

Background: Molecular mechanisms of plasticity at GABAergic synapses are presently unclear.
Results: ERK phosphorylates gephyrin at Ser-268 to regulate size of gephyrin postsynaptic scaffold and strength of GABAergic transmission. Ser-268 phosphorylation by ERK is functionally coupled to Ser-270 phosphorylation by GSK3β to determine calpain action on gephyrin.
Conclusion: Multiple signaling cascades regulate gephyrin postsynaptic clustering.
Significance: Dynamic modulation of gephyrin clustering by phosphorylation regulates GABAergic synaptic transmission.
Keywords: ERK, GABA Receptors, Glycogen Synthase Kinase 3, Phosphorylation, Protease, Inhibitory Neurotransmission, Postsynaptic Density
Abstract
Molecular mechanisms of plasticity at GABAergic synapses are currently poorly understood. To identify signaling cascades that converge onto GABAergic postsynaptic density proteins, we performed MS analysis using gephyrin isolated from rat brain and identified multiple novel phosphorylation and acetylation residues on gephyrin. Here, we report the characterization of one of these phosphoresidues, Ser-268, which when dephosphorylated leads to the formation of larger postsynaptic scaffolds. Using a combination of mutagenesis, pharmacological treatment, and biochemical assays, we identify ERK as the kinase phosphorylating Ser-268 and describe a functional interaction between residues Ser-268 and Ser-270. We further demonstrate that alterations in gephyrin clustering via ERK modulation are reflected by amplitude and frequency changes in miniature GABAergic postsynaptic currents. We unravel novel mechanisms for activity- and ERK-dependent calpain action on gephyrin, which are likely relevant in the context of cellular signaling affecting GABAergic transmission and homeostatic synaptic plasticity in pathology.
Introduction
Long term plasticity of glutamatergic synapses, such as Long Term Potentiation (LTP) and Long Term Depression (LTD), is widely accepted as a cellular mechanism underlying learning and memory. These forms of plasticity involve synaptic scaling and structural remodeling, enabling stable changes in information processing in specific neuronal circuits. Proteins of the postsynaptic density play a key role in these adaptive changes, especially by regulating trafficking and function of structural and signaling molecules (1, 3). Two major signaling cascades involved in long lasting synaptic plasticity are the mTOR (mammalian target of rapamycin) and the MAP kinase pathways (4), with ERK1/2 being essential for early structural adaptations and changes in gene expression underlying multiple forms of learning and memory.
At GABAergic synapses, studies of plasticity mechanisms have focused mainly on the modulation of GABAA receptors (GABAAR) (5, 6).3 Several protein kinases are known to modulate GABAergic transmission by affecting GABAAR channel gating properties or cell surface trafficking (7, 8). Similar mechanisms also operate at glycinergic synapses, where, for instance, phosphorylation of a specific residue in the glycine receptor β subunit affects surface mobility and synaptic function (9). With a focus on the scaffolding molecule gephyrin, we have recently demonstrated that regulation of a single gephyrin residue (Ser-270) via GSK3β-mediated phosphorylation induces the formation of new, functional GABAergic synapses (2). Although several studies identified gephyrin as a target for serine/threonine-directed phosphorylation (10–12), a recent study detailed 18 different phosphorylation residues on gephyrin (13), demonstrating the importance of gephyrin phosphorylation for its varied functions. Structural changes affecting the postsynaptic scaffold organized by gephyrin have started gaining attention, with studies demonstrating neuronal activity-dependent dynamics of gephyrin clustering in vivo (14–16). Given that phosphorylation and intracellular Ca2+ rises make gephyrin susceptible to proteolysis by calpain (2), neuronal activity-driven gephyrin dynamics could very likely be phosphorylation-dependent.
Hence, to uncover novel signaling pathways converging on gephyrin and regulating calpain 1 action on gephyrin, we performed MS analysis of gephyrin immunoprecipitated from rat brain homogenate and identified novel phosphorylation and acetylation sites on gephyrin. Site-directed mutagenesis and screening of different gephyrin mutants for alterations in postsynaptic clustering phenotypes allowed us to identify the Ser-268 residue as a novel phosphorylation site on gephyrin important for scaling (up or down) GABAergic transmission. Using a multidisciplinary approach, we demonstrate that ERK modulates the Ser-268 phosphorylation status, thereby influencing GSK3β-mediated phosphorylation of the closely related Ser-270 residue to regulate gephyrin cluster size and density, respectively. Furthermore, our data shows how neuronal activity-dependent down-regulation of gephyrin clusters is orchestrated via ERK and calpain activation, providing mechanistic insights into plasticity-related changes at GABAergic synapses.
EXPERIMENTAL PROCEDURES
Plasmids
eGFP-gephyrin P1 variant has been described earlier (17). eGFP-gephyrin (S268A, S268E, S268A/S270A, S268E/S270E, and S268A/S270E) mutants were created in eGFP-gephyrin template using site-directed mutagenesis PCR and sequence-confirmed. Gephyrin 3′-UTR shRNA and the control shRNA-3m (18) have been described before. pCR3-FLAG-gephyrin (FLAG-G, FLAG-GC, and FLAG-E) has been described earlier (2). pCR3-FLAG-gephyrin (S268A, S268E, S270A, and S270E) mutants were created using site-directed mutagenesis and sequence-confirmed. pMT-HA-ERK1 (Addgene no. 12656) and pcDNA3-HA-ERK2 WT (Addgene no. 8974) were obtained from Addgene. Myc-CAST, HA-calpain 1, and bacterial expression of STREP-Gephyrin have been described earlier (2).
Primary Neuron Culture Treatments and Staining
All animal experiments were approved by the cantonal veterinary office of Zurich. Primary hippocampal mixed cultures were prepared from embryonic day 17 rat embryos and maintained in medium containing minimum essential medium, Nu serum (15%), B27 supplement, HEPES (pH 7.1; 15 mm), glucose (0.45%), sodium pyruvate, and l-glutamine. The cells were transfected after 11 DIV with 1 μg of total plasmid concentration using a combination of Lipofectamine 2000 and CombiMag (OZ Biosciences) as described (19). The transfected cells were fixed in 4% paraformaldehyde for 10 min at room temperature, permeabilized with 0.1% Triton X-100 in PBS containing 10% normal goat serum, and stained using desired primary antibodies for 60 min at room temperature, followed by three 5-min 1- PBS washes; goat anti-Alexa 488 (Molecular Probes), goat anti-Cy3, and goat anti-Cy5 (Jackson Immunoresearch) were used to label the proteins for immunofluorescence either 4 or 7 days post-transfection (referred to as 11 + 4 or 11 + 7 DIV). α2-GABAAR subunit staining was done live for 90 min before proceeding with fixation and permeabilization of cells and proceeding with staining for gephyrin and/or synapsin-1. Pharmacological treatment using 25 μm ERK1/2 inhibitor PD98059 (Calbiochem) or vehicle control Me2SO (equal volume) were performed overnight, or for 3 h, followed by fixation and staining. GSK3β inhibition was done using 25 μm inhibitor GSK3βIX (Calbiochem) overnight, and calpain was inhibited using 30 μm inhibitor MDL28170 (Calbiochem) overnight. Neurons were stimulated using 40 mm KCl for 4 min prior to fixation and staining.
Mouse anti-gephyrin (1:1000; clones mAb7a or 3B11; Synaptic Systems) and rabbit anti-Ser(P)-268 antibody (1:500) were raised against the phosphopeptide DTASLSTTPsESPR (Genscript Corporation) and affinity-purified; rabbit anti-synapsin 1 (1:3000; Synaptic Systems), rabbit anti-VGAT (1:3000; Synaptic Systems), mouse anti-phospho-ERK1/2 (1:10000; Sigma), mouse anti-Myc (1:5000; Roche Applied Science) were used in the present study. Guinea pig antibody anti-α2-GABAAR subunit (1:10000) was made in house as described earlier (20).
HEK-293 Cell Cultures
HEK293-T cells were cultured at 37 °C in 5% CO2 in DMEM supplemented with 10% FBS. The HEK cells were transfected 16 h after plating with 1 μg of each plasmid using the transfection agent polyethylenimine (Polysciences Inc.) as suggested by the vendor. The transfected cells were lysed for biochemical analysis 24 h post-transfection using EBC buffer.
To increase the levels of phosphoERK1/2 in HEK cells, transfection of FLAG-gephyrin S268A was performed as described above, and 16 h post-transfection the cells were serum-starved for 4 h and stimulated with 20% FBS-containing medium. The phospho-ERK1/2 levels and FLAG-gephyrin S268A phosphorylation were analyzed at the indicated time points.
Immunoprecipitation and Western Blot Analysis
Immunoprecipitation and Western blot (WB) protocols were performed using whole cell lysate from HEK cells or rat brain in EBC buffer (50 mm Tris, pH 8.0, 120 mm NaCl,0.5% Nonidet P-40; cOmplete mini protease inhibitor mixture) (Roche) and phosphatase inhibitor mixture 1 and 3 (Sigma). 1 μg of desired antibody (mouse anti-FLAG, Sigma; mouse IgG, Jackson Immunoresearch) was used for immunoprecipitation, and the complexes were collected using 25 μl of protein A-agarose (Calbiochem). The complexes were washed three times in ice-cold EBC buffer to remove nonspecific interactions before denaturation in 2× SDS loading buffer at 90 °C for 3 min. The immunoprecipitated complexes were analyzed using WB assay, with rabbit anti-HA (Santa Cruz, 1:1000) and HRP-conjugated mouse or rabbit secondary.
In Vitro Kinase Assay
In vitro kinase assay was performed using purified full-length STREP-gephyrin expressed in bacteria (21). The purified gephyrin was phosphorylated in kinase buffer (50 mm MOPS, pH 6.5, 100 μm ATP, 10 mm MgCl2, 1 mm EGTA, and H2O to a final volume of 50 μl) and purified with activated ERK 0.5 μl (Calbiochem) at 30 °C for 30 min. The reaction was stopped with the addition of 2× SDS loading buffer and boiling the samples at 90 °C for 3 min. WB to detect gephyrin phosphorylation was performed using either mouse monoclonal antibody 3B11 to detect total gephyrin or affinity-purified rabbit anti-phospho-gephyrin Ser-268 (2 μg/ml).
MS Analysis
Gephyrin immunoprecipitated from rat whole brain lysate using the monoclonal antibody 3B11 was separated on SDS-PAGE. Coomassie-stained protein bands were subjected to reduction, alkylation, and in-gel tryptic digestion as described previously (22). The digests were analyzed by nanoflow LC-MS/MS using an LTQ Orbitrap Velos mass spectrometer coupled to an EASY-nLC II liquid chromatograph (Thermo Fisher Scientific). A database search was performed against the Swiss-Prot (Rattus norvegicus) using Mascot 2.4 (Matrix Science) via Proteome Discoverer 1.3 (Thermo Fisher Scientific).
Image Analysis and Quantification
Confocal laser scanning microscope (LSM 510 Meta; Carl Zeiss) was used for acquiring images in sequential mode over the full dynamic range of the photodetectors with a 100× objective lens (NA 1.4). The pinhole was set at 1 Airy unit, and pixel size was set at 90 nm. At least nine cells from three independent batches per condition were used to acquire images as a z-stack (three optical sections, 0.5-μm step size) for analysis. Image processing and analysis was done using the software ImageJ, for which maximum intensity projections were created from the z-stacks for analysis.
Gephyrin cluster size and density analysis has been described earlier (2). Unless indicated, only gephyrin clusters apposed to synapsin-1 or VGAT-positive puncta were considered for analysis. Total dendritic length in image analysis area was calculated using the line tool in ImageJ, and cluster density per 20-μm length of dendrite was calculated from this value. The integrated density values were used to calculate the cluster area in μm2.
Electrophysiology
Whole cell patch clamp recordings were made from transfected hippocampal neurons (11 + 4 DIV) at room temperature and at a holding potential of −60 mV. Mock (nontransfected) transfected cells on the same coverslip were used as controls. Recordings were performed as described previously (2) with a HEKA EPC-7 amplifier and Patch Master v2.11 software (HEKA Elektronik). Spontaneous GABAergic miniature postsynaptic currents (mIPSCs) were recorded in the presence of Tetredotoxin (TTX) (0.1 μm; Tocris) and isolated pharmacologically using 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX) (2 μm; Tocris) and AP-5 (50 μm; Tocris). At least 175 individual events in a total of three neurons per culture were analyzed for each condition. For the average amplitude calculation, only cells with more than 25–30 events in the recording period were included. Neurons that did not display GABAergic mIPSCs were excluded from the statistical analyses.
Statistical Analyses
Statistical analysis of morphological data (gephyrin cluster size and density) was performed either pair-wise (mutant versus WT) using unpaired Student's t test or Kolmogorov-Smirnov (K/S) test, or using one-way ANOVA followed by Bonferroni post hoc test when multiple groups were compared. Amplitude and interevent interval distributions of mIPSCs were analyzed pair-wise using K/S test. p ≤ 0.05 was considered as being statistically significant.
RESULTS
Phosphopeptide Analysis of Gephyrin from Rat Brain Homogenate
We have reported the identification of Ser-270 as a phosphorylation residue and characterized the functional significance of this residue for GABAergic synapse formation (2). In this report, we also described gephyrin as a substrate for calpain and demonstrated that phosphorylation at Ser-270 promotes gephyrin processing by calpain. Given the importance of gephyrin for GABAergic transmission, we hypothesized that this regulation by calpain might involve activation of more than one signaling pathway. To identify additional kinase pathways and determine their regulation of gephyrin, we immunoprecipitated gephyrin from rat brain homogenate and subjected it to MS phosphopeptide analysis. In this process, we identified 10 phosphorylated and 10 acetylated residues on gephyrin, many of which are novel and have not been described previously (Table 1). Although there is a report identifying gephyrin acetylation at Lys-148 (23), the functional significance of acetylation on gephyrin is presently unknown. Recently, a detailed phosphopeptide analysis using recombinant gephyrin purified from SF9 cells listed 18 phosphorylation residues (13). Our analysis identified Thr-266, Ser-268, Ser-270, Ser-280, and Ser-295 residues in common with this report; in addition we also identified Ser-262, Ser-286, Ser-303, Ser-305, and Ser-319 as phosphoresidues in gephyrin from rat brain. Consistent identification of residues phosphorylated across different cell types and organisms suggests that gephyrin phosphorylation is well conserved, and convergence of different signals onto gephyrin might orchestrate its spatial and temporal functional dynamics via the phosphorylation of subset of residues.
TABLE 1.
Phosphorylation and acetylation sites of gephyrin from rat brain identified by LC-MS/MS
| Amino acids | Sequencea | Modifications | Mascot ion scoreb |
|
|---|---|---|---|---|
| CID-ITc | HCD-FTd | |||
| 2–16 | aTEGmILTNHDHQIR | N-terminal (acetyl) | 37 | 32 |
| 137–148 | GkTLIINLPGSK | Lys-138 (acetyl) | 44 | |
| 139–148 | TLIINLPGsK | Ser-147 (acetyl) | 36 | 27 |
| 244–251 | IPDsIISR | Ser-247 (acetyl) | 31 | |
| IPDSIIsR | Ser-250 (acetyl) | 36 | 37 | |
| 259–272 | DTAsLSTTPSESPR | Ser-262 (phospho) | 55 | 58 |
| DTAsLSTTPSESPR | Ser-262 (acetyl) | 51 | 47 | |
| DTASLStTPSESPR | Thr-265 (acetyl) | 57 | ||
| DTASLSTtPSESPR | Thr-266 (phospho) | 103 | 67 | |
| DTASLSTtPSESPR | Thr-266 (acetyl) | 59 | ||
| DTASLSTTPsESPR | Ser-268 (phospho) | 75 | ||
| DTASLSTTPsESPR | Ser-268 (acetyl) | 74 | 61 | |
| 259–278 | DTASLSTTPSEsPRAQATSR | Ser-270 (phospho) | 44 | |
| 279–288 | LsTAScPTPK | Ser-280 (phospho) | 40 | |
| LSTAScPtPK | Thr-286 (phospho) | 40 | 39 | |
| 293–301 | cSsKENILR | Ser-295 (phospho) | 59 | |
| 302–311 | AsHSAVDITK | Ser-303 (phospho) | 39 | |
| ASHsAVDITK | Ser-305 (phospho) | 45 | ||
| 318–328 | msPFPLTSMDK | Ser-319 (phospho) | 40 | |
| 435–446 | EsDDGTEELEVR | Ser-436 (acetyl) | 48 | 93 |
a Lowercase letters represent modified amino acid residues: m, oxidized methionine; c, carbamidomethylated cysteine.
b Mascot ions scores for peptides identified with expectation values less than 0.05 and manual inspection of MS/MS spectra.
c Collision-induced dissociation (CID) MS/MS spectra were acquired in the LTQ ion trap (IT) analyzer.
d Higher energy collisional dissociation (HCD) MS/MS spectra were acquired in the Orbitrap Fourier transform (FT) analyzer.
Among the many phosphorylation residues identified in our own MS analysis, the Ser-268 site drew our attention, because of its proximity to the Ser-270 residue, which we have shown previously to be targeted by GSK3β, and the fact that it does not appear to be phosphorylated in the C3-gephyrin splice variant, expressed mainly in non-neuronal cells (13). Hence, we proceeded to understand the biological significance of Ser-268 phosphorylation site on gephyrin, focusing on postsynaptic clustering.
Ser-268 Mutation Affects Gephyrin Clustering Phenotype
To investigate the relevance of Ser-268 phosphorylation, we mutated Ser-268 in eGFP-gephyrin to create eGFP-S268A and eGFP-S268E gephyrin mutants for transfection in primary hippocampal neurons. Cultures were transfected by magnetofection after 11 DIV and analyzed 7 days later (11 + 7 DIV) using immunofluorescence staining and confocal microscopy. In this preparation, eGFP-tagged gephyrin forms clusters at postsynaptic sites identified by apposition to axon terminals labeled for a presynaptic marker (synapsin-1 or VGAT) or alternatively by co-localization with the GABAAR α2 subunit (2). Therefore, changes in size and density of fluorescent clusters formed by eGFP-gephyrin S268A and S268E mutants were quantified in comparison with WT eGFP-gephyrin (Fig. 1, A–C). As a control, we verified that eGFP-gephyrin S268A and S268E clusters co-localized with GABAAR, visualized by α2 subunit immunofluorescence (Fig. 1, D and D′), confirming their postsynaptic localization.
FIGURE 1.

Morphological analysis of gephyrin phosphorylation site mutants in primary neuron cultures. A, A′, and A′, typical examples are illustrated for WT eGFP-gephyrin, S268A, and S268E constructs; postsynaptic clustering is demonstrated in the middle row by apposition of eGFP-gephyrin clusters (green) to synapsin-1-positive terminals (red). The distribution of eGFP-gephyrin alone is depicted in the bottom row. B, distribution histograms of postsynaptic cluster density for WT, S268A, and S268E mutants on dendrites of transfected neurons (normalized to 20-μm dendritic length). A marked increase is evident for the S268A mutant (see main text for statistics). C, average cluster size of WT and mutant gephyrin constructs (means ± S.E.; Bonferroni post hoc test; **, p < 0.01). D and D′, clusters formed by eGFP-gephyrin S268A and S268E are co-localized with GABAAR α2 subunit immunofluorescence (red), confirming postsynaptic localization. E, E′, and E′, representative images of WT eGFP-gephyrin, S268A, and S268E constructs (green) co-transfected with GPHN shRNA 3′-UTR (not shown) to deplete endogenous gephyrin. Staining with antibodies to gephyrin reveals the clusters formed by the eGFP-constructs, as well as endogenous gephyrin clusters in nontransfected cells; presynaptic terminals are stained with synapsin-1 (blue). F, histograms of cluster density distribution per 20-μm dendritic length shows that down-regulation of endogenous gephyrin differentially alters the distribution of clusters formed by the S268A and S268E mutant constructs (see main text for statistics). G, quantification of cluster size (means ± S.E.; Bonferroni post hoc test; ***, p < 0.001) shows enlargement of eGFP-gephyrin S268A clusters and shrinkage of eGFP-gephyrin S268E clusters, respectively. Scale bars: A, A′, and A′, top panel, 10 μm, and bottom panels, 5 μm; D, E, E′, and E′, 5 μm.
Expression of eGFP-gephyrin phospho-mutant constructs significantly altered the density and size of postsynaptic clusters (one-way ANOVA; density: F2,88 = 17.43; p < 0.0001; size: F2,1783 = 29.4; p < 0.0001). Neurons expressing the eGFP-gephyrin S268A phospho-deficient mutant had an increased density of postsynaptic gephyrin clusters compared with WT eGFP-gephyrin (10.5 ± 1 clusters versus 7 ± 0.5 per 20-μm dendrite segment; Bonferroni post hoc test, p < 0.05; Fig. 1, A′ and B); furthermore, the S268A clusters were also significantly increased in size (Fig. 1C). In contrast, analysis of eGFP-gephyrin S268E phospho-mimicking mutant showed no significant change in density (5.3 ± 0.3 per 20-μm dendrite; p = 0.08; Fig. 1, A′ and B) and size (Fig. 1C). These observations suggested that constitutive dephosphorylation of Ser-268 promotes formation and growth of gephyrin clusters at postsynaptic sites.
In our previous study, we had demonstrated that expression of eGFP-gephyrin S270A in cultured neurons was independent of endogenous gephyrin expression (2). Hence, we wanted to make sure that the phenotypes of eGFP-gephyrin S268A and S268E were not being influenced by the presence of endogenous gephyrin. Neurons were co-transfected with an shRNA targeting the GPHN mRNA 3′-UTR to silence endogenous gephyrin without affecting the expression of eGFP-constructs (which lack the 3′-UTR). Efficiency of this construct to selectively deplete endogenous gephyrin has been documented earlier (2). The cells were analyzed for eGFP-gephyrin postsynaptic clustering after 11 + 7 DIV (Fig. 1E). Consistent with our earlier observations, depletion of endogenous gephyrin did not affect the WT eGFP-gephyrin phenotype; however, silencing endogenous gephyrin expression changed the phenotypes of eGFP-S268A and S268E mutants (Fig. 1, E′ and E′), as confirmed by quantitative analysis (one-way ANOVA; density: F2,66 = 18.06; p < 0.0001; size: F2,1368 = 45.15; p < 0.001). The density of eGFP-S268A clusters was reduced (Bonferroni post hoc test, p < 0.01; Fig. 1F), but their size was markedly larger compared with WT gephyrin (p < 0.001; Fig. 1G). Expression of eGFP-S268E in the absence of endogenous gephyrin resulted in increased density compared with eGFP-gephyrin (11 ± 1.2 versus 7.2 ± 0.4 per 20-μm dendrite; p < 0.05; Fig. 1F), whereas their size was decreased (p < 0.001; Fig. 1G). The observation that Ser-268 mutant phenotypes are affected by the presence or absence of endogenous gephyrin underscores the fact that sequence alteration at Ser-268 was not the cause for the S268A and S268E phenotypes. Furthermore, these results suggest a primary role of Ser-268 phosphorylation in regulating gephyrin cluster size, whereas the effect of S268A or S268E mutation on cluster density reflects the interaction with another post-translational modification of gephyrin (the difference in S268A and S268E cluster density in the presence or absence of endogenous gephyrin is characterized better in Fig. 5).
FIGURE 5.

Gephyrin phosphorylation sites at Ser-270 and Ser-268 are regulated by GSK3β and ERK, respectively. A–E, representative images of eGFP-Gephyrin WT, S268A, S268A/S270A, S268A/S270E, and S268E/S270E mutant constructs, co-transfected with GPHN shRNA 3′-UTR (not shown) to silence endogenous gephyrin. F, histograms of cluster density distribution (normalized per 20-μm dendritic length) showing the higher density of S268A/S270A mutant clusters (see main text for statistics). G, quantification of cluster size (means ± S.E.; Bonferroni post hoc test; **, p < 0.001), demonstrating the role of Ser-268 phosphorylation for regulating cluster size. H, K, and N, representative images of eGFP-S268E and S268E/S270E treated overnight with either GSK3β IX or PD98059 (25 μm). I and J, histograms of cluster density distribution (normalized to 20-μm dendritic length) and cluster size (means ± S.E.), showing the increased density, but not size, of S268E mutant following GSK3β IX treatment (see main text for statistical analysis). L, M, O, and P, quantification of cluster density and size of S268E/S270E double mutant construct, demonstrating that clustering of this mutant gephyrin is not influenced by blockade of GSK3β or ERK. Scale bars, 5 μm. DMSO, dimethyl sulfoxide.
ERK Regulates Ser-268 Site on Gephyrin
To identify a suitable kinase to phosphorylate the Ser-268 residue, we performed a bioinformatics search for potential conserved kinase domains around Ser-268 using a published algorithm (24) and identified ERK as a potential kinase that might phosphorylate this residue. We therefore tested the ERK inhibitor PD98059 (25 μm) in transfected neurons. After overnight treatment we observed increased eGFP-gephyrin cluster density (15.4 ± 1.0 versus 9.4 ± 0.5; Student's t test, t61 = 8.63; p < 0.0001) and size (Mann Whitney test, p < 0.0001; Fig. 2, A–C), mimicking the phenotype of the S268A mutant. As a control, we treated cells expressing eGFP-S268E construct with PD98059 and did not observe any increase in the cluster size (Fig. 2, A′ and C; Student's t test, t1552 = 0.52; p = 0.6); however, we saw a significant increase in cluster density (17.3 ± 0.9 versus 6.8 ± 0.4; Student's t test, t52 = 6.98; p < 0.0001; Fig. 2B), like the change seen upon depletion of endogenous gephyrin (Fig. 1F). These findings suggested that ERK might be the kinase modulating Ser-268 phosphorylation status to modulate gephyrin cluster size; further, they confirm that the regulation of gephyrin cluster density is not primary mediated by phosphorylation of Ser-268.
FIGURE 2.
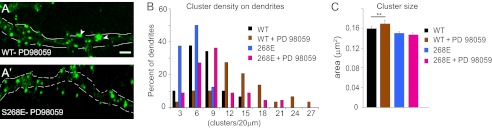
ERK modulates gephyrin clustering via Ser-268. A and A′, overnight exposure to PD98059 (25 μm) enhances eGFP-gephyrin cluster size (arrowheads) and density in 11 + 7 DIV transfected neurons, unlike in neurons expressing the S268E phospho-mimicking construct. B and C, histograms of gephyrin cluster density distribution (normalized to 20-μm segments) and size (means ± S.E.), showing that PD98059 treatment enhanced the density of WT and S268E gephyrin clusters and the size of S268E clusters (**, p < 0.01) (see main text for statistics). Scale bars: A and A′, 5 μm.
Gephyrin Is a Direct Substrate for ERK Phosphorylation
To confirm the role of ERK in phosphorylating gephyrin at Ser-268, we generated an antibody against the Ser(P)-268 peptide and performed in vitro kinase assay using purified ERK1 and PKA. First, we tested the presence of native gephyrin phosphorylated at Ser-268 in the rat brain by WB, using whole brain homogenate fractionated into soluble cytosolic fraction and insoluble membrane-bound fraction (Fig. 3A). The protein levels were controlled using the monoclonal antibody 3B11 (lower panels). The Ser(P)-268 antibody showed a more intense signal in the membrane-bound fraction of gephyrin compared with the cytosolic fraction. The specificity of the antibody was confirmed by preincubation with excess phosphopeptide prior to WB (right top panel). Next, we proceeded with the in vitro kinase assay using bacterially expressed STREP-gephyrin and purified kinase (Fig. 3B). Our Ser(P)-268 antibody detected phosphorylated gephyrin only in the presence of ERK1 and ATP (fourth lane), but not when we performed the same assay in the absence of ATP or presence of λ protein phosphatase or purified PKA (fifth and sixth lanes). The amount of STREP-gephyrin used in our assay is shown (bottom panel) using simple Coomassie staining of the membrane after the WB with Ser(P)-268 antibody.
FIGURE 3.

Gephyrin is a direct substrate for ERK phosphorylation. A, detection of Ser(P)-268 in gephyrin pulled down from rat whole brain extract. Cyto and Memb represent the soluble cytosolic fraction and insoluble membrane-associated fraction. The nitro-cellulose membrane was separated into two, and each of the gephyrin immunoprecipitation lanes were probed with either Ser(P)-268 antibody (S268, left panel) or Ser(P)-268 antibody blocked with Ser(P)-268 peptide to demonstrate selective epitope recognition (right panel). Ab, antibody. Each of the two blots were stripped and probed again with 3B11 mouse anti-gephyrin to show that equal levels of gephyrin was precipitated in all the lanes (lower panels). B, in vitro phosphorylation of gephyrin using purified ERK1 and detected using a polyclonal antibody against gephyrin Ser(P)-268. The gephyrin band was detected only in the presence of ERK1 and ATP (fourth lane) and not in the presence of λ protein phosphatase (λPP) or PKA (fifth and sixth lanes). Staining the blot with Coomassie showed equal amounts of full-length gephyrin in all lanes (lower panel). C, FLAG-gephyrin (geph) interaction with HA-ERK1 and HA-ERK2 in HEK293 cells. Immunoprecipitation (IP) for FLAG-gephyrin using mouse anti-FLAG antibody followed by WB against HA showed that HA-ERK1 interacts with FLAG-gephyrin (top blot, second lane) and FLAG-GC domain (fifth lane). Loading controls demonstrate the presence of FLAG and HA constructs as indicated. The scheme depicts gephyrin domain organization. D, bacterial purified STREP-gephyrin immobilized using STREP-tactin beads and incubated with HEK293 cell extracts overexpressing HA-ERK1 and HA-ERK2 shows an interaction between STREP-gephyrin and ERK1, as well as a weak interaction with HA-ERK2.
Next, to confirm that gephyrin is a direct substrate for ERK1, we looked for potential interactions between HA-ERK1 or HA-ERK2 (25) and FLAG-gephyrin overexpressed in HEK293 cells (Fig. 3C). Pulldown for FLAG-gephyrin followed by WB for HA-ERK showed an interaction between HA-ERK1 and FLAG-gephyrin (Fig. 3C, top panel, second lane). We also tested individual gephyrin domains FLAG-G, FLAG-GC, and FLAG-E for ERK-1 interaction (Fig. 3C, top panel, fourth, fifth, and sixth lanes) and found a robust interaction with the N-terminal GC domain of gephyrin but not the E-domain.
We additionally confirmed this interaction using a bacterially overexpressed STREP-gephyrin. We pulled down HA-ERK from lysates of HEK293 overexpressing HA-ERK1 or HA-ERK2 (Fig. 3D, bottom panel) using bacterial STREP-gephyrin. WB for HA confirmed the interaction between gephyrin and HA-ERK1; in addition we could also see a weak interaction with HA-ERK2. Collectively, these results confirmed ERK as a candidate enzyme phosphorylating gephyrin at Ser-268.
Functional Characterization of Gephyrin Ser-268 Mutants
To assess the functional relevance of Ser-268 phosphorylation, whole cell patch clamp recordings of pharmacologically isolated GABAergic mIPSCs were performed in 11 + 4 DIV hippocampal neurons. Compared with mock transfected cells present in the same cultures, overexpression of WT eGFP-gephyrin did not influence mIPSC amplitudes or interevent intervals (Fig. 4 and Table 2). In contrast, the average amplitude of mIPSC recorded in neurons expressing S268A was 34% larger than control (Fig. 4, B and C; K/S test, p < 0.01), and the interevent intervals were shortened by 45% (Fig. 4, D and E, and Table 2; K/S test, p < 0.001), suggesting increased size and density of functional GABAergic synapses in S268A-expressing neurons. Cells expressing eGFP-gephyrin S268E mutant showed mIPSC similar to WT or mock transfected cells (Fig. 4, A–E). Analyses of rise and decay kinetics of GABAergic mIPSC showed only minor differences in S268A or S268E mutants (Fig. 4, F and G). Thus, constitutive blockade of gephyrin Ser-268 phosphorylation facilitates GABAergic synaptic transmission by increasing GABAergic mIPSC amplitude and by the formation of additional GABAergic release sites.
FIGURE 4.

Effects of eGFP-gephyrin WT and Ser-268 mutants expression on GABAergic mIPSCs in cultured hippocampal neurons. A, representative current traces show pharmacologically isolated GABAergic mIPSCs recorded at 11 + 4 DIV for the constructs tested and nontransfected neurons from the same coverslips (mock). B and C, cumulative probability distribution and histograms of the average amplitude of mIPSCs. D and E, cumulative probability distribution and histograms of the average interevent intervals of mIPSCs. F and G, average rise time and decay time constants of mIPSCs (see Table 2 for statistical analysis; ***, p < 0.001).
TABLE 2.
Properties of mIPSCs recorded
Transfected neurons were used at DIV 11 + 4 for recording the mIPSCs as described under “Experimental Procedures.” The values are given as the means ± S.E. Statistical analysis was done using unpaired Student's t test and ANOVA. Neurons were treated with vehicle (saline) or PD98059 (25 μm) for 3 h at DIV 15 prior to recording the mIPSC currents.
| Events | Amplitude | Interevent interval | Rise time | Decay time | |
|---|---|---|---|---|---|
| pA | ms | ms | ms | ||
| In hippocampal neurons transfected with eGFP-gephyrin constructs | |||||
| WT | 329 | 36.8 ± 0.7 | 2181 ± 143 | 4.1 ± 0.3 | 22.9 ± 1.2 |
| Mock | 240 | 38.4 ± 0.8 | 1996 ± 129 | ||
| S268A | 479 | 49.3 ± 0.7a,c | 1198 ± 64a,b | 3.9 ± 0.3 | 22.5 ± 0.79 |
| S268E | 161 | 40.3 ± 0.8a | 2079 ± 222 | 3.4 ± 0.4 | 21.7 ± 1.1 |
| In hippocampal neurons treated with PD98059 | |||||
| Vehicle | 1599 | 47.4 ± 0.8 | 860 ± 28 | ||
| PD98059 | 1250 | 60.6 ± 1.1c | 1353 ± 50c |
a p < 0.001 between S268A and S268E.
b p < 0.001 between WT and S268A or S268E.
c p < 0.001, unpaired Student's t test.
Phosphorylation at Ser-268 and Ser-270 Regulates Distinct Aspects of Gephyrin Clustering
A potential cross-talk between two phosphorylation residues on gephyrin became apparent, as PD98059 treatment of S268E caused an increase in cluster density and the down-regulation of endogenous gephyrin in S268E-expressing neurons using GPHN shRNA also influenced postsynaptic cluster density. Given the identification of GSK3β as the kinase targeting Ser-270 (2) and the proximity of the Ser-268 site, we speculated that these two sites are functionally coupled and influence each other's phosphorylation status. Hence, we wanted to test whether gephyrin cluster size was under the control of Ser-268 residue and cluster density under the control of Ser-270 residue. We generated S268A/S270A, S268A/S270E, and S268E/S270E eGFP-gephyrin double mutants and co-expressed them in neurons along with GPHN 3′-UTR shRNA to silence endogenous gephyrin (Fig. 5, A–E). Quantification of the mutant phenotypes confirmed the idea that these two sites are functionally coupled (Fig. 5, F and G; one-way ANOVA, cluster density: F3,72 = 19.31, p < 0.0001; cluster size: F3,3269 = 5.44; p = 0.001). As predicted, S268A/S270A mutant showed an increase in both density (Bonferroni post-test, p < 0.01; Fig. 5F) and size (p < 0.01; Fig. 5G) of postsynaptic gephyrin clusters compared with WT gephyrin, whereas expression of S268A/S270E showed increased size (p < 0.01; Fig. 5G) but no change in density of gephyrin clusters (Fig. 5F). Finally, the S268E/S270E mutant showed a phenotype similar to the WT in terms of cluster density and size (Fig. 5, E–G). These findings indicate that dephosphorylation at Ser-268 promotes cluster size increase, whereas dephosphorylation at Ser-270 promotes increase in cluster density. However, they do not yet explain why treating neurons expressing eGFP-gephyrin S268E with PD98059 results in increased cluster density.
Therefore, we tested the respective roles for the Ser-268 and Ser-270 phosphorylation status on gephyrin clustering using pharmacological inhibitors. First, we treated neurons co-transfected with eGFP-gephyrin S268E and 3′-UTR-gephyrin shRNA with the GSK3β inhibitor, GSK3β IX, or vehicle control (Fig. 5H). As per our initial expectations, we found an increase in the density of clusters (Fig. 5I; Student's t test, t13 = 2.289, p = 0.0394) but no change in size (Fig. 5J) compared with cells treated with vehicle. To conclusively demonstrate the interaction between the Ser-268 and Ser-270 sites, we treated cultures expressing eGFP-gephyrin S268E/S270E mutant with PD98059 or GSK3β IX (Fig. 5, K and N). As expected, both inhibitors had no effect on the density (PD98059: t14 = 1.128 p = 0.269; GSK3β IX: t14 = 0.3337, p = 0.7436) (Fig. 5, L and O) or size (Fig. 5, M and P) of gephyrin S268E/S270E double mutant clusters. Collectively, the results confirm that the Ser-270 and Ser-268 phosphorylation sites are functionally coupled via pathways controlling GSK3β and ERK activity, linking gephyrin clustering to GABAergic synaptic plasticity. Further, they suggest that PD98059 application favors dephosphorylation of Ser-270, possibly by blocking the GSK3β pathway in addition to the ERK1/2 pathway.
Short Term Effects of ERK Signaling on Gephyrin Clustering
Studies on cellular signaling have demonstrated that transient or prolonged signaling can produce distinct functional changes at synaptic sites (26). Our data showed that long term pharmacological block in ERK signaling using PD98059 (Fig. 2A) increases the size and density of eGFP-gephyrin clusters at GABAergic synapses, producing a phenotype similar to S268A/S270A double mutant (Fig. 5C). Hence, we wanted to investigate the short term effects of ERK signaling at GABAergic synapses and evaluate its significance compared with long term effects. To this end, we blocked ERK signaling using PD98059 for 3 h in cultures of eGFP-gephyrin-transfected neurons (DIV 11 + 4) (Fig. 6, A and A′). Quantitative image analysis did not reveal a significant reduction in cluster density compared with vehicle (Me2SO)-treated controls (4.9 ± 0.4 versus 3.7 ± 0.4; Student's t test, t18 = 1.265, p = 0.222), but these clusters showed a significant increase in size (0.29 ± 0.01 versus 0.38 ± 0.01 μm2; t906 = 3.393, p = 0.0007) (Fig. 6, B and C). To check whether 3 h of exposure to PD98059 affected endogenous gephyrin clustering in a similar way, we treated DIV 15 neurons and stained for endogenous gephyrin using the gephyrin monoclonal antibody mAb7a (Fig. 6, D and D′). This treatment caused a significant reduction in cluster density (7 ± 0.5 versus 4.9 ± 0.4 clusters/20-μm dendrite; t16 = 2.972, p = 0.009); however, an increase in size was not evident (Fig. 6, E and F).
FIGURE 6.

Effects of short term ERK inhibition on gephyrin clustering and GABAergic mIPSC. A and A′, representative images of eGFP-gephyrin-transfected neurons (11 + 4 DIV) treated with either vehicle (dimethyl sulfoxide (DMSO)) or ERK inhibitor PD98059 (25 μm) for 3 h before fixation and staining. Boxed areas are enlarged below the main picture to demonstrate postsynaptic localization of eGFP-gephyrin clusters (green) apposed tot VGAT-positive terminals (red). B and C, histograms of cluster density distribution and size, showing that short term blockade of ERK increases eGFP-gephyrin cluster size (means ± S.E.; ***, p < 0.001) but not density (see main text for statistics). D and D′, 15 DIV neurons treated with dimethyl sulfoxide or PD98059 for 3 h before fixation and staining for endogenous gephyrin (green) and GABAergic presynaptic terminals VGAT (red). E and F, quantification of endogenous gephyrin cluster density (normalized per 20-μm dendrite) revealed a significant decrease but no change in size (means ± S.E.; see main text for statistics). G–J, interevent intervals and amplitude of GABAergic mIPSC recorded in 15 DIV neurons following 3 h of exposure to dimethyl sulfoxide and PD98059. Cumulative probability distribution analysis revealed significantly increased interevent intervals and amplitude, as depicted also with histograms (means ± S.E.; **, p < 0.01; see Table 2 for statistical analysis). Scale bars: A and D, 10 μm. Ctrl, control; PD, PD98059.
To determine the functional implications of short term blockade of ERK signaling on GABAergic transmission, we measured the amplitude and frequency of GABAergic mIPSC in nontransfected 15 DIV hippocampal neurons after a 3-h ERK inhibition with PD98059 or vehicle. This short treatment with PD98059 resulted in increased interevent intervals by 57% (Fig. 6, G and H, and Table 2), in line with the morphological data (Fig. 6, E and F). Interestingly, this loss of functional synapses was compensated by a modest increase in mIPSC amplitude by 27% (Fig. 6, I and J, and Table 2). These alterations in GABAergic signaling reflected the morphology of S268A/S270E gephyrin mutant. Hence, it is conceivable that the S268A/S270E mutant depicts one possible physiological state wherein gephyrin phosphorylation at Ser-270 facilitates down-regulation of synaptic number to balance the excitability and/or network activity of the cell.
Neuronal Activity-dependent Regulation of Gephyrin Clusters by Calpain
Activity-dependent dynamics of gephyrin clusters at GABAergic synapses in vivo have been reported recently (14). We have shown earlier that gephyrin is a substrate for calpain, a Ca2+-dependent protein that can be activated by ERK signaling. Hence, we wanted to test whether neuronal activity-dependent alteration of gephyrin clusters was dependent on ERK and calpain activation. ERK activation was achieved by depolarizing cultured hippocampal neurons transfected with eGFP-gephyrin using 40 mm KCl for 4 min before fixation and staining. Calpain activation was prevented using co-transfection of its endogenous inhibitor, calpastatin. As expected, this mild stimulation paradigm led to a robust increase in the levels of phospho-ERK1/2 immunofluorescence (Fig. 7, A and A′). Despite its brief duration, it caused a significant decrease in eGFP-gephyrin cluster size (0.46 ± 0.01 versus 0.35 ± 0.01 μm2) without affecting their density (6.5 ± 1 versus 4.8 ± 0.4 clusters/20-μm dendrite) (Fig. 7, B and B′). However, KCl treatment had no observable effect on gephyrin clustering density (5.8 ± 0.6 versus 4.7 ± 1.1 clusters/20-μm dendrite) and size (0.47 ± 0.2 versus 0.48 ± 0.2 μm2) when endogenous calpain activity was blocked using Myc-Calpastatin (Cast) (Fig. 7, C and C′). These changes in morphology were confirmed by statistical analysis (one-way ANOVA; density: F3,56 = 1.015, p = 0.393; size: F3,1707 = 6.329, p = 0.0003; Fig. 7, D and E). These findings reveal that activity-dependent gephyrin dynamics at GABAergic synapses involves ERK activation and calpain action on gephyrin.
FIGURE 7.

Increase in neuronal activity leads to ERK activation and calpain-mediated clipping of gephyrin clusters. A and A′, induction of neuronal activity by KCl (40 mm; 4 min) leads to elevation of phospho-ERK1/2 immunoreactivity in cultured neurons. B and B′, neurons transfected with eGFP-gephyrin (8 + 7 DIV) show a reduction in cluster size upon ERK1/2 activation in the presence of KCl. C and C′, inhibiting calpain via the co-transfection of Myc-Cast prevents the reduction of eGFP-gephyrin cluster size upon KCl treatment. D and E, histograms of cluster density distribution (normalized to 20-μm dendrites) and size (means ± S.E.; Bonferroni post hoc test; **, p < 0.01) confirming the calpain-dependent reduction of gephyrin cluster size in the presence of KCl (see main text for statistics). Scale bars: A, 15 μm; B, 5 μm.
Ser-268 Phosphorylation Restricts Gephyrin Cluster Size via Calpain 1 Proteolysis
Having established that neuronal activity can activate both ERK and calpain to influence gephyrin clustering, we wanted to test whether the cluster size reduction seen in cells expressing eGFP-gephyrin S268E mutant (Fig. 1, E and G) was because of susceptibility of gephyrin to calpain action. We constitutively inhibited calpain in neurons by co-transfecting eGFP-S268E and Myc-Cast. Strikingly, this constitutive inhibition of calpain resulted in increased size (0.16 ± 0.04 versus 0.2 ± 0.08 μm2; Student's t test, t13 = 3.448, p = 0.0006) and density (5.3 ± 0.3 versus 9.4 ± 0.7 clusters/20-μm dendrite; t12 = 5.893, p < 0.0001) of eGFP-S268E clusters (11 + 7 DIV) to a level comparable with the S268A mutant (Fig. 8, A–C). To ensure that the reversal of S268E phenotype in cells co-expressing Myc-Cast was not due to other functions of Cast than calpain inhibition, we treated eGFP-S268E-expressing neurons (8 + 7 DIV) with the calpain inhibitor MDL28170 (30 μm) overnight and observed for clustering changes (Fig. 8, D and D′). Quantification of these images showed a significant increase in cluster size (0.33 ± 0.01 versus 0.46 ± 0.02 μm2; t693 = 3.981, p < 0.0001) but not density (3.3 ± 0.4 versus 3.4 ± 0.3 clusters/20-μm dendrite; t19 = 1.254, p = 0.225) (Fig. 8, E and F). As a control, we also tested whether pharmacological inhibition of calpain would affect eGFP-gephyrin clustering (Fig. 8, G and G′). Quantification of these images likewise showed a significant increase in cluster size (0.32 ± 0.01 versus 0.41 ± 0.02 μm2; t759 = 2.193, p = 0.0286) but no significant change in cluster density (3.2 ± 0.1 versus 4 ± 0.2 clusters/20-μm dendrite; t18 = 0.058, p = 0.9544) (Fig. 8, H and I). These results suggest that even under physiological conditions, eGFP-gephyrin cluster size is regulated via calpain activity, with phosphorylation of Ser-268 favoring its proteolytic cleavage.
FIGURE 8.
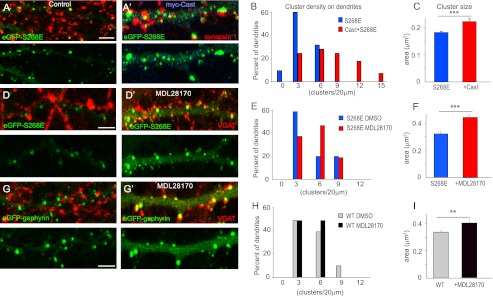
Calpain activity influences eGFP-gephyrin clustering. A and A′, co-expression of Myc-Cast (blue) with eGFP-gephyrin S268E construct in cultured hippocampal neurons (11 + 7 DIV) increases postsynaptic gephyrin clustering to the same level as seen in cells expressing S268A (see Fig. 1). Representative panels of transfected dendrites stained with synapsin 1 (red) to confirm that postsynaptic localization of eGFP-gephyrin clusters are shown. B and C, histograms of cluster density distribution (normalized per 20-μm dendritic length) and cluster size (means ± S.E.; ***, p < 0.001), confirming the increased density and size. D and D′, direct effect of calpain on S268E mutant (11 + 7 DIV, double-stained for VGAT) was confirmed by blocking calpain with MDL28170 (30 μm) overnight. E and F, quantification of cluster density showed no significant change, whereas the cluster area was significantly increased (***, p < 0.001). G and G′, MDL28170 treatment in neurons transfected with WT eGFP-gephyrin and its effect on clustering. H and I, quantification of WT eGFP-gephyrin clusters after MDL28170 treatment showed no change in cluster density but a significant increase in cluster overall size (means ± S.E.; **, p < 0.01; see main text for statistical analyses). Scale bar, 5 μm.
DISCUSSION
Only little data are available on the functional characterization of the diverse phosphorylation sites that have been identified on gephyrin (9, 11, 27, 28). Given the sheer number of sites potentially modified by diverse kinases, it is likely that multiple functional interactions take place between them, and that some of these sites act as “master switches” controlling the phosphorylation status of downstream phosphoresidues. Our results identify Ser-268 as a phosphorylation site on gephyrin targeted by ERK signaling to modulate GABAergic synaptic function by regulating the size of postsynaptic gephyrin clusters. Further, we unravel a functional coupling between Ser-268 with Ser-270, which are targeted by ERK and GSK3β, respectively, allowing for changes in gephyrin cluster size and density that are mirrored by changes in amplitude and frequency of GABAergic mIPSCs. Although direct evidence for the phosphorylation state-dependent co-regulation between the sites Ser-268 and Ser-270 has been hard to obtain because of technical difficulties, the indirect evidence provided by the mutational analysis of these sites suggests that this is likely the mechanism of regulation. Therefore, gephyrin phosphorylation status directly impacts on GABAergic synaptic function, presumably by allowing formation of synapses (2) and recruitment (or stabilization) of GABAAR to the postsynaptic density. Further, by extending our earlier work, we also demonstrate that activation of calpain is a general mechanism restricting postsynaptic gephyrin clustering in a phosphorylation-dependent manner. Therefore, multiple signaling cascades converge onto gephyrin to modify its scaffolding properties at GABAergic postsynaptic density and influence synapse function and homeostasis in the CNS.
Our data do not provide any evidence of position hierarchy for Ser-268 or Ser-270 residues within the context of other identified gephyrin phosphorylation sites. However, given the importance of ERK1 and GSK3β in neuronal function and synaptic plasticity, Ser-268 and Ser-270 appear more likely to represent downstream sites involved in fine-tuning of gephyrin (and GABAergic) function rather than “upstream switches.” The importance of acetylation on serine and threonine residues for protein regulation is still unclear, but the identification of acetylation at Ser-268 suggests that protein acetylation might add another regulatory step to prevent “mistaken” phosphorylation at Ser-268 by ERK and subsequent down-regulation of GABAergic transmission. The observation that the S268E/S270E gephyrin double mutant exhibits a clustering phenotype similar to WT instead of a complete loss of clusters is a further indication that Ser-268 and Ser-270 likely are downstream effector sites rather than upstream modulators.
A recent report describing a shRNA screen to knock down signaling pathways that affect gephyrin clustering identified ERK, GSK3β, and mTOR as effectors of gephyrin scaffolds (29). In this report, shRNA-mediated knockdown of ERK resulted in reduced density of gephyrin clusters. Our data also show a reduced gephyrin cluster density when ERK action of gephyrin is blocked either with S268A mutation (Fig. 1F) or using PD98059 (Fig. 6E). In addition, our functional data show that ERK-mediated reduction in gephyrin clusters is coupled to prolongation of GABAergic mIPSC interevent intervals, apparently compensated for by increased mIPSC amplitude (Fig. 6, G–J). Our molecular dissection of this phenomenon shows that phosphorylation at Ser-268 and Ser-270 is controlled by ERK and GSK3β signaling pathways regulating distinct aspects of gephyrin clustering. In particular, we show that treating neurons expressing eGFP-S268E with PD98059 does not alter gephyrin cluster size but increases their density, and this increase in density is facilitated by GSK3β regulation of the Ser-270 site. It is possible that blocking ERK signaling likely affects GSK3β phosphorylation of Ser-270, thereby allowing the formation of small, supernumerary postsynaptic gephyrin clusters. Furthermore, the specificity of the effects seen by mutagenesis and pharmacological treatment argues against the possibility that alterations in gephyrin S268A and S268E clustering are due to changes in its primary sequence.
By investigating the role of calpain in modulation of gephyrin clustering, our study provides new insight into the underlying molecular mechanism. The present observation that co-expression of Cast with eGFP-S268E mutant increases postsynaptic gephyrin cluster size and density to the levels seen in neurons expressing gephyrin S268A unambiguously demonstrates a role for calpain in restricting gephyrin clustering. In contrast, we have shown previously that Cast co-expression with eGFP-S270E has no effect on cluster size of this gephyrin mutant (2), suggesting that regulation of cluster size by calpain is dependent specifically on phosphorylation of Ser-268. Based on this evidence, one can speculate that the Ser-270 residue dominates over the Ser-268 site.
The main insight into the role of calpain for regulating gephyrin clustering comes from our experiments demonstrating activity-dependent regulation of ERK and calpain to reduce the size of gephyrin clusters upon chronic neuronal depolarization by KCl (Fig. 7). The fact that Cast co-expression prevents the cluster size reduction induced by KCl exposure directly demonstrates the involvement of calpain in this process, most likely in concert with ERK-mediated Ser-268 phosphorylation.
A recent report showed that Ser-270 phosphorylation affects the epitope recognized by the monoclonal antibody mAb7a (30), reflecting a change in gephyrin conformation (28). This structural change might explain why the presence of endogenous gephyrin affects the phenotype of Ser-268 phospho-mutants (Fig. 1) but not that of Ser-270 phospho-mutants (2). Furthermore, this conformational change could be the underlying basis for the Ser-270 site dominating over the Ser-268 residue to determine calpain action on gephyrin. In light of the complex interplay between the phosphorylation status of Ser-268 and Ser-270 on gephyrin cluster size and density, we have summarized our main results schematically (Fig. 9).
FIGURE 9.

Model summarizing the regulation of Ser-268 and Ser-270 for gephyrin cluster dynamics. Step 1, under basal conditions both Ser-268 and Ser-270 are phosphorylated, and this prevents the recruitment of new gephyrin molecules to the clusters. Step 2, upon inactivation of ERK and GSK3β signaling (or activation of unidentified phosphatases), the Ser-268 and Ser-270 residues allow fresh gephyrin molecule recruitment into the new clusters, as well as formation of new clusters. This dephosphorylated gephyrin is also insensitive to calpain regulation. Step 3, phosphorylation of either Ser-268 or Ser-270 residue renders gephyrin sensitive to calpain regulation, leading to reduced cluster size or reduced density. This graded response to calpain sensitivity is facilitated by changes in gephyrin conformation, depending on the Ser-270 phosphorylation status.
In neurons, ERK controls several forms of long term plasticity, including NMDA-dependent and independent forms of LTP, and sustained ERK activity is required for structural remodeling of dendritic spines underlying learning and memory (31). Although it is easy to envision how alterations at glutamatergic synapses have to be paralleled by concomitant changes at GABAergic synapses to prevent hyperexcitability or silencing of the network, our present results suggest a more complex scenario, by which ERK activity constitutively limits the strength of GABAergic synapses, as seen by the marked increase in mIPSC amplitude in hippocampal neurons upon 3 h of PD98059 exposure. This effect of ERK is likely permissive for potentiation of glutamatergic synapses, whereas homeostatic regulation of GABAergic transmission by gephyrin likely involves protein phosphatases or deacetylases to counteract the action of ERK. Hence, further characterizations of such additional signal transduction pathways converging on gephyrin are likely to shed light on the mechanistic adaptability of GABAergic inhibition to fluctuations within an existing neuronal network.
Acknowledgments
We thank Corinne Sidler, Giovanna Bosshard, and Cornelia Schwerdel for help with the neuronal cultures and experimental set for this manuscript. We are grateful to Prof. Angel De Blas (University of Connecticut, Storrs) for providing the GPHN 3′-UTR shRNA constructs.
This work was supported by grants from the Forschungskredit of the University of Zurich, the EMDO Foundation, and the Hartmann Müller Foundation (to S. K. T.) and from the Swiss National Science Foundation (to J. M. F.).
- GABAAR
- GABAA receptor
- Cast
- calpastatin
- DIV
- days in vitro
- VGAT
- vesicular GABA transporter
- WB
- Western blot
- ANOVA
- analysis of variance
- eGFP
- enhanced GFP
- mIPSC
- miniature inhibitory postsynaptic current
- K/S
- Kolmogorov-Smirnov.
REFERENCES
- 1. Siddiqui T. J., Craig A. M. (2011) Synaptic organizing complexes. Curr. Opin. Neurobiol. 21, 132–143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Tyagarajan S. K., Ghosh H., Yévenes G. E., Nikonenko I., Ebeling C., Schwerdel C., Sidler C., Zeilhofer H. U., Gerrits B., Muller D., Fritschy J.-M. (2011) Regulation of GABAergic synapse formation and plasticity by GSK3β-dependent phosphorylation of gephyrin. Proc. Natl. Acad. Sci. U.S.A. 108, 379–384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Sturgill J. F., Steiner P., Czervionke B. L., Sabatini B. L. (2009) Distinct domains within PSD-95 mediate synaptic incorporation, stabilization, and activity-dependent trafficking. J. Neurosci. 29, 12845–12854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Stoica L., Zhu P. J., Huang W., Zhou H., Kozma S. C., Costa-Mattioli M. (2011) Selective pharmacogenetic inhibition of mammalian target of rapamycin complex I (mTORC1) blocks long-term synaptic plasticity and memory storage. Proc. Natl. Acad. Sci. U.S.A. 108, 3791–3796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Jacob T. C., Wan Q., Vithlani M., Saliba R. S., Succol F., Pangalos M. N., Moss S. J. (2009) GABAA receptor membrane trafficking regulates spine maturity. Proc. Natl. Acad. Sci. U.S.A. 106, 12500–12505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Luscher B., Fuchs T., Kilpatrick C. L. (2011) GABAA receptor trafficking-mediated plasticity of inhibitory synapses. Neuron 70, 385–409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Houston C. M., Hosie A. M., Smart T. G. (2008) Distinct regulation of β2 and β3 subunit-containing cerebellar synaptic GABAA receptors by calcium/calmodulin-dependent protein kinase II. J. Neurosci. 28, 7574–7584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kittler J. T., Moss S. J. (2003) Modulation of GABAA receptor activity by phosphorylation and receptor trafficking: implications for the efficacy of synaptic inhibition. Curr. Opin. Neurobiol. 13, 341–347 [DOI] [PubMed] [Google Scholar]
- 9. Specht C. G., Grünewald N., Pascual O., Rostgaard N., Schwarz G., Triller A. (2011) Regulation of glycine receptor diffusion properties and gephyrin interactions by protein kinase C. EMBO J. 30, 3842–3853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lundby A., Secher A., Lage K., Nordsborg N. B., Dmytriyev A., Lundby C., Olsen J. V. (2012) Quantitative maps of protein phosphorylation sites across 14 different rat organs and tissues. Nat. Commun. 3, 876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zita M. M., Marchionni I., Bottos E., Righi M., Del Sal G., Cherubini E., Zacchi P. (2007) Post-phosphorylation prolyl isomerisation of gephyrin represents a mechanism to modulate glycine receptors function. EMBO J. 26, 1761–1771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Beausoleil S. A., Villén J., Gerber S. A., Rush J., Gygi S. P. (2006) A probability-based approach for high-throughput protein phosphorylation analysis and site localization. Nat. Biotechnol. 24, 1285–1292 [DOI] [PubMed] [Google Scholar]
- 13. Herweg J., Schwarz G. (2012) Splice-specific glycine receptor binding, folding, and phosphorylation of the scaffolding protein gephyrin. J. Biol. Chem. 287, 12645–12656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. van Versendaal D., Rajendran R., Saiepour M. H., Klooster J., Smit-Rigter L., Sommeijer J.-P., De Zeeuw C. I., Hofer S. B., Heimel J. A., Levelt C. N. (2012) Elimination of inhibitory synapses is a major component of adult ocular dominance plasticity. Neuron 74, 374–383 [DOI] [PubMed] [Google Scholar]
- 15. Chen J. L., Villa K. L., Cha J. W., So P. T., Kubota Y., Nedivi E. (2012) Clustered dynamics of inhibitory synapses and dendritic spines in the adult neocortex. Neuron 74, 361–373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Vlachos A., Reddy-Alla S., Papadopoulos T., Deller T., Betz H. (2012) Homeostatic regulation of gephyrin scaffolds and synaptic strength at mature hippocampal GABAergic postsynapses. Cerebral Cortex, in press [DOI] [PubMed] [Google Scholar]
- 17. Lardi-Studler B., Smolinsky B., Petitjean C. M., Koenig F., Sidler C., Meier J. C., Fritschy J.-M., Schwarz G. (2007) Vertebrate-specific sequences in the gephyrin E-domain regulate cytosolic aggregation and postsynaptic clustering. J. Cell Sci. 120, 1371–1382 [DOI] [PubMed] [Google Scholar]
- 18. Yu W., Jiang M., Miralles C. P., Li R. W., Chen G., de Blas A. L. (2007) Gephyrin clustering is required for the stability of GABAergic synapses. Mol. Cell. Neurosci. 36, 484–500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Buerli T., Pellegrino C., Baer K., Lardi-Studler B., Chudotvorova I., Fritschy J. M., Medina I., Fuhrer C. (2007) Efficient transfection of DNA or shRNA vectors into neurons using magnetofection. Nat. Protoc. 2, 3090–3101 [DOI] [PubMed] [Google Scholar]
- 20. Panzanelli P., Gunn B. G., Schlatter M. C., Benke D., Tyagarajan S. K., Scheiffele P., Belelli D., Lambert J. J., Rudolph U., Fritschy J.-M. (2011) Distinct mechanisms regulate GABAA receptor and gephyrin clustering at perisomatic and axo-axonic synapses on CA1 pyramidal cells. J. Physiol. 589, 4959–4980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Tyagarajan S. K., Ghosh H., Harvey K., Fritschy J.-M. (2011) Collybistin splice variants differentially interact with gephyrin and Cdc42 to regulate gephyrin clustering at GABAergic synapses. J. Cell Sci. 124, 2786–2796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Imanishi S. Y., Kochin V., Ferraris S. E., de Thonel A., Pallari H. M., Corthals G. L., Eriksson J. E. (2007) Reference-facilitated Phosphoproteomics: Fast and reliable phosphopeptide validation by LC-ESI-Q-TOF MS/MS. Mol. Cell. Proteomics 6, 1380–1391 [DOI] [PubMed] [Google Scholar]
- 23. Schwer B., Eckersdorff M., Li Y., Silva J. C., Fermin D., Kurtev M. V., Giallourakis C., Comb M. J., Alt F. W., Lombard D. B. (2009) Calorie restriction alters mitochondrial protein acetylation. Aging Cell 8, 604–606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Xue Y., Ren J., Gao X., Jin C., Wen L., Yao X. (2008) GPS 2.0, a tool to predict kinase-specific phosphorylation sites in hierarchy. Mol. Cell. Proteomics 7, 1598–1608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Dimitri C. A., Dowdle W., MacKeigan J. P., Blenis J., Murphy L. O. (2005) Spatially separate docking sites on ERK2 regulate distinct signaling events in vivo. Curr. Biol. 15, 1319–1324 [DOI] [PubMed] [Google Scholar]
- 26. Fischer A., Sananbenesi F., Pang P. T., Lu B., Tsai L.-H. (2005) Opposing roles of transient and prolonged expression of p25 in synaptic plasticity and hippocampus-dependent memory. Neuron 48, 825–838 [DOI] [PubMed] [Google Scholar]
- 27. Tyagarajan S. K., Fritschy J.-M. (2010) GABA(A) receptors, gephyrin and homeostatic synaptic plasticity. J. Physiol. 588, 101–106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kuhse J., Kalbouneh H., Schlicksupp A., Mükusch S., Nawrotzki R., Kirsch J. (2012) Phosphorylation of gephyrin in hippocampal neurons by cyclin-dependent kinase CDK5 at Ser-270 is dependent on collybistin. J. Biol. Chem. 287, 30952–30966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wuchter J., Beuter S., Treindl F., Herrmann T., Zeck G., Templin M. F., Volkmer H. (2012) A Comprehensive Small Interfering RNA Screen Identifies Signaling Pathways Required for Gephyrin Clustering. J. Neurosci. 32, 14821–14834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Prior P., Schmitt B., Grenningloh G., Pribilla I., Multhaup G., Beyreuther K., Maulet Y., Werner P., Langosch D., Kirsch J. (1992) Primary structure and alternative splice variants of gephyrin, a putative glycine receptor-tubulin linker protein. Neuron 8, 1161–1170 [DOI] [PubMed] [Google Scholar]
- 31. Thomas G. M., Huganir R. L. (2004) MAPK cascade signalling and synaptic plasticity. Nat. Rev. Neurosci. 5, 173–183 [DOI] [PubMed] [Google Scholar]