

Background: Chymotrypsin C (CTRC) targets specific regulatory cleavage sites within trypsinogens and procarboxypeptidases.
Results: The crystal structure of CTRC reveals the structural basis of substrate specificity.
Conclusion: Long-range electrostatic and hydrophobic complementarity drives CTRC association with preferred substrates.
Significance: The observations reveal the mechanistic basis for CTRC selectivity in digestive enzyme activation and degradation.
Keywords: Crystal Structure, Molecular Modeling, Protease Inhibitor, Protein-Protein Interactions, Serine Protease, Molecular Recognition, Substrate Specificity, Zymogen Activation
Abstract
Human chymotrypsin C (CTRC) is a pancreatic serine protease that regulates activation and degradation of trypsinogens and procarboxypeptidases by targeting specific cleavage sites within their zymogen precursors. In cleaving these regulatory sites, which are characterized by multiple flanking acidic residues, CTRC shows substrate specificity that is distinct from that of other isoforms of chymotrypsin and elastase. Here, we report the first crystal structure of active CTRC, determined at 1.9-Å resolution, revealing the structural basis for binding specificity. The structure shows human CTRC bound to the small protein protease inhibitor eglin c, which binds in a substrate-like manner filling the S6-S5′ subsites of the substrate binding cleft. Significant binding affinity derives from burial of preferred hydrophobic residues at the P1, P4, and P2′ positions of CTRC, although acidic P2′ residues can also be accommodated by formation of an interfacial salt bridge. Acidic residues may also be specifically accommodated in the P6 position. The most unique structural feature of CTRC is a ring of intense positive electrostatic surface potential surrounding the primarily hydrophobic substrate binding site. Our results indicate that long-range electrostatic attraction toward substrates of concentrated negative charge governs substrate discrimination, which explains CTRC selectivity in regulating active digestive enzyme levels.
Introduction
Digestive proteases are synthesized and secreted by the pancreas as inactive zymogens. Physiological activation takes place in the duodenum, where enteropeptidase initiates an activation cascade by specifically activating trypsinogens, which in turn activate chymotrypsinogens, proelastases, procarboxypeptidases, and other digestive enzymes (1). Premature activation of trypsin within the pancreas is understood to be a major initiating factor in chronic pancreatitis, and chymotrypsin C (CTRC)4 is a significant player in this process. In approximately half of the families affected by autosomal dominant hereditary pancreatitis, the disease is caused by mutations in the cationic trypsinogen gene PRSS1 that result in either enhanced trypsinogen activation or resistance to degradation (2–4). CTRC possesses the unique capacity to impact trypsinogen activation and stability via two opposing mechanisms: it can cleave cationic trypsinogen either at Phe18-Asp19 within the trypsinogen activation peptide, leading to enhanced autoactivation (5), or at Leu81-Glu82 within the Ca2+-binding loop, resulting in degradation (6). A number of disease-causing cationic trypsinogen mutations exert their effect in part through accelerating cleavage by CTRC at Phe18-Asp19 or through diminishing cleavage by CTRC at Leu81-Glu82 (4). The p.A16V mutation, which accounts for a small percentage of hereditary pancreatitis kindreds and is also associated with idiopathic chronic pancreatitis (7, 8), appears to exert its pathological effect solely by increasing the vulnerability of the cationic trypsinogen activation peptide to cleavage by CTRC (4). Finally, several mutations in the CTRC gene itself that lead to loss or impairment of protein function are significantly associated with chronic pancreatitis (9–12).
Both cationic trypsin and CTRC are members of the chymotrypsin family of serine peptidases, which share a common two β-barrel-fold, a famous triad of catalytic residues Ser, His, and Asp, and a conserved catalytic mechanism for nucleophilic cleavage of peptide bonds. Although these enzymes all catalyze the same reaction, they are differentiated by the distinct substrate sequences that they recognize through a series of subsites located within the active site cleft between the two β-barrels (13, 14). CTRC has 50–66% sequence identity with human pancreatic elastase isoforms, which have broad specificity for cleavage after hydrophobic P1 residues,5 and ∼40% sequence identity with other human chymotrypsin isoforms, which typically cleave after aromatic P1 residues. CTRC distinguishes itself from these other family members by uniquely targeting the regulatory cleavage sites involved in trypsinogen activation (5) and degradation (6), and also serves as a co-activator, with trypsin, of procarboxypeptidases CPA1 and CPA2 (15). The inability of other chymotrypsin and elastase isoforms to target these regulatory sequences (5, 15, 16) points to the existence of unique elements of CTRC specificity, as does a recent study in which we have identified selective inhibitors of CTRC using phage display (17).
The mechanisms by which CTRC, confronted with multiple potential substrates and cleavage sites, selects from among them, is critical to understanding its protective role in the pancreas and its pathological role in disease. Given the opposing effects of trypsinogen cleavage by CTRC within the activation peptide versus the Ca2+-binding loop, it is apparent that the hierarchy of CTRC selectivity toward these competing sites has been carefully titrated by nature. Remarkably, even a very subtle mutation within a cleavage site can shift the activity of CTRC and tip the balance toward a disease state.
In the absence of a crystal structure for CTRC, our efforts to understand this specificity have made use of inhibitor phage display selection and mutagenesis of the natural substrate cationic trypsinogen, with somewhat contradictory results (16, 17). Inhibitor studies suggest strong preference for Leu followed by Met at the P1 position (17), whereas substrate mutagenesis suggests that CTRC is much more permissive of alternative P1 residues (16). Another potential specificity feature is suggested by the presence of multiple Asp and Glu residues within favored CTRC target sequences (Table 1); these acidic residues appear consistently in the P4′ position but are otherwise non-uniformly situated. Inhibitor phage display confirms a strong preference for acidic residues at the P4′ position (17), but mutagenesis studies again show only moderate effects upon alteration of individual charged residues within the cationic trypsinogen Ca2+-binding loop (16).
TABLE 1.
Specific substrate/inhibitor sequences recognized by CTRC
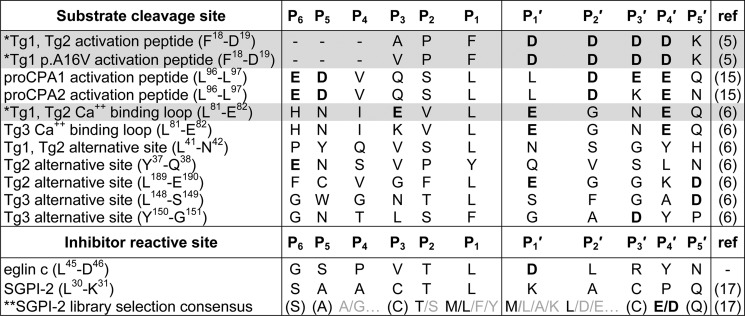
The abbreviations used in the table are: Tg1, human cationic trypsinogen; Tg2, human anionic trypsinogen; Tg3, human mesotrypsinogen; proCPA1, human procarboxypeptidase A1; proCPA2, human procarboxypeptidase A2; SGPI-2, Schistocerca gregaria proteinase inhibitor-2. *, substrate peptides shaded in gray have been modeled in the CTRC active site as described in the text, structural coordinates for the models are provided as supplemental Models S1–S3. **, sequence shown is the consensus of multiple clones selected from a phage-displayed SGPI-2 library that was diversified at P4, P2, P1, P1′, P2′, and P4′. Residues shown in parentheses were at positions that were not diversified in the library.
Here, the first structure of active CTRC reveals a familiar fold with distinctive electrostatic features surrounding the substrate binding cleft. The CTRC active site is occupied by the inhibitor eglin c bound in a substrate-like conformation, offering insights into the structural basis for the unique substrate specificity of CTRC toward key physiological and pathological substrate sequences. Analysis of the structure suggests that whereas the bulk of binding energy derives from burial of the P1 residue and several other hydrophobic side chains, specificity may derive largely from the exaggerated role of long-range electrostatic interactions, from a moderate preference for Leu at the P1 position, and may also be influenced by local sequence-dependent backbone conformational tendencies.
EXPERIMENTAL PROCEDURES
Protein Expression and Purification
Human chymotrypsinogen C bearing a C-terminal His10 tag was expressed using transiently transfected HEK 293T cells, purified using metal chelation chromatography, dialyzed against 50 mm sodium phosphate (pH 8.0) and 300 mm NaCl, concentrated to 20 mg/ml, and activated by cleavage with cationic trypsin as previously described (4).
A construct for recombinant bacterial expression of eglin c from Hirudo medicinalis was a generous gift from Professor Robert S. Fuller, University of Michigan Medical School. Eglin c was expressed in Escherichia coli host strain BL21(DE3) and purified to homogeneity from periplasmic extract by SP-Sepharose chromatography essentially as previously described (18). Eglin c was dialyzed into 10 mm (NH4)2OAc at pH 6.0, lyophilized, and stored at −80 °C until use, when it was reconstituted with H2O.
Crystallization
A 1:1 (mol/mol) mixture of CTRC and eglin c was concentrated to achieve a protein concentration of 4–5 mg/ml. Crystallization employed the hanging drop vapor diffusion method. Diffraction quality crystals were grown at room temperature from droplets containing 0.2 m lithium sulfate monohydrate, 0.1 m Tris-HCl (pH 8.5), and 30% (w/v) PEG 4000. Crystals grew over the course of 3 weeks to 0.1 × 0.2 × 0.2 mm. Crystals were soaked in a cryoprotectant solution (0.2 m lithium sulfate monohydrate, 0.1 m Tris-HCl (pH 8.5), and 30% (w/v) PEG 4000, and 15% glycerol) and cryocooled in liquid N2.
Crystals were screened for diffraction and data were collected at 100 K at beamline X29 at the National Synchrotron Light Source, Brookhaven National Laboratory. The best crystals diffracted to 1.9-Å resolution. We identified crystals of CTRC/eglin c belonging to orthorhombic space group P212121, with unit cell dimensions a = 56.27, b = 76.25, c = 81.82, and containing one complex in the asymmetric unit. The data were merged and scaled using DENZO/SCALEPACK (19).
Structure Determination
The x-ray structure of chymotrypsin C in complex with eglin c was solved by molecular replacement using Phaser (20) supported by CCP4. The elastase chain from the previously solved structure of a porcine elastase-inhibitor complex (PDB code 1EAI, chain A) (21) and eglin c from bovine α-chymotrypsin-eglin c (PDB 1ACB, chain I) (22) were used as search models. ARP/wARP was employed after molecular replacement for automated rebuilding of the CTRC-eglin c complex structure (23, 24). A test set of 5% of the total reflections was excluded from rebuilding and refinement of the model. Refmac5 (25) was used to carry out refinement and to place water molecules into difference peaks (Fo − Fc) greater than 3σ; manual rebuilding was done using COOT (26). A 10-residue activation peptide chain that was not liberated following proteolytic activation of CTRC due to a disulfide link with the enzyme (chain Q) was manually built into electron density maps (2Fo − Fc) using COOT. Phosphate ions were also added using COOT. The final stage of the restrained refinement included water molecules with peaks greater than 1σ and within acceptable H-bonding distances from neighboring protein atoms and three phosphate ions. Surprisingly, we were unable to build either the His10 tag or the N-linked glycosyl groups of CTRC due to the absence of electron density. The final R/Rfree was 0.157/0.210. Figs. 1, 2, 4A, and 5 were generated using PyMOL (Schrödinger, LLC).
FIGURE 1.

Crystal structure of the CTRC-eglin c complex. A, structural overview of the complex. CTRC is shown in blue, with catalytic triad residues Ser195, His57, and Asp102 in red and disulfide links in yellow. Eglin c is displayed in green. B, view into the substrate binding cleft of CTRC. CTRC is shown with a semitransparent gray surface. Eglin c binding loop residues 40–50 are rendered in stick representation, filling (from left to right) CTRC S6-S1 and S1′-S5′ subsites. C, retained activation peptide of CTRC. Residues 1–10 of the chymotrypsin C activation peptide are tethered to the activated enzyme through a disulfide link between Cys1 and Cys122. The activation peptide is depicted in cyan in stick representation, with a 2Fo − Fc electron density map contoured at 1.6σ. Strong density around the Leu10 carboxyl terminus confirms that residues 11–13 of the activation peptide are not disordered but have been proteolytically removed.
FIGURE 2.

Eglin c inhibitory interaction with CTRC. A, stabilization of the inhibitory loop of eglin c. The eglin c binding loop assumes a substrate-like canonical conformation of the peptide backbone, stabilized on the nonprimed side by hydrophobic interactions of Leu37, Val43, and Phe55, and on the primed side by an H-bond network involving Thr44, Asp46, the Arg48 amide nitrogen, Arg51, Arg53, and C-terminal Gly70. The side chain of Arg48 is omitted for clarity. B, the CTRC-eglin c complex resembles an enzyme-substrate Michaelis complex. The Leu45-Asp46 reactive site peptide bond of eglin c, linking the P1 and P1′ residues, lies in proper orientation for attack by the catalytic Ser195 of CTRC. The 2Fo − Fc electron density map is shown contoured at 2.0σ. C, key CTRC-eglin c binding interactions. The eglin c P1 residue Leu45 fills the S1 pocket bordered by CTRC Ala190, Val213, and Val226. P4 residue Pro42 fills a hydrophobic concavity formed by CTRC Leu99 and Phe215. P2′ residue Leu47 fills a pocket formed by CTRC Arg143 and Ile151. Multiple backbone H-bonds orient the inhibitor, indicated by black dotted lines. D, positively charged P6 pocket displaces eglin c backbone to bind phosphate. Basic side chains of CTRC Arg175, Arg218, and Lys224 coordinate a phosphate ion, displacing eglin c from the orientation in which it is found in complex with bovine α-chymotrypsin (shown in semitransparent white stick representation; PDB code 1ACB). The 2Fo − Fc electron density map shown for the phosphate ion is contoured at 1.6σ.
FIGURE 4.

Cationic trypsinogen Ca2+-binding loop targeted for cleavage by CTRC. A, the Ca2+-binding loop of human cationic trypsinogen contains multiple acidic residues (orange), with additional acidic residues located in nearby neighboring loops. The bound Ca2+ is shown in purple; the Leu81-Glu82 peptide bond represents the preferred cleavage site targeted by CTRC. Structure coordinates are from PDB code 2RA3 (55). B, the electrostatic surface potential calculated for cationic trypsin reveals a negative charge surrounding the site of bound Ca2+, indicated by the large black arrow. Molecular surfaces were generated using the Molecular Surface module of Schrodinger 2012 as described under “Experimental Procedures”; the electrostatic potential color ramp was set from a minimum of −0.35 to a maximum of 0.15. C, Ca2+-binding loop cleavage sequence of cationic trypsinogen is shown modeled into the active site cleft of CTRC. The hydrophobic side chains of Ile78 and Leu81 occupy the complementary S4 and S1 subsites, respectively. Acidic residues are shown in orange; Glu79 at the P2 position and Glu85 at the P4′ position are near CTRC regions of positive surface potential.
FIGURE 5.

Structural modeling of CTRC-substrate interactions. A, trypsinogen Ca2+-binding loop substrate positioning relative to nearby CTRC residues illustrates H-bonds formed between enzyme and substrate as black dotted lines. None of substrate acidic residues Glu79, Glu82, or Glu85 lie in close enough proximity to the CTRC basic side chains to form direct salt bridges. B, a similar view showing a model of trypsinogen activation peptide cleavage sequence bound to CTRC. This shorter alternative substrate forms one fewer non-primed side hydrogen bond with the enzyme, and lacks the hydrophobic stabilization that would be provided by a P4 substrate residue. However, a direct salt bridge is formed between P2′ residue Asp20 and CTRC Arg143. C and D, minimal differences are present in models of trypsinogen activation peptide (C) and p.A16V mutant (D) bound to CTRC. Where the Ala16 N-terminal amine is predicted to hydrogen bond with the CTRC Gly216 carbonyl (C), rotation of the Val16 (magenta) eliminates this bond but the bulkier side chain forms additional hydrophobic contacts with the Pro17 ring and the β-carbon of Arg217A (D).
Homology Modeling of Human Elastase and Chymotrypsin Isoforms
Homology models of human elastases (ELA2A, ELA3A, and ELA3B) and chymotrypsins (CTRB1, CTRB2, and CTRL1) were constructed using the SWISS-MODEL workspace (27). Homology models were constructed using porcine elastase bound to an Ascaris chymotrypsin/elastase inhibitor (PDB code 1EAI, chain A) (21) or our model of active CTRC bound to eglin c (PDB code 4H4F, chains A and Q) as templates with 40–50% amino acid identity to the modeled proteins. Molecular surfaces were generated using the Molecular Surface module of Schrödinger 2012 (Schrödinger, LLC). Molecular surfaces were based upon high-resolution settings (0.3 Å grid, probe radius 1.4 Å, and van der Waals radius scale of 1.0 Å). All surfaces were rendered in red - white - blue color scale using PB electrostatic potential from calculated charges at pH 8.5 with the color ramp set to a minimum of −0.35 and maximum of 0.15 for the structures shown in Figs. 3 and 4; the figures were generated using the Maestro all-purpose molecular modeling environment, version 9.3.023 from the Schrödinger Suite 2012.
FIGURE 3.

Distinct highly charged electrostatic surface of CTRC. The electrostatic surface potential of CTRC (top) shows intense concentration of a positive charge (blue) surrounding the active site cleft. Positions of substrate-binding subsites are indicated by black arrows. Smaller panels below show for comparison the electrostatic surface potentials generated for homology models of human elastase isoforms (left) and chymotrypsin isoforms (right). Molecular surfaces were generated using the Molecular Surface module of Schrodinger 2012 as described under “Experimental Procedures”; the electrostatic potential color ramp was set from a minimum of −0.35 to a maximum of 0.15.
Molecular Modeling of CTRC Substrate Binding
The x-ray structure for CTRC was imported into the Protein-Preparation-Wizard graphical user interface of Schrödinger with Maestro 2012 version 9.3.5 (Schrödinger, LLC) for adaption to the OPLS2005 force field. Bond orders were assigned, zero-order bonds to metals were determined, disulfide bonds were created, and all hydrogens were generated for every residue. Hydrogen bond assignment was based on sampling water orientations and taking into account crystallographic waters. Protonation states were predicted for pH 8.5 using PROPKA (28, 29). Steric clashes were resolved with convergence of a root mean square deviation to 0.3 Å using the OPLS2005 force field within the Schrödinger interface.
For modeling and molecular docking of peptide substrate sequences, starting conformations of substrates were obtained by Polak-Ribiere Conjugate Gradient energy minimization (30) with the OPLS 2005 force field for 5000 steps, or until the energy difference between subsequent structures was <0.001 kJ/mol Å (28). Force field minimization used a water-based solvent, generating charges with an extended cutoff (van der Waals 8.0 Å, electrostatic 20 Å, H-bond 4.0 Å). We placed soft restraints on all residues >6 Å from the modeled substrate by using harmonic restraints at 100 kcal/mol, and allowed the residues within the 6-Å cutoff to move freely during Polak-Ribiere Conjugate Gradient energy minimization over 500 iterations with repetition as necessary to converge upon a gradient threshold of <0.05.
We have previously described the methodology used for substrate docking (31); briefly, the binding site was generated via overlapping grids based on the x-ray structure with a default rectangular box centered on the target substrate. Substrates were docked into the binding site of CTRC using Glide extra precision (XP) (Glide, version 5.6, Schrödinger, LLC); molecular conformations were sampled using methods described previously (32). A structure-based pharmacophore score was generated from the optimized, best scoring pose for each substrate ligand based on the descriptors from the Glide XP score using an established approach (31, 33, 34). The energetic value assigned to each pharmacophore feature was calculated using Phase (Phase, version 3.2, Schrödinger, LLC) as the sum of the Glide XP contributions of the atoms comprising the site. Overall dockings at the active site were quantified and ranked on the basis of these energetic terms (33, 34). To account for protein flexibility and lessen the effects of minor steric clashes, excluded volumes spheres corresponding to 80% of the van der Waals atomic radii were created for all CTRC atoms within 6 Å of each substrate or mutagenized residue modeled. A minimum of two poses per substrate, chosen for a combination of best-scoring features, was selected for visual and energetic comparison (31, 33, 34).
RESULTS
Structure Solution and Refinement
The structure of active human CTRC bound to inhibitor eglin c was determined to 1.9-Å resolution in space group P212121 with one bimolecular complex per asymmetric unit. The structure was solved by molecular replacement using as search models the previously reported structures of porcine pancreatic elastase (PDB 1EAI) (21), featuring 52% sequence identity to human CTRC, and eglin c (PDB 1ACB) (22). A structure of the bovine CTRC precursor chymotrypsinogen C, possessing 80% sequence identity to human CTRC, has been reported previously (PDB 1PYT) (35); however, the elastase structure was judged to offer a better search model as a result of substantial conformational changes that take place upon protease activation (36). The model was rebuilt and refined to a final Rcryst (Rfree) of 15.7% (21.0%); crystallographic statistics are summarized in Table 2.
TABLE 2.
Crystallographic data collection and refinement statistics for CTRC-eglin c complex
| PDB ID 4H4F | |
|---|---|
| Complexes per ASU | 1 |
| Space group | P212121 |
| Unit cell, Å | 56.27, 76.25, 81.82 |
| 90°, 90°, 90° | |
| Resolution, Å | 1.9 |
| Unique reflections | 27,661 |
| Completeness (%) | 97.34 (76.8)a |
| Multiplicity | 6.8 (4.8)a |
| Mean I/ (σ) | 51.3 (9.0)a |
| R-merge | 0.043 (0.146)a |
| Rsym | 0.043 (0.146)a |
| Rcryst/Rfree (%) | 0.157/0.210 |
| Root mean square deviation bonds, Å | 0.027 |
| Root mean square deviation angles (degree) | 2.066 |
| Protein atoms | 2469 |
| Ions | 3 (PO4−2) |
| Water molecules | 320 |
| Φ,Ψ angle distributionb | |
| In favored regions | 209 (87.8%) |
| In additionally allowed regions | 28 (11.8%) |
| In generously allowed regions | 1 (0.4%) |
a Values in parentheses are for the highest resolution shell (1.93–1.9 Å).
b Ramachandran distribution is reported as defined by PROCHECK/PDB validation.
Overall Structure of the CTRC-Eglin c Complex
Like other serine proteases of the chymotrypsin family (37), CTRC is comprised of two β-barrels, at the interface of which is located the active site containing the catalytic triad of Ser195 (Ser216),6 His57 (His74), and Asp102 (Asp121) (Fig. 1A). The substrate binding cleft between the β-barrels is occupied by bound inhibitor eglin c, a 70-amino acid protein protease inhibitor originally isolated from the leech H. medicinalis (38). As has been described previously for eglin c (22, 39, 40) and for the structurally related chymotrypsin inhibitor 2 (41, 42), the inhibitor is a wedge-shaped molecule featuring a hydrophobic core formed by a helix and a small β-sheet, from which protrudes the inhibitory canonical loop, forming the thin edge of the wedge, which fits into the substrate binding cleft of the enzyme (Fig. 1, A and B). The canonical loop of eglin c, comprised of residues 40–50, binds to the active site of CTRC in the substrate-like fashion typical of canonical serine protease inhibitors (43–45) (Fig. 1B).
CTRC residues 1–10 of the cleaved activation peptide chain (CGVPSFPPNL) are retained by the activated enzyme due to a disulfide link between Cys1 (Cys17) of the activation peptide and Cys122 (Cys141), located in the linker between the two β-barrels on the enzyme face distal from the active site (Fig. 1C). The disulfide bonding pattern and consequent retention of the activation peptide, conserved among other chymotrypsins and the elastase 2A isoform, has been demonstrated to stabilize the enzyme against denaturation and proteolytic digestion by pepsin (46). In addition to the covalent link, the activation peptide association is stabilized by two backbone-backbone H-bonds of Gly2 (Gly18), an H-bond between the carbonyl of Pro7 (Pro23) and the side chain of Arg23 (Arg37), and substantial hydrophobic interactions of Pro4 (Pro20), Phe6 (Phe22), Pro8 (Pro24), and Leu10 (Leu26) (Fig. 1C). Clear density is observed for the C-terminal Leu10 (Leu26) of the activation peptide chain, revealing that residues Ser11-Ala12-Arg15 (Ser27-Ala28-Arg29) of the activation peptide have been proteolytically removed. Given the binding preference of CTRC for Leu at the P1 position (17), it is probable that removal of the tripeptide is accomplished through autoproteolytic cleavage in trans, as suggested previously (47).
Although the recombinant CTRC possessed a His10 tag, none of the residues of the tag were visible in the electron density, suggesting that either the tag was disordered or had been proteolytically removed post-purification by autoproteolysis. Likewise, although CTRC has been shown to be glycosylated on Asn36B (Asn52), a modification required for efficient folding and secretion (48), we did not observe density for the glycosyl group. The side chain of Asn36B (Asn52) was poorly defined and it is likely that the sugar at this site was present but disordered.
Insights into Chymotrypsinogen C Activation
Comparison of the human CTRC structure with the earlier reported structure of bovine chymotrypsinogen C (PDB 1PYT) (35) shows that upon cleavage of the activation loop at Arg15-Val16 (Arg29-Val30), the new N-terminal residue Val16 (Val30) becomes buried, forming a salt bridge with Asp194 (Asp215) and an H-bond with the carbonyl oxygen of Arg143 (Arg162). This reorganization has little impact on the structure of the first β-barrel domain, but results in major conformational alterations of the second β-barrel domain, particularly within loops 1 and 2, which shape the S1 specificity pocket and oxyanion hole, and loop D, which helps to shape the primed side subsites. Although the catalytic triad residues are already roughly positioned in chymotrypsinogen C, small conformational adjustments in all three members of the triad bring them into appropriate alignment for catalysis, reducing the Ser195 Oγ-His57 Nϵ2 distance from 3.83 to 2.88 Å, and shortening the His57 Nδ1-Asp102 Oδ2 H-bond from 2.95 to 2.55 Å. These alterations are typical of the conserved mechanism of activation of chymotrypsinogen family members (13).
Details of the Inhibitory Interaction
The inhibitory loop of eglin c is stabilized in the distinctive canonical conformation by a hydrophobic “mini-core” (49) comprised of Leu37, Val43, and Phe55, and by a H-bond network involving Thr44, Asp46, the Arg48 backbone N, Arg51, Arg53, and the C-terminal carboxyl group of Gly70 (Fig. 2A). This H-bond network is critical for maintaining protease affinity and resistance to proteolysis of the inhibitor (50–54).
The mode of protease binding of canonical inhibitors like eglin c has been shown to very closely mimic the reactive Michaelis complex with a true peptide substrate (45). As is typical of such complexes, the Leu45-Asp46 reactive site peptide bond of eglin c is appropriately positioned for nucleophilic attack by the catalytic Ser195 (Ser216) of CTRC (Fig. 2B), the initial step in enzyme-catalyzed proteolysis. The largely hydrophobic substrate binding cleft of CTRC is fully occupied by eglin c residues 40–50, which mimic non-primed side substrate residues P1-P6 and primed side residues P1′-P5′. The non-primed side residues are positioned by antiparallel backbone-backbone H-bonds between the P3 residue Val43 and CTRC Gly216 (Gly238), and between the P1 residue Leu45 and CTRC Ser214 (Ser236) (Fig. 2C). The Leu45 carbonyl oxygen is positioned to interact with the CTRC oxyanion hole amide nitrogens of Ser195 (Ser216) and Gly193 (Gly214). On the primed side, additional H-bonds are formed between P2′ residue Leu47 and CTRC Thr41 (Thr58) (Fig. 2C).
The S1 primary specificity pocket of CTRC, occupied in the complex by the side chain of eglin c Leu45, is shaped by the hydrophobic side chains of CTRC residues Ala190 (Ala211), Val213 (Val235), and Val226 (Val250) (Fig. 2C). A more shallow hydrophobic depression in the substrate binding cleft is shaped by the side chains of CTRC Leu99 (Leu118) and Phe215 (Phe237), which form a binding pocket for P4 residue Pro42. On the primed side, the S2′ subsite, a pocket bordered by the basic side chain of CTRC Arg143 (Arg162) and the hydrophobic side chain of Ile151 (Ile169), is filled by the hydrophobic P2′ residue Leu47 (Fig. 2C).
The backbone conformation of eglin c residues 43–49 bound to CTRC closely parallels that seen in the previously reported structure of eglin c with bovine α-chymotrypsin (PDB 1ACB (22)). By contrast, eglin c residues 39–42 are shifted compared with the bovine α-chymotrypsin-eglin c complex to lie ∼3 Å further removed from a basic patch formed by CTRC residues Arg175 (Arg195), Arg218 (Arg241), and Lys224 (Lys248) (Fig. 2D). This basic pocket forms the S6 subsite of CTRC, which is occupied in the complex by a coordinated phosphate ion in the absence of a side chain on the eglin c P6 residue Gly40 (Fig. 2D).
Structural Insights into CTRC Substrate Specificity
CTRC has been shown to act as a regulator of pancreatic zymogen activation by targeting a specific set of substrate cleavage sites not recognized by other chymotrypsin or elastase-like digestive proteases (Table 1) (5, 6, 15, 16). One element of this specificity is more highly efficient cleavage after Leu residues when compared with other chymotrypsin and elastase isoforms (16). By contrast, the elastase isoforms show broad P1 specificity but comparatively low catalytic efficiency on short peptide substrates, whereas chymotrypsins prefer aromatic residues Phe, Tyr, or Trp at P1 (16). The position occupied in CTRC by Val226 (Val250), one of the hydrophobic residues shaping the S1 pocket (Fig. 2C), is in other chymotrypsins filled by Gly or Ala, and the bulkier Val residue at this position is likely to be responsible for the modest binding selectivity of CTRC for Leu in preference to Met, Phe, or Tyr at the P1 position, as identified by phage display selection and inhibitor binding studies (17) and by Km values for cleavage of tetrapeptide substrates (16). Nevertheless, the S1 subsite of CTRC is still capable of accommodating aromatic residues, and in fact, CTRC catalytic rates are slightly enhanced for cleavage after these bulkier residues, resulting in comparable catalytic efficiencies for cleavage after Leu, Met, Phe, or Tyr (16). This result is also consistent with the identification of natural CTRC cleavage sites within protein substrates after Phe, Leu, and Tyr (Table 1).
Another distinctive feature shared by the natural target sites of CTRC is an unusual clustering of acidic residues. Asp or Glu appear very frequently at the P4′ position, an element of specificity corroborated by phage display selection (17). Acidic residues can also be found at P1′, P2′, P3′, and P5′ on the primed side of the cleavage site, and at P3, P5, and P6 on the non-primed side of the cleavage site (Table 1). To gain insight into the probable electrostatic contribution to this unusual substrate specificity, we calculated the predicted electrostatic surface potential of the CTRC structure, and for comparison we generated homology models and calculated electrostatic surfaces for other human chymotrypsin and elastase isoforms, which do not target the same regulatory cleavage sites (Fig. 3). We observed a particularly striking concentration of positive charge in a ring surrounding the substrate binding cleft of CTRC (Fig. 3, top). The most intense concentrations of positive charge are created by one cluster of basic residues on the non-primed side of the cleft in the region that makes contact with P5 and P6 substrate residues, and a second cluster of basic residues on the primed side bordering the subsites that recognize P2′, P3′, and P4′ substrate residues. Notably, this charge distribution contrasts markedly with the predicted electrostatic surfaces of other elastase and chymotrypsin isoforms (Fig. 3, lower panels). The human elastases feature small patches of both positive and negative charge in roughly equal proportions, whereas human chymotrypsins possess substrate binding clefts lined with primarily negative charge. Thus, the unusual concentration of positive charge surrounding the CTRC substrate binding cleft is likely to be a major determinant driving specificity for substrate sequences of net negative charge.
Modeling of CTRC-Substrate Complexes
Selection by CTRC among several potential cleavage sites within human cationic trypsinogen has profound health implications. Whereas cleavage in the trypsinogen Ca2+-binding loop is a normal mechanism for CTRC to apply the brakes to a premature cascade of digestive enzyme activation in the pancreas (6), an alternative cleavage within the activation peptide has the potential to accelerate this activation cascade (5). To serve its protective function, CTRC must significantly favor the former cleavage over the latter in the pancreas environment. To gain insight into the interactions of competing cleavage sites with CTRC, we examined the structural context of the Ca2+-binding loop site in our previously reported structure of human cationic trypsin (55), and we also used the new CTRC structure as a starting point to generate models of CTRC bound to specific substrate sequences.
In the cationic trypsin structure, Ca2+ is coordinated within the loop by the side chains of residues Glu75 and Glu85, which anchor the base of the loop, and by carbonyl oxygens of Asn77 and Val80 (Fig. 4A).7 CTRC targets the Leu81-Glu82 peptide bond for cleavage. With Ca2+ bound, Leu81 is exposed and accessible to CTRC; however, the peptide backbone stretching away from the cleavage site is unable to assume the canonical conformation required for cleavage (56). Cationic trypsinogen possesses a large number of acidic residues in this region; in addition to Glu75 and Glu85, there are two additional glutamates in the loop (Glu79 and Glu82), as well as three acidic residues in neighboring loops (Glu32, Asp156, and Glu157; Fig. 4A). Thus, even with Ca2+ present to neutralize the charge of Glu75 and Glu85, there is a strongly negative electrostatic potential covering this region of the molecule (Fig. 4B). This potential will generate macroscopic electrostatic complementarity between the Ca2+-binding loop of cationic trypsin or trypsinogen and the substrate-binding site of CTRC. It is anticipated that when Ca2+ is released, this loop becomes more flexible and accessible to CTRC, able to assume a productive binding orientation, and that electrostatic attraction may increase further due to exposure of Glu75 and Glu85.
Because eglin c binds to CTRC in the canonical orientation of an ideal substrate (45), we were able to model substrate sequences into the binding cleft of CTRC using eglin c as a template and then to optimize the interactions through energy minimization. Models were generated for CTRC bound to the human cationic trypsinogen activation peptide (APFDDDDK) and the CTRC-labile site within the cationic trypsinogen Ca2+-binding loop (HNIEVLEGNEQ). The resulting “global” total enthalpies (ΔH) from energy minimization were −70,572 and −71,057 kcal/mol, respectively. Following modeling and minimization, we conducted substrate docking for each substrate with CTRC, giving docking scores of −10.16 and −17.72 kcal/mol, respectively. The cationic trypsinogen Ca2+-binding loop, which contacts the greater number of non-primed side subsites, was predicted to be the more preferred substrate based on overall lowest energy from docking/binding with CTRC.
The docked model of preferred substrate HNIEVLEGNEQ shows the positioning of substrate residues relative to the electrostatic features of the CTRC surface (Fig. 4C); residues Asn77-Ile78-Glu79-Val80-Leu81-Glu82-Gly83-Asn84-Glu85 fill the largely hydrophobic cleft between the flanking clusters of positive charge, with the Leu81 side chain embedded in the hydrophobic S1 subsite. Surprisingly, none of the acidic side chains of the substrate, Glu79, Glu82, or Glu85, which fill the P3, P1′, and P4′ positions, respectively, form direct salt bridges with the clustered basic side chains of CTRC in the energy minimized docked model (Fig. 5A). Instead, it would appear that the complex stabilization attributable to charge complementarity derives from longer range electrostatic interactions. Trypsinogen Glu82 in the P1′ position is stabilized by CTRC Arg62A via an interaction bridged by H-bonds with the P3′ side chain of trypsinogen Asn84. The trypsinogen Glu85 P4′ side chain is positioned equidistant from the guanidinium groups of CTRC Arg39 and Arg143 (about 6 Å from each). The major favorable close interactions in the complex are comprised primarily of hydrophobic and van der Waals interactions, and a series of hydrogen bonds tethering the substrate peptide backbone within the binding cleft (Fig. 5A).
The docked model of the competing substrate cleavage site, APFDDDDK (the trypsinogen activation peptide), shows that this shorter substrate fills only the S1-S3 subsites on the non-primed side of the cleft, forming fewer H-bonds and hydrophobic interactions as compared with the longer substrate (Fig. 5B). In this model, Phe18 fills the hydrophobic S1 subsite, and the substrate does form a single direct salt bridge between Asp20 at the P2′ position and Arg143 (Arg162) of CTRC (Fig. 5B). However, as was the case for the Ca2+-binding loop site, electrostatic stabilization conferred by the Asp residues at the P1′, P3′, and P4′ positions is apparently mediated through longer-range interactions with CTRC.
Mutation of a single residue in the trypsinogen activation peptide, where Val is substituted for Ala16 at the P3 position, predisposes carriers for development of pancreatitis, apparently by altering the balance of the CTRC substrate selectivity in favor of the activation peptide (4, 7, 8). To explore the structural basis for this shift in selectivity, we modeled the complex of CTRC with the p.A16V mutant sequence. The docked model of the p.A16V mutant trypsinogen activation peptide VPFDDDDK very closely resembles that of the wild-type sequence, with significant differences confined to the mutated residue itself. The N-terminal amine of Ala16 H-bonds to the CTRC Gly216 carbonyl in the model of the wild-type sequence (Fig. 5C), whereas in the mutant model this H-bond is disrupted by slight rotation of Val16 to optimize hydrophobic contacts with the Pro17 ring and with the β-carbon of Arg217A (Fig. 5D). The impact of the mutation on binding interactions with CTRC suggested by these models would not appear to be sufficient to explain the functional importance of the mutation in predisposing carriers to pancreatitis.
Consistent with these minimal structural differences, the calculated binding energy for the p.A16V mutant sequence was very close to that of the wild-type sequence (−8.811 versus −10.16 kcal/mol, respectively) and did not suggest enhanced binding upon mutation as might be predicted by functional studies (4, 5). One possible explanation for this apparent discrepancy is that the energy calculations do not take into account entropic components of binding energy, which may differ between the substrate sequences. This explanation is highly plausible, because entropic factors involved in binding are anticipated to differ between the wild-type and mutant activation peptides as detailed further under “Discussion.” Thus, it is probable that the mutation critically influences the conformational ensemble and dynamics of the activation peptide in its unbound form, resulting in differential entropic contributions to formation of the CTRC-substrate complex and thereby altering cleavage selectivity.
DISCUSSION
On one hand, biochemical studies with purified proteins show that CTRC, like most digestive proteases, is capable of proteolytic cleavage of multiple target sequences revealing no highly conserved recognition motif (Table 1). CTRC appears to be relatively insensitive to a variety of mutations of the trypsinogen Ca2+-binding loop cleavage site (16), further suggesting fairly promiscuous activity. On the other hand, incontrovertible genetic evidence demonstrates that a subtle mutation of the CTRC cleavage site within the cationic trypsinogen activation peptide predisposes to development of chronic pancreatitis (7, 8), as does loss of function of CTRC itself (9–12); these observations reveal a specific regulatory role for the enzyme.
To reconcile these contrasting views of CTRC specificity, we consider how the protease selects its substrates in vivo. Steady state enzyme kinetics studies using individual substrates can reveal the thermodynamic stability of the Michaelis complex (Km) and the overall catalytic rate of the reaction (kcat) from which the specificity constant kcat/Km is derived. However, in the scenario in which multiple candidate substrates are in direct competition, kinetic rather than thermodynamic control may prevail (57, 58). Protein-protein association is a multistep process, with different molecular forces influencing the rates of each step, and electrostatic interactions provide the dominant long-range force capable of accelerating the rate of initial molecular collision (58, 59). The intense concentration of positive charge surrounding the CTRC active site, yet apparently not positioned for optimal formation of tight salt bridges in the final Michaelis complex, suggests that this funnel-like ring of surface charge may instead be optimized to kinetically favor the initial attraction of polyacidic substrate sequences, akin to a molecular tractor beam.
Nonspecific long-range electrostatic attraction will be a dominant driving force as CTRC and its substrate first approach each other, at a stage when they may not yet be correctly rotationally oriented. The stabilization conferred by electrostatic attraction, combined with the viscosity of the surrounding solution, may also extend the lifespan of the low affinity transient complex, allowing the two proteins to rotate and sample multiple trajectories of approach in repeated microcollisions (58). Thus, an electrostatically complementary substrate will have enhanced probability of finding a productive binding conformation, achieving the Michaelis complex, and becoming proteolyzed. A similar influence of electrostatic potential on substrate specificity was observed previously for the hepatitis C virus NS3 protease using pre-steady state kinetics, where clusters of positively charged residues near the active site, complementary to clusters of negative charge on the substrate, were found to drive very rapid association (60). The influence of these long-range interactions on substrate selection in vivo may be underestimated in comparisons of overall substrate affinity, which depend upon the relative rates of formation and dissociation of the high affinity Michaelis complex. The later stages of binding that mark the progression from transient to Michaelis complex are likely to be slower, due to the radical alterations of local substrate conformation often required (56).
Following formation of a roughly aligned encounter complex, the next stage of CTRC-substrate association is likely to be docking of the large hydrophobic P1 “anchor” residue within the S1 subsite (57). As with electrostatic charge, the kinetic importance of this interaction and its impact on specificity under conditions of direct substrate competition may be underestimated in studies with single purified substrates. For example, CTRC binding studies with tight binding inhibitors revealed a clear preference for P1 Leu over Met, both of which were represented much more highly than Phe or Tyr in a pool of inhibitors selected by phage display (17). By contrast, CTRC appeared insensitive to mutation of the P1 Leu in the trypsinogen Ca2+-binding loop to Met, Tyr, or Phe (16). We would speculate that in the case of the rigidly structured inhibitors, the overall rate of association reflects the rate of P1-S1 docking, which is fastest for Leu due to optimal complementarity with the S1 pocket. By contrast, with highly flexible or natively unstructured substrate sequences, the slower rate with which flanking residues conform to adjacent subsites through induced fit (57) may mask the importance of P1 specificity.
CTRC serves a protective function in the pancreas, as demonstrated by the association of CTRC loss of function with chronic pancreatitis (9–12), and by the finding that pathological activation of hereditary pancreatitis-causing cationic trypsinogen mutants is dependent on CTRC activity (4). We have found that CTRC can cleave cationic trypsinogen at alternative cleavage sites; proteolysis within the trypsinogen activation peptide leads to enhanced autoactivation of trypsin (5), whereas proteolysis within the Ca2+-binding loop leads to trypsinogen degradation (6). The protective function of CTRC would suggest that the dominant activity of CTRC activated in the pancreas must be the degradation of cationic trypsinogen, initiated by cleavage within the Ca2+-binding loop.
Our modeling results are consistent with the idea that when sterically accessible, the Ca2+-binding loop is the more preferred site of cleavage. This site fills a greater number of nonprimed subsites, burying a greater solvent accessible surface area, it possesses the preferred Leu at the anchor P1 position, and it is calculated to have a thermodynamically more favorable binding energy. In the absence of bound Ca2+, the Ca2+-binding loop may be preferred by CTRC from the perspective of long-range electrostatic interactions as well, because it possesses four Glu residues within the primary sequence of the loop and an additional three exposed acidic residues on nearby loops. In comparison, the unstructured activation peptide possesses four tandem Asp residues, but the negative charge of these side chains would be offset by an adjacent Lys residue and by the N-terminal amine.
It is more difficult to explain from a structural perspective why the p.A16V mutant trypsinogen activation peptide is improved as a CTRC substrate, to the extent of significantly diminishing the protective function of CTRC in the pancreas. However, it may be as a result of the impact of this substitution on the conformational ensemble represented by the activation peptide and on its dynamics. The activation peptide is at the N terminus of trypsinogen, and is unstructured in the crystal structures of bovine and rat trypsinogens (61–63). In this unstructured state, the Ala16-Pro17 (or Val16-Pro17) peptide bond exists as an equilibrium mixture of cis and trans isomers (64, 65), and the bulkier Val16 will greatly reduce the rate of cis-trans isomerization (66). Perhaps more importantly, the steric bulk of the proline ring restricts the conformations available to the preceding residue (67), and the β-branched Val16 will encounter further restrictions than the smaller Ala16 (68). To bind to the CTRC active site and undergo proteolysis, the Pro peptide bond must assume the trans configuration (69), and both residues must conform to the idealized binding mode of a canonical loop, in which the P3 residue main chain assumes an antiparallel β-strand conformation and the P2 residue main chain assumes a polyproline II conformation (43). We speculate that the steric restrictions and reduced mobility of the p.A16V mutant have the effect of locking the activation peptide into a more substrate-like conformation, or of biasing the conformational ensemble toward a more substrate-like conformation. The impact would be to minimize the unfavorable entropic contribution to the binding energy, rendering the p.A16V mutant trypsinogen activation peptide a more favorable substrate.
In conclusion, the molecular structure and analyses of CTRC presented here offer mechanistic insight into the striking selectivity of the enzyme for regulatory sites in trypsinogens and procarboxypeptidases. Long-range electrostatic interactions between enzyme and substrate, rather than specific charge pairing, underlie substrate discrimination. In addition, the structure of CTRC establishes a framework that may enable interpretation of the functional effects of other clinically significant trypsinogen mutations that impact CTRC substrate selectivity, as well as mutations within CTRC itself that modify risk for development of chronic pancreatitis.
Acknowledgments
Diffraction data were measured at beamline X29 of the National Synchrotron Light Source, which is supported by the Offices of Biological and Environmental Research and Basic Energy Sciences of the United States Department of Energy, and the National Center for Research Resources of the National Institutes of Health.
This work was supported, in whole or in part, by National Institutes of Health Grants R01 DK082412-S2 (ARRA), R01 DK082412, R01 DK058088, and R01 DK095753 (to M. S.-T.) and Florida Department of Health Postdoctoral Fellowship 1BD-01 (to J. B.).

This article contains supplemental Models S1–S3.
Substrate residues surrounding the cleavage site are designated by the nomenclature of Schechter and Berger (70): starting from the scissile bond, substrate residues are numbered P1, P2, P3, etc. in the direction of the N terminus (collectively the non-primed residues), and P1′, P2′, P3′, etc. in the direction of the C terminus (collectively the primed residues). Corresponding enzyme subsites are numbered S1, S2, S3, etc.
The CTRC residue numbering used in this report and in the crystal structure coordinates is derived by homology to bovine chymotrypsin, the archetypal member of this peptidase family, for consistency with structural literature in the serine protease field. Designations in parentheses are the corresponding residue numbers based on sequential numbering of the CTRC precursor.
The human cationic trypsinogen residue numbering used here is based on sequential numbering of the trypsinogen precursor as is conventionally used in designating natural polymorphisms, e.g. p.A16V.
- CTRC
- chymotrypsin C
- PDB
- Protein Data Bank.
REFERENCES
- 1. Rinderknecht H. (1986) Activation of pancreatic zymogens. Normal activation, premature intrapancreatic activation, protective mechanisms against inappropriate activation. Dig. Dis. Sci. 31, 314–321 [DOI] [PubMed] [Google Scholar]
- 2. Whitcomb D. C., Gorry M. C., Preston R. A., Furey W., Sossenheimer M. J., Ulrich C. D., Martin S. P., Gates L. K., Jr., Amann S. T., Toskes P. P., Liddle R., McGrath K., Uomo G., Post J. C., Ehrlich G. D. (1996) Hereditary pancreatitis is caused by a mutation in the cationic trypsinogen gene. Nat. Genet. 14, 141–145 [DOI] [PubMed] [Google Scholar]
- 3. Teich N., Rosendahl J., Tóth M., Mössner J., Sahin-Tóth M. (2006) Mutations of human cationic trypsinogen (PRSS1) and chronic pancreatitis. Hum. Mutat. 27, 721–730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Szabó A., Sahin-Tóth M. (2012) Increased activation of hereditary pancreatitis-associated human cationic trypsinogen mutants in presence of chymotrypsin C. J. Biol. Chem. 287, 20701–20710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Nemoda Z., Sahin-Tóth M. (2006) Chymotrypsin C (caldecrin) stimulates autoactivation of human cationic trypsinogen. J. Biol. Chem. 281, 11879–11886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Szmola R., Sahin-Tóth M. (2007) Chymotrypsin C (caldecrin) promotes degradation of human cationic trypsin. Identity with Rinderknecht's enzyme Y. Proc. Natl. Acad. Sci. U.S.A. 104, 11227–11232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Witt H., Luck W., Becker M. (1999) A signal peptide cleavage site mutation in the cationic trypsinogen gene is strongly associated with chronic pancreatitis. Gastroenterology 117, 7–10 [DOI] [PubMed] [Google Scholar]
- 8. Grocock C. J., Rebours V., Delhaye M. N., Andrén-Sandberg A., Weiss F. U., Mountford R., Harcus M. J., Niemczyck E., Vitone L. J., Dodd S., Jørgensen M. T., Ammann R. W., Schaffalitzky de Muckadell O., Butler J. V., Burgess P., Kerr B., Charnley R., Sutton R., Raraty M. G., Devière J., Whitcomb D. C., Neoptolemos J. P., Lévy P., Lerch M. M., Greenhalf W., European Registry of Hereditary Pancreatitis and Pancreatic Cancer (2010) The variable phenotype of the p.A16V mutation of cationic trypsinogen (PRSS1) in pancreatitis families. Gut 59, 357–363 [DOI] [PubMed] [Google Scholar]
- 9. Rosendahl J., Witt H., Szmola R., Bhatia E., Ózsvári B., Landt O., Schulz H. U., Gress T. M., Pfützer R., Löhr M., Kovacs P., Blüher M., Stumvoll M., Choudhuri G., Hegyi P., te Morsche R. H., Drenth J. P., Truninger K., Macek M., Jr., Puhl G., Witt U., Schmidt H., Büning C., Ockenga J., Kage A., Groneberg D. A., Nickel R., Berg T., Wiedenmann B., Bödeker H., Keim V., Mössner J., Teich N., Sahin-Tóth M. (2008) Chymotrypsin C (CTRC) variants that diminish activity or secretion are associated with chronic pancreatitis. Nat. Genet. 40, 78–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Masson E., Chen J. M., Scotet V., Le Maréchal C., Férec C. (2008) Association of rare chymotrypsinogen C (CTRC) gene variations in patients with idiopathic chronic pancreatitis. Hum. Genet. 123, 83–91 [DOI] [PubMed] [Google Scholar]
- 11. Zhou J., Sahin-Tóth M. (2011) Chymotrypsin C mutations in chronic pancreatitis. J. Gastroenterol. Hepatol. 26, 1238–1246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Beer S., Zhou J., Szabó A., Keiles S., Chandak G. R., Witt H., Sahin-Tóth M. (September 1, 2012) Comprehensive functional analysis of chymotrypsin C (CTRC) variants reveals distinct loss-of-function mechanisms associated with pancreatitis risk. Gut 10.1136/gutjnl-2012-303090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hedstrom L. (2002) Serine protease mechanism and specificity. Chem. Rev. 102, 4501–4524 [DOI] [PubMed] [Google Scholar]
- 14. Perona J. J., Craik C. S. (1995) Structural basis of substrate specificity in the serine proteases. Protein Sci. 4, 337–360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Szmola R., Bence M., Carpentieri A., Szabó A., Costello C. E., Samuelson J., Sahin-Tóth M. (2011) Chymotrypsin C is a co-activator of human pancreatic procarboxypeptidases A1 and A2. J. Biol. Chem. 286, 1819–1827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Szabó A., Sahin-Tóth M. (2012) Determinants of chymotrypsin C cleavage specificity in the calcium-binding loop of human cationic trypsinogen. FEBS J. 279, 4283–4292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Szabó A., Héja D., Szakács D., Zboray K., Kékesi K. A., Radisky E. S., Sahin-Tóth M., Pál G. (2011) High affinity small protein inhibitors of human chymotrypsin C (CTRC) selected by phage display reveal unusual preference for P4′ acidic residues. J. Biol. Chem. 286, 22535–22545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Komiyama T., Fuller R. S. (2000) Engineered eglin c variants inhibit yeast and human proprotein processing proteases, Kex2 and furin. Biochemistry 39, 15156–15165 [DOI] [PubMed] [Google Scholar]
- 19. Otwinowski Z., Minor W. (1997) in Macromolecular Crystallography, Part A (Carter C. W., Jr., Sweet R. M., eds) pp. 307–326, Academic Press, New York [Google Scholar]
- 20. McCoy A. J., Grosse-Kunstleve R. W., Storoni L. C., Read R. J. (2005) Likelihood-enhanced fast translation functions. Acta Crystallogr. D Biol. Crystallogr. 61, 458–464 [DOI] [PubMed] [Google Scholar]
- 21. Huang K., Strynadka N. C., Bernard V. D., Peanasky R. J., James M. N. (1994) The molecular structure of the complex of Ascaris chymotrypsin/elastase inhibitor with porcine elastase. Structure 2, 679–689 [DOI] [PubMed] [Google Scholar]
- 22. Frigerio F., Coda A., Pugliese L., Lionetti C., Menegatti E., Amiconi G., Schnebli H. P., Ascenzi P., Bolognesi M. (1992) Crystal and molecular structure of the bovine α-chymotrypsin-eglin c complex at 2.0-Å resolution. J. Mol. Biol. 225, 107–123 [DOI] [PubMed] [Google Scholar]
- 23. Perrakis A., Harkiolaki M., Wilson K. S., Lamzin V. S. (2001) ARP/wARP and molecular replacement. Acta Crystallogr. D Biol. Crystallogr. 57, 1445–1450 [DOI] [PubMed] [Google Scholar]
- 24. Perrakis A., Morris R., Lamzin V. S. (1999) Automated protein model building combined with iterative structure refinement. Nat. Struct. Biol. 6, 458–463 [DOI] [PubMed] [Google Scholar]
- 25. Murshudov G. N., Vagin A. A., Dodson E. J. (1997) Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 53, 240–255 [DOI] [PubMed] [Google Scholar]
- 26. Emsley P., Cowtan K. (2004) Coot. Model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 27. Arnold K., Bordoli L., Kopp J., Schwede T. (2006) The SWISS-MODEL workspace. A web-based environment for protein structure homology modelling. Bioinformatics 22, 195–201 [DOI] [PubMed] [Google Scholar]
- 28. Sondergaard C. R., Olsson M. H. M., Rostkowski M., Jensen J. H. (2011) Improved treatment of ligands and coupling effects in empirical calculation and rationalization of pKa values. J. Chem. Theory Comput. 7, 2284–2295 [DOI] [PubMed] [Google Scholar]
- 29. Li H., Robertson A. D., Jensen J. H. (2005) Very fast empirical prediction and rationalization of protein pK29a values. Proteins 61, 704–721 [DOI] [PubMed] [Google Scholar]
- 30. Polak E., Ribière G. (1969) Note sur la convergence de méthodes de directions conjuguées. Rev. Fr. d'informatique et de Recherche Opérationnelle 16, 35–43 [Google Scholar]
- 31. Caulfield T., Medina-Franco J. L. (2011) Molecular dynamics simulations of human DNA methyltransferase 3B with selective inhibitor nanaomycin A. J. Struct. Biol. 176, 185–191 [DOI] [PubMed] [Google Scholar]
- 32. Caulfield T., Devkota B. (2012) Motion of transfer RNA from the A/T state into the A-site using docking and simulations. Proteins 80, 2489–2500 [DOI] [PubMed] [Google Scholar]
- 33. Loving K., Salam N. K., Sherman W. (2009) Energetic analysis of fragment docking and application to structure-based pharmacophore hypothesis generation. J. Comput. Aided Mol. Des. 23, 541–554 [DOI] [PubMed] [Google Scholar]
- 34. Salam N. K., Nuti R., Sherman W. (2009) Novel method for generating structure-based pharmacophores using energetic analysis. J. Chem. Inf. Model 49, 2356–2368 [DOI] [PubMed] [Google Scholar]
- 35. Gomis-Rüth F. X., Gómez M., Bode W., Huber R., Avilés F. X. (1995) The three-dimensional structure of the native ternary complex of bovine pancreatic procarboxypeptidase A with proproteinase E and chymotrypsinogen C. EMBO J. 14, 4387–4394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Birktoft J. J., Kraut J., Freer S. T. (1976) A detailed structural comparison between the charge relay system in chymotrypsinogen and in α-chymotrypsin. Biochemistry 15, 4481–4485 [DOI] [PubMed] [Google Scholar]
- 37. Page M. J., Di Cera E. (2008) Serine peptidases. Classification, structure and function. Cell. Mol. Life Sci. 65, 1220–1236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Seemüller U., Meier M., Ohlsson K., Müller H. P., Fritz H. (1977) Isolation and characterization of a low molecular weight inhibitor (of chymotrypsin and human granulocytic elastase and cathepsin G) from leeches. Hoppe-Seylers Z. Physiol. Chem. 358, 1105–1107 [DOI] [PubMed] [Google Scholar]
- 39. Bode W., Papamokos E., Musil D., Seemueller U., Fritz H. (1986) Refined 1.2 A crystal structure of the complex formed between subtilisin Carlsberg and the inhibitor eglin c. Molecular structure of eglin and its detailed interaction with subtilisin. EMBO J. 5, 813–818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. McPhalen C. A., Schnebli H. P., James M. N. (1985) Crystal and molecular structure of the inhibitor eglin from leeches in complex with subtilisin Carlsberg. FEBS Lett. 188, 55–58 [DOI] [PubMed] [Google Scholar]
- 41. McPhalen C. A., James M. N. (1988) Structural comparison of two serine proteinase-protein inhibitor complexes. Eglin-c-subtilisin Carlsberg and CI-2-subtilisin Novo. Biochemistry 27, 6582–6598 [PubMed] [Google Scholar]
- 42. McPhalen C. A., Svendsen I., Jonassen I., James M. N. (1985) Crystal and molecular structure of chymotrypsin inhibitor 2 from barley seeds in complex with subtilisin Novo. Proc. Natl. Acad. Sci. U.S.A. 82, 7242–7246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Bode W., Huber R. (1992) Natural protein proteinase inhibitors and their interaction with proteinases. Eur. J. Biochem. 204, 433–451 [DOI] [PubMed] [Google Scholar]
- 44. Krowarsch D., Cierpicki T., Jelen F., Otlewski J. (2003) Canonical protein inhibitors of serine proteases. Cell Mol. Life Sci. 60, 2427–2444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Radisky E. S., Koshland D. E., Jr. (2002) A clogged gutter mechanism for protease inhibitors. Proc. Natl. Acad. Sci. U.S.A. 99, 10316–10321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kardos J., Bódi A., Závodszky P., Venekei I., Gráf L. (1999) Disulfide-linked propeptides stabilize the structure of zymogen and mature pancreatic serine proteases. Biochemistry 38, 12248–12257 [DOI] [PubMed] [Google Scholar]
- 47. Wang H., Yuan D., Xu R., Chi C. W. (2011) Purification, cDNA cloning, and recombinant expression of chymotrypsin C from porcine pancreas. Acta Biochim. Biophys. Sin. 43, 568–575 [DOI] [PubMed] [Google Scholar]
- 48. Bence M., Sahin-Tóth M. (2011) Asparagine-linked glycosylation of human chymotrypsin C is required for folding and secretion but not for enzyme activity. FEBS J. 278, 4338–4350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Otzen D. E., Rheinnecker M., Fersht A. R. (1995) Structural factors contributing to the hydrophobic effect. The partly exposed hydrophobic minicore in chymotrypsin inhibitor 2. Biochemistry 34, 13051–13058 [DOI] [PubMed] [Google Scholar]
- 50. Heinz D. W., Hyberts S. G., Peng J. W., Priestle J. P., Wagner G., Grütter M. G. (1992) Changing the inhibitory specificity and function of the proteinase inhibitor eglin c by site-directed mutagenesis. Functional and structural investigation. Biochemistry 31, 8755–8766 [DOI] [PubMed] [Google Scholar]
- 51. Lu W., Randal M., Kossiakoff A., Kent S. B. (1999) Probing intermolecular backbone H-bonding in serine proteinase-protein inhibitor complexes. Chem. Biol. 6, 419–427 [DOI] [PubMed] [Google Scholar]
- 52. Lu W. Y., Starovasnik M. A., Dwyer J. J., Kossiakoff A. A., Kent S. B., Lu W. (2000) Deciphering the role of the electrostatic interactions involving Gly70 in eglin C by total chemical protein synthesis. Biochemistry 39, 3575–3584 [DOI] [PubMed] [Google Scholar]
- 53. Radisky E. S., King D. S., Kwan G., Koshland D. E., Jr. (2003) The role of the protein core in the inhibitory power of the classic serine protease inhibitor, chymotrypsin inhibitor 2. Biochemistry 42, 6484–6492 [DOI] [PubMed] [Google Scholar]
- 54. Radisky E. S., Lu C. J., Kwan G., Koshland D. E., Jr. (2005) Role of the intramolecular hydrogen bond network in the inhibitory power of chymotrypsin inhibitor 2. Biochemistry 44, 6823–6830 [DOI] [PubMed] [Google Scholar]
- 55. Salameh M. A., Soares A. S., Hockla A., Radisky E. S. (2008) Structural basis for accelerated cleavage of bovine pancreatic trypsin inhibitor (BPTI) by human mesotrypsin. J. Biol. Chem. 283, 4115–4123 [DOI] [PubMed] [Google Scholar]
- 56. Hubbard S. J., Campbell S. F., Thornton J. M. (1991) Molecular recognition. Conformational analysis of limited proteolytic sites and serine proteinase protein inhibitors. J. Mol. Biol. 220, 507–530 [DOI] [PubMed] [Google Scholar]
- 57. Rajamani D., Thiel S., Vajda S., Camacho C. J. (2004) Anchor residues in protein-protein interactions. Proc. Natl. Acad. Sci. U.S.A. 101, 11287–11292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Schreiber G., Haran G., Zhou H. X. (2009) Fundamental aspects of protein-protein association kinetics. Chem. Rev. 109, 839–860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Grünberg R., Leckner J., Nilges M. (2004) Complementarity of structure ensembles in protein-protein binding. Structure 12, 2125–2136 [DOI] [PubMed] [Google Scholar]
- 60. Koch U., Biasiol G., Brunetti M., Fattori D., Pallaoro M., Steinkühler C. (2001) Role of charged residues in the catalytic mechanism of hepatitis C virus NS3 protease. Electrostatic precollision guidance and transition-state stabilization. Biochemistry 40, 631–640 [DOI] [PubMed] [Google Scholar]
- 61. Pasternak A., Ringe D., Hedstrom L. (1999) Comparison of anionic and cationic trypsinogens. The anionic activation domain is more flexible in solution and differs in its mode of BPTI binding in the crystal structure. Protein Sci. 8, 253–258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Fehlhammer H., Bode W., Huber R. (1977) Crystal structure of bovine trypsinogen at 1–8 Å resolution. II. Crystallographic refinement, refined crystal structure and comparison with bovine trypsin. J. Mol. Biol. 111, 415–438 [DOI] [PubMed] [Google Scholar]
- 63. Kossiakoff A. A., Chambers J. L., Kay L. M., Stroud R. M. (1977) Structure of bovine trypsinogen at 1.9-Å resolution. Biochemistry 16, 654–664 [DOI] [PubMed] [Google Scholar]
- 64. Grathwohl C., Wüthrich K. (1981) NMR studies of the rates of proline cis–trans isomerization in oligopeptides. Biopolymers 20, 2623–2633 [Google Scholar]
- 65. Wedemeyer W. J., Welker E., Scheraga H. A. (2002) Proline cis-trans isomerization and protein folding. Biochemistry 41, 14637–14644 [DOI] [PubMed] [Google Scholar]
- 66. Brandts J. F., Halvorson H. R., Brennan M. (1975) Consideration of the possibility that the slow step in protein denaturation reactions is due to cis-trans isomerism of proline residues. Biochemistry 14, 4953–4963 [DOI] [PubMed] [Google Scholar]
- 67. MacArthur M. W., Thornton J. M. (1991) Influence of proline residues on protein conformation. J. Mol. Biol. 218, 397–412 [DOI] [PubMed] [Google Scholar]
- 68. Schimmel P. R., Flory P. J. (1968) Conformational energies and configurational statistics of copolypeptides containing l-proline. J. Mol. Biol. 34, 105–120 [DOI] [PubMed] [Google Scholar]
- 69. Fischer G. (1994) Peptidyl-prolyl cis/trans isomerases and their effectors. Angew. Chem. Int. Ed. Engl. 33, 1415–1436 [Google Scholar]
- 70. Schechter I., Berger A. (1967) On the size of the active site in proteases. I. Papain. Biochem. Biophys. Res. Commun. 27, 157–162 [DOI] [PubMed] [Google Scholar]