

Background: ER stress plays a critical role in the pathogenesis of type 2 diabetes, and HHcy induces insulin resistance in adipose tissue.
Results: Hcy induced ER stress markers in adipose tissue both in vivo and in vitro.
Conclusion: HHcy inhibited adipose insulin sensitivity by inducing ER stress, promoting proinflammatory cytokine production, and facilitating macrophage infiltration.
Significance: This work reveals a new mechanism of HHcy-induced insulin resistance.
Keywords: Adipose Tissue, Diabetes, Endoplasmic Reticulum Stress, G Protein-coupled receptors (GPCR), Homocysteine, Inflammation
Abstract
Type 2 diabetes is a chronic inflammatory metabolic disease, the key point being insulin resistance. Endoplasmic reticulum (ER) stress plays a critical role in the pathogenesis of type 2 diabetes. Previously, we found that hyperhomocysteinemia (HHcy) induced insulin resistance in adipose tissue. Here, we hypothesized that HHcy induces ER stress, which in turn promotes insulin resistance. In the present study, the direct effect of Hcy on adipose ER stress was investigated by the use of primary rat adipocytes in vitro and mice with HHcy in vivo. The mechanism and the effect of G protein-coupled receptor 120 (GPR120) were also investigated. We found that phosphorylation or expression of variant ER stress markers was elevated in adipose tissue of HHcy mice. HHcy activated c-Jun N-terminal kinase (JNK), the downstream signal of ER stress in adipose tissue, and activated JNK participated in insulin resistance by inhibiting Akt activation. Furthermore, JNK activated c-Jun and p65, which in turn triggered the transcription of proinflammatory cytokines. Both in vivo and in vitro assays revealed that Hcy-promoted macrophage infiltration aggravated ER stress in adipose tissue. Chemical chaperones PBA and TUDCA could reverse Hcy-induced inflammation and restore insulin-stimulated glucose uptake and Akt activation. Activation of GPR120 reversed Hcy-induced JNK activation and prevented inflammation but not ER stress. Therefore, HHcy inhibited insulin sensitivity in adipose tissue by inducing ER stress, activating JNK to promote proinflammatory cytokine production and facilitating macrophage infiltration. These findings reveal a new mechanism of HHcy in the pathogenesis of insulin resistance.
Introduction
Type 2 diabetes is a chronic inflammatory metabolic disorder. Insulin resistance disturbs the functions of insulin target organs, such as adipose tissue, which is related to glucose metabolism, lipogenesis, and adipokine secretion. Intensive work has demonstrated an association of obesity and insulin resistance (1, 2), and inflammation is one of the novel reasons for insulin resistance in adipose tissue (3). Proinflammatory cytokines, such as tumor necrosis factor α (TNF-α) and interleukin 6 (IL-6), can activate c-Jun N-terminal kinase (JNK), which in turn inhibits Akt activity and impairs the insulin signaling pathway (4).
Recently, various data have suggested that endoplasmic reticulum (ER)4 stress could be involved in insulin resistance (5–7). The ER functions in modifying, folding, and transporting proteins, especially those expressed on the cellular membrane and secreted from cells (8–11). Therefore, a functionally disordered ER significantly impairs the physiological function of adipose tissue (5, 7, 12), which is treated as an important endocrine organ.
Homocysteine (Hcy) is a sulfur-containing amino acid formed during the metabolism of methionine. Hyperhomocysteinemia (HHcy) has been implicated as an independent risk factor of atherosclerosis (13). We have found that Hcy induces the expression and secretion of proinflammatory factors, such as MCP-1 (macrophage chemoattractant protein 1) and IL-8 in human monocytes (14). Epidemiological literature suggests that HHcy is associated with insulin resistance, considered a chronic inflammatory status (15). Previously, we have shown that HHcy, as a potent proinflammatory factor, promotes insulin resistance in mice by inducing the expression and secretion of resistin, a proinflammatory adipokine, from adipose tissue (16). A genetic HHcy animal model, the Tg-I278T cystathionine β-synthase knock-out mouse, has exhibited strong biological phenotypes, including ER stress in the liver and kidney (17). Although Hcy can induce protein damage in the form of Hcy-thiolactone (18) and induce ER stress in endothelial cells (19, 20), cardiomyocytes (21), and atherosclerotic lesion (22), whether HHcy can induce insulin resistance in adipose tissue by provoking ER stress is still unknown. Furthermore, the relationship of the proinflammatory effect of Hcy and ER stress requires investigation.
GPR120 (G protein-coupled receptor 120), a member of the rhodopsin family of G protein-coupled receptors, functions as a receptor for long-chain free fatty acids and mediates the anti-inflammatory and insulin-sensitizing effects of ω-3 fatty acids (23). It may be a potential therapeutic target for metabolic diseases, such as diabetes mellitus and obesity. Because it is highly expressed in adipose tissue (24), activation of GPR120 by its ligand GW9508 may result in similar functions as the unsaturated fatty acids and prevent the effect of HHcy on ER stress and inflammation.
Here we have reported that HHcy induces insulin resistance in vivo in mouse adipose tissue and in vitro in adipocytes by provoking ER stress and inflammation. GW9508 prevents the Hcy effect by inhibiting adipose inflammation rather than ER stress.
EXPERIMENTAL PROCEDURES
Materials
The antibodies (all rabbit source) of anti-phospho-Akt (Ser-473), anti-Akt, anti-phospho-double-stranded RNA-activated protein kinase-like endoplasmic reticulum kinase (PERK) (Thr-980), anti-Bip, anti-phospho-eukaryotic initiation factor 2α (eIF2α), anti-eIF2α, anti-ATF6 (activating transcription factor 6), anti-phospho-eukaryotic initiation factor 5 (eIF5), anti-phospho-JNK, anti-JNK, anti-phospho-c-Jun, anti-c-Jun, and anti-nuclear factor (NF)-κB p65 were from Cell Signaling Technology (Beverly, MA). The antibodies of mouse anti-β-actin, rabbit anti-IκB-α, and rabbit anti-Mac-3 were from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA). IRDyeTM-conjugated affinity-purified anti-rabbit and -mouse IgGs were from Rockland (Gilbertsville, PA). 2-Deoxy-d-[1-3H]glucose was from GE Healthcare. SP600125, 4-phenylbutyric acid (PBA), and taurine-conjugated derivative ursodeoxycholic acid (TUDCA) were from Calbiochem. All other chemicals and drugs were from Sigma.
Animals and Treatments
Male C57BL/6J mice, 6 weeks old, were fed normal mouse chow with or without 1.8 g/liter dl-Hcy (which is more stable than l-Hcy in solution) added to the drinking water for 4 weeks as we described previously (16, 25). For the oral glucose tolerance tests, mice were fasted for 12 h and then were fed glucose (3 g/kg body weight) by gavage. For the insulin tolerance test, mice were fasted for 4 h, and then 2 IU/kg insulin was injected intraperitoneally. The blood was drawn from a cut at the tip of the tail at 0, 30, 60, 90, or 120 min, and blood glucose concentrations were detected immediately. After 3 days' adaptation, mice were sacrificed; blood and epididymal fat pads were taken. All animal protocols were approved by the Animal Care and Use Committee of Peking University.
Plasma Hcy Measurement
Total Hcy level in plasma was quantified by gas chromatography-mass spectrometry (16, 25). The HHcy animal model was confirmed by a plasma Hcy level of 17.59 ± 0.5 μm as compared with 4.49 ± 0.12 μm (Fig. 1A) in control mice.
FIGURE 1.

HHcy promoted insulin resistance in vivo. HHcy animal models were developed by feeding C57BL/6J mice with supplement of Hcy (1.8 g/liter) in drinking water for 4 weeks. A, blood Hcy concentrations; B, body weight; C, ratio of epididymal fat pad and body weight; D, food intake in normal and HHcy mice. E, oral glucose tolerance test. Mice were fed 3 g/kg body weight glucose by gavage. Blood glucose concentrations were detected at 0, 30, 60, 90, and 120 min. F, insulin tolerance test. 1 IU/kg insulin was injected intraperitoneally into each mouse. Blood glucose concentrations were detected at 0, 30, 60, 90, and 120 min. Data are means ± S.E. (error bars) (n = 8). *, p < 0.05 versus normal control mice at the same state.
Analysis of Proinflammatory Cytokines in Plasma
A bead-based immunoassay (BDTM cytometric bead array, BD Biosciences) was used for analysis of soluble cytokines in plasma. Bead populations with distinct fluorescence intensities were coated with antibodies against MCP-1, TNF-α, IL-6, IFN-γ, IL-10, and IL-12 and then mixed together to form the cytometric bead array, which was resolved by flow cytometry (BD Biosciences). We analyzed the concentration in a sample file based on the known concentration values in the set of standards.
Immunohistochemical Analysis
Mouse epididymal fat pads were quickly removed and postfixed in 4% paraformaldehyde, dehydrated, embedded in wax, and sectioned at 8 μm. Paraffin-embedded sections were dewaxed, rehydrated, and rinsed in phosphate-buffered saline (PBS). After being boiled for 10 min in 0.01 mol/liter sodium citrate buffer (pH 6.0), sections were blocked in 5% goat preimmune serum in PBS for 1 h at room temperature and then incubated overnight with rabbit anti-Mac-3 antibody (1:100) and then horseradish peroxidase-conjugated goat anti-rabbit secondary antibody at 37 °C for 1 h and 3,3-diaminobenzidine at room temperature for 10 min. Sections were counterstained with hematoxylin.
Peritoneal Macrophage Isolation
Peritoneal macrophages were obtained from mice by peritoneal lavage with 8 ml of cold PBS with 1 mm EDTA and 10% fetal bovine serum (FBS). Cells were plated at 1.0 × 106 cells/ml RPMI 1640 with 10% FBS. After incubation for 3 h at 37 °C, non-adherent cells were washed away, and adherent cells were collected.
Migration of Murine Peritoneal Macrophages
For a modified Boyden chamber cell migration assay, murine peritoneal macrophages were suspended at 4 × 105/ml in Dulbecco's modified Eagle's medium (DMEM) plus 0.5% FBS. A 50-μl suspension with macrophages was placed in the upper chamber of chemotaxis chambers (Neuroprobe, Pleasanton, CA), and 27 μl of DMEM plus 5% FBS was placed in the lower chamber. After incubation, the cells on the upper surface were removed, and the cells on the underside were fixed and stained. The mean number of migrated cells was counted from five randomly chosen fields under light microscopy in three independent experiments.
Isolation and Culture of Primary Rat Adipocytes
Mature adipocytes were isolated from epididymal fat pads of 8-week-old male Sprague-Dawley rats (180–200 g) as described (16, 26). Packed adipocytes were diluted in serum-free DMEM to generate a 10% (v/v) cell suspension. After being incubated at 37 °C for 1 h, adipocytes were treated with dl-Hcy (500 μm) for 24 h. For inhibition experiments, primary rat adipocytes were pretreated with the indicated inhibitors for 1 h before stimulation with Hcy at 500 μm for 24 h.
Differentiation of Rat Preadipocytes
Adipose precursor cells were isolated from epididymal fat pads of 8-week-old male Sprague-Dawley rats (180–200 g) and differentiated into adipocytes for 3 days in serum-free DMEM plus F-12 (1:1) supplemented with 5 μg/ml insulin, 33 μmol/liter biotin, and 200 pmol/liter triiodothyronine (26). Differentiated adipocytes were then incubated in serum-free DMEM plus F-12 (1:1) for another 2 days before treatment.
Measurement of 2-Deoxy-d-[1-3H]glucose Uptake
Treated differentiated rat adipocytes in a 96-well plate were glucose-starved for 30 min. Insulin (100 nm) was then added for 30 min, and then glucose (0.33 μCi of 2-deoxy-d-[1-3H]glucose/ml) was added in the continued presence of insulin for another 2 h. After terminating the reaction with three washes of ice-cold PBS, the plate was set in a FilterMate harvester (PerkinElmer Life Sciences), and tritium counts were obtained.
Quantitative Real-time PCR Analysis
Total RNA from primary rat adipocytes or mouse epididymal adipose tissue was isolated with the use of TRIzol reagent (Promega, Madison, WI). Total RNA (2 μg) was reverse transcribed with use of a reverse transcription system (Promega). 1 μl of the reaction mixture underwent PCR. The PCR products in each cycle were evaluated by SYBR Green I fluorescence. Primers for mouse or rat MCP-1, TNF-α, IL-6, PAI-1 (plasminogen activator inhibitor 1), and β-actin are listed in Table 1. All amplification reactions involved the Mx3000 multiplex quantitative PCR system (Stratagene, La Jolla, CA). Results were analyzed with use of Stratagene Mx3000 software, and target mRNA levels were normalized to levels of β-actin.
TABLE 1.
List and sequence of primers
| Upstream primer (5′-3′) | Downstream primer (5′-3′) | GenBankTM code | |
|---|---|---|---|
| Mouse MCP-1 | TCTTCCTCCACCACCATGC | TTTGGGACACCTGCTGCTG | NM 011333.3 |
| Rat MCP-1 | TCTGTGCTGACCCCAATAAGG | AAGTGCTTGAGGTGGTTGTGG | NM 031530.1 |
| Mouse TNF-α | CGTCGTAGCAAACCACCAAG | GAGATAGCAAATCGGCTGACG | NM 013693 |
| Rat TNF-α | CCAGGTTCTCTTCAAGGGACA | GTACTTGGGCAGGTTGACCTC | X66539 |
| Mouse IL-6 | AGTTGTGCAATGGCAATTCTG | GGAAATTGGGGTAGGAAGGAC | NM 031168 |
| Mouse PAI-1 | CCTCACCAACATCTTGGATGCT | TGCAGTGCCTGTGCTACAGAGA | M33960 |
| Mouse β-actin | ATCTGGCACCACACCTTC | AGCCAGGTCCAGACGCA | NM 007393 |
| Rat β-actin | GAGACCTTCAACACCCCAGCC | TCGGGGCATCGGAACCGCTCA | NM 031144 |
Preparation of Cytosolic and Nuclear Proteins
Treated primary rat adipocytes were packed and homogenized in ice-cold fractionation buffer. The cell lysates were incubated on ice for 15 min and then centrifuged at 20,000 × g for 30 min at 4 °C. The cytosolic fraction was collected. Nuclear proteins were extracted with the use of NE-PER reagents (Pierce). Protein concentration was determined by use of the BCA protein assay kit (Pierce).
Western Blot Analysis
Proteins underwent SDS-PAGE with 10% running gel and then were transferred to a polyvinylidene fluoride membrane, which was incubated with 0.1% bovine serum albumin in Tris/Tween 20-buffered saline at room temperature for 1 h and then different antibodies at 4 °C for 12 h and then IRDyeTM-conjugated secondary antibody for 1 h. Immunofluorescence bands were detected by the Odyssey infrared imaging system (LI-COR Biosciences, Lincoln, NE).
Statistical Analysis
Data are expressed as mean ± S.E. and were analyzed by use of GraphPad Prism. One-way analysis of variance, Student-Newman-Keul's test (comparisons between multiple groups), or unpaired Student's t test (between two groups) was used as appropriate. p < 0.05 was considered statistically significant.
RESULTS
HHcy Induced Adipose ER Stress in Vivo and in Vitro
Our previous work has demonstrated that HHcy induces insulin resistance in mice (16), which is also confirmed in the present study (Fig. 1, E and F, and Table 2). Consistent with our previous study, the body weight, food intake, adipose tissue content, and other biochemical indexes showed no difference between HHcy mice and control (Fig. 1, B–D, and Table 2). Here, we addressed whether ER stress could be triggered by HHcy in adipose tissue and lead to insulin resistance. We found that the phosphorylation of ER stress markers, such as PERK and eIF2α, was higher in mouse adipose tissue than in controls with HHcy (Fig. 2A). Similar results were observed in primary cultured rat adipocytes. Hcy (500 μm) time-dependently (Fig. 2, B and C) and at 30–500 μm concentration-dependently increased the phosphorylation of PERK and eIF2α (Fig. 2, E and F). Consistently, the chaperone protein Bip was increased with Hcy (Fig. 2, A, D, and G). Hcy also induced ATF6 protein level (Fig. 2, H–J) but not XBP-1 (X box protein 1) splicing (data not shown). Therefore, HHcy induced ER stress in mouse adipose tissue.
TABLE 2.
Biometric and biochemistry indexes for normal and HHcy mice
Data are means ±S.E. (n = 7). TG, triglyceride; TC, total cholesterol; HDL-C, high density lipoprotein cholesterol; LDL-C, low density lipoprotein cholesterol.
| Normal mice | HHcy mice | |
|---|---|---|
| TG (mg/dl) | 81.12 ± 11.21 | 71.36 ± 7.57 |
| TC (mg/dl) | 65.43 ± 2.00 | 66.84 ± 2.38 |
| HDL-C (mg/dl) | 41.36 ± 2.48 | 41.67 ± 3.53 |
| LDL-C (mg/dl) | 12.73 ± 0.61 | 12.63 ± 1.45 |
| Insulin (μIU/ml) | 5.87 ± 0.52 | 9.17 ± 1.03a |
a p < 0.05 compared with control.
FIGURE 2.

HHcy induced adipose ER stress in vivo and in vitro. Shown are the protein level of Bip and phosphorylation (p-) of PERK, eIF2α (A), and ATF6 (H) in adipose tissue of normal and HHcy mice. Right-hand panels show the quantification of protein level. Relative protein levels were normalized to levels for normal mice. Data are means ± S.E. (n = 8). *, p < 0.05 versus control mice. B, C, D, and I, time course effect of Hcy (500 μm); E, F, G, and J, dose-response effect of Hcy on phosphorylation of PERK, eIF2α, ATF6, and Bip in primary cultured adipocytes. Bottom panels show quantification of protein level. Relative protein levels were normalized to that for non-Hcy-treated cells. Data are means ± S.E. (error bars) from four separate experiments. *, p < 0.05 versus Hcy-untreated cells.
HHcy Induced Adipose Inflammation in Vivo
Infiltration of macrophages is involved in the progress of inflammation and insulin resistance in adipose tissue (27, 28). Our previous work has demonstrated that Hcy can induce MCP-1 expression and aggravate atherosclerosis in mice (14, 25). In the present study, we found more Mac-3-positive macrophages in adipose tissue from HHcy than control mice (Fig. 3A). As well, mRNA levels of both MCP-1 and TNF-α in the plasma and adipose tissue (Fig. 3, B and C) were significantly elevated in HHcy mice, with no change in the expression of IL-6 and PAI-1; these results are consistent with those for macrophage infiltration. Finally, the phosphorylation of JNK, the inflammatory signaling and possible ER-stress downstream molecule, was significantly greater in HHcy than control adipose tissue, as was the phosphorylation of p65 and c-Jun, the subunits of inflammatory transcriptional factors NF-κB and AP-1 (Fig. 3D). Therefore, HHcy induced chronic inflammation in mouse adipose tissue.
FIGURE 3.

HHcy induced adipose inflammation in vivo. A, immunohistochemical staining of Mac-3-positive macrophages in epididymal adipose tissue in HHcy mice (right) and normal control mice (left). B, RT-PCR analysis of mRNA levels of MCP-1, TNF-α, IL-6, and PAI-1 in adipose tissue of HHcy and control mice. C, plasma MCP-1 and TNF-α levels in HHcy and control mice. D, phosphorylation (p-) of JNK, p65, and c-Jun in adipose tissue of normal and HHcy mice. Right, quantification of phosphorylation. Relative protein levels were normalized to levels for control mice. Data are means ± S.E. (error bars) (n = 8). *, p < 0.05 versus control mice.
Inhibition of ER Stress Reversed Hcy-induced Adipose Inflammation
Because adipose inflammation plays an important role in insulin resistance and might be the putative downstream pathway of ER stress, we investigated the inhibitory effect of chemical chaperones, PBA (5 mm) and TUDCA (200 μm), on Hcy-induced inflammatory cytokine production. Inhibiting ER stress with PBA and TUDCA or inhibiting JNK expression with SP600125 (20 μm) reversed MCP-1 and TNF-α production (Fig. 4, A and B), JNK activation and IκB degradation induced by Hcy in adipocytes (Fig. 4C). Therefore, HHcy promoted adipose inflammation via inducing ER stress and JNK activation.
FIGURE 4.

Inhibition of ER stress reversed Hcy-induced adipose inflammation. A and B, attenuation of Hcy (500 μm)-induced MCP-1 and TNFα expression by PBA (5 mm), TUDCA (200 μm), and SP600125 (20 μm) in primary rat adipocytes. Relative mRNA levels were normalized to levels for untreated cells. Data are means ± S.E. (error bars) from four separate experiments. C, attenuation of Hcy (500 μm)-stimulated JNK phosphorylation (p-) and IκB degradation by PBA, TUDCA, and SP60125 in primary rat adipocytes. The bottom panels show quantification of protein level. Relative protein levels were normalized to that for non-treated cells. *, p < 0.01 versus untreated cells; #, p < 0.01 versus Hcy treatment alone.
Macrophage Infiltration Aggravated Hcy-induced Adipose ER Stress
Infiltrating macrophages can aggravate local inflammation in adipose tissue (27, 28). Therefore, we investigated whether infiltrating macrophages aggravated Hcy-induced ER stress. Macrophages and primary isolated adipocytes were co-cultured with or without Hcy stimulation (500 μm), and then ER stress markers in adipocytes were detected. The data showed that Hcy-induced phosphorylation of PERK and eIF2α was enhanced in adipocytes co-cultured with macrophages (Fig. 5). Thus, infiltrating macrophages might aggravate Hcy-induced ER stress in the microenvironment (niche) of adipose tissue.
FIGURE 5.
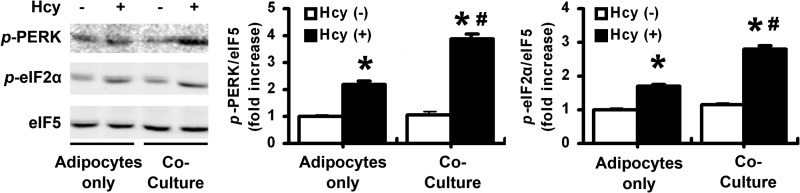
Macrophage infiltration aggravated Hcy-induced adipose ER stress. Effect of Hcy on phosphorylation (p-) of PERK and eIF2α in primary cultured adipocytes co-cultured with or without peritoneal macrophages. The right-hand panels show quantification of phosphorylation. Relative protein levels were normalized to that for non-Hcy-treated adipocytes. Data are means ± S.E. (error bars) from four separate experiments. *, p < 0.05 versus Hcy-untreated cells; #, p < 0.01 versus adipocytes only.
Inhibiting ER Stress or Inflammation Reversed Hcy-induced Insulin Resistance
Because we demonstrated that HHcy induced insulin resistance and ER stress in mice, we wondered whether insulin resistance results from HHcy-induced ER stress in adipose tissue. To address that question, we used the chemical chaperones PBA (5 mm) and TUDCA (200 μm) to determine whether inhibiting ER stress would reverse Hcy-impaired insulin signaling. The results showed that Hcy (500 μm) impaired insulin (100 nm)-stimulated Akt phosphorylation, which could be reversed by preadministration with PBA or TUDCA (Fig. 6A). Consistently, PBA and TUDCA reversed Hcy-impaired glucose uptake with insulin stimulation in rat adipocytes (Fig. 6C). Administration of SP600125 (20 μm), the inhibitor of JNK signaling, gave results similar to those with chemical chaperones (Fig. 6, B and D). Therefore, Hcy induced insulin resistance in adipose tissue, at least in part by inducing ER stress and inflammation.
FIGURE 6.

Inhibiting ER stress or inflammation reversed Hcy-induced insulin resistance. A and C, reversal of Hcy-impaired insulin-stimulated Akt phosphorylation (p-) by PBA (5 mm), TUDCA (200 μm), and SP600125 (20 μm) in primary rat adipocytes. The bottom panels show quantification of phosphorylation. Relative protein levels were normalized to that for non-insulin- and non-Hcy-treated cells. B and D, reversal of Hcy-impaired insulin-stimulated glucose uptake by PBA, TUDCA, and SP60125 in primary rat adipocytes. Data are means ± S.E. (error bars) from four separate experiments. *, p < 0.01 versus insulin treatment alone; #, p < 0.01 versus Hcy treatment alone.
Activation of GPR120 Reversed Hcy-induced Insulin Resistance by Antagonizing Hcy-induced Inflammation but Not ER Stress
According to above data, Hcy induced adipose inflammation and promoted insulin resistance. Recently, GPR120 has been found as a new receptor of long-chain free fatty acids, and it is highly expressed in adipose tissue. Furthermore, activation of GPR120 has anti-inflammatory and insulin-sensitizing effects (23, 24). Therefore, we detected whether the effect of Hcy could be reversed by GPR120. Not surprisingly, the activation of GPR120 by GW9508 (10–100 μm) reversed Hcy-impaired Akt activation and glucose uptake (Fig. 7, A and B). As well, activation of GPR120 with GW9508 inhibited the Hcy-augmented JNK activation and IκB degradation (Fig. 7C) as well as mRNA expression of MCP-1 and TNF-α in adipocytes (Fig. 7, D and E), which suggests to us that activation of GPR120 reverses Hcy-induced inflammation in adipocytes. Consistent with this, by Boyden chamber assay, we found that compared with Hcy treatment only, administration of GW9508 (100 μm) significantly reduced the number of migrating macrophages to the adipocytes (Fig. 7F). Therefore, activation of GPR120 reversed Hcy-induced insulin resistance and inflammation.
FIGURE 7.

Activation of GPR120 reversed Hcy-induced insulin resistance by antagonizing Hcy-induced inflammation but not ER stress. A, reversal of Hcy-impaired insulin-stimulated Akt phosphorylation (p-) by GW9508 in primary rat adipocytes. The bottom panel shows quantification of phosphorylation. Relative protein levels were normalized to that for non-insulin- and non-Hcy-treated cells. B, reversal of Hcy-impaired, insulin-stimulated glucose uptake by GW9508 in primary rat adipocytes. Data are means ± S.E. (error bars) from four separate experiments. *, p < 0.01 versus insulin treatment alone; #, p < 0.01 versus Hcy treatment alone. C, attenuation of Hcy-stimulated JNK phosphorylation and IκB degradation by GW9508 in primary rat adipocytes. The bottom panels show quantification of protein level. Relative protein levels were normalized to that for non-treated cells. D and E, attenuation of Hcy-induced MCP-1 and TNFα expression by GW9508 in primary rat adipocytes. Relative mRNA levels were normalized to levels for untreated cells. Data are means ± S.E. from four separate experiments. *, p < 0.01 versus untreated cells; #, p < 0.01 versus Hcy treatment alone. F, modified Boyden chamber assay of migration of peritoneal macrophages co-cultured with adipocytes under Hcy with or without GW9508. Right, results of macrophage migration. *, p < 0.01 versus macrophages only; #, p < 0.01 versus Hcy-untreated cells; &, p < 0.01 versus Hcy treatment alone. G, effect of PBA and TUDCA on Hcy-stimulated JNK phosphorylation in primary rat adipocytes. H, effect of SP600125 on Hcy-stimulated eIF2α phosphorylation in primary rat adipocytes. I, effect of TUDCA and GW9508 on Hcy-stimulated JNK and eIF2α phosphorylation in primary rat adipocytes. The bottom panels show quantification of phosphorylation. Relative protein levels were normalized to that for non-treated cells. *, p < 0.01 versus untreated cells; #, p < 0.01 versus Hcy treatment alone.
Subsequently, we addressed the target of GPR120 activation in the signaling pathway and confirmed the up- and downstream relation of Hcy-induced ER stress and inflammation. Pretreatment with SP600125 (20 μm) or GW9508 (100 μm) did not reverse the Hcy-induced phosphorylation of eIF2α (Fig. 7, H and I), whereas TUDCA and GW9508 pretreatment reversed the Hcy-augmented JNK activation (Fig. 7, G and I). Thus, Hcy-induced ER stress was upstream of JNK activation, and activation of GRP120 improved insulin sensitivity by resisting the Hcy effect through inhibiting inflammation but not ER stress.
DISCUSSION
We demonstrate that Hcy promotes insulin resistance by inducing adipose ER stress and downstream inflammation in mice. This conclusion is supported by the following observations: 1) HHcy induces ER stress in vivo in mouse adipose tissue and in vitro in adipocytes; 2) Hcy activates inflammatory signal molecules in mouse adipose tissue; 3) Hcy facilitates the migration of macrophages into adipose tissue to enhance inflammation; 4) inhibition of ER stress reverses the Hcy-induced insulin resistance; and 5) activation of GPR120 reverses the Hcy-induced insulin resistance by inhibiting inflammation but not Hcy-induced ER stress.
As we have reported previously (16), HHcy induces insulin resistance in vivo and in vitro. Here, we have shown that this Hcy effect is via inducing ER stress because inhibitors of ER stress can recover the insulin-stimulated activation of Akt and glucose uptake, impaired by Hcy (Fig. 6, A and B). Both in vivo and in vitro assays show that ER stress markers, such as phospho-PERK, phospho-eIF2α, and the chaperone protein Bip are significantly up-regulated by Hcy (Fig. 2). Other pathways can be triggered by the unfolded protein response (29, 30), and we have found that Hcy can also influence ATF6 pathways (Fig. 2, H–J) but not the iron-response element 1α (IRE1α)-XBP-1 pathway. Here, we have focused on the PERK-eIF2α pathway because Hcy induces acute responses in cells, and the PERK-eIF2α pathway is the fastest responding to the unfolded protein response (31, 32). In addition, Hcy can induce oxidative stress through the generation of reactive oxygen species (16, 25), which is commonly thought to be the upstream signal of ER stress. We have also found that elimination of reactive oxygen species mitigated Hcy-induced ER stress in adipocytes.5
Many studies have demonstrated that Hcy is a potent proinflammatory factor that aggravates the pathogenesis of chronic inflammatory diseases, such as atherosclerosis (14). Because a number of studies have shown the link between inflammation and insulin resistance (4, 33, 34), we investigated whether Hcy induces inflammation in adipose tissue as a result of Hcy-induced ER stress. We have found the mRNA expression of MCP-1 and TNF-α elevated in adipose tissue (Fig. 3B) and in circulation (Fig. 3C). ER stress inhibitors reverse the Hcy-increased expression of MCP-1 and TNF-α (Fig. 4, A and B). Consistent with Hcy-increased MCP-1 expression, Mac-3-positive macrophages are increased in adipose tissue (Fig. 3A). An in vitro assay demonstrated that Hcy facilitates adipocyte-mediated migration of macrophages (Fig. 7F), and the migrated macrophages aggravate Hcy-induced ER stress in adipocytes (Fig. 5).
Infiltration of macrophages into lesions contributes to many pathogenic processes of inflammatory diseases (35, 36). A number of studies have demonstrated two phenotypes of macrophage polarization: M1 and M2 macrophages (37, 38). M1 macrophages are characterized by high expression of MCP-1 and TNF-α, and M2 macrophages are characterized by expression of IL-4 and IL-10. This specific expression pattern agrees with the functions of M1 macrophages aggravating but M2 macrophages relieving inflammation. During pathogenesis, tissue damage causes the release of cytokines, which recruit different phenotypes of macrophages (35). For example, MCP-1 recruits M1 macrophages because they express CCR2, the MCP-1 receptor. Although we did not find Hcy directly participating in macrophage polarization, co-culture with adipocytes significantly increases M1 macrophage markers in rat adipocytes (data not shown), which indicates that adipocytes promote macrophage polarization into M1, which requires further investigation.
JNK is a crucial molecule involved in inflammation and cytokine production. It activates the transcription factor AP-1 and then triggers the transcription of inflammatory cytokines (39). This observation is consistent with our finding that Hcy induces JNK activation (Figs. 3D and 4C) and MCP-1 and TNF-α expression in adipose tissue. The increased inflammatory cytokine expression is via Hcy-induced JNK activation (Fig. 4, A and B). Furthermore, JNK contributes to insulin resistance by impairing insulin signaling (4, 40) because inhibition of JNK reversed insulin-stimulated Akt activity and glucose uptake in Hcy-treated adipocytes (Fig. 6, C and D). ER stress is usually considered the result of inflammatory stimulation; we have found that inhibiting ER stress abolished Hcy-induced JNK phosphorylation (Fig. 7G), so Hcy-induced ER stress activated JNK, which then triggers inflammation and insulin resistance, as was found previously (5–7).
Hcy, the intermediate product of methionine, induces insulin resistance through adipose ER stress and inflammation, so finding an intrinsic antagonist of Hcy is important. One of the putative candidates is long-chain free fatty acids, which induce various cellular responses by inducing GPR120. Free fatty acid induction of GPR120 resulted in an elevated level of [Ca2+]i and activation of the ERK cascade (23). The high expression of GPR120 found in mature adipocytes and macrophages indicates that GPR120 might have an important role in these cell types. Long-chain free fatty acids and synthetic agonists, such as GW9508, have potent anti-inflammatory effects through GPR120. Also, β-arrestin 2 is associated with ligand-stimulated GPR120 and participates in the downstream signaling mechanisms. The mechanism of GPR120-mediated anti-inflammation involves inhibiting TAK1 (transforming growth factor-β activated kinase 1), upstream of JNK and NF-κB, through an effect dependent on β-arrestin 2/TAB1 (TAK1-binding protein 1). Because Hcy inhibits the expression of β-arrestin 2 in primary cultured adipocytes,5 GPR120 might exert its anti-Hcy effects by restoring the β-arrestin 2 level, which requires further clarification. Although the activation of GPR120 abolishes Hcy-induced adipose inflammation and insulin resistance (Fig. 7, A–F), the ER stress induced by Hcy is not released (Fig. 7I), so ER stress might be upstream of inflammation in Hcy-induced insulin resistance in adipose tissue. The adipose ER stress may further cause the release of free fatty acids and inflammatory adipokines, inducing systemic insulin resistance, which requires further investigation.
In conclusion, we have demonstrated that HHcy induces ER stress and then provokes adipose inflammation to affect insulin sensitivity. GPR120 reverses Hcy-induced insulin resistance by antagonizing Hcy-induced inflammation. These findings reveal a new mechanism of HHcy in the pathogenesis of insulin resistance and provide further evidence of the anti-inflammatory effect of GPR120 in metabolic disorders.
This work was supported by National Natural Science Foundation of China Grants 81070683, 81121061, and 31230035 (to X. W.) and 81170795 and 30971085 (to Y. L.); Major National Basic Research Program of China Grants 2011CB503904 and 2010CB912504 (to X. W.); Program for New Century Excellent Talents in University Grant NCET-10-0183 (to Y. L.); and Beijing Natural Science Foundation Grant 7112080 (to Y. L.).
Y. Li, H. Zhang, C. Jiang, M. Xu, Y. Pang, J. Feng, X. Xiang, W. Kong, G. Xu, Y. Li, and X. Wang, unpublished data.
- ER
- endoplasmic reticulum
- Hcy
- homocysteine
- HHcy
- hyperhomocysteinemia
- PERK
- double-stranded RNA-activated protein kinase-like endoplasmic reticulum kinase
- eIF2α
- eukaryotic initiation factor 2α
- PBA
- 4-phenylbutyric acid
- TUDCA
- taurine-conjugated derivative ursodeoxycholic acid.
REFERENCES
- 1. Vandanmagsar B., Youm Y. H., Ravussin A., Galgani J. E., Stadler K., Mynatt R. L., Ravussin E., Stephens J. M., Dixit V. D. (2011) The NLRP3 inflammasome instigates obesity-induced inflammation and insulin resistance. Nat. Med. 17, 179–188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Tan C. K., Leuenberger N., Tan M. J., Yan Y. W., Chen Y., Kambadur R., Wahli W., Tan N. S. (2011) Smad3 deficiency in mice protects against insulin resistance and obesity induced by a high-fat diet. Diabetes 60, 464–476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Mehta N. N., McGillicuddy F. C., Anderson P. D., Hinkle C. C., Shah R., Pruscino L., Tabita-Martinez J., Sellers K. F., Rickels M. R., Reilly M. P. (2010) Experimental endotoxemia induces adipose inflammation and insulin resistance in humans. Diabetes 59, 172–181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hirosumi J., Tuncman G., Chang L., Görgün C. Z., Uysal K. T., Maeda K., Karin M., Hotamisligil G. S. (2002) A central role for JNK in obesity and insulin resistance. Nature 420, 333–336 [DOI] [PubMed] [Google Scholar]
- 5. Ozcan U., Yilmaz E., Ozcan L., Furuhashi M., Vaillancourt E., Smith R. O., Görgün C. Z., Hotamisligil G. S. (2006) Chemical chaperones reduce ER stress and restore glucose homeostasis in a mouse model of type 2 diabetes. Science 313, 1137–1140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kaneto H., Matsuoka T. A., Nakatani Y., Kawamori D., Miyatsuka T., Matsuhisa M., Yamasaki Y. (2005) Oxidative stress, ER stress, and the JNK pathway in type 2 diabetes. J. Mol. Med. 83, 429–439 [DOI] [PubMed] [Google Scholar]
- 7. Ozcan U., Cao Q., Yilmaz E., Lee A. H., Iwakoshi N. N., Ozdelen E., Tuncman G., Görgün C., Glimcher L. H., Hotamisligil G. S. (2004) Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science 306, 457–461 [DOI] [PubMed] [Google Scholar]
- 8. Buchberger A., Bukau B., Sommer T. (2010) Protein quality control in the cytosol and the endoplasmic reticulum. Brothers in arms. Mol. Cell 40, 238–252 [DOI] [PubMed] [Google Scholar]
- 9. Bukau B., Weissman J., Horwich A. (2006) Molecular chaperones and protein quality control. Cell 125, 443–451 [DOI] [PubMed] [Google Scholar]
- 10. Hampton R. Y. (2000) ER stress response: getting the UPR hand on misfolded proteins. Curr. Biol. 10, R518–521 [DOI] [PubMed] [Google Scholar]
- 11. Kaufman R. J., Scheuner D., Schröder M., Shen X., Lee K., Liu C. Y., Arnold S. M. (2002) The unfolded protein response in nutrient sensing and differentiation. Nat. Rev. Mol. Cell Biol. 3, 411–421 [DOI] [PubMed] [Google Scholar]
- 12. Deng J., Liu S., Zou L., Xu C., Geng B., Xu G. (2012) Lipolysis response to endoplasmic reticulum stress in adipose cells. J. Biol. Chem. 287, 6240–6249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Robinson K., Gupta A., Dennis V., Arheart K., Chaudhary D., Green R., Vigo P., Mayer E. L., Selhub J., Kutner M., Jacobsen D. W. (1996) Hyperhomocysteinemia confers an independent increased risk of atherosclerosis in end-stage renal disease and is closely linked to plasma folate and pyridoxine concentrations. Circulation 94, 2743–2748 [DOI] [PubMed] [Google Scholar]
- 14. Zeng X., Dai J., Remick D. G., Wang X. (2003) Homocysteine-mediated expression and secretion of monocyte chemoattractant protein-1 and interleukin-8 in human monocytes. Circ. Res. 93, 311–320 [DOI] [PubMed] [Google Scholar]
- 15. Buysschaert M., Dramais A. S., Wallemacq P. E., Hermans M. P. (2000) Hyperhomocysteinemia in type 2 diabetes. Relationship to macroangiopathy, nephropathy, and insulin resistance. Diabetes Care 23, 1816–1822 [DOI] [PubMed] [Google Scholar]
- 16. Li Y., Jiang C., Xu G., Wang N., Zhu Y., Tang C., Wang X. (2008) Homocysteine upregulates resistin production from adipocytes in vivo and in vitro. Diabetes 57, 817–827 [DOI] [PubMed] [Google Scholar]
- 17. Gupta S., Kühnisch J., Mustafa A., Lhotak S., Schlachterman A., Slifker M. J., Klein-Szanto A., High K. A., Austin R. C., Kruger W. D. (2009) Mouse models of cystathionine β-synthase deficiency reveal significant threshold effects of hyperhomocysteinemia. FASEB J. 23, 883–893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Jakubowski H. (2003) Homocysteine-thiolactone and S-nitroso-homocysteine mediate incorporation of homocysteine into protein in humans. Clin. Chem. Lab. Med. 41, 1462–1466 [DOI] [PubMed] [Google Scholar]
- 19. Roybal C. N., Yang S., Sun C. W., Hurtado D., Vander Jagt D. L., Townes T. M., Abcouwer S. F. (2004) Homocysteine increases the expression of vascular endothelial growth factor by a mechanism involving endoplasmic reticulum stress and transcription factor ATF4. J. Biol. Chem. 279, 14844–14852 [DOI] [PubMed] [Google Scholar]
- 20. Hossain G. S., van Thienen J. V., Werstuck G. H., Zhou J., Sood S. K., Dickhout J. G., de Koning A. B., Tang D., Wu D., Falk E., Poddar R., Jacobsen D. W., Zhang K., Kaufman R. J., Austin R. C. (2003) TDAG51 is induced by homocysteine, promotes detachment-mediated programmed cell death, and contributes to the cevelopment of atherosclerosis in hyperhomocysteinemia. J. Biol. Chem. 278, 30317–30327 [DOI] [PubMed] [Google Scholar]
- 21. Wei H., Zhang R., Jin H., Liu D., Tang X., Tang C., Du J. (2010) Hydrogen sulfide attenuates hyperhomocysteinemia-induced cardiomyocytic endoplasmic reticulum stress in rats. Antioxid. Redox. Signal. 12, 1079–1091 [DOI] [PubMed] [Google Scholar]
- 22. Zhou J., Werstuck G. H., Lhoták S., de Koning A. B., Sood S. K., Hossain G. S., Møller J., Ritskes-Hoitinga M., Falk E., Dayal S., Lentz S. R., Austin R. C. (2004) Association of multiple cellular stress pathways with accelerated atherosclerosis in hyperhomocysteinemic apolipoprotein E-deficient mice. Circulation 110, 207–213 [DOI] [PubMed] [Google Scholar]
- 23. Hirasawa A., Tsumaya K., Awaji T., Katsuma S., Adachi T., Yamada M., Sugimoto Y., Miyazaki S., Tsujimoto G. (2005) Free fatty acids regulate gut incretin glucagon-like peptide-1 secretion through GPR120. Nat. Med. 11, 90–94 [DOI] [PubMed] [Google Scholar]
- 24. Oh D. Y., Talukdar S., Bae E. J., Imamura T., Morinaga H., Fan W., Li P., Lu W. J., Watkins S. M., Olefsky J. M. (2010) GPR120 is an ω-3 fatty acid receptor mediating potent anti-inflammatory and insulin-sensitizing effects. Cell 142, 687–698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Dai J., Li W., Chang L., Zhang Z., Tang C., Wang N., Zhu Y., Wang X. (2006) Role of redox factor-1 in hyperhomocysteinemia-accelerated atherosclerosis. Free Radic. Biol. Med. 41, 1566–1577 [DOI] [PubMed] [Google Scholar]
- 26. He J., Jiang H., Tansey J. T., Tang C., Pu S., Xu G. (2006) Calyculin and okadaic acid promote perilipin phosphorylation and increase lipolysis in primary rat adipocytes. Biochim. Biophys. Acta 1761, 247–255 [DOI] [PubMed] [Google Scholar]
- 27. Shoelson S. E., Lee J., Goldfine A. B. (2006) Inflammation and insulin resistance. J. Clin. Invest. 116, 1793–1801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wellen K. E., Hotamisligil G. S. (2003) Obesity-induced inflammatory changes in adipose tissue. J. Clin. Invest. 112, 1785–1788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Yoshida H., Matsui T., Yamamoto A., Okada T., Mori K. (2001) XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell 107, 881–891 [DOI] [PubMed] [Google Scholar]
- 30. Lee K., Tirasophon W., Shen X., Michalak M., Prywes R., Okada T., Yoshida H., Mori K., Kaufman R. J. (2002) IRE1-mediated unconventional mRNA splicing and S2P-mediated ATF6 cleavage merge to regulate XBP1 in signaling the unfolded protein response. Genes Dev. 16, 452–466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Yamaguchi Y., Larkin D., Lara-Lemus R., Ramos-Castañeda J., Liu M., Arvan P. (2008) Endoplasmic reticulum (ER) chaperone regulation and survival of cells compensating for deficiency in the ER stress response kinase, PERK. J. Biol. Chem. 283, 17020–17029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wolfson J. J., May K. L., Thorpe C. M., Jandhyala D. M., Paton J. C., Paton A. W. (2008) Subtilase cytotoxin activates PERK, IRE1 and ATF6 endoplasmic reticulum stress-signalling pathways. Cell Microbiol. 10, 1775–1786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Yang H., Youm Y. H., Vandanmagsar B., Ravussin A., Gimble J. M., Greenway F., Stephens J. M., Mynatt R. L., Dixit V. D. (2010) Obesity increases the production of proinflammatory mediators from adipose tissue T cells and compromises TCR repertoire diversity. Implications for systemic inflammation and insulin resistance. J. Immunol. 185, 1836–1845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Arkan M. C., Hevener A. L., Greten F. R., Maeda S., Li Z. W., Long J. M., Wynshaw-Boris A., Poli G., Olefsky J., Karin M. (2005) IKK-β links inflammation to obesity-induced insulin resistance. Nat. Med. 11, 191–198 [DOI] [PubMed] [Google Scholar]
- 35. Kanda H., Tateya S., Tamori Y., Kotani K., Hiasa K., Kitazawa R., Kitazawa S., Miyachi H., Maeda S., Egashira K., Kasuga M. (2006) MCP-1 contributes to macrophage infiltration into adipose tissue, insulin resistance, and hepatic steatosis in obesity. J. Clin. Invest. 116, 1494–1505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Bourlier V., Bouloumie A. (2009) Role of macrophage tissue infiltration in obesity and insulin resistance. Diabetes Metab. 35, 251–260 [DOI] [PubMed] [Google Scholar]
- 37. Mantovani A., Sica A., Locati M. (2005) Macrophage polarization comes of age. Immunity 23, 344–346 [DOI] [PubMed] [Google Scholar]
- 38. Lumeng C. N., Bodzin J. L., Saltiel A. R. (2007) Obesity induces a phenotypic switch in adipose tissue macrophage polarization. J. Clin. Invest. 117, 175–184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Morse D., Pischke S. E., Zhou Z., Davis R. J., Flavell R. A., Loop T., Otterbein S. L., Otterbein L. E., Choi A. M. (2003) Suppression of inflammatory cytokine production by carbon monoxide involves the JNK pathway and AP-1. J. Biol. Chem. 278, 36993–36998 [DOI] [PubMed] [Google Scholar]
- 40. Masharani U. B., Maddux B. A., Li X., Sakkas G. K., Mulligan K., Schambelan M., Goldfine I. D., Youngren J. F. (2011) Insulin resistance in non-obese subjects is associated with activation of the JNK pathway and impaired insulin signaling in skeletal muscle. PLoS One 6, e19878. [DOI] [PMC free article] [PubMed] [Google Scholar]