

Background: Phospholipase D is a signaling protein whose expression is elevated in transformed cells.
Results: The tyrosine kinases JAK and Fes are responsible for high PLD activity and expression and high cell proliferation.
Conclusion: A new signaling pathway (JAK-Fes-PLD) has been found whose operation is heightened in cancer cells.
Significance: Modulating this new pathway could control the highly invasive phenotype of transformed cells.
Keywords: Breast Cancer, Leukocyte, Phosphorylation, Protein Kinases, Signal Transduction, Tyrosine Protein Kinase (Tyrosine Kinase), Phospholipids, Proliferation
Abstract
The products of the oncogene Fes and JAK3 are tyrosine kinases, whose expressions are elevated in tumor growth, angiogenesis, and metastasis. Phosphatidic acid, as synthesized by phospholipase D (PLD), enhances cancer cell survival. We report a new signaling pathway that integrates the two kinases with the lipase. A new JAK3-Fes-PLD2 axis is responsible for the highly proliferative phenotype of MDA-MB-231 breast cancer cells. Conversely, this pathway is maintained at a low rate of expression and activity levels in untransformed cells such as MCF10A. We also deciphered the inter-regulation that exists between the two kinases (JAK3 and the oncogene Fes) and between these two kinases and the lipase (PLD2). Whereas JAK3 and Fes marginally activate PLD2 in non-transformed cells, these kinases greatly enhance (>200%) PLD activity following protein-protein interaction through the SH2 domain and the Tyr-415 residue of PLD2. We also found that phosphatidic acid enhances Fes activity in MDA-MB-231 cells providing a positive activation loop between Fes and PLD2. In summary, the JAK3, Fes and PLD2 interactions in transformed cells maintain PLD2 at an enhanced level that leads to abnormal cell growth. Modulating this new JAK3-Fes-PLD2 pathway could be important to control the highly invasive phenotype of breast cancer cells.
Introduction
Breast cancer is one of the most common forms of cancer in the world and accounts for one-fourth of all cancers in women. Breast cancer is particularly dangerous due to its ability to metastasize to other parts of the body such as the lungs, which leads to high rates of mortality (1). One of the means to abrogate the rampant metastasis problem is through disruption of cell proliferation, and much research is being conducted in this regard.
Our laboratory and others have sought to study the role of PLD22 in cancer cell invasion and metastasis. There are two classic mammalian isoforms of PLD, PLD1 (2) and PLD2 (3), which share 52% identity. PLD1 is a 1072-amino acid, 120-kDa protein, whereas PLD2 is a 933-amino acid, 103-kDa protein. Both isoforms require phosphatidylinositol 4,5-bisphosphate (PIP2) as a co-factor for activity and release structurally identical PA species in mammalian cells.
A participation of PLD in cell migration was initially documented in leukocytes (4, 5). Subsequently, it has been explained in other cell types that PLD2 induces cell migration and invasion in human cancer cells (6), phagocytes (4, 5, 7, 8), fibroblasts (9, 10) and epithelial cells (11, 12). S6K (10), Rac2 (13), Fer (14, 15) and phosphocofilin (16) are also involved in PLD2-mediated invasion. PLD also exerts its influence over cellular signaling primarily through generation of the phospholipid PA, and this activity is implicated in a wide variety of signaling pathways due to its ability to influence the membrane localization of proteins (17, 18).
In this study, we selected the human breast cancer cell line, MDA-MB-231, as an ideal model due to their high rate of proliferation and the impact of PLD2 on these cells (6). MDA-MB-231 cells possess the WNT7B oncogene, which leads to a disruptive expression of several key growth promoting genes (19). The MDA-MB-231 cell line is highly proliferative and metastatic and also bears high PLD2 enzymatic activity (20) for reasons not well understood. Therefore, we hypothesized that PLD2 plays an essential role in maintaining the high levels of proliferation that are observed in breast cancer cells.
We report the existence of a new signaling pathway involving PLD2, JAK3 and Fes that is heightened in transformed cells. As we also observed that cell proliferation is dependent on these three molecules and their inter-regulation, we concluded that this new pathway could be crucial to promote abnormally high cell proliferation in breast cancer cells.
EXPERIMENTAL PROCEDURES
Materials
Dulbecco's modified Eagle's medium (DMEM) and Iscove's modified Eagle's medium were from Mediatech (Manassas, VA); mammary epithelial cell growth medium was from Cell Applications, Inc. (San Diego, CA); Lipofectamine and Plus reagent were from Invitrogen; siQuest transfection reagent was from Mirus (Madison, WI); apigenin was from Sigma; protein G-agarose beads, mouse anti-PLD2 IgG antibody (SKB2), purified recombinant human JAK3, purified recombinant human Fes, and the Fes substrate peptide were from Millipore (Billerica, MA); JAK3tide synthetic peptide substrate was from Anaspec (Fremont, CA); [3H]butanol was from American Radiolabeled Chemicals (St. Louis, MO); [γ-32P]ATP was from PerkinElmer Life Sciences; rabbit anti-JAK3 and rabbit anti-Fes IgG antibodies were from Cell Signaling (Danvers, MA); donkey anti-rabbit tetramethylrhodamine isothiocyanate IgG antibody were from Santa Cruz Biotechnology (Santa Cruz, CA); and si-Negative, siFES, and siJAK3 were from Applied Biosystems (Foster City, CA).
Cells and Cell Culture
MDA-MB-231, COS-7, and HL-60 cells were obtained from ATCC. MCF-7, MDA-MB-231, H1299, and COS-7 cells were cultured in DMEM supplemented with 10% (v/v) fetal bovine serum (FBS), whereas MCF10A cells were cultured in mammary epithelial cell growth with 0.4% (v/v) bovine pituitary extract, 10−7% (w/v) epidermial growth factor, 5 × 10−4% (w/v) insulin and 5 × 10−5% (w/v) hydrocortisone. Promyelocytic leukemic HL-60 cells and AML-3D10 cells were grown at 37 °C in a 5% CO2 incubator in Iscove's modified Eagle's medium + 20% (v/v) heat-inactivated fetal bovine serum, 2 mm l-glutamine. Cell density was maintained between 0.1 and 1.0 × 106/ml.
Plasmid Constructs
The plasmids used in these experiments were as follows: pcDNA3.1-mycPLD2-WT, pcDNA3.1-mycPLD2-Y169F, pcDNA3.1-mycPLD2-Y179F, pcDNA3.1-mycPLD2-Y415F, pCMV6-XL4-Fes-WT (Origene, Rockville, MD) and pME1S-JAK3-WT.
Generation of JAK3/PLD2/Fes Mutants
Mutant forms of JAK3, Fes and PLD2 were designed in the lab and they were generated (Fig. 2A) by site-directed mutagenesis by Mutagenex (Hillsborough, NJ). The catalytic sites were targeted on each kinase to generate kinase-dead mutants JAK3-K855E (JAK3-KD) and Fes-K590E (Fes-KD). Also, SH2 binding-deficient forms of JAK3 (JAK3-SH2i) and Fes (Fes-SH2i), which target residues Arg-402/Arg-403 in the SH2-like domain of JAK3 and residues Glu-469/Glu-472 in the SH2 domain of Fes. Regarding PLD2, two tyrosine residues Tyr-169 and Tyr-179 situated within the YxN motif that is recognized by SH2-bearing proteins and Tyr-415, which is essential for JAK3 binding (21), were targeted to generate SH2 binding-deficient mutants PLD-Y169F and PLD-Y179F and JAK3 binding-deficient mutant PLD-Y415F.
FIGURE 2.

Analysis of wild type and mutant JAK3, Fes and PLD2 proteins. A, the sites of mutagenesis in JAK3 are indicated, including Arg-402 and Arg-403, which are important for ligand binding, and Lys-855, which is important for kinase activity. Fes has Tyr kinase and Src homology 2 (SH2) domains that bind to ligands and enhance kinase activity. The sites of mutagenesis in Fes are indicated, including Glu-469 and Glu-472, important for ligand binding, and Lys-590, essential for kinase activity. The sites of mutagenesis in PLD2 are indicated. B–D, Western blot (W.B.) analyses of expression of recombinant wild type and mutant JAK3, Fes and PLD2 proteins. Shown are kinase and lipase activities in vitro of cell lysates that overexpress the three proteins of interest (JAK3, Fes and PLD2). Results in this figure are the means + S.E. from at least three independent experiments conducted in duplicate. F-BAR, FER/CIP4 homology-Bin/Amphyphysin/Rvs domain; FX, F-BAR extension domain; PX, Phox homology (PX) domain; PH, Pleckstrin-homology domain; HKDI and HKD2, phospholipase D signature motifs/catalytic sites.
Cell Transfection and Gene Silencing
Transfections were performed using 2 μg of plasmid DNA, 5 μl of Lipofectamine (Invitrogen), and 5 μl of Plus reagent (Invitrogen) in Opti-MEM medium (Invitrogen), per the manufacturer's instructions. MDA-MB-231 or MCF10A cells were transfected for 3 h, washed, refed with prewarmed complete medium and maintained for 36 h. Fes and JAK3 expression in cells was knocked down using small interfering RNA (200 nm siRNA) in combination with the siQuest transfection reagent (6 μl). Double-stranded RNA (dsRNA) was from Applied Biosystems (Foster City, CA). For JAK3, we used a siRNA that targeted exon 19 (locus s7653, 5′-GUAUCGUGGUGUCAGCUAUd(TT)-3′ (sense)). For Fes, siRNA-targeted exons 16 and 18 (locus s5113, 5′-CCUCAGCAAUCAGCAGACAd(TT)-3′ (sense)). A negative control for siRNA was from Applied Biosystems.
The GFP-based PA Sensor
The PA sensor plasmid takes advantage of a 40-amino acid sequence found in Spo20 that binds to cell membrane phospholipids, particularly PA. Spo20 (sporulation-specific protein 20) is a yeast protein required for the fusion of exocytic vesicles with the plasma membrane during yeast sporulation through its interactions with the SNARE complex (22–24). Spo has an inhibitory region that sequesters the protein in the nucleus (24, 25) and a positive regulatory region that binds to phospholipids (including PA) in the cell membrane. Cloning of the PABD in pEGFPC1 vector (Clontech) leading to pEGFP-Spo20PABD-WT for use in microscopy of mammalian cells was performed in Ref. 26. The PA sensor was used in mammalian cells (27), and the restriction digestion analysis is documented in (28), indicating that the construct is cut by ApaLI at the pUC origin (in addition, AgeI and DraII are one cut, Pst is a two-cut, and NcoI is a four-cut restriction enzyme).
Detection of Intracellular PA in Cancer Cells
MCF10A or MDA-MB-231 cells were transfected with 1.5 μg of wild type GFP-based PA sensor DNA and/or a combination of 1.5 μg of Fes-WT DNA and/or 1.5 μg of PLD2-WT DNA and were grown on glass coverslips inside six-well tissue cultures plates. Two days post-transfection, cells were analyzed and fixed with 4% paraformaldehyde, permeabilized in 0.5% Triton X-100 in phosphate-buffered saline (PBS), blocked in 10% fetal calf serum (FCS) in 0.1% Triton X-100 in PBS, (if relevant, probed with either rabbit anti-Fes or rabbit anti-PLD2 (H-133) IgG antibodies and then donkey anti-rabbit TRITC IgG antibody). Cells were examined on a Nikon Eclipse 50i fluorescence microscope.
Cell Proliferation Assay
Cells were plated into 24-well plates prior to use. Duplicate wells were untreated or treated with 300 nm siRNA for several time points (up to 72 h) or were untransfected or transfected with 1.5 μg of various PLD2, Fes, or JAK3 plasmid constructs for 60 h. After incubation, cells were washed 2× with PBS and trypsinized, and the final volume was brought up to 1 ml/well using complete DMEM containing 10% FCS. Fifty μl of trypan blue was added to each cell sample, and viable cells/ml were counted.
Co-immunoprecipitation and Western Blot Analyses
After transfection, cells were harvested and lysed with special lysis buffer (5 mm HEPES, pH 7.8, 100 μm sodium orthovanadate and 0.1% Triton X-100). The lysates were sonicated and treated with 1 μl of monoclonal antibody for the respective protein and 20 μl of protein G-agarose beads and incubated at 4 °C for 4 h. After washing, resulting pellets were then analyzed using SDS-PAGE and Western blot analyses.
PLD Activity Assay
Cell lysates were processed for PLD activity (29) in PC8 (1,2-dioctanoyl-sn-glycero-3-phosphocholine) liposomes and [3H]butanol beginning with the addition of the following reagents (final concentrations): 3.5 mm PC8 phospholipid, 1 μm PIP2, 45 mm HEPES (pH 7.8) and 1.0 μCi of [3H]butanol in a liposome form, as indicated in Ref. 29 to accomplish the transphosphatidylation reaction of PLD (30). The amount of [3H]phosphatidylbutanol that co-migrated with authentic phosphatidylbutanol standards (Rf = 0.45–0.50) was measured by scintillation spectrometry.
JAK3 Kinase Assay
Cells (2 × 106) were sedimented, washed and finally lysed via sonication in 20 μl of special lysis buffer (5 mm HEPES, pH 7.8, 100 μm sodium orthovanadate and 0.1% Triton X-100) containing protease inhibitors. Lysates were incubated in the presence of the following final concentration of each of the following: 4 mm MOPS, pH 7.0, 15 mm MgCl2, 1 mm EGTA, 0.2 mm sodium orthovanadate, 0.2 mm DTT, 1 μCi of [γ-32P]ATP, 100 μm cold ATP and 42 μm JAK3tide substrate to yield a 40-μl total kinase reaction volume. Reactions were incubated at 30 °C for 20 min (and stopped by spotting 20-μl reactions onto 2.5 × 2.5 cm2 pieces of P81 Whatman filter paper for duplicate determinations. Filters were washed and counted in a Beckman LS 6000TA liquid scintillation.
Fes Kinase Assay
The phosphoacceptor peptide substrate was the Fes substrate peptide (poly(Glu4-Tyr) biotin-conjugated (Millipore)) in freshly prepared kinase buffer (8 mm MOPS, pH 7.2, 9 mm MgOAc, 30 μm Na2VO3, 5 mm p-nitrophenyl phosphate, 1 mm EDTA, 2 μm cAMP-dependent kinase inhibitor, 0.42 μCi [γ-32P]ATP (7 nm) and 100 μm unlabeled ATP. To initiate the phosphotransferase reaction, aliquots (20 μl) of kinase buffer containing the Fes substrate peptide were mixed 1:2 (v/v) with the anti-Fes immunoprecipitates. The reaction was carried out at 37 °C for 10 min and terminated by adding 5 μl of 3% phosphoric acid and blotting 30 μl of the reaction mixture onto SAM-2 biotin capture membranes (Promega). Membrane squares were extensively washed with methanol and then water, dried and counted for radioactivity. Positive controls used recombinant fully active Fes (Millipore). Negative controls were run in parallel with no Fes substrate peptide.
PA- and PIP2 Liposome Preparation
The lipids utilized in this study were a cell membrane PA-soluble form, 1,2-dioleoyl-sn-glycero-3-phosphate and PIP2 (Avanti Polar Lipids, Alabaster, AL) and were prepared by resuspending 1 mg of lipid powder in 1.4 ml of liposome buffer (1× PBS plus 0.5% BSA, pH 7.2). The solutions were diluted in HBSS buffer and then sonicated on ice.
Statistical Analysis
Data are presented as mean ± S.E. The difference between means was assessed by the single factor analysis of variance test. Probability of p < 0.05 indicated a significant difference.
RESULTS
Higher Enzymatic Activities of Fes, JAK3 and PLD2 Were Found in Transformed Versus Untransformed Cells
We measured the endogenous activity of JAK3, Fes and PLD2 in nontransformed (MCF10A epithelial cells) and transformed cells (MDA-MB-231 breast cancer cells) and found that the latter possess greater endogenous JAK3, Fes and PLD activities when compared with the nontransformed MCF10A cells (Fig. 1, A–C). A higher activity was also found in other transformed cells (HL-60, AML4 leukemic cells) versus other untransformed cells (COS-7 or RAW264.7). We also found that JAK3 and PLD2 protein expression levels are significantly higher in the cancer cells than in MCF10A cells (Fig. 1, D–F). Therefore, the endogenous higher protein levels and intrinsic enzymatic activities are present in transformed cells. Why this was the case warranted further examination.
FIGURE 1.
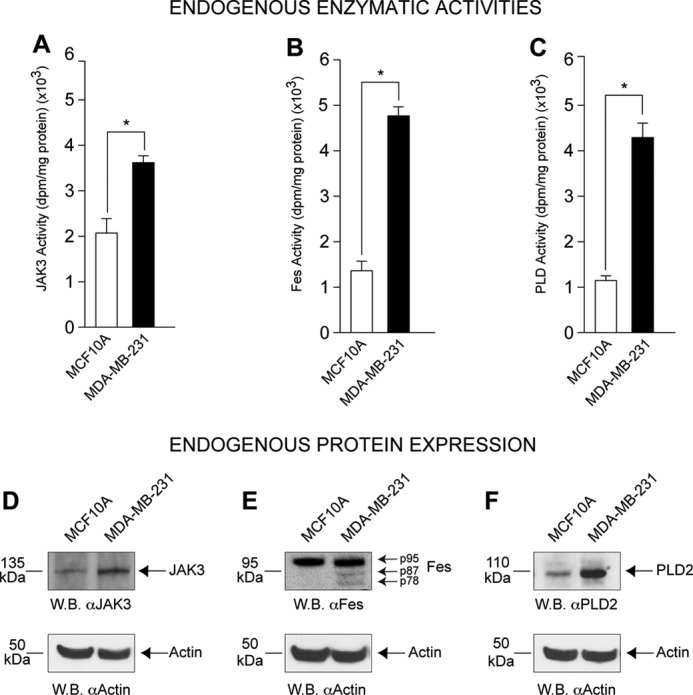
Endogenous JAK3, FES, and PLD2 activity levels in untransformed and transformed cells. JAK3 (A), Fes (B), and PLD2 (C) activities were measured in unstimulated and untransfected MCF10A and MDA-MB-231 cell lysates. The experiments started from similar number of cells per sample and then adjusted for similar protein levels per sample, so proper comparison can be made. Results in this figure are the means + S.E. from at least three independent experiments conducted in duplicate. The asterisk denotes statistically significant (p < 0.05) differences (increases) between samples and controls. Western blot (W.B.) analysis of endogenous JAK3 (D), Fes (E), and PLD2 (F) proteins (100 μg/lane, similar amount for each cell type) in non-transformed MCF10A and transformed MDA-MB-231 cancer cell lines with actin expression included as equal protein loading controls.
Design and Characterization of JAK3, Fes, and PLD2 Mutants
An array of biological tools to study the three molecules (JAK3, Fes and PLD2) was developed for this study. Schematics of different shape and color (Fig. 2A) represent JAK3, Fes and PLD2 and are used consistently throughout this study. We generated kinase-dead and SH2-deficient mutants and overexpressed them in transformed cells. The left panels of Fig. 2, B–D, indicate similar expression levels. Kinase-dead mutations abrogate both JAK3 and Fes activities (Fig. 2, B and C, right panels), whereas kinase SH2 mutations and PLD2 YF mutations do not significantly affect enzymatic activities (Fig. 2, B–D, right panels), which made these mutants very useful for subsequent experiments.
JAK3 Kinase Regulates PLD2 in Both Nontransformed and Transformed Cells
We investigated whether PLD2 would be under control of either kinase (Fes or JAK3) and concentrated on JAK first. JAK3 expression was effectively silenced (Fig. 3A, left panel). Fig. 3A (right panel) indicates that JAK has a positive role on PLD activity in transformed MDA-MB-231 cells but a negative role in MCF10A cells. Fig. 3B shows the effect of overexpression of JAK3 on PLD activity in vivo. Cells thus overexpressing JAK3 had somewhat decreased lipase activity in non-transformed MCF10A cells, which was significantly increased in MDA-MB-231 cells.
FIGURE 3.
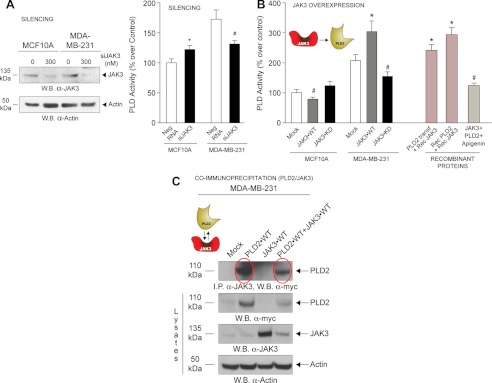
Effect of JAK3 on PLD2 activity in non-transformed versus transformed cells. A, left, evidence of JAK3 silencing using short interfering RNA by Western blots (W.B.). Right, effect of silencing JAK3 on endogenous PLD activity in MCF10A and MDA-MB-231 cells. Results in this figure are the means + S.E. from at least three independent experiments conducted in duplicate. The asterisk and # denote statistically significant (p < 0.05) differences (increases or decreases, respectively) between samples and controls. B, JAK3-WT or JAK3-KD were each overexpressed in MCF10A and MDA-MB-231 cells, and PLD activity was measured from the resulting cell lysates. Recombinant purified JAK3 was also used in some samples as positive controls to measure PLD2 activities in vitro. C, JAK3 and PLD2 form a protein-protein complex as detected using co-immunoprecipitation. Immunoprecipitation was performed with rabbit anti-JAK3 IgG antibodies bound to protein G-agarose beads, and after SDS-PAGE, the resulting Western blots were probed with rabbit anti-Myc IgG antibodies, which bound to the Myc-tagged PLD2 (top panel). PLD2 was detected at the native molecular mass of ∼110 kDa (second panel from top). JAK3 was detected at the native molecular mass of ∼135 kDa (second panel from bottom). Equal protein loading controls were detected using rabbit α-actin antibody (bottom panels). Neg RNA, scramble RNA (negative control).
Overexpression of JAK3-KD significantly reduced PLD2 activity, suggesting that a catalytically active JAK3 is necessary for PLD2 activation in transformed cells. As positive controls, recombinant purified JAK3 was also utilized in parallel reactions, which significantly increased PLD2 activity and was reversed by a tyrosine kinase inhibitor (apigenin).
In Fig. 3C, co-immunoprecipitation indicates that PLD2 and JAK3 form a protein-protein complex in MDA-MB-231 transformed cells (JAK3 was immunoprecipitated from lysates, and then the subsequent Western blot was probed with α-Myc antibodies, which interacted with the Myc-tagged PLD2, top panel). However, a fainter band in the combinations PLD2+JAK3 with respect to either alone indicates that such a complex is not very strong and that co-expression is negatively affected (top panel, rightmost lanes).
Fes Differentially Modulates PLD2 Activity in Both Cell Types
Having studied JAK3-PLD2 regulation, we next investigated a putative Fes-PLD2 regulation. Fig. 4A (left panel) is a control that demonstrates effective silencing of Fes. Fig. 4A (right panel) indicates the negative effect of Fes silencing on PLD activity in both cell lines, although it was greater in MDA-MB-231 cells. Thus, Fes is a positive regulator especially in these cells.
FIGURE 4.

Effect of Fes on PLD2 activity in non-transformed versus transformed cells. A, left, Western blots (W.B.) were performed to determine that Fes was effectively silenced. Right, effect of silencing Fes on endogenous PLD activity in MCF10A and MDA-MB-231 cells. B and C, PLD2 activity in MCF10A and MDA-MB-231 cells overexpressing a combination of Fes and/or PLD2 proteins. D, PLD2 and Fes form a protein-protein complex as detected by immunoprecipitations. Anti-PLD2 antibodies were used for immunoprecipitations, and anti-Fes antibodies were used to probe the Western blots (top panels). Negative controls using IgG antibody for co-immunoprecipitations are included for each cell line (second panel from top). PLD2 was detected at the native molecular mass of ∼105 kDa.
We present in Fig. 4B that PLD2 activity in MDA-MB-231 cells is negatively affected by loss of the SH2 and the kinase catalytic domains in Fes. PLD2 in MCF10A cells was likewise inhibited by Fes-KD but not by the SH2 mutant.
Our laboratory has previously demonstrated phosphorylation of PLD2 at Tyr-415 following cell stimulation (31). As the modular architecture of Fes indicates (Fig. 2A), Fes has a SH2 domain. We reasoned that the Fes SH2 domain could interact with PLD2 at Y-169 and Y-179, and possibly with Y-415. Fig. 4C indicates that PLD2 interacts with Fes at Tyr-169 and Tyr-179 but not with Tyr-415 in transformed cells. Conversely, MCF10A cells rely on Tyr-415. Thus, a different pattern of PLD2-Fes regulation exists between untransformed and transformed cells.
Fig. 4D indicates that PLD2 and Fes form a protein-protein complex in both cell types (red ovals, top panels). In MDA-MB-231, the interaction is through the SH2 domain of Fes.
Fes Activity, but Not JAK3, Is Modulated by Phospholipids
After studying the effects of kinases on PLD, we asked whether the regulation would operate conversely, i.e. from the lipase to the kinases, logically through PA, the product of PLD activity. We transfected a GFP-based PA sensor into both cell lines (Fig. 5, A and B). Fig. 5A documents more endogenous PA in MDA-MB-231 cells than in MCF10A cells. PA is localized in or around the nucleus in MCF10A cells and MDA-MB-231 cells in the absence of EGF stimulation (Fig. 5A, upper panels). After EGF stimulation, PA translocated to the cytoplasm in both cell types (Fig. 5A, lower panels). A significant increase in endogenous PA was seen following Fes co-transfection in MDA-MB-231 cells and a more moderate increase was evident in MCF10A cells (Fig. 5B, upper panels).
FIGURE 5.

Interplay between PA and Fes and JAK3 kinases. A, MCF10A and MDA-MB-231 cells were plated onto glass coverslips and then transfected with 1 μg of Fes plasmid in combination with 1 μg of the PA sensor (EGFP-based) plasmid. Green denotes the EGFP-tagged PA sensor. Red denotes the TRITC-tagged Fes. Blue denotes DAPI staining of the nucleus. Shown is a staining of the PA sensor (FITC) and nuclei (DAPI) only in cells in the absence (upper row) or presence (lower row) of EGF. B, similar to above, this time in cells co-expressing the PA sensor and Fes (TRITC). C, effect of 100 nm lipids (PA, PIP2, or PC) on recombinant JAK3 and Fes activities in vitro. D, differential effect of PA (1,2-dioleoyl-sn-glycero-3-phosphate, PA in a di-oleoyl or membrane-soluble form) on endogenous Fes activity.
As elevation in endogenous PA correlates to a change in Fes localization in MDA-MB-231 cells, we next investigated the effect of PA on Fes and JAK3 kinase activities using either cell lysates or whole cells (Fig. 5, C and D). PA significantly increased Fes activity but not JAK3 activity (Fig. 5C). Although the PA-binding domain of the PA sensor does recognize other anionic phospholipids, it has a preference for PA, which emphasizes its role in PLD signaling. Additionally, we observed that PIP2 also had a positive effect on Fes (probably mediated by phosphatidylinositol 5-kinase (32).
In summary, PA has a positive effect on Fes (but not on JAK3), especially in transformed cells, indicating that a feedback loop is established in these cells such that Fes activates the lipase, and the kinase activity is upregulated by the lipase. This could explain the high activity turnover of enzymatic activities observed in Fig. 1.
JAK3 and Fes Regulate Each Other
A final possible interaction in the new JAK3-Fes-PLD2 axis that was left to investigate is an inter-regulation between the two kinases, JAK3 and Fes. First, we silenced JAK3 and determined the effect on Fes activity in a broad range of cell lines. As shown in Fig. 6A, all cell lines except COS-7 cells showed that silencing JAK3 protein expression has a negative effect on Fes activity. Also, Fes activity was augmented in both cell lines following JAK3 overexpression (Fig. 6B).
FIGURE 6.

Interaction between JAK3 and Fes kinases. A, Fes activity in non-transformed (MCF10A and COS-7) and transformed (MDA-MB-231, MCF-7, AML-3D10 and dHL-60) cells following silencing of JAK3. B, Fes activity following transfection with either Fes or JAK3 plasmid DNA. (The Fes plasmid was used as a positive control for Fes kinase activity.) C, JAK3 activity of MCF10A and MDA-MB-231 cells following transfection with either Fes or JAK3 plasmid DNA. (The JAK3 plasmid was used as a positive control for JAK3 kinase activity.) D, JAK3 and Fes form a protein-protein complex as detected using co-immunoprecipitation. Immunoprecipitation (I.P.) was performed with rabbit anti-JAK3 IgG antibodies bound to protein G-agarose beads, and after SDS-PAGE, the resulting Western blots (W.B.) were probed with rabbit anti-Fes IgG antibodies (top panel). Negative controls using IgG antibody for co-immunoprecipitations are included for each cell line (second panel from top). Fes was detected at the native molecular mass of ∼95 kDa (second panel from bottom).
Fig. 6C indicates the effect of Fes on JAK3 in both cell lines, JAK3 activity is downregulated by Fes expression but to a lesser extent in the untransformed MCF10A cells. All this suggests that both kinases influenced the kinase activity of the other kinase with JAK3 having a positive impact on Fes in both cell types and Fes having a negative impact on both cell lines.
Next, we investigated whether a JAK3-Fes protein-protein interaction could be detected by co-immunoprecipitation. In Fig. 6D (red ovals, top panels), Fes and JAK3 interact with each other when recombinant Fes-WT is overexpressed. The JAK3 interaction with Fes is weaker in MDA-MB-231 cancer cells when compared with that of MCF10A cells. In summary, even though Fes affects JAK3 activity, its negative effect is stronger in MCF10A cells than in MDA-MB-231 cells as a result of stronger interaction in the non-transformed MCF10A cells.
The Physiological Significance of a New JAK3-Fes-PLD2 Pathway
We next examined the biological significance high enzymatic activity has on JAK3, Fes and PLD2 in cell proliferation. PLD2 and Fes accelerated cell proliferation in both non-transformed cells and transformed cells (Fig. 7A). JAK3 overexpression had a slight positive effect on MCF10A cell proliferation and a more significant effect on MDA-MB-231 cells. Interestingly, the level of cell proliferation of basal MDA-MB-231 cells is equivalent to PLD2 overexpressed in MCF10A cells, indicating that simply by overexpressing PLD2, untransformed cells can become highly proliferative.
FIGURE 7.

The effect of PLD2, Fes or JAK3 overexpression or silencing on cell proliferation in non-transformed versus transformed cells. A, cell proliferation of MCF10A or MDA-MB-231 cells in the presence of overexpressed PLD2, Fes or JAK3 60 h post-transfection. B, left, cell proliferation of MCF10A cells at varying time points using control siRNA, siJAK3, siFes or siJAK3+siFes+siPLD2 (200 nm each). Right, cell proliferation of MDA-MB-231 cells with similar conditions as in left. no sig, not significant. Transf, transfection; No sig, not significant.
As shown in Fig. 7B, silencing PLD2 and Fes slowed cell growth in the transformed MDA-MB-231 cells (right panel) and to a lesser extent in the untransformed MCF10A cells (left panel). When all three proteins are simultaneously silenced, proliferation is decreased in both cell lines with a more pronounced negative impact evident in the MDA-MB-231 cancer cells. This highlights the crucial importance of this new JAK3-Fes-PLD2 axis in cell proliferation.
DISCUSSION
We have demonstrated the existence of a new cell signaling pathway that includes PLD2 and that is very active in transformed cells and is crucial to the higher levels of cell proliferation. The reason for this heightened proliferation is due to increased protein expression and increased catalytic activity, as well as the inter-regulation between the kinases, JAK3 and Fes, and the lipase. This is further confirmed by a positive feedback of PA on Fes that forms a very active loop.
Phosphorylation of tyrosine residues on signaling molecules plays a pivotal role in a wide range of eukaryotic cellular processes (33, 34), which also includes phosphorylation/activation of PLD by epidermal growth factor receptor (35, 36) or by protein kinase Cα (PKCα) (37). We observed the presence of the tyrosine kinases Fes and JAK3 in MCF10A (as a model of an untransformed cell line) and MDA-MB-231 (as a model of transformed cells) and determined how these kinases regulate one another and also how they regulate PLD enzymatic activity. This inter-regulation is qualitatively and quantitatively different in the two cell lines investigated. The overall picture that emerged from the experiments described here is that in untransformed cells both the protein expression levels and the enzymatic activity of the three signaling molecules (PLD2, JAK and Fes) are maintained at a low level.
A complex inter-regulation equilibrium composed of activations and inhibitions keep PLD and the synthesis of PA in balance and at basal levels. The situation is very different in transformed cells; both the protein expression levels and the activity of PLD2, JAK and Fes are upregulated. The kinases activate PLD that then synthesizes higher amounts of PA. Furthermore, this PA feeds back positively on Fes that upon binding to the phospholipid is further activated. Fes negatively feeds back onto JAK3 in normal cells; yet this role is diminished in cancer cells, where binding between the two proteins appears to be somewhat abrogated. Previously, it has been documented that PLD activity levels are elevated in MDA-MB-231 breast cancer cells, which provides a survival signal in these transformed cells when deprived of serum growth factors (20, 38).
Fes interacts with PLD2 through the Tyr-415 residue in MCF10A cells, wherein it phosphorylates that residue and induces activation. In MDA-MB-231 cells, the SH2 domain becomes more important, and interaction between Fes and PLD2 ceases when this domain is disrupted. In MCF10A cells, this domain is inconsequential, and interaction can still occur, whereas the converse is true in MDA-MB-231 cells with the Tyr-415 mutant. The SH2 domain binding of PLD2 increases PLD2 activity in cancer cells and also contributes to heightened Fes activity due to the fact that binding of the Fes SH2 domain to its target ligand/protein stabilizes the kinase activity of Fes (39) and creates a positive feedback loop which restimulates PLD2.
PLD2 has been previously shown to be abundant in cancer cells that promote cell growth (6, 20). Targeting PLD2 enzyme lipase activity with selective inhibitors has also been shown to have large effects on decreasing metastasis and invasion (40). We have indicated here that PLD2 is controlled through both the JAK3 and Fes tyrosine kinases. JAK3 acts differentially in cancer cells, where it acts as a positive regulator through the SH2 domain. Taken together, these differential interactions indicate why PLD2 is upregulated and helps to explain why the cancerous phenotype of MDA-MB-231 cells exists.
The Existence of a New Signaling Pathway (JAK3 → Fes → PLD2 and the Mechanism of Inter-regulation)
The results of this study led to the construction of the model in Fig. 8, indicating that transformed cells have a heightened JAK3 → Fes → PLD2 pathway with respect to untransformed cells.
FIGURE 8.

Model for the new PLD2-Fes-Jak3 pathway. In MCF10A cells (left), both the protein expression levels and the enzymatic activity of the three signaling molecules (PLD2, JAK and Fes) are maintained at a low level. A complex inter-regulation equilibrium composed of activations and inhibitions keep PLD and the synthesis of PA in balance and at a basal level. In MDA-MB-231 cells (right), the situation is very different; both the protein expression levels and the activity of PLD2, JAK and Fes are upregulated. The kinases activate PLD that then synthesizes higher amounts of PA. Furthermore, this PA feeds back positively on Fes that, upon binding to the phospholipid, is further activated. PC, phosphatidylcholine; HKD, phospholipase D signature motif.
In MCF10A untransformed cells (Fig. 8, left), Fes has an inhibitory effect on JAK3 so that JAK3 activity is at basal level. Fes also competitively binds to PLD2. Due to the lower activities of these two tyrosine kinases JAK3 and Fes, PLD2 activity is kept in balance that is less abundant in untransformed MCF10A cells.
The reasons for elevated PLD2 activity in transformed MDA-MB-231 cells (Fig. 8, right) appear to be multiple and can be thought out as follows: 1) activation leads to robust PA generation; 2) parallel to this event, Fes autophosphorylates and also phosphorylates JAK3, which increases Fes activity; 3) PA generated by PLD2 upregulates Fes for its further activation and, in turn, Fes interacts with PLD2 through the SH2 binding motif and stimulates it; and 4) JAK3 activation further contributes to higher PLD2 activity (PA production) and abnormal cell proliferation in transformed cells.
In summary, this study shows for the first time a novel signaling pathway involving JAK3, Fes and PLD2 in vivo, which is crucial for abnormal cell proliferation of transformed cells. This pathway was tested in a panel of normal, untransformed cells and transformed, highly proliferative breast cancer cells. These novel mechanisms of PLD2 signaling present potential therapeutic targets to prevent cancer progression.
Acknowledgments
We thank Dr. Michael Frohman (SUNY Stony Brook) for providing us with the GFP-based Spo20 PA sensor; Dr. John O'Shea (National Institutes of Health/NIAMS) for providing the pME1S-JAK3 plasmid; Dr. Steven Berberich (Wright State University) for providing MCF-7 cells; and Dr. Michael Baumann (Wright State University) for providing AML-3D10 leukemic cells.
This work was supported by National Institutes of Health Grant HL056653, Boonshoft School of Medicine Grant 229102, and the State of Ohio Research Incentive Grant 668372 (to J. G. C.).
- PLD2
- phospholipase D 2
- PIP2
- phosphatidylinositol 4,5-bisphosphate
- KD
- kinase-dead
- EGFP
- enhanced GFP.
REFERENCES
- 1. Walker-Nasir E., Codington J. F., Jahnke M. R., Fuller T. C., Jeanloz R. W. (1982) Isolation and partial characterization of surface components of cell line MDA-MB-231 derived from a human metastatic breast carcinoma. J. Natl. Cancer Inst. 69, 371–380 [PubMed] [Google Scholar]
- 2. Hammond S. M., Altshuller Y. M., Sung T. C., Rudge S. A., Rose K., Engebrecht J., Morris A. J., Frohman M. A. (1995) Human ADP-ribosylation factor-activated phosphatidylcholine-specific phospholipase D defines a new and highly conserved gene family. J. Biol. Chem. 270, 29640–29643 [DOI] [PubMed] [Google Scholar]
- 3. Colley W. C., Sung T. C., Roll R., Jenco J., Hammond S. M., Altshuller Y., Bar-Sagi D., Morris A. J., Frohman M. A. (1997) Phospholipase D2, a distinct phospholipase D isoform with novel regulatory properties that provokes cytoskeletal reorganization. Curr. Biol. 7, 191–201 [DOI] [PubMed] [Google Scholar]
- 4. Gomez-Cambronero J., Di Fulvio M., Knapek K. (2007) Understanding phospholipase D (PLD) using leukocytes: PLD involvement in cell adhesion and chemotaxis. J. Leukoc. Biol. 82, 272–281 [DOI] [PubMed] [Google Scholar]
- 5. Lehman N., Di Fulvio M., McCray N., Campos I., Tabatabaian F., Gomez-Cambronero J. (2006) Phagocyte cell migration is mediated by phospholipases PLD1 and PLD2. Blood 108, 3564–3572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Zheng Y., Rodrik V., Toschi A., Shi M., Hui L., Shen Y., Foster D. A. (2006) Phospholipase D couples survival and migration signals in stress response of human cancer cells. J. Biol. Chem. 281, 15862–15868 [DOI] [PubMed] [Google Scholar]
- 7. Carrigan S. O., Pink D. B., Stadnyk A. W. (2007) Neutrophil transepithelial migration in response to the chemoattractant fMLP but not C5a is phospholipase D-dependent and related to the use of CD11b/CD18. J. Leukoc. Biol. 82, 1575–1584 [DOI] [PubMed] [Google Scholar]
- 8. Nagasaki A., Inotsume K., Kanada M., Uyeda T. Q. (2008) Phospholipase D is essential for keratocyte-like migration of NBT-II cells. Cell Struct. Funct. 33, 27–33 [DOI] [PubMed] [Google Scholar]
- 9. Pilquil C., Dewald J., Cherney A., Gorshkova I., Tigyi G., English D., Natarajan V., Brindley D. N. (2006) Lipid phosphate phosphatase-1 regulates lysophosphatidate-induced fibroblast migration by controlling phospholipase D2-dependent phosphatidate generation. J. Biol. Chem. 281, 38418–38429 [DOI] [PubMed] [Google Scholar]
- 10. Knapek K., Frondorf K., Post J., Short S., Cox D., Gomez-Cambronero J. (2010) The molecular basis of phospholipase D2-induced chemotaxis: elucidation of differential pathways in macrophages and fibroblasts. Mol. Cell Biol. 30, 4492–4506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Mazie A. R., Spix J. K., Block E. R., Achebe H. B., Klarlund J. K. (2006) Epithelial cell motility is triggered by activation of the EGF receptor through phosphatidic acid signaling. J. Cell Sci. 119, 1645–1654 [DOI] [PubMed] [Google Scholar]
- 12. Block E. R., Klarlund J. K. (2008) Wounding sheets of epithelial cells activates the epidermal growth factor receptor through distinct short- and long-range mechanisms. Mol. Biol. Cell 19, 4909–4917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Chae Y. C., Kim J. H., Kim K. L., Kim H. W., Lee H. Y., Heo W. D., Meyer T., Suh P. G., Ryu S. H. (2008) Phospholipase D activity regulates integrin-mediated cell spreading and migration by inducing GTP-Rac translocation to the plasma membrane. Mol. Biol. Cell 19, 3111–3123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Itoh T., Hasegawa J., Tsujita K., Kanaho Y., Takenawa T. (2009) The tyrosine kinase Fer is a downstream target of the PLD-PA pathway that regulates cell migration. Sci. Signal 2, ra52. [DOI] [PubMed] [Google Scholar]
- 15. Nishikimi A., Fukuhara H., Su W., Hongu T., Takasuga S., Mihara H., Cao Q., Sanematsu F., Kanai M., Hasegawa H., Tanaka Y., Shibasaki M., Kanaho Y., Sasaki T., Frohman M. A., Fukui Y. (2009) Sequential regulation of DOCK2 dynamics by two phospholipids during neutrophil chemotaxis. Science 324, 384–387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Han L., Stope M. B., de Jesús M. L., Oude Weernink P. A., Urban M., Wieland T., Rosskopf D., Mizuno K., Jakobs K. H., Schmidt M. (2007) Direct stimulation of receptor-controlled phospholipase D1 by phospho-cofilin. EMBO J. 26, 4189–4202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kraft C. A., Garrido J. L., Fluharty E., Leiva-Vega L., Romero G. (2008) Role of phosphatidic acid in the coupling of the ERK cascade. J. Biol. Chem. 283, 36636–36645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zhang Y., Du G. (2009) Phosphatidic acid signaling regulation of Ras superfamily of small guanosine triphosphatases. Biochim. Biophys. Acta 1791, 850–855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Matsuda Y., Schlange T., Oakeley E. J., Boulay A., Hynes N. E. (2009) WNT signaling enhances breast cancer cell motility and blockade of the WNT pathway by sFRP1 suppresses MDA-MB-231 xenograft growth. Breast Cancer Res. 11, R32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hui L., Zheng Y., Yan Y., Bargonetti J., Foster D. A. (2006) Mutant p53 in MDA-MB-231 breast cancer cells is stabilized by elevated phospholipase D activity and contributes to survival signals generated by phospholipase D. Oncogene 25, 7305–7310 [DOI] [PubMed] [Google Scholar]
- 21. Henkels K. M., Farkaly T., Mahankali M., Segall J. E., Gomez-Cambronero J. (2011) Cell invasion of highly metastatic MTLn3 cancer cells is dependent on phospholipase D2 (PLD2) and Janus kinase 3 (JAK3). J. Mol. Biol. 408, 850–862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Neiman A. M. (1998) Prospore membrane formation defines a developmentally regulated branch of the secretory pathway in yeast. J. Cell Biol. 140, 29–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Neiman A. M., Katz L., Brennwald P. J. (2000) Identification of domains required for developmentally regulated SNARE function in Saccharomyces cerevisiae. Genetics 155, 1643–1655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Nakanishi H., de los Santos P., Neiman A. M. (2004) Positive and negative regulation of a SNARE protein by control of intracellular localization. Mol. Biol. Cell 15, 1802–1815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Weimbs T., Low S. H., Chapin S. J., Mostov K. E., Bucher P., Hofmann K. (1997) A conserved domain is present in different families of vesicular fusion proteins: a new superfamily. Proc. Natl. Acad. Sci. U.S.A. 94, 3046–3051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zeniou-Meyer M., Zabari N., Ashery U., Chasserot-Golaz S., Haeberlé A. M., Demais V., Bailly Y., Gottfried I., Nakanishi H., Neiman A. M., Du G., Frohman M. A., Bader M. F., Vitale N. (2007) Phospholipase D1 production of phosphatidic acid at the plasma membrane promotes exocytosis of large dense-core granules at a late stage. J. Biol. Chem. 282, 21746–21757 [DOI] [PubMed] [Google Scholar]
- 27. Su W., Yeku O., Olepu S., Genna A., Park J. S., Ren H., Du G., Gelb M. H., Morris A. J., Frohman M. A. (2009) 5-Fluoro-2-indolyl des-chlorohalopemide (FIPI), a phospholipase D pharmacological inhibitor that alters cell spreading and inhibits chemotaxis. Mol. Pharmacol. 75, 437–446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Frondorf K., Henkels K. M., Frohman M. A., Gomez-Cambronero J. (2010) Phosphatidic acid is a leukocyte chemoattractant that acts through S6 kinase signaling. J. Biol. Chem. 285, 15837–15847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Di Fulvio M., Lehman N., Lin X., Lopez I., Gomez-Cambronero J. (2006) The elucidation of novel SH2 binding sites on PLD2. Oncogene 25, 3032–3040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Andrews B., Bond K., Lehman J. A., Horn J. M., Dugan A., Gomez-Cambronero J. (2000) Direct inhibition of in vitro PLD activity by 4-(2-aminoethyl)-benzenesulfonyl fluoride. Biochem. Biophys. Res. Commun. 273, 302–311 [DOI] [PubMed] [Google Scholar]
- 31. Henkels K. M., Peng H. J., Frondorf K., Gomez-Cambronero J. (2010) A comprehensive model that explains the regulation of phospholipase D2 activity by phosphorylation-dephosphorylation. Mol. Cell Biol. 30, 2251–2263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kantonen S., Hatton N., Mahankali M., Henkels K. M., Park H., Cox D., Gomez-Cambronero J. (2011) A novel phospholipase D2-Grb2-WASp heterotrimer regulates leukocyte phagocytosis in a two-step mechanism. Mol. Cell Biol. 31, 4524–4537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Natarajan V., Scribner W. M., Vepa S. (1996) Regulation of phospholipase D by tyrosine kinases. Chem. Phys. Lipids 80, 103–116 [DOI] [PubMed] [Google Scholar]
- 34. Lee C. S., Kim K. L., Jang J. H., Choi Y. S., Suh P. G., Ryu S. H. (2009) The roles of phospholipase D in EGFR signaling. Biochim. Biophys. Acta 1791, 862–868 [DOI] [PubMed] [Google Scholar]
- 35. Slaaby R., Jensen T., Hansen H. S., Frohman M. A., Seedorf K. (1998) PLD2 complexes with the EGF receptor and undergoes tyrosine phosphorylation at a single site upon agonist stimulation. J. Biol. Chem. 273, 33722–33727 [DOI] [PubMed] [Google Scholar]
- 36. Kim Y. R., Cha H. Y., Lim K., Hwang B. D., Hoe K. L., Namgung U., Park S. K. (2003) Activation of epidermal growth factor receptor is responsible for pervanadate-induced phospholipase D activation. Exp. Mol. Med. 35, 118–124 [DOI] [PubMed] [Google Scholar]
- 37. Singer W. D., Brown H. A., Jiang X., Sternweis P. C. (1996) Regulation of phospholipase D by protein kinase C is synergistic with ADP-ribosylation factor and independent of protein kinase activity. J. Biol. Chem. 271, 4504–4510 [DOI] [PubMed] [Google Scholar]
- 38. Cross M. J., Roberts S., Ridley A. J., Hodgkin M. N., Stewart A., Claesson-Welsh L., Wakelam M. J. (1996) Stimulation of actin stress fibre formation mediated by activation of phospholipase D. Curr. Biol. 6, 588–597 [DOI] [PubMed] [Google Scholar]
- 39. Filippakopoulos P., Kofler M., Hantschel O., Gish G. D., Grebien F., Salah E., Neudecker P., Kay L. E., Turk B. E., Superti-Furga G., Pawson T., Knapp S. (2008) Structural coupling of SH2-kinase domains links Fes and Abl substrate recognition and kinase activation. Cell 134, 793–803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Scott S. A., Selvy P. E., Buck J. R., Cho H. P., Criswell T. L., Thomas A. L., Armstrong M. D., Arteaga C. L., Lindsley C. W., Brown H. A. (2009) Design of isoform-selective phospholipase D inhibitors that modulate cancer cell invasiveness. Nat. Chem. Biol. 5, 108–117 [DOI] [PMC free article] [PubMed] [Google Scholar]