

Background: The development of antibodies against coagulation factor VIII (fVIII) is a serious complication of hemophilia A.
Results: Small angle x-ray scattering reveals a molecular envelope solution structure of two inhibitor antibodies bound to the C2 domain of fVIII.
Conclusion: Multiple inhibitor antibodies can bind to the fVIII C2 domain simultaneously, and modeling suggests the localization of key epitopes.
Significance: Understanding fVIII-inhibitor interactions is crucial for developing more effective hemophilia A therapies.
Keywords: Antibodies, Blood Coagulation Factors, Factor VIII, Structural Biology, X-ray Scattering
Abstract
The most significant complication for patients with severe cases of congenital or acquired hemophilia A is the development of inhibitor antibodies against coagulation factor VIII (fVIII). The C2 domain of fVIII is a significant antigenic target of anti-fVIII antibodies. Here, we have utilized small angle x-ray scattering (SAXS) and biochemical techniques to characterize interactions between two different classes of anti-C2 domain inhibitor antibodies and the isolated C2 domain. Multiple assays indicated that antibodies 3E6 and G99 bind independently to the fVIII C2 domain and can form a stable ternary complex. SAXS-derived numerical estimates of dimensional parameters for all studied complexes agree with the proportions of the constituent proteins. Ab initio modeling of the SAXS data results in a long kinked structure of the ternary complex, showing an angle centered at the C2 domain of ∼130°. Guided by biochemical data, rigid body modeling of subunits into the molecular envelope of the ternary complex suggests that antibody 3E6 recognizes a C2 domain epitope consisting of the Arg2209–Ser2216 and Leu2178–Asp2187 loops. In contrast, antibody G99 recognizes the C2 domain primarily through the Pro2221–Trp2229 loop. These two epitopes are on opposing sides of the fVIII C2 domain, are consistent with the solvent accessibility in the context of the entire fVIII molecule, and provide further structural detail regarding the pathogenic immune response to fVIII.
Introduction
Hemophilia A is an X-linked bleeding disorder that affects 1 in 5000 males worldwide and that is caused by loss of function of blood coagulation factor VIII (fVIII),2 usually as the result of a genetic mutation. Currently, the most effective treatment for hemophilia A patients is fVIII replacement therapy, which involves infusions of functional fVIII (either recombinant or plasma-derived) (1–3). The most significant complication to this treatment is the development of an immune response to the infused fVIII, occurring in ∼30% of hemophilia patients that receive treatment (4–7). Additionally, antibodies against fVIII develop in the non-hemophiliac population, resulting in acquired hemophilia A (8). Although immune tolerance induction has shown clinical success for the eradication of inhibitor antibodies in many cases, approximately one in four patients fail immune tolerance induction therapy (9). Additionally, immune tolerance induction can be a prohibitively expensive treatment due to the large quantities of fVIII required.
fVIII is a large 2332-residue glycoprotein cofactor within the intrinsic pathway of blood coagulation. The domain architecture of unprocessed fVIII is A1-A2-B-A3-C1-C2 (10, 13). The three A domains form a trimeric structure homologous to ceruloplasmin, and the two C domains are distant homologs to the discoidin protein fold, including galactose oxidase and lactadherin (14). After secretion, fVIII circulates as an A1-A2-B/A3-C1-C2 heterodimer bound to von Willebrand factor (vWF) (15–17). Upon proteolytic activation by either fXa or thrombin, fVIII is converted to “activated” fVIII (fVIIIa), which forms an A1/A2/A3-C1-C2 heterotrimer that dissociates from vWF and binds to activated platelet surfaces (PS) via stereoselective recognition of exposed l-phosphatidylserine headgroups (12, 18). Upon binding PS in the presence of calcium, fVIII interacts with fIXa (forming the intrinsic “tenase” complex), increasing the fIXa-catalyzed activation of fX by 200,000-fold (10–12, 19).
The immune response against therapeutic doses of plasma-derived or recombinant fVIII results in antibody responses wherein the majority of epitopes are found within the A2 and C2 domains (20). Antibodies with epitopes localized to the C2 domain can inhibit the activity of fVIII by a variety of mechanisms, including 1) blocking the ability of fVIII to bind vWF and/or PS, 2) inhibiting the proteolytic activation of fVIII by thrombin or fXa, or 3) directly inhibiting the cofactor function of fVIIIa (21–26). fVIII inhibition behavior generally falls within one of two distinct kinetic regimes referred to as types I and II. Type I inhibitor antibodies obey second-order kinetics and result in full inhibition of fVIII, whereas type II inhibitors exhibit more complex kinetics and do not fully inactivate fVIII, even at saturating concentrations (27).
Initial characterization of “classical” anti-C2 inhibitor antibodies showed interference with the ability of fVIII to bind PS and vWF (21, 24–26). The binding regions for PS and vWF have been shown to at least partially overlap, as binding to PS and vWF is mutually exclusive (28–30). More recent studies have described the development of “non-classical” inhibitor antibodies that block the proteolytic activation of fVIII by thrombin or fXa in both the presence and absence of vWF (22, 31). Moreover, additional studies suggest that the anti-C2 immune response is largely dominated by non-classical inhibitors (22). This class of anti-C2 antibodies often possesses type II kinetics with inhibitory titers above 10,000 Bethesda units/mg of IgG and have been shown to be pathogenic (32, 33).
The C2 domain of fVIII makes a direct contribution to the binding of vWF and PS and is essential for the cofactor function of fVIIIa (11, 14). Some studies also suggest a role for the C2 domain in interactions of fVIII with thrombin and fXa, although more studies are needed to fully elaborate the details of these proposed C2-protease interactions (31, 34, 35). Various x-ray crystal structures of the C2 domain, accompanied by biochemical data, have provided a putative model for the mechanism of membrane binding by fVIII, in which surface-exposed hydrophobic residues protruding from the ends of two β-hairpin turns within the C2 domain embed within the nonpolar lipid bilayer of PS (14, 36, 37). Directly above the β-hairpin turns is a ring of positively charged basic residues that plausibly interact electrostatically with the negative charge of the phosphatidylserine headgroup. The 2.0 Å x-ray crystal structure of the C2 domain bound to a classical antibody inhibitor (BO2C11) shows that these residues are completely sequestered at the protein-protein interface, thereby completely blocking the ability of fVIII to bind PS, as well as vWF (37).
To investigate the structural details of interactions between the fVIII C2 domain and classical/non-classical inhibitor antibodies, we employed small angle x-ray scattering (SAXS). The SAXS technique has been successfully utilized for the low resolution structural characterization of a wide range of biomolecular systems, from discrete proteins to complex assemblies (40). Despite the low resolution limitation of SAXS, a major advantage is that protein complexes can be analyzed in solution, under physiological conditions, with negligible perturbation of native tertiary structure.
Epitope mapping studies have recently shown that various classical and non-classical antibody pairings have discrete binding regions, allowing for more than one antibody to bind fVIII simultaneously (22). Here, we present the SAXS-derived solution structure model of the fVIII C2 domain in a ternary complex with the Fab fragments of monoclonal antibodies (mAbs) 3E6 and G99, which have been previously characterized as classical and non-classical antibody inhibitors, respectively (22). Preliminary evidence suggests that the 3E6 and G99 antibody pair displays positive cooperativity.3 Previous descriptions of antibody cooperativity have been limited to studies of antibody-based HIV-1 vaccine studies and Fc-dependent mechanisms (38, 39).
The SAXS-derived molecular envelope for the ternary complex indicates an extended bent structure, which, along with biochemical data, allows for modeling of epitopes on the surface of the fVIII C2 domain for mAbs 3E6 and G99. To our knowledge, this study represents the first use of SAXS for the determination of antibody epitopes to the level of specific exposed loops on the antigenic surface. These results contribute to the understanding of the complex pathogenic humoral response to fVIII.
EXPERIMENTAL PROCEDURES
Wild-type and K2183A Mutant fVIII C2 Domain Expression and Purification
The fVIII C2 domain was subcloned into an ampicillin-resistant pET15b vector containing an N-terminal His6 tag adjacent to a thrombin cleavage sequence as described previously (41, 42). The introduction of the K2183A mutation into the plasmid encoding the C2 domain was accomplished using the QuikChange Lightning mutagenesis kit (Agilent, Santa Clara, CA) following the manufacturer's recommendations. Sequencing of the K2183A mutant plasmid was performed by Nevada Genomics (Reno, NV). The expression constructs were transformed into Escherichia coli BL21(DE3) cells and subsequently grown in LB medium in the presence of 50 μg/ml ampicillin. Single colonies were used for inoculating 10-ml LB/ampicillin cultures, which were grown overnight with shaking at 37 °C and then added to 1 liter of LB/ampicillin and shaken at 37 °C until the absorbance at 600 nm reached a value of 0.6–0.8. Protein growth was induced by the addition of isopropyl β-d-thiogalactopyranoside to 500 μm, and the temperature was adjusted to 15 °C for overnight induction.
Following harvest by centrifugation at 8000 rpm for 10 min at 4 °C (FIBERLite F10-6x500y rotor, Thermo Fisher Scientific, Waltham, MA), cells were resuspended in lysis buffer (300 mm NaCl, 20 mm Tris-HCl (pH 7.0), 10 mm imidazole, 0.01% (v/v) Triton X-100, and 2.5% (v/v) glycerol), 1 mm PMSF, and 0.5–0.8 mg/ml lysozyme for 1 h at 4 °C. Resuspended cells were sonicated on ice using a 1/2-inch titanium horn attached to a Branson 450 sonifier (50% duty cycle) and then clarified by centrifugation at 16,000 rpm for 30 min at 4 °C (FIBERLite F21-8×50y rotor, Thermo Fisher Scientific), followed by filtration with 0.8-μm cellulose sterile syringe filters. TALON-immobilized cobalt affinity resin (Clontech, Mountain View, CA) was equilibrated in lysis buffer and incubated with filtered lysate for 1 h at 4 °C. The resin was rinsed with lysis buffer; with 300 NaCl, 20 mm Tris-HCl (pH 7.2), 10 mm imidazole, and 2.5% (v/v) glycerol; and with the same buffer but with 150 mm NaCl. His6-tagged C2 was eluted with 150 mm NaCl, 20 mm Tris-HCl (pH 7.2), 150 mm imidazole, and 2.5% (v/v) glycerol (yield of ∼2.5 mg/1 liter growth).
The eluate was exchanged into storage buffer containing 50 mm NaCl and 25 mm Tris-HCl (pH 7.2) and concentrated to 1–4 mg/ml using a Mr 10,000 cutoff concentrator (Millipore). Next, ion exchange chromatography was performed using Macro-PrepTM strong cation exchange support (Bio-Rad). Elution was achieved using a salt gradient from 50 to 500 mm NaCl. Eluted His6-C2 was concentrated to 6–8 mg/ml in storage buffer (100 mm NaCl and 25 mm Tris-HCl (pH 7.2)) and further purified by size exclusion chromatography with a Superdex 75 column (GE Healthcare) equilibrated in storage buffer. Elution fractions containing pure C2 (assessed by SDS-PAGE to be >95% pure) were pooled, concentrated to 5–10 mg/ml, flash-frozen in liquid nitrogen, and stored at −80 °C. For C2 preparations to be analyzed by SAXS, the hexahistidine tag was removed as follows. His6-C2 was mixed with 1 unit of thrombin/100 μg of His6-C2, diluted with thrombin cleavage buffer (50 mm NaCl, 20 mm Tris-HCl (pH 8.0), 2.5 mm CaCl2, and 2.5% (v/v) glycerol) to a final volume of 100–200 μl, and incubated overnight at 15 °C. Thrombin was removed with benzamidine-Sepharose 6B resin (GE Healthcare), and residual His-tagged C2 was removed via a second TALON affinity column. Finally, cleaved C2 was purified by size exclusion chromatography, concentrated, and stored as described above.
Antibody Production and Fab Purification
Murine anti-fVIII C2 domain monoclonal hybridomas (3E6 and G99) were developed as described previously (22). Large-scale antibody production was performed by Elizabeth Wayner (Antibody Production Facility, Fred Hutchinson Cancer Research Center, Seattle, WA). mAbs were purified from hybridoma supernatant with a NAbTM Protein A Plus spin column (Thermo Scientific, Rockford, IL) according to the manufacturer's instructions. The Fab fragments of 3E6- and G99-purified IgG were generated using immobilized papain (Thermo Scientific) following the manufacturer's cleavage protocol. Fab fragments were then isolated from the resultant Fab/Fc mixture via secondary passage over a Protein A column and collection of the initial flow-through. Fab fractions were purified further by size exclusion chromatography with a Superdex 75 column in storage buffer. Purified Fab was concentrated to 10–20 mg/ml, frozen in liquid nitrogen, and stored at −80 °C.
Complex Preparation and Size Exclusion Chromatography Analysis
To prepare C2-Fab complexes, a 1.5 molar excess of the purified C2 domain was incubated with Fab fragments at 37 °C for 30 min, followed by adjustment of the sample volume to 2 ml and injection onto a Superdex 75 size exclusion column equilibrated in storage buffer. Elution peaks corresponding to C2-Fab complexes were collected and concentrated to 5 mg/ml. For the relative comparison of complex masses, elution profiles of identically prepared samples consisting of C2, Fab, or C2-Fab complexes were employed.
C2 Domain Affinity Pulldown Assay
Human His6-C2 (50 μm) was incubated with 3E6 or G99 Fab (50 μm) in 100 μl of binding buffer (25 mm Tris (pH 7.2), 100 mm NaCl, and 5 mm imidazole) for 20 min at room temperature. Samples were then applied to 100 μl of equilibrated nickel-nitrilotriacetic acid (Ni-NTA) resin for an additional 20 min with gentle rocking. Next, the Ni-NTA resin was pelleted via centrifugation at 13,000 rpm for 1 min, followed by removal of the supernatant, which was stored for SDS-PAGE analysis. The resin was then washed three times with 200 μl of binding buffer by mixing, incubated for 1 min, and centrifuged at 13,000 rpm for 1 min, and the supernatant was removed. Finally, the resin was washed with 200 μl of elution buffer (25 mm Tris (pH 7.2), 100 mm NaCl, and 150 mm imidazole), and the supernatant was subjected to SDS-PAGE analysis. Control samples consisted of C2 or Fab alone.
For analysis of ternary complex formation, the same procedure was employed with minor alterations. Ni-NTA-bound His6-C2 was incubated with a mixture consisting of 3E6 Fab and G99 IgG or, conversely, 3E6 IgG and G99 Fab. After washing the resin, samples were eluted and analyzed on nonreducing SDS-polyacrylamide gels, which allowed for the unambiguous identification of 3E6 and G99 within individual samples.
Enzyme-linked Immunosorbent Assays
All ELISA experiments utilized NuncTM MaxiSorp 96-well ELISA plates (Fisher). Incubation periods were carried out at 37 °C for 45–60 min except where indicated, and sample wells were rinsed three times with 150 μl of TBS buffer (25 mm Tris-HCl (pH 7.4), 150 mm NaCl, and 2 mm KCl) between each incubation step.
For the measurement of C2 (WT or mutant) binding to 3E6 or G99 IgG, plates were first coated via incubation with 100 μl of 100 nm IgG in TBS buffer. Plates were then blocked by incubation with 175 μl of 2% BSA in TBS buffer. Next, 100 μl of His6-C2 in 1% BSA/TBS was added at the concentrations indicated in the figure legends, followed by incubation for 90 min at 37 °C. To bind exposed His tags, ELISA wells were then incubated with 100 μl of a 1:2000 dilution of HisDetectorTM Ni-NTA-alkaline phosphatase conjugate (KPL, Gaithersburg, MD) in 1% BSA/TBS. Finally, for colorimetric detection, 100 μl of 1 mg/ml p-nitrophenyl phosphate (pNPP; MP Biomedicals, Santa Ana, CA) in 0.1 m 2-amino-2-methyl-1,3-propanediol (pH 10.3) was added, the reaction was allowed to proceed for 10–20 min, and well absorbances at 405 nm were read in a BioTek Epoch plate reader. A one-site saturation binding model was used to fit the normalized ELISA data from at least three separate experiments, and estimates for the disassociation constants (Kd) for each interaction were calculated using GraphPad Prism software.
For ELISA-based detection of C2–3E6 Fab-G99 IgG ternary complex formation, a modified procedure was employed. Plates were first coated with 100 μl of 100 nm 3E6 Fab, followed by secondary coating with 100 nm C2 domain. After blocking, 100 μl of G99 IgG was added at varying concentrations, followed by detection using 100 μl of a 1:1000 dilution of goat anti-mouse IgG (Invitrogen) in 1% BSA/TBS, incubation with 1 mg/ml pNPP, and absorbance measurement as described above.
SASX Data Collection and Processing
SAXS data were collected remotely at the Advanced Light Source at Lawrence Berkeley National Laboratory (Berkeley, CA) on SIBYLS beamline 12.3.1. Data were collected for each sample at three different protein concentrations with x-ray exposure times of 0.5, 1, and 6 s. X-ray scattering data were processed, and buffer scattering was subtracted according to Advanced Light Source beamline specifications. Scattering curves for each protein and/or complex were merged and subsequently used for Guinier and Kratky analyses using the program PRIMUS (43). The maximum particle dimensions (Dmax) for each data set were determined with Perl scripts that iteratively calculate the real-space pairwise distance distributions (P(r)) for varying Dmax values with the GNOM program (44, 45). Radii of gyration (Rg) were calculated with default settings from both the Guinier approximation and P(r) distributions. Ab initio modeling was performed with both DAMMIF and GASBOR software (46, 47). Fifty independent runs of each simulation were averaged together with the DAMAVER software (48). Bead models of the molecular envelopes for each structure were converted to surfaces with Situs and Chimera following the developers' specifications (49, 50). Rigid body modeling of known structural components (C2 domain and Fab scaffolds) into SAXS-derived molecular envelopes was manually performed using PyMOL (DeLano Scientific LLC).
Bethesda Assays for Inhibition of fVIII
The effect of 3E6/G99 whole IgG or Fab on inhibition of fVIII in a one-stage clotting assay was determined as described previously (22). Full fVIII activity corresponded to the response of pooled normal human plasma (George King Bio-Medical, Inc., Overland Park, KS). The resulting inhibition curve was fitted using a four-parameter logistic equation (22).
RESULTS
The anti-fVIII inhibitor antibodies 3E6 and G99 were initially characterized in a competition ELISA study employing full-length fVIII (22). To confirm the specificity of these antibodies for the fVIII C2 domain, we first employed an affinity capture assay using the hexahistidine-tagged C2 domain as bait. After preincubation of the C2 domain with each Fab fragment of mAbs 3E6 and G99, the putative binary complexes were bound to Ni-NTA-immobilized metal affinity resin, washed, and then eluted with high concentrations of imidazole. The resultant fractions were analyzed by SDS-PAGE (Fig. 1). The flow-through from each incubation indicated that both the purified C2 domain alone and each Fab fragment in the presence of the C2 domain did not immediately elute from the column. In contrast, the control samples lacking C2, composed of both the G99 and 3E6 Fab fragments, were detected in the initial flow-through, indicating that neither Fab fragment possessed affinity for the Ni-NTA resin. Upon addition of imidazole, the isolated C2 domain, as well as each binary complex, eluted as expected (Fig. 1). These results indicate that Fab fragments derived from both antibodies G99 and 3E6 recognize the C2 domain independently. Additionally, a variation of this method provided evidence of ternary complex formation; immobilized C2 formed stable complexes with Fab/IgG pairs (i.e. 3E6 Fab + G99 IgG), which could be identified by nonreducing SDS-PAGE via size discrimination (Fig. 2).
FIGURE 1.
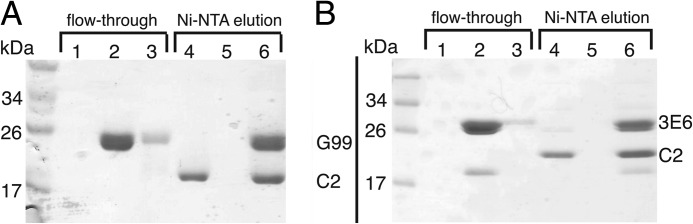
Formation of C2-Fab binary complexes. Shown is the stable binding of Fab fragments to His6-C2 as measured by retention on Ni-NTA resin. Flow-through fractions are defined as fractions unbound by resin, and the Ni-NTA elution fractions were recovered by the addition of high imidazole concentrations. A, C2-G99 Fab binary complex. B, C2–3E6 Fab binary complex. Lanes 1 and 4, fVIII C2 domain; lanes 2 and 5, Fab alone; lanes 3 and 6, C2-Fab binary complex.
FIGURE 2.

Ternary complex formation. An assay similar to that described in the legend to Fig. 1 was performed, except that samples were analyzed under nonreducing conditions by SDS-PAGE. A, C2–3E6 IgG-G99 Fab ternary complex. B, C2–3E6 Fab-G99 IgG ternary complex. Lanes 1 and 5, C2; lanes 2 and 6, IgG and Fab alone; lanes 3 and 7, C2-Fab-IgG ternary complex; lane 4, molecular mass ladder.
Size exclusion chromatography elution profiles provided further quantification of C2-Fab complex formation (Fig. 3). An excess of the C2 domain was incubated with 3E6 and/or G99 Fab and injected onto a Superdex 75 column. As shown in Fig. 2, the relative elution volumes for the isolated C2 domain, each Fab fragment, each C2-Fab binary complex, and the ternary complex decreased, consistent with the expected increase in molecular mass of each complex. Unexpectedly, the isolated 3E6 Fab fragment exhibited a smaller retention volume than the G99 Fab fragment, despite the near-identical masses of these two components. Upon inspection of the SDS-polyacrylamide gel of purified 3E6 Fab, we observed a small protein contaminant that co-purified with 3E6. Following formation of the C2–3E6 Fab binary complex, however, the majority of the small protein contamination was removed, as the elution volumes of both binary complexes were identical. Upon analysis of the putative ternary complex, we observed a single high molecular mass peak at a reduced retention volume, relative to the binary complexes, which supports the conclusion that a stable ternary complex is formed. Finally, a peak corresponding to the isolated C2 domain was observed for each complex due to the molar excess of the C2 domain used in the formation of each complex to assist in minimizing residual unbound Fab (the presence of which could present difficulties in chromatographic separation). Upon quantification of the C2 domain present in these excess peaks, we found equivalency between the mass of C2 collected and the molar excess of C2 added during complex preparation, providing further validation of our preparative techniques.
FIGURE 3.

Size exclusion chromatography of proteins and complexes. Relative elution volumes for each protein and/or complex are overlaid with normalized absorption values. Dashed lines, uncomplexed protein components (C2 domain, G99 Fab, and 3E6 Fab); solid lines, binary and ternary protein complexes (C2-G99 Fab binary, C2–3E6 Fab binary, and C2–3E6 Fab-G99 Fab ternary). An excess of the fVIII C2 domain was present in each complex to ensure saturation of Fab-binding sites.
The binding of His6-C2 to immobilized 3E6 or G99 IgG was analyzed using ELISA methods (Fig. 4A). Upon titration of 3E6 or G99 IgG-coated wells with His6-C2 and subsequent detection of His6-C2 with the Ni-NTA-alkaline phosphatase/pNPP system, we observed saturation binding behavior, which, upon fitting a classical one-site binding model, yielded apparent Kd estimates of 3.2 ± 0.8 nm for the C2–3E6 complex and 8.1 ± 1.9 nm for the C2-G99 complex. The binding of His6-C2 to blocked control wells lacking IgG was negligible (data not shown).
FIGURE 4.

ELISA of C2-mAb interactions. A, binding of His6-C2 to immobilized 3E6 IgG (●) and G99 IgG (○). Bound His6-C2 was detected using the Ni-NTA-alkaline phosphatase/pNPP system. B, ternary complex formation. G99 IgG was incubated at various concentrations under the following conditions: ELISA wells coated with 3E6 Fab, blocked, and then coated with C2 (●); wells coated with 3E6 Fab and blocked (○); and wells blocked and then incubated with C2 domain only (■). Bound G99 IgG was detected using goat anti-mouse IgG-alkaline phosphatase. Error bars represent the mean ± S.D. of three to six independent measurements.
An additional ELISA experiment provided further evidence for ternary complex formation. ELISA plate wells were coated with 3E6 Fab, blocked, and then incubated with the C2 domain to bind immobilized 3E6 Fab. Titration of these wells with G99 IgG and detection with goat anti-mouse IgG-alkaline phosphatase/pNPP revealed binding of G99 IgG to 3E6 Fab-immobilized C2 (apparent Kd of ∼5 nm), indicative of ternary complex formation (Fig. 4B). The initial coating of plates with 3E6 Fab, instead of whole IgG, was performed to ensure that only bound G99 IgG would be detected, as the goat anti-mouse IgG-alkaline phosphatase conjugate used selectively recognizes the Fc region of murine IgG, which is absent in Fab preparations. G99 IgG did not bind to wells coated only with 3E6 Fab or to blocked wells that were subsequently incubated with C2. The complementary experiment, coating with G99 Fab and C2, followed by 3E6 IgG titration, did not yield the expected binding results due to poor binding of G99 Fab to the ELISA plate wells (data not shown).
To further characterize the solution behavior of the C2 domain and its complexes with mAbs 3E6 and G99, we employed SAXS (Figs. 5–7). The merged scattering curves for all proteins and complexes are shown in Fig. 5. Initial characterization of the fVIII C2 domain, the 3E6 and G99 Fab fragments, both binary complexes, and the C2–3E6-G99 ternary complex indicated that each protein and/or complex was folded (by Kratky plot analysis) (supplemental Fig. 2), and no indication of protein aggregation was present in all samples (by Guinier approximation analysis) (supplemental Fig. 1). Additionally, the Rg and the extrapolated intensity at zero scattering angle (I(0) were approximated from each scattering curve (Rgrecip) and calculated with the GNOM program (Rgreal). Both independent determinations of Rg were consistent with each other, as shown in Table 1. Additionally, the Dmax and P(r) values for each complex were calculated with the GNOM program (Fig. 6 and Table 1).
FIGURE 5.

Merged SAXS scattering curves. Scattering curves for each complex were background-corrected and merged for three different protein concentrations and three different x-ray exposure times. A, fVIII C2 domain. B, 3E6 Fab. C, G99 Fab. D, C2–3E6 Fab binary complex. E, C2-G99 Fab binary complex. F, C2–3E6 Fab-G99 Fab ternary complex.
FIGURE 6.

SAXS-derived pairwise distance distributions. Pairwise distributions were calculated with Dmax values. Blue, fVIII C2 domain; green, 3E6 Fab; yellow, G99 Fab; red, C2–3E6 Fab binary complex; purple, C2-G99 Fab binary complex; black, C2–3E6 Fab-G99 Fab ternary complex.
FIGURE 7.

Ab initio-derived solution structures and rigid body modeling. Molecular envelopes were generated with 50 independent GASBOR ab initio structure calculations that were class-averaged with DAMAVER. Surfaces were calculated from bead models with Situs and displayed with Chimera. A, superposition of the ternary envelope (gray mesh) with molecular envelopes of the C2 domain (magenta), 3E6 Fab (violet), and G99 Fab (cyan). B, superposition of the ternary envelope (gray mesh) with molecular envelopes of the C2–3E6 Fab binary complex (violet) and the C2-G99 Fab binary complex (cyan; the overlap represents the fVIII C2 domain location). C, rigid body modeling of known structural components into the molecular envelope of the C2–3E6 Fab-G99 Fab ternary complex. Magenta, C2 domain; cyan, 3E6 Fab; yellow, G99 Fab.
TABLE 1.
SAXS-derived structural parameters
| Complex | Rgrecip | Rgreal | I(0) | Dmax |
|---|---|---|---|---|
| Å | Å | Å | ||
| C2 domain | 16.6 ± 0.053 | 16.52 ± 0.011 | 80.3 ± 0.052 | 50 |
| 3E6 Fab | 27.3 ± 0.174 | 27.31 ± 0.045 | 288 ± 0.36 | 95 |
| G99 Fab | 26.6 ± 0.076 | 26.93 ± 0.028 | 286 ± 0.23 | 90 |
| C2–3E6 | 30.2 ± 0.132 | 31.86 ± 0.043 | 340 ± 0.41 | 115 |
| C2-G99 | 31.6 ± 0.137 | 32.34 ± 0.058 | 315 ± 0.43 | 120 |
| C2–3E6-G99 | 47.8 ± 0.649 | 48.85 ± 0.154 | 491 ± 1.5 | 170 |
As expected, the C2 domain, 3E6 Fab, and G99 Fab each possessed the smallest Rg values: 16.52, 27.31, and 26.93 Å, respectively. The C2–3E6 and C2-G99 binary complexes displayed similar dimensions, with Rg values of 31.86 and 32.34 Å and Dmax values of 115 and 120 Å, respectively. Finally, the C2–3E6-G99 ternary complex possessed the largest dimensions, with an Rg of 47.8 Å and a Dmax of 170 Å (Table 1). The significantly larger dimensions for the ternary complex are consistent with a long extended structure in which the C2 domain is flanked by the two respective antibodies binding to opposing sides.
Following GNOM calculation of P(r) functions from scattering data for each protein or complex, low resolution structural models were calculated ab initio using the programs DAMMIF and GASBOR. For each SAXS data set, 50 independent runs were performed with both programs, which were subsequently averaged with the DAMAVER program. Bead and dummy residue models from the DAMMIF and GASBOR programs, respectively, were transformed into molecular envelopes with the programs Situs and Chimera (Fig. 7).
The ab initio molecular envelope models of the C2 domain, each Fab fragment, and each binary complex all displayed the dimensions expected for each protein or complex. However, the ternary complex displayed a molecular envelope that is elongated with an angle of ∼130°. Alignment of the molecular envelopes for the C2 domain, 3E6 Fab, and G99 Fab in the ternary complex (Fig. 7A) and alignment of the two binary complexes (Fig. 7B) indicated the molecular arrangement of each protein within the ternary structure. As shown in Fig. 7A, the C2 domain is flanked by molecular envelopes for each of the respective anti-Fab antibodies. Additionally, alignment of the two binary complexes indicated an overlap of the two molecular envelopes that coincides with the size of the C2 domain (Fig. 7B).
With the aid of prior epitope mapping data based on competition ELISAs and mutagenesis (22), structures for two prototypical Fab models (Protein Data Bank code 1IQD) as well as the 1.5 Å x-ray crystal structure of the fVIII C2 domain (code 1D7P), were modeled as rigid bodies into the ternary complex ab initio molecular envelope (Fig. 7C). Previous studies had localized the G99 epitope to Lys2227 of the fVIII C2 domain (22). With this region of the C2 domain docked to the hypervariable region of one Fab model, the opposing Fab fragment for antibody 3E6 was docked in accordance with the ab initio molecular envelope for the ternary complex. In the resulting model, the epitope for antibody G99 is centered upon the Pro2221–Trp2229 loop. By contrast, the modeled epitope for antibody 3E6, on the opposing exposed face of the C2 domain, was inferred to include loops Arg2209–Ser2216 and Leu2178–Asp2187.
To substantiate the proposed 3E6 epitope, we were able to generate a soluble C2 mutant (K2183A), which we analyzed via ELISA for 3E6 and G99 IgG binding (Fig. 8). In agreement with our SAXS-based model, the K2183A mutant exhibited significantly decreased affinity for 3E6 (Fig. 8A), with a >20-fold reduction in its apparent Kd value (71 nm for K2183A compared with 3.2 nm for WT C2). However, this K2183A mutation did not have an observed effect on G99 IgG binding (Fig. 8B). These data support our ab initio model and our assignment of the 3E6 epitope. Additionally, it was observed previously that mAb 3E6 competes with murine antibody ESH4 for binding to fVIII, which has been determined, using porcine/human fVIII hybrid constructs, to interact with C2 domain residues, including Lys2181–Met2199 and Thr2207–Val2243, regions that encompass the vicinity of our proposed 3E6 epitope.3
FIGURE 8.

ELISA of K2183A-mAb interactions. A, His6-K2183A binding to immobilized 3E6 IgG (○) compared with His6-WT C2 binding (●). B, His6-K2183A binding to immobilized G99 IgG (○) compared with His6-WT C2 binding (●). For clarity, the fitted saturation binding curve for K2183A-G99 binding in B is shown as a dashed line. Error bars represent the mean ± S.D. of at least three independent measurements.
DISCUSSION
For ∼30% of hemophilia A patients, therapeutic infusions of fVIII result in the development of anti-fVIII inhibitor antibodies, which are the major impediment to effective therapy, especially for patients that show poor response to immune tolerance induction therapy. To develop more effective therapies for hemophilia A, a more thorough understanding of fVIII-antibody interactions is crucial. In this work, we used the emergent SAXS technique coupled with biochemical characterization to investigate complexes formed between the human fVIII C2 domain and Fab fragments from two anti-human C2 inhibitor mAbs, 3E6 and G99, derived from hemophilia A mice that were infused with human fVIII. Plasma derived from human hemophilia A patients has been shown to harbor inhibitor antibodies that compete with these mAbs for binding to C2, indicating that human inhibitor antibodies can recognize equivalent epitopes on the C2 domain (32). Therefore, analysis of the interaction between these murine mAbs and the human C2 domain can likely be directly applicable to the understanding of the human immune response to fVIII.
We sought to further establish the specific interactions between mAbs 3E6 and G99 with the isolated fVIII C2 domain. Three lines of biochemical evidence presented herein confirmed and extended the prior competition ELISA analysis of fVIII interactions with mAbs 3E6 and G99. Results from binding assays employing purified His6-C2 immobilized on Ni-NTA resin (Figs. 1 and 2), size exclusion chromatography analysis (Fig. 3), and ELISA experiments (Fig. 4) indicate that 3E6 and G99 target the C2 domain and, furthermore, can simultaneously bind to C2 to form a stable ternary complex. On the basis of these results, we were able to characterize a C2-specific ternary complex with two Fab fragments, which was essential for the interpretation of SAXS data.
Preliminary analysis of SAXS data provided additional corroborating evidence for the formation of stable C2–3E6, C2-G99, and ternary complexes. First, analysis of the linearity of Guinier plots derived from scattering intensity profiles of all complexes indicates the absence of any significant aggregation, in support of the proposition that the apparent shift in size exclusion chromatography retention volumes is due to complex formation as opposed to being attributed to nonspecific aggregation. Calculated Rg values, derived from the pairwise distribution function of all analyzed components, also agree with the formation of C2–3E6/C2-G99 binary and C2–3E6-G99 ternary complexes (Table 1).
Upon calculation of SAXS-based molecular envelope structures of the C2–3E6/C2-G99 binary and C2–3E6-G99 ternary complexes, multiple insightful morphological details could be discerned. Previously determined x-ray crystal structures of the C2 domain, as well as a canonical Fab fragment lacking complementarity-determining region loops, were readily modeled into the molecular envelopes. Interestingly, the ternary complex forms an elongated, moderately bent structure, with C2 at its central point and wherein each Fab fragment recognizes a distinct opposing face of the C2 domain. There appears to be no overlap between bound Fab fragments, in support of the existence of such interactions existing in vivo without any steric clashing between Fab fragments. Curiously, the proposed membrane-binding hydrophobic loops of the bound C2 domain appear not to be directly involved in binding interactions with either Fab fragment. This is in contrast to the previously determined structure of C2 bound to the classical inhibitor BOC211, derived from a hemophilia A patient and in which the C2 membrane-binding region is fully enveloped by the Fab fragment (37). It is possible that mAb 3E6 binds proximal to the membrane association surface of C2 and blocks membrane binding via steric interactions arising from the peripheral IgG structure, especially considering that a Fab fragment composes only ∼33% of the full-length bound inhibitor antibody. However, preliminary analyses of fVIII inhibition behavior using a one-stage clotting assay, comparing the Fab and whole IgG forms of 3E6, showed that the 3E6 Fab fragment exhibits essentially identical inhibition kinetics to whole IgG (Fig. 9A). Therefore, further explorations into the mechanism of fVIII inhibition by classical type I inhibitor antibodies is warranted.
FIGURE 9.

Bethesda assays for inhibition of fVIII activity in plasma. Pooled normal human plasma was successively diluted with IgG or Fab, and one-stage coagulation activity was measured. A, inhibition of fVIII activity by 3E6 whole IgG (○) and 3E6 Fab (●). B, inhibition of fVIII activity by G99 whole IgG (○) and G99 Fab (●). 100% activity corresponds to the coagulation activity of pooled normal human plasma.
Determination of the epitopes for both mAbs 3E6 and G99, combined with previous analysis of modes of inhibition by these antibodies, may provide further insight into the regions of binding of the fVIII C2 domain to PS and vWF, as well as the proteases that activate fVIII, thrombin, and fXa. The proposed epitope for mAb 3E6 suggests that the Ala2209–Ser2216 and Leu2178–Asp2187 loops may contribute to PS and/or vWF binding. The G99 epitope suggests that thrombin and/or fXa binds to the opposing face, which likely includes the Pro2221–Trp2229 loop. However, initial functional comparison of fVIII inhibitory activity by G99 Fab and IgG showed that G99 Fab reduced fVIII activity by only ∼40% compared with ∼80% by whole G99 IgG (Fig. 9B). The dependence of fVIII inhibition activity on the presence of the full IgG molecule suggests that this particular epitope may not represent a binding site for thrombin or fXa per se, but that the presence of the large full IgG may sterically inhibit the interaction of these proteases with other proximal interaction sites outside, but adjacent to, the C2 domain. The implications of this variability in fVIII inhibition are currently being examined further.
The interaction of C2 with classical inhibitors has been well studied both biochemically and, in the case of the human-derived mAb BO2C11, structurally (37). However, the SAXS-derived molecular models presented here provide the first low resolution glimpse of the binding of a non-classical type II inhibitor to C2, as well as an additional binding mode for a classical type I inhibitor. To our knowledge, this is the first example of the use of SAXS to identify antibody epitopes at the level of specific residues. Additionally, this is the first direct structural evidence of multiple Fab fragments binding simultaneously to the C2 domain. These observations help expand the understanding of the complex fVIII immune response and may assist in the development of more robust strategies for overcoming the formulation of inhibitors. Elaboration of these results with high resolution x-ray crystallographic mapping of epitopes could lead to the design of recombinant fVIII variants that display reduced immunogenicity. Future efforts will be directed toward unambiguously determining whether these mAbs exhibit cooperative behavior, investigating their mechanism of inhibition, and determining a high resolution x-ray crystal structure of the C2–3E6-G99 ternary complex.
Acknowledgments
We thank Elizabeth Wayner for production of monoclonal antibodies, the staff at SIBYLS beamline 12.3.1 (Advance Light Source) for collecting SAXS data, Nathan Baird for technical assistance, and Barry Stoddard and Betty Shen for assistance in data collection.
This work was supported, in whole or in part, by National Institutes of Health Grant R15 HL103518; Grant U54 HL112309 (to S. L. M. and to P. L.); Grant K08 HL102262 (to S. L. M.); and Grants R01 HL082609 and R01 HL040921 (to P. L.). This work was also supported by National Science Foundation Grant CHE-1062722, a project development award from Western Washington University (to P. C. S.), and Hemophilia of Georgia, Inc. (to S. L. M. and P. L.).

This article contains supplemental Figs. 1 and 2.
P. Lollar, personal communication.
- fVIII
- coagulation factor VIII
- vWF
- von Willebrand factor
- PS
- activated platelet surfaces
- SAXS
- small angle x-ray scattering
- mAb
- monoclonal antibody
- Ni-NTA
- nickel-nitrilotriacetic acid
- pNPP
- p-nitrophenyl phosphate.
REFERENCES
- 1. Hoots W. K. (2001) The future of plasma-derived clotting factor concentrates. Haemophilia 7, 4–9 [DOI] [PubMed] [Google Scholar]
- 2. Mauser-Bunschoten E. P., van der Bom J. G., Bongers M., Twijnstra M., Roosendaal G., Fischer K., van den Berg H. M. (2001) Purity of factor VIII product and incidence of inhibitors in previously untreated patients with haemophilia A. Haemophilia 7, 364–368 [DOI] [PubMed] [Google Scholar]
- 3. Scharrer I., Bray G. L., Neutzling O. (1999) Incidence of inhibitors in haemophilia A patients–a review of recent studies of recombinant and plasma-derived factor VIII concentrates. Haemophilia 5, 145–154 [DOI] [PubMed] [Google Scholar]
- 4. Bray G. L., Gomperts E. D., Courter S., Gruppo R., Gordon E. M., Manco-Johnson M., Shapiro A., Scheibel E., White G., 3rd, Lee M. (1994) A multicenter study of recombinant factor VIII (recombinate): safety, efficacy, and inhibitor risk in previously untreated patients with hemophilia A. The Recombinate Study Group. Blood 83, 2428–2435 [PubMed] [Google Scholar]
- 5. Kreuz W., Ettingshausen C. E., Zyschka A., Oldenburg J., Saguer I. M., Ehrenforth S., Klingebiel T. (2002) Inhibitor development in previously untreated patients with hemophilia A: a prospective long-term follow-up comparing plasma-derived and recombinant products. Semin. Thromb. Hemost. 28, 285–290 [DOI] [PubMed] [Google Scholar]
- 6. Lusher J. M., Arkin S., Abildgaard C. F., Schwartz R. S. (1993) Recombinant factor VIII for the treatment of previously untreated patients with hemophilia A. Safety, efficacy, and development of inhibitors. Kogenate Previously Untreated Patient Study Group. N. Engl. J. Med. 328, 453–459 [DOI] [PubMed] [Google Scholar]
- 7. Lusher J. M., Lee C. A., Kessler C. M., Bedrosian C. L. (2003) The safety and efficacy of B domain-deleted recombinant factor VIII concentrate in patients with severe haemophilia A. Haemophilia 9, 38–49 [DOI] [PubMed] [Google Scholar]
- 8. Franchini M., Lippi G. (2008) Acquired factor VIII inhibitors. Blood 112, 250–255 [DOI] [PubMed] [Google Scholar]
- 9. Hay C. R., DiMichele D. M. (2012) The principal results of the International Immune Tolerance Study: a randomized dose comparison. Blood 119, 1335–1344 [DOI] [PubMed] [Google Scholar]
- 10. Gitschier J., Wood W. I., Goralka T. M., Wion K. L., Chen E. Y., Eaton D. H., Vehar G. A., Capon D. J., Lawn R. M. (1984) Characterization of the human factor VIII gene. Nature 312, 326–330 [DOI] [PubMed] [Google Scholar]
- 11. Kane W. H., Davie E. W. (1988) Blood coagulation factors V and VIII: structural and functional similarities and their relationship to hemorrhagic and thrombotic disorders. Blood 71, 539–555 [PubMed] [Google Scholar]
- 12. van Dieijen G., Tans G., Rosing J., Hemker H. C. (1981) The role of phospholipid and factor VIIIa in the activation of bovine factor X. J. Biol. Chem. 256, 3433–3442 [PubMed] [Google Scholar]
- 13. Toole J. J., Knopf J. L., Wozney J. M., Sultzman L. A., Buecker J. L., Pittman D. D., Kaufman R. J., Brown E., Shoemaker C., Orr E. C. (1984) Molecular cloning of a cDNA encoding human antihaemophilic factor. Nature 312, 342–347 [DOI] [PubMed] [Google Scholar]
- 14. Shen B. W., Spiegel P. C., Chang C. H., Huh J. W., Lee J. S., Kim J., Kim Y. H., Stoddard B. L. (2008) The tertiary structure and domain organization of coagulation factor VIII. Blood 111, 1240–1247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Foster P. A., Fulcher C. A., Marti T., Titani K., Zimmerman T. S. (1987) A major factor VIII binding domain resides within the amino-terminal 272 amino acid residues of von Willebrand factor. J. Biol. Chem. 262, 8443–8446 [PubMed] [Google Scholar]
- 16. Hill-Eubanks D. C., Parker C. G., Lollar P. (1989) Differential proteolytic activation of factor VIII-von Willebrand factor complex by thrombin. Proc. Natl. Acad. Sci. U.S.A. 86, 6508–6512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lenting P. J., van Mourik J. A., Mertens K. (1998) The life cycle of coagulation factor VIII in view of its structure and function. Blood 92, 3983–3996 [PubMed] [Google Scholar]
- 18. Lollar P., Parker C. G. (1989) Subunit structure of thrombin-activated porcine factor VIII. Biochemistry 28, 666–674 [DOI] [PubMed] [Google Scholar]
- 19. Davie E. W. (1995) Biochemical and molecular aspects of the coagulation cascade. Thromb. Haemost. 74, 1–6 [PubMed] [Google Scholar]
- 20. Prescott R., Nakai H., Saenko E. L., Scharrer I., Nilsson I. M., Humphries J. E., Hurst D., Bray G., Scandella D. (1997) The inhibitor antibody response is more complex in hemophilia A patients than in most nonhemophiliacs with factor VIII autoantibodies. Recombinate and Kogenate Study Groups. Blood 89, 3663–3671 [PubMed] [Google Scholar]
- 21. Jacquemin M. G., Desqueper B. G., Benhida A., Vander Elst L., Hoylaerts M. F., Bakkus M., Thielemans K., Arnout J., Peerlinck K., Gilles J. G., Vermylen J., Saint-Remy J. M. (1998) Mechanism and kinetics of factor VIII inactivation: study with an IgG4 monoclonal antibody derived from a hemophilia A patient with inhibitor. Blood 92, 496–506 [PubMed] [Google Scholar]
- 22. Meeks S. L., Healey J. F., Parker E. T., Barrow R. T., Lollar P. (2007) Antihuman factor VIII C2 domain antibodies in hemophilia A mice recognize a functionally complex continuous spectrum of epitopes dominated by inhibitors of factor VIII activation. Blood 110, 4234–4242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Saenko E. L., Shima M., Gilbert G. E., Scandella D. (1996) Slowed release of thrombin-cleaved factor VIII from von Willebrand factor by a monoclonal and a human antibody is a novel mechanism for factor VIII inhibition. J. Biol. Chem. 271, 27424–27431 [DOI] [PubMed] [Google Scholar]
- 24. Scandella D., Gilbert G. E., Shima M., Nakai H., Eagleson C., Felch M., Prescott R., Rajalakshmi K. J., Hoyer L. W., Saenko E. (1995) Some factor VIII inhibitor antibodies recognize a common epitope corresponding to C2 domain amino acids 2248–2312, which overlap a phospholipid-binding site. Blood 86, 1811–1819 [PubMed] [Google Scholar]
- 25. Shima M., Nakai H., Scandella D., Tanaka I., Sawamoto Y., Kamisue S., Morichika S., Murakami T., Yoshioka A. (1995) Common inhibitory effects of human anti-C2 domain inhibitor alloantibodies on factor VIII binding to von Willebrand factor. Br. J. Haematol. 91, 714–721 [DOI] [PubMed] [Google Scholar]
- 26. Shima M., Scandella D., Yoshioka A., Nakai H., Tanaka I., Kamisue S., Terada S., Fukui H. (1993) A factor VIII neutralizing monoclonal antibody and a human inhibitor alloantibody recognizing epitopes in the C2 domain inhibit factor VIII binding to von Willebrand factor and to phosphatidylserine. Thromb. Haemost. 69, 240–246 [PubMed] [Google Scholar]
- 27. Gawryl M. S., Hoyer L. W. (1982) Inactivation of factor VIII coagulant activity by two different types of human antibodies. Blood 60, 1103–1109 [PubMed] [Google Scholar]
- 28. Andersson L. O., Brown J. E. (1981) Interaction of factor VIII-von Willebrand factor with phospholipid vesicles. Biochem. J. 200, 161–167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lajmanovich A., Hudry-Clergeon G., Freyssinet J. M., Marguerie G. (1981) Human factor VIII procoagulant activity and phospholipid interaction. Biochim. Biophys. Acta 678, 132–136 [DOI] [PubMed] [Google Scholar]
- 30. Saenko E. L., Scandella D. (1995) A mechanism for inhibition of factor VIII binding to phospholipid by von Willebrand factor. J. Biol. Chem. 270, 13826–13833 [DOI] [PubMed] [Google Scholar]
- 31. Nogami K., Shima M., Hosokawa K., Nagata M., Koide T., Saenko E. L., Tanaka I., Shibata M., Yoshioka A. (2000) Factor VIII C2 domain contains the thrombin-binding site responsible for thrombin-catalyzed cleavage at Arg1689. J. Biol. Chem. 275, 25774–25780 [DOI] [PubMed] [Google Scholar]
- 32. Meeks S. L., Healey J. F., Parker E. T., Barrow R. T., Lollar P. (2008) Nonclassical anti-C2 domain antibodies are present in patients with factor VIII inhibitors. Blood 112, 1151–1153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Meeks S. L., Healey J. F., Parker E. T., Barrow R. T., Lollar P. (2009) Non-classical anti-factor VIII C2 domain antibodies are pathogenic in a murine in vivo bleeding model. J. Thromb. Haemost. 7, 658–664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Nogami K., Shima M., Hosokawa K., Suzuki T., Koide T., Saenko E. L., Scandella D., Shibata M., Kamisue S., Tanaka I., Yoshioka A. (1999) Role of factor VIII C2 domain in factor VIII binding to factor Xa. J. Biol. Chem. 274, 31000–31007 [DOI] [PubMed] [Google Scholar]
- 35. Saenko E. L., Shima M., Rajalakshmi K. J., Scandella D. (1994) A role for the C2 domain of factor VIII in binding to von Willebrand factor. J. Biol. Chem. 269, 11601–11605 [PubMed] [Google Scholar]
- 36. Pratt K. P., Shen B. W., Takeshima K., Davie E. W., Fujikawa K., Stoddard B. L. (1999) Structure of the C2 domain of human factor VIII at 1.5 Å resolution. Nature 402, 439–442 [DOI] [PubMed] [Google Scholar]
- 37. Spiegel P. C., Jr., Jacquemin M., Saint-Remy J. M., Stoddard B. L., Pratt K. P. (2001) Structure of a factor VIII C2 domain-immunoglobulin G4κ Fab complex: identification of an inhibitory antibody epitope on the surface of factor VIII. Blood 98, 13–19 [DOI] [PubMed] [Google Scholar]
- 38. Greenspan N. S., Dacek D. A., Cooper L. J. (1989) Cooperative binding of two antibodies to independent antigens by an Fc-dependent mechanism. FASEB J. 3, 2203–2207 [DOI] [PubMed] [Google Scholar]
- 39. Montefiori D., Sattentau Q., Flores J., Esparza J., Mascola J. (2007) Antibody-based HIV-1 vaccines: recent developments and future directions. PLoS Med. 4, e348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Mertens H. D., Svergun D. I. (2010) Structural characterization of proteins and complexes using small-angle x-ray solution scattering. J. Struct. Biol. 172, 128–141 [DOI] [PubMed] [Google Scholar]
- 41. Spiegel P. C., Kaiser S. M., Simon J. A., Stoddard B. L. (2004) Disruption of protein-membrane binding and identification of small-molecule inhibitors of coagulation factor VIII. Chem. Biol. 11, 1413–1422 [DOI] [PubMed] [Google Scholar]
- 42. Spiegel P. C., Murphy P., Stoddard B. L. (2004) Surface-exposed hemophilic mutations across the factor VIII C2 domain have variable effects on stability and binding activities. J. Biol. Chem. 279, 53691–53698 [DOI] [PubMed] [Google Scholar]
- 43. Konarev P. V., Volkov V. V., Sokolova A. V., Koch M. H. J., Svergun D. I. (2003) PRIMUS: a Windows PC-based system for small-angle scattering data analysis. J. Appl. Crystallogr. 36, 1277–1282 [Google Scholar]
- 44. Lipfert J., Das R., Chu V. B., Kudaravalli M., Boyd N., Herschlag D., Doniach S. (2007) Structural transitions and thermodynamics of a glycine-dependent riboswitch from Vibrio cholerae. J. Mol. Biol. 365, 1393–1406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Svergun D. I. (1992) Determination of the regularization parameter in indirect-transform methods using perceptual criteria. J. Appl. Crystallogr. 25, 495–503 [Google Scholar]
- 46. Franke D., Svergun D. I. (2009) DAMMIF, a program for rapid ab initio shape determination in small-angle scattering. J. Appl. Crystallogr. 42, 342–346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Svergun D. I., Petoukhov M. V., Koch M. H. J. (2001) Determination of domain structure of proteins from x-ray solution scattering. Biophys. J. 80, 2946–2953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Volkov V. V., Svergun D. I. (2003) Uniqueness of ab initio shape determination in small-angle scattering. J. Appl. Crystallogr. 36, 860–864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Sanner M. F., Olson A. J., Spehner J. C. (1996) Reduced surface: an efficient way to compute molecular surfaces. Biopolymers 38, 305–320 [DOI] [PubMed] [Google Scholar]
- 50. Wriggers W., Chacon P. (2001) Using Situs for the registration of protein structures with low-resolution bead models from x-ray solution scattering. J. Appl. Crystallogr. 34, 773–776 [Google Scholar]