+/+ mice (Fig. 8C).
FIGURE 8.
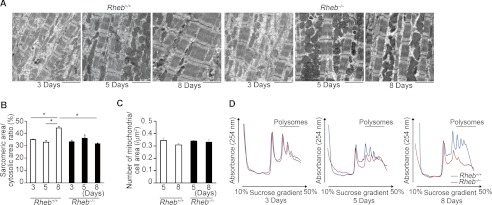
Sarcomeric maturation and protein metabolism in Rheb−/− hearts. A, electron microscopic analysis. Scale bar, 2.5 μm. B, average of sarcomeric area to cytosolic area ratios on electron micrograph. Open and closed bars represent Rheb+/+ and Rheb−/− mice, respectively. Values are expressed as the mean ± S.E. (n = 3–5 per group). *, p < 0.05. C, average number of mitochondria/cell area. Values are expressed as the mean ± S.E. (n = 3 per group). D, mRNA translation in Rheb−/− hearts. Heart lysates were separated on a sucrose gradient. The absorbance at 254 nm is shown as a function of gradient depth. Actively translated mRNA is associated with high molecular weight polysomes deep in the gradient.
It has been reported that mTORC1 promotes cell proliferation through enhanced translation of mRNAs mediated through 4E-BP1 (24). We thus examined the translation activity by estimating the association of ribosomes with mRNA at various time points after birth (Fig. 8D). In this assay, the number of ribosomes within the polysomal fraction of mRNA is a reflection of de novo protein synthesis (25). Rheb ablation caused a decrease in polysomal fractions from postnatal day 5, indicating a reduction in translation in Rheb−/− hearts.
Autophagy Was Not Involved in the Development of Cardiomyopathy in Rheb−/− Hearts
Determinants for cell size should be the levels of protein synthesis and degradation. There are two protein degradation systems in mammalian cells, namely the autophagy and ubiquitin/proteasome systems. We have previously reported that constitutive autophagy is critical for the maintenance of cardiac function, whereas inducible autophagy plays an important role to protect hearts from hemodynamic stress (21). mTORC1 is known to be an important negative regulator of autophagy in mammalian cells (10). We investigated the autophagic activity in Rheb−/− hearts by immunoblot for microtubule-associated protein light chain 3 (LC3) and p62 at postnatal day 8 (Fig. 9A). Conversion of LC3-I to LC3-II is an essential step for autophagosome formation. There was no significant difference in the ratio of LC3-II to α-tubulin between in Rheb−/− and Rheb+/+ hearts, whereas the ratio of LC3-II to LC3-I increased in Rheb−/− hearts. There was no significant difference in the level of p62, a marker for autophagy flux, between Rheb−/− and Rheb+/+ hearts. These suggest that autophagy flux was not enhanced in Rheb−/− hearts compared with Rheb+/+ hearts. We detected no significant difference in levels of ubiquitinated proteins between Rheb−/− and Rheb+/+ hearts (Fig. 9B).
FIGURE 9.

Protein degradation systems are not involved in the cardiac phenotypes in Rheb−/− mice. A, Western blot analysis of Rheb−/− hearts using anti-p62 or LC3 antibody. Lower panels show densitometric analysis. The average value for Rheb+/+ mice was set equal to 1. Values are expressed as the mean ± S.E. (n = 3). *, p < 0.05. B, Western blot analysis of Rheb−/− hearts using anti-ubiquitin antibody. C, analysis of Rheb, Atg5 double knock-out mice. Upper panel shows Western blot analysis of Rheb−/−;Atg5−/− hearts using anti-LC3 antibody. Lower panel shows survival curve of Rheb−/−;Atg5+/+ and Rheb−/−;Atg5−/− mice after birth. Open and closed circles represent Rheb−/−;Atg5+/+ (n = 4) and Rheb−/−;Atg5−/− (n = 5) mice, respectively.
To exclude the contribution of autophagy to cardiomyopathic phenotypes observed in Rheb−/− mice, we generated cardiac-specific double knock-out mice of Rheb and Atg5, which is an essential molecule for autophagy. Ablation of Atg5 resulted in a decrease in the protein level of LC3-II in Rheb−/− hearts (Fig. 9C). However, ablation of Atg5 had no effect on the survival of Rheb−/− mice, suggesting that autophagy was not related to the premature death of Rheb−/− mice (Fig. 9C).
Ablation of Eif4ebp1 Improved Survival, Cardiac Function, and Hypertrophic Growth in Rheb−/− Mice
Because it has been reported that deletion of S6 kinases did not attenuate pathological, physiological, or insulin-like growth factor 1 receptor-phosphoinositide 3-kinase-induced cardiac hypertrophy (26), we examined the contribution of the decrease in 4E-BP1 phosphorylation to the observed phenotypes in Rheb−/− mice by generating Rheb−/−;Eif4ebp1−/− mice. Ablation of Eif4ebp1 resulted in a significant improvement of survival in Rheb−/− mice (Fig. 10A). Echocardiographic analysis on mice at postnatal day 8 showed larger diastolic interventricular septum thickness and posterior wall thickness, smaller end diastolic and systolic LV dimensions, and higher fractional shortening in Rheb−/−;Eif4ebp1−/− mice than those in Rheb−/−;Eif4ebp1+/+ mice (Fig. 10, B and C). Furthermore, ablation of Eif4ebp1 led to increases in heart weight, the ratio of heart weight to body weight, hematoxylin/eosin stainability, and cross-sectional area (Fig. 10, D–F). Rheb−/−;Eif4ebp1−/− mice showed wider and more mature sarcomeres compared with control Rheb−/−;Eif4ebp1+/+ mice at postnatal day 8 (Fig. 10G). Eif4ebp1 ablation caused an increase in polysomal fraction at postnatal day 8 in Rheb−/− hearts (Fig. 10H).
FIGURE 10.

Ablation of Eif4ebp1 rescued the cardiac phenotypes in Rheb−/− mice. A, survival ratio of Rheb−/−;Eif4ebp1−/− and Rheb−/−;Eif4ebp1+/+ mice after birth. Open and closed circles represent Rheb−/−;Eif4ebp1+/+ (n = 9) and Rheb−/−;Eif4ebp1−/− mice (n = 16), respectively. *, p < 0.05. B, representative M-mode echocardiographic tracings at postnatal day 8. Scale bars, 0.1 s and 1 mm, respectively. In C, D, and F, open and closed bars represent Rheb−/−;Eif4ebp1+/+ and Rheb−/−;Eif4ebp1−/− mice, respectively, and values are expressed as the mean ± S.E. *, p < 0.05. C, echocardiographic parameters of Rheb−/−;Eif4ebp1−/− (n = 5) and Rheb−/−;Eif4ebp1+/+ mice (n = 3) at postnatal day 8. IVSd indicates diastolic interventricular septum thickness; PWThd, diastolic left ventricular posterior wall thickness; LVIDd, end diastolic left ventricle internal dimension; LVIDs, end systolic left ventricle internal dimension; FS, left ventricle fractional shortening; HR, heart rate. D, physiological parameters of Rheb−/−;Eif4ebp1−/− (n = 3) and Rheb−/−;Eif4ebp1+/+ mice (n = 3) at postnatal day 8. BW and HW indicate body weight and heart weight, respectively. E, hematoxylin-eosin-stained heart sections. Scale bar, 20 μm. F, cross-sectional area of cardiomyocytes (n = 3). G, electron microscopic analysis. Scale bar, 1 μm. H, mRNA translation. Heart lysates were separated on a sucrose gradient. The absorbance at 254 nm is shown as a function of gradient depth.
DISCUSSION
In this study, we analyzed the in vivo function of Rheb, a vital regulator of mTORC1 signaling, in the heart by conditionally deleting Rheb from cardiomyocytes. We showed that Rheb is not essential for embyronic and early postnatal heart development but essential for heart development beyond postnatal day 5. Conventional Rheb-deficient mice died around midgestation due to impaired development of the cardiovascular system (12). Our results indicate that this impaired development of the cardiovascular system could be the secondary consequences of Rheb deletion. Rheb-dependent mTORC1 activation is indispensable for cardiac hypertrophic growth from postnatal day 5. However, we observed the activation of S6 kinase and 4E-BP1 at postnatal day 3 in Rheb−/− mice. Thus, Rheb is not essential for the mTORC1 activation during neonatal cardiac development soon after birth. Possibly, an isofom of Rheb such as RhebL1 (27) partially compensates for the loss of Rheb. Alternatively, mTORC1 may exhibit Rheb-independent activity in in vivo hearts. Our experiments using rapamycin indicate that Rheb-independent mTORC1 pathway exists at postnatal days 3 and 8 in the heart (Fig. 7A). Those also indicate that mTORC1 is involved in both proliferation and protein synthesis during early postnatal cardiac development. Although Rheb-dependent mTORC1 activation is required for cardiac hypertrophic growth at postnatal day 8, Rheb-independent mTORC1 pathway plays an important role in cardiac development at postnatal day 3 (Fig. 7B). The contribution of the Rheb-independent mTORC1 pathway in cardiac growth at postnatal day 8 remains to be elucidated. mTORC1 has been reported to be activated by several Rheb-independent pathways (3). Akt activation by growth factors can activate mTORC1 in a TSC1/2-independent manner by promoting the phosphorylation and dissociation of PRAS40 from mTORC1 (28, 29). In fact, we detected an increase in the phosphorylation level of PRAS40 in Rheb−/− hearts at postnatal day 3 (Fig. 6F). The energy status of the cell is signaled to mTORC1 through AMP-activated protein kinase 1. AMP-activated protein kinase 1 reduces mTORC1 activity by phosphorylating TSC2 (30) or directly phosphorylating Raptor (31). However, we detected no significant difference in phosphorylation level of AMP-activated protein kinase 1 between Rheb−/− and Rheb+/+ mice at postnatal day 8 (data not shown). Amino acids, especially leucine, activate mTORC1 in a TSC1/2-independent manner (32). Leucine administration slightly improved the survival of Rheb−/− mice (age when all mice died; postnatal day 10 and 11 for control and leucine-treated Rheb−/− mice, respectively) (data not shown). However, we cannot exclude a possibility that the extension of survival is due to nonspecific improvement of nutritional status. The Rheb-independent mTORC1 signaling pathway in the neonatal period soon after birth remains to be investigated.
It has been reported that cardiomyocyte cell volume remains constant in rat hearts during the first 3 days of age, whereas cell number increases between days 1 and 3 after birth (33). After day 3, the number of cardiomyocytes in the heart remains relatively constant, and the volume of cardiomyocyte begins to increase. Thus, it is possible that the Rheb-mTORC1 signaling pathway is necessary for heart development after transition of cardiomyocytes from proliferation to hypertrophic growth after early postnatal period. In this study, the cardiomyocyte surface area in the heart was dramatically increased between postnatal days 5 and 8 in Rheb+/+ mice, whereas it remained constant during this period in Rheb−/− mice.
There may be several possible mechanisms underlying the development of dilated cardiomyopathy in Rheb−/− mice. Recently, it has been reported that ablation of Mtor in the adult mouse myocardium results in fatal, dilated cardiomyopathy that is characterized by an increase in apoptosis, induction of autophagy, altered mitochondrial structure, and accumulation of 4E-BP1 (34). Ablation of Eif4ebp1 improved cardiac function and survival with a decreased level of apoptosis in Mtor−/− mice, whereas overexpression of a non-phosphorylatable form of 4E-BP1 in isolated cardiomyocytes induced apoptosis. The authors suggested that increased apoptosis might be a cause for the development of cardiomyopathy in Mtor−/− mice. However, we did not observe an increase in apoptosis in Rheb−/− mice. One possible explanation for the discrepancy between our results and those obtained with the use of Mtor−/− mice may be due to the fact that mTOR deletions affect both mTOR complexes mTORC1 and -2. Another explanation is that the involvement of mTOR in apoptosis may be mediated through a Rheb-independent signaling pathway. It is also possible that the role of mTORC1 may be different between adulthood and neonatal periods.
It has been reported that mTORC1 suppresses autophagy (10). However, we did not detect an increase in autophagic activity in Rheb−/− mice, and ablation of Atg5 had no effect on survival of Rheb−/− mice, suggesting that autophagy was not related to the premature death of Rheb−/− mice. Rheb-dependent mTORC1 activation might not be a major signaling pathway to regulate autophagy in the heart.
Activation of mTORC1 stimulates mRNA translation via its downstream substrates 4E-BP1 and S6 kinase (9). In this study, we found that Rheb ablation led to an increase in the nonphosphorylated form of 4E-BP1, a decrease in S6 kinase activity and reduction in translation. The rescue experiment using Rheb−/−;Eif4ebp1−/− mice indicates that the mTORC1–4E-BP1 axis plays an important role in the maintenance of cardiac function and structure during the neonatal period after transition from proliferation to hypertrophic growth. The development of dilated cardiomyopathy in Rheb−/− mice appears to result from the lack of cardiac hypertrophic growth due to the reduction in translation. In this experiment, ablation of Eif4ebp1 was able to improve survival of Rheb−/− mice but failed to accomplish complete rescue of survival. Because we observed chamber dilatation in dead Rheb−/−;Eif4ebp1−/− mice, heart failure appears to be the cause of death in these animals (data not shown). We did not identify the molecular mechanism underlying the premature death in Rheb−/−;Eif4ebp1−/− mice. Some pathway other than the mTORC1–4E-BP1 signaling pathway such as mTORC1-S6 kinase and mTORC1–4E-BP2 may be involved in the premature death in Rheb−/−;Eif4ebp1−/− mice.
In conclusion, the Rheb signaling pathway is essential for normal heart development from postnatal day 5. In this period, the Rheb-mTORC1–4E-BP1 signaling cascade plays a pivotal role in mRNA translation and protein synthesis and thereby cardiac hypertrophic growth.
Acknowledgments
We thank professor Noboru Mizushima (Tokyo Medical and Dental University) for a gift of Atg5flox/flox mice and Kana Takada and Sachie Koyama for technical assistance.
This work was supported by Grant-in-aid for Scientific Research CH/11/3/29051 from the Ministry of Education, Culture, Sports, and Science, Japan to Osaka University and Grant RG/11/12/29052 from the British Heart Foundation to King's College London.
- mTORC1
- mammalian target of rapamycin complex 1
- TSC
- tuberous sclerosis complex
- 4E-BP
- 4E-binding protein
- αMHC-Cre mice
- α-myosin heavy chain promoter driven Cre recombinase transgenic mice
- LV
- left ventricular
- PCNA
- proliferating cell nuclear antigen
- LC3
- light chain 3.
REFERENCES
- 1. Ahuja P., Sdek P., MacLellan W. R. (2007) Cardiac myocyte cell cycle control in development, disease, and regeneration. Physiol. Rev. 87, 521–544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Shiojima I., Walsh K. (2006) Regulation of cardiac growth and coronary angiogenesis by the Akt/PKB signaling pathway. Genes Dev. 20, 3347–3365 [DOI] [PubMed] [Google Scholar]
- 3. Laplante M., Sabatini D. (2009) mTOR signaling at a glance. J. Cell Sci. 122, 3589–3594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Gromov P. S., Madsen P., Tomerup N., Celis J. E. (1995) A novel approach for expression cloning of small GTPases: identification, tissue distribution and chromosome mapping of the human homolog of rheb. FEBS Lett. 377, 221–226 [DOI] [PubMed] [Google Scholar]
- 5. Yamagata K., Sanders L. K., Kaufmann W. E., Yee W., Barnes C. A., Nathans D., Worley P. F. (1994) rheb, a growth factor- and synaptic activity-regulated gene, encodes a novel Ras-related protein. J. Biol. Chem. 269, 16333–16339 [PubMed] [Google Scholar]
- 6. Inoki K., Li Y., Xu T., Guan K. L. (2003) Rheb GTPase is a direct target of TSC2 GAP activity and regulates mTOR signaling. Genes Dev. 17, 1829–1834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Gingras A. C., Raught B., Sonenberg N. (2001) Regulation of translation initiation by FRAP/mTOR. Genes Dev. 15, 807–826 [DOI] [PubMed] [Google Scholar]
- 8. Richter J. D., Sonenberg N. (2005) Regulation of cap-dependent translation by eIF4E inhibitory proteins. Nature 433, 477–480 [DOI] [PubMed] [Google Scholar]
- 9. Magnuson B., Ekim B., Fingar D. C. (2012) Regulation and function of ribosomal protein S6 kinase (S6K) within mTOR signalling networks. Biochem. J. 441, 1–21 [DOI] [PubMed] [Google Scholar]
- 10. Ravikumar B., Vacher C., Berger Z., Davies J. E., Luo S., Oroz L. G., Scaravilli F., Easton D. F., Duden R., O'Kane C. J., Rubinsztein D. C. (2004) Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat. Genet. 36, 585–595 [DOI] [PubMed] [Google Scholar]
- 11. Kim J., Kundu M., Viollet B., Guan K. L. (2011) AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 13, 132–141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Goorden S. M., Hoogeveen-Westerveld M., Cheng C., van Woerden G. M., Mozaffari M., Post L., Duckers H. J., Nellist M., Elgersma Y. (2011) Rheb Is essential for murine development. Mol. Cell. Biol. 31, 1672–1678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zou J., Zhou L., Du X. X., Ji Y., Xu J., Tian J., Jiang W., Zou Y., Yu S., Gan L., Luo M., Yang Q., Cui Y., Yang W., Xia X., Chen M., Zhao X., Shen Y., Chen P. Y., Worley P. F., Xiao B. (2011) Rheb1 is required for mTORC1 and myelination in postnatal brain development. Dev. Cell 20, 97–108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wang Y., Huang B. P., Luciani D. S., Wang X., Johnson J. D., Proud C. G. (2008) Rheb activates protein synthesis and growth in adult rat ventricular cardiomyocytes. J. Mol. Cell Cardiol. 45, 812–820 [DOI] [PubMed] [Google Scholar]
- 15. Sciarretta S., Zhai P., Shao D., Maejima Y., Robbins J., Volpe M., Condorelli G., Sadoshima J. (2012) Rheb is a critical regulator of autophagy during myocardial ischemia: Pathophysiological implications in obesity and metabolic syndrome. Circulation 125, 1134–1146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Liu P., Jenkins N. A., Copeland N. G. (2003) A highly efficient recombineering-based method for generating conditional knockout mutations. Genome Res. 13, 476–484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Nishida K., Yamaguchi O., Hirotani S., Hikoso S., Higuchi Y., Watanabe T., Takeda T., Osuka S., Morita T., Kondoh G., Uno Y., Kashiwase K., Taniike M., Nakai A., Matsumura Y., Miyazaki J., Sudo T., Hongo K., Kusakari Y., Kurihara S., Chien K. R., Takeda J., Hori M., Otsu K. (2004) p38α mitogen-activated protein kinase plays a critical role in cardiomyocyte survival but not in cardiac hypertrophic growth in response to pressure overload. Mol. Cell. Biol. 24, 10611–10620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Tsukiyama-Kohara K., Poulin F., Kohara M., DeMaria C. T., Cheng A., Wu Z., Gingras A. C., Katsume A., Elchebly M., Spiegelman B. M., Harper M. E., Tremblay M. L., Sonenberg N. (2001) Adipose tissue reduction in mice lacking the translational inhibitor 4E-BP1. Nat. Med. 7, 1128–1132 [DOI] [PubMed] [Google Scholar]
- 19. Hara T., Nakamura K., Matsui M., Yamamoto A., Nakahara Y., Suzuki-Migishima R., Yokoyama M., Mishima K., Saito I., Okano H., Mizushima N. (2006) Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 441, 885–889 [DOI] [PubMed] [Google Scholar]
- 20. Hikoso S., Yamaguchi O., Higuchi Y., Hirotani S., Takeda T., Kashiwase K., Watanabe T., Taniike M., Tsujimoto I., Asahi M., Matsumura Y., Nishida K., Nakajima H., Akira S., Hori M., Otsu K. (2004) Pressure overload induces cardiac dysfunction and dilation in signal transducer and activator of transcription 6-deficient mice. Circulation 110, 2631–2637 [DOI] [PubMed] [Google Scholar]
- 21. Nakai A., Yamaguchi O., Takeda T., Higuchi Y., Hikoso S., Taniike M., Omiya S., Mizote I., Matsumura Y., Asahi M., Nishida K., Hori M., Mizushima N., Otsu K. (2007) The role of autophagy in cardiomyocytes in the basal state and in response to hemodynamic stress. Nat. Med. 13, 619–624 [DOI] [PubMed] [Google Scholar]
- 22. Dowling R. J., Zakikhani M., Fantus I. G., Pollak M., Sonenberg N. (2007) Metformin inhibits mammalian target of rapamycin-dependent translation initiation in breast cancer cells. Cancer Res. 67, 10804–10812 [DOI] [PubMed] [Google Scholar]
- 23. Lyons G. E., Schiaffino S., Sassoon D., Barton P., Buckingham M. (1990) Developmental regulation of myosin gene expression in mouse cardiac muscle. J. Cell Biol. 111, 2427–2436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Dowling R. J., Topisirovic I., Alain T., Bidinosti M., Fonseca B. D., Petroulakis E., Wang X., Larsson O., Selvaraj A., Liu Y., Kozma S. C., Thomas G., Sonenberg N. (2010) mTORC1-mediated cell proliferation, but not cell growth, controlled by the 4E-BPs. Science 328, 1172–1176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Koritzinsky M., Magagnin M. G., van den Beucken T., Seigneuric R., Savelkouls K., Dostie J., Pyronnet S., Kaufman R. J., Weppler S. A., Voncken J. W., Lambin P., Koumenis C., Sonenberg N., Wouters B. G. (2006) Gene expression during acute and prolonged hypoxia is regulated by distinct mechanisms of translational control. EMBO J. 25, 1114–1125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. McMullen J. R., Shioi T., Zhang L., Tarnavski O., Sherwood M. C., Dorfman A. L., Longnus S., Pende M., Martin K. A., Blenis J., Thomas G., Izumo S. (2004) Deletion of ribosomal S6 kinases does not attenuate pathological, physiological, or insulin-like growth factor 1 receptor-phosphoinositide 3-kinase-induced cardiac hypertrophy. Mol. Cell. Biol. 24, 6231–6240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Yuan J., Shan Y., Chen X., Tang W., Luo K., Ni J., Wan B., Yu L. (2005) Identification and characterization of RHEBL1, a novel member of Ras family, which activates transcriptional activities of NF-κB. Mol. Biol. Rep. 32, 205–214 [DOI] [PubMed] [Google Scholar]
- 28. Sancak Y., Thoreen C. C., Peterson T. R., Lindquist R. A., Kang S. A., Spooner E., Carr S. A., Sabatini D. M. (2007) PRAS40 is an insulin-regulated inhibitor of the mTORC1 protein kinase. Mol. Cell 25, 903–915 [DOI] [PubMed] [Google Scholar]
- 29. Vander Haar E., Lee S. I., Bandhakavi S., Griffin T. J., Kim D. H. (2007) Insulin signalling to mTOR mediated by the Akt/PKB substrate PRAS40. Nat. Cell Biol. 9, 316–323 [DOI] [PubMed] [Google Scholar]
- 30. Inoki K., Zhu T., Guan K. L. (2003) TSC2 mediates cellular energy response to control cell growth and survival. Cell 115, 577–590 [DOI] [PubMed] [Google Scholar]
- 31. Gwinn D. M., Shackelford D. B., Egan D. F., Mihaylova M. M., Mery A., Vasquez D. S., Turk B. E., Shaw R. J. (2008) AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol. Cell 30, 214–226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Nobukuni T., Joaquin M., Roccio M., Dann S. G., Kim S. Y., Gulati P., Byfield M. P., Backer J. M., Natt F., Bos J. L., Zwartkruis F. J., Thomas G. (2005) Amino acids mediate mTOR/raptor signaling through activation of class 3 phosphatidylinositol 3OH-kinase. Proc. Natl. Acad. Sci. U.S.A. 102, 14238–14243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Li F., Wang X., Capasso J. M., Gerdes A. M. (1996) Rapid transition of cardiac myocytes from hyperplasia to hypertrophy during postnatal development. J. Mol. Cell. Cardiol. 28, 1737–1746 [DOI] [PubMed] [Google Scholar]
- 34. Zhang D., Contu R., Latronico M. V., Zhang J., Zhang J. L., Rizzi R., Catalucci D., Miyamoto S., Huang K., Ceci M., Gu Y., Dalton N. D., Peterson K. L., Guan K. L., Brown J. H., Chen J., Sonenberg N., Condorelli G. (2010) MTORC1 regulates cardiac function and myocyte survival through 4E-BP1 inhibition in mice. J. Clin. Invest. 120, 2805–2816 [DOI] [PMC free article] [PubMed] [Google Scholar]