

Background: The inflammatory caspase-1 shows positive cooperativity not seen for the apoptotic caspase-3.
Results: Substrate binding increases the dimerization affinity and activity of caspase-1 but not for caspase-3.
Conclusion: Caspase-1 is regulated by concentration with substrates, whereas caspase-3 is not.
Significance: Subcellular co-localization of caspase-1 with substrates in inflammosomes may explain its more restricted family of substrates observed.
Keywords: Caspase, Cooperativity, Inflammation, Kinetics, Protease, Allostery, Dimerization, Positive Cooperativity, Steady-state Kinetic Model
Abstract
Caspases are intracellular cysteine-class proteases with aspartate specificity that is critical for driving processes as diverse as the innate immune response and apoptosis, exemplified by caspase-1 and caspase-3, respectively. Interestingly, caspase-1 cleaves far fewer cellular substrates than caspase-3 and also shows strong positive cooperativity between the two active sites of the homodimer, unlike caspase-3. Biophysical and kinetic studies here present a molecular basis for this difference. Analytical ultracentrifugation experiments show that mature caspase-1 exists predominantly as a monomer under physiological concentrations that undergoes dimerization in the presence of substrate; specifically, substrate binding shifts the KD for dimerization by 20-fold. We have created a hemi-active site-labeled dimer of caspase-1, where one site is blocked with the covalent active site inhibitor, benzyloxycarbonyl-Val-Ala-Asp-fluoromethylketone. This hemi-labeled enzyme is about 9-fold more active than the apo-dimer of caspase-1. These studies suggest that substrate not only drives dimerization but also, once bound to one site in the dimer, promotes an active conformation in the other monomer. Steady-state kinetic analysis and modeling independently support this model, where binding of one substrate molecule not only increases substrate binding in preformed dimers but also drives the formation of heterodimers. Thus, the cooperativity in caspase-1 is driven both by substrate-induced dimerization as well as substrate-induced activation. Substrate-induced dimerization and activation seen in caspase-1 and not in caspase-3 may reflect their biological roles. Whereas caspase-1 cleaves a dramatically smaller number of cellular substrates that need to be concentrated near inflammasomes, caspase-3 is a constitutively active dimer that cleaves many more substrates located diffusely throughout the cell.
Introduction
Many oligomeric enzymes show positive cooperativity where substrate binding induces a transition from a less active to a more active state (for recent reviews, see Refs. 1–3). The structural basis for this phenomenon can derive from the substrate inducing oligomerization or an allosteric change in the oligomer or both. For example, B-Raf, a kinase important in cell proliferation, has recently been shown to undergo a substrate-induced monomer-to-dimer transition that leads to a highly active form of the enzyme (4). By contrast, glycogen phosphorylase shows positive cooperativity through binding of substrate, leading to a conformational switch that induces a more active dimer (5).
Caspases, a family of aspartate-specific dimeric cysteine proteases important in inflammation and apoptosis, are also known to undergo allosteric transitions (for reviews, see Refs. 6 and 7). The most dramatic is caspase-1, which shows a Hill coefficient of 1.4 for substrate activation (8). Caspase-1 was first discovered as the enzyme that processes IL-1β (9, 10) and was later isolated, cloned, and identified as a heterodimeric cysteine protease (11, 12). Interestingly, upon dilution it was found to lose activity, leading to speculation that the tetramer may dissociate into catalytically inactive subunits at low concentrations (12).
The first x-ray structure confirmed that it is a dimeric cysteine-class protease (13). Crystal structures have been solved of caspase-1 in the presence (10) and absence (11) of active site inhibitors that show dimeric structures in these crystals with substantial differences in the active site region (Fig. 1). The apo-like structure has also been trapped with allosteric inhibitors (9) at the dimer interface ∼15 Å from either active site, showing that the apo-state can be trapped in an inactive form. These studies suggest that dimeric caspase-1 is capable of substrate-induced activation through an allosteric transition within the dimer. However, pro-caspase-1 exists as an inactive and monomeric precursor and thus is subject to dimerization after proteolytic maturation. Also, cross-linking studies suggest that mature caspase-1 at low concentration can exist in a monomeric form (14). We wished to quantify the dimerization constant of the mature enzyme in the presence and absence of substrate or active site inhibitors to understand how binding influences dimerization and to determine the relative importance of these processes to substrate activation and cooperativity that we observe (8).
FIGURE 1.

Structures of caspase-1 with the active site inhibitor z-VAD-fmk (PDB coordinates 2HBQ; Ref. 10) (panel A), with no ligand (PDB coordinates 1SC1; Ref. 11) (Panel B), or with the allosteric inhibitor (PDB coordinates 2FQQ; ref. 9) (Panel C). Ligands are shown as space-filled models. The large subunits (p20) are colored blue and orange, and the small subunits (p10) are colored green and red. As shown, the blue and green chains comprise the left half of the caspase-1 dimer, whereas the red and orange chains form the right half. The loops near the active sites (top left and bottom right of structures) undergo marked changes between conformations.
Here, biophysical and kinetic measurements show that binding of either inhibitor or substrate to the active site of caspase-1 shifts the monomer-dimer equilibrium constant in caspase-1 by greater than 20-fold. We also show that binding at one site enhances by 10-fold the catalytic efficiency at the second site. An integrated kinetic model predicts that at physiological concentrations caspase-1 exists predominantly as a monomer that undergoes both substrate-induced dimerization as well as substrate- or inhibitor-induced activation that can account for the significant positive cooperativity observed. In contrast, we find that caspase-3, which is a constitutive dimer at physiologic conditions, lacks positive cooperativity and shows a very weak substrate or inhibitor-induced activation. These data suggest that the changes in oligomer state upon substrate binding in caspase-1 but not caspase-3 can account for the differences in positive cooperativity and have important implications for different biological functions of these two enzymes.
EXPERIMENTAL PROCEDURES
Expression and Purification of Caspase-1
Recombinant caspase-1 was prepared by expression in Escherichia coli (E. coli) as insoluble inclusion bodies followed by refolding (15, 16). The p20 (residues 120–297) and p10 (residues 317–404) subunits of wild-type human caspase-1 were cloned into NdeI and EcoRI restriction endonuclease sites of the pRSET plasmid (Invitrogen). Site-directed mutagenesis was performed to construct the C285A active site null mutant.
Caspase-1 subunits were expressed separately in E. coli BL21(DE3) Star cells (Invitrogen). Cells were harvested after induction of a log phase culture with 1 mm isopropyl β-d-1-thiogalactopyranoside for 4 h at 37 °C and then disrupted with a microfluidizer. The inclusion body pellets were isolated by centrifugation of lysate for 20 min at 4 °C. Pellets were washed once with 50 mm HEPES (pH 8.0), 300 mm NaCl, 1 m guanidine-HCl, 5 mm DTT, and 1% Triton X-100 and washed 2 more times with the same buffer without the detergent. The washed inclusion body pellets were solubilized in 6 m guanidine-HCl and 20 mm DTT and stored frozen at −80 °C.
Refolding of caspase-1 was performed by combining guanidine-HCl-solubilized large and small subunits (10 mg of large subunit and 20 mg of small subunit) in a 250-ml beaker followed by rapid dilution with 100 ml of 50 mm HEPES (pH 8.0), 100 mm NaCl, 10% sucrose, 1 m nondetergent sulfobetaine 201 (NDSB-201), and 10 mm DTT. Renaturation proceeded at room temperature for 6 h. Samples were centrifuged at 16,000 × g for 10 min to remove precipitate and then dialyzed overnight at 4 °C against 50 mm sodium acetate (pH 5.9), 25 mm NaCl, 5% glycerol, and 4 mm DTT. Dialyzed protein was purified by cation exchange chromatography using a prepacked 5-ml HiTrap SP HP column (GE Healthcare). Protein was eluted using a linear gradient of 0–1.0 m NaCl over 20 min in a buffer containing 50 mm sodium acetate (pH 5.9) and 5% glycerol. Peak fractions were pooled, and 2-mercaptoethanol was added to a concentration of 1 mm before samples were stored frozen at −80 °C.
For the kinetic analyses at varying enzyme and substrate concentrations for wild-type caspase-1, the cation exchange peak fractions were concentrated using Millipore Ultrafree-15 devices with a molecular weight cutoff of 10,000 Da and further purified using size exclusion chromatography using a Superdex 200 16/60 column in 25 mm Tris (pH 8.0), 50 mm NaCl, 5% glycerol, and 1 mm DTT. For the analytical ultracentrifugation (AUC)5 experiments, cation exchange peak fractions were concentrated and further purified using size exclusion chromatography using a Superdex 200 16/60 column in a buffer containing 50 mm HEPES (pH 8.0), 50 mm KCl, and 200 mm NaCl and then frozen at −80 °C.
Analytical Ultracentifrugation
Sedimentation equilibrium experiments were performed on a Beckman XL-I analytical ultracentrifuge at 20 °C at rotor speeds of 10,000, 14,000, and 20,000 rpm. For the C285A active site null construct, samples were centrifuged at 90,000 rpm for 10 min to remove the remaining aggregate before measurement. To obtain initial absorbance values of between 0.2 and 0.8 absorbance units, loading concentrations were 16, 12, and 8 μm for C285A caspase-1 and 19.5 μm for double-labeled wild-type caspase-1. The lower equilibrium concentrations observed in the AUC experiment was chosen to avoid some aggregates that formed and were subsequently spun to the bottom of the cell. Data analysis was performed using SEDFIT (17) and SEDPHAT (18). For global fitting, a monomer-dimer equilibrium model was chosen, mass conservation was employed, and the meniscus, bottom, local concentrations, and log association constant were floated as parameters.
Active Site Titrations
Functional protein concentration for enzyme kinetic analysis was determined by active site titration (19); caspase was incubated in assay buffer for 2 h at room temperature with a titration from a 0- to 2-fold stoichiometric ratio using the irreversible active site inhibitor z-VAD-fmk. The protein was diluted to an enzyme concentration of 50 nm, and activity was determined using fluorogenic tetrapeptide substrate (Enzo Life Sciences) at 25 μm for caspase-1 or 50 μm for caspase-3. The substrates used were Ac-WEHD-AFC for caspase-1 constructs and Ac-DEVD-AFC for caspase-3 constructs (20–22).
Expression and Purification of Half-labeled Caspase Constructs
The expression of hybrid caspase-1 and caspase-3 constructs follows the protocol described above. Caspase-1 (residues 120–297) and caspase-3 (residues 29–175) large subunits containing an N-terminal His6 or Strep affinity tags were generated by designing 5′-primers with the appropriate sequence for the affinity tag and using polymerase chain reaction to generate dsDNA inserts. The affinity-tag amino acid sequences were MRGSHHHHHHSAG- for the His6-tagged construct and MWSHPQFEKSAG- for the Strep-tagged construct. The Strep tag is an eight-amino acid peptide that binds with high selectivity to the streptavidin variant Strep-Tactin (IBA GmbH, Germany). These inserts were subcloned into NdeI and EcoRI restriction endonuclease sites of the pRSET plasmid (Invitrogen). The p10 small subunit of caspase-1 (residues 317–404) and the p17 small subunit of caspase-3 (residues 176–277) were also cloned into the pRSET plasmid. These constructs were then expressed and purified as inclusion body pellets as described above. In addition, full-length caspase-3 constructs (residues 1–277) containing N-terminal affinity tags were generated using the above 5′-primer and then subcloned into NdeI and EcoRI restriction endonuclease sites of the pET-23b plasmid (Novagen). This construct was used for soluble expression of caspase-3.
Expression and Purification of Half-labeled Caspase-1
To generate the hemi-labeled caspase-1 construct, active caspase-1 was first generated by refolding a His6- or Strep-tagged large subunit with the small subunit to generate the active caspase dimer as described above. After refolding, samples were centrifuged at 16,000 × g for 10 min to remove precipitate, diluted 2-fold into 50 mm sodium acetate (pH 5.9) buffer, and then centrifuged once more at 16,000 × g for 10 min. Samples were filtered and purified by cation exchange chromatography.
The tagged caspase-1 construct was then labeled with the irreversible active site inhibitor z-VAD-fmk in labeling buffer containing 50 mm HEPES (pH 7.4), 50 mm KCl, 200 mm NaCl, and 10 mm DTT overnight at 4 °C. Complete labeling of the tagged p20 subunit was verified by liquid chromatography-mass spectrometry (LC-MS; Waters, Milford, MA) and complete inhibition of catalytic activity. Excess inhibitor was removed using a Superdex 200 10/300 gel filtration column (GE Healthcare) in buffer containing 25 mm Tris (pH 8.0), 50 mm NaCl, 5% glycerol, and 1 mm DTT. The VAD-fmk-labeled tagged caspase-1 was then concentrated using Millipore Ultrafree-15 devices with a molecular weight cutoff of 10,000 Da and then denatured in 6 m guanidine. This sample was then refolded in the presence of the other tagged p20 subunit and excess p10 subunit. Refolding and purification by cation exchange chromatography were performed as described above. The three affinity-tagged dimeric caspase-1 species were then separated using sequential 1-ml HisTrap and 1-ml StrepTrap columns for affinity purification (GE Healthcare). The final half-labeled caspase-1 construct was purified by size exclusion chromatography using a Superdex 200 16/60 column in 25 mm Tris (pH 8.0), 50 mm NaCl, 5% glycerol, and 1 mm DTT. Final verification of sample purity as being properly half-labeled was performed by LC-MS.
The control heterodimer caspase-1 with His6 or Strep affinity tags, but no labeling with the active site inhibitor z-VAD-fmk was generated in a similar fashion as above. Three caspase-1 constructs, the His6-tagged p20, Strep-tagged p20, and p10 subunits from inclusion bodies were refolded together, then purified as above using cation exchange chromatography, sequential His6 and Strep tag affinity-based purification, and finally size exclusion chromatography.
Expression and Purification of Half-labeled Caspase-3
To generate the half-labeled caspase-3 construct, active caspase-3 was first generated by soluble expression in E. coli BL21(DE3)pLysS cells (Stratagene). Cells were grown in 2xYT media containing 200 μg/ml ampicillin and 50 μg/ml chloramphenicol at 37 °C to an A600 nm of 0.8–1.0. Overexpression of caspase-3 was induced with 200 μm isopropyl β-d-1-thiogalactopyranoside at 37 °C for 3 h. Cells were harvested and resuspended in 100 mm Tris (pH 8.0) and 100 mm NaCl for lysis by microfluidization (Microfluidics). The cell lysate was spun at 45,000 × g for 30 min at 4 °C. Caspase-3 with an N-terminal His6-affinity tag was isolated using a 1-ml HisTrap HP Ni-NTA affinity column (GE Healthcare) eluted with buffer containing 200 mm imidazole. The eluted protein was diluted 2-fold with buffer containing 20 mm Tris, pH 8.0, and then purified by anion-exchange chromatography (HiTrap Q HP, GE Healthcare) with a 30-column volume gradient from 0 to 0.5 m NaCl.
The His6-affinity tagged caspase-3 construct was then labeled and purified with the irreversible active site inhibitor z-VAD-fmk as described above for caspase-1. After refolding with Strep affinity-tagged p17 large subunit and p12 small subunit, samples were dialyzed overnight at 4 °C against 20 mm Tris (pH 5.9), 1 mm DTT. Dialyzed protein was purified by anion-exchange chromatography as described above. The three affinity-tagged caspase-3 species were then separated using sequential 1-ml HisTrap and 1-ml StrepTrap affinity purification (GE Healthcare), and the final half-labeled caspase-3 construct was purified using a Superdex 200 16/60 gel filtration in 20 mm Tris (pH 8.0), 50 mm NaCl, and half-labeling was verified by LC-MS.
The control heterodimer caspase-3 with His6 or Strep affinity tags but no labeling with the active site inhibitor z-VAD-fmk was generated in a similar fashion as above. Three caspase-3 constructs, the His6-tagged p17, Strep-tagged p17, and p12 subunits from inclusion bodies were refolded together and then purified as above using anion exchange chromatography, sequential His6- and Strep-tag affinity-based purification, and finally size exclusion chromatography.
Enzyme Kinetic Analysis
Kinetic analysis of caspase-1 was performed in a buffer containing 50 mm HEPES (pH 8.0), 50 mm KCl, 200 mm NaCl, 10 mm DTT, 0.1% CHAPS, and NaOH was added dropwise to correct the pH to 8.0. Kinetic analysis of caspase-3 was performed in buffer containing 50 mm HEPES (pH 7.4), 50 mm KCl, 0.1 mm EDTA, 1 mm DTT, and 0.1% CHAPS. Steady-state kinetic analysis was done by titrating enzyme with fluorogenic tetrapeptide substrate (Ac-WEHD-AFC for caspase-1 constructs and Ac-DEVD-AFC for caspase-3 constructs, Enzo Life Sciences). Kinetic data were collected for a 10-min time course using a Spectramax M5 microplate reader (Molecular Devices, Sunnyvale, CA) with excitation, emission, and cutoff filters set to 365, 495, and 435 nm, respectively.
For the tagged caspase-1 and caspase-3 constructs, the Vmax, Km, and the Hill coefficient (nHill) were calculated using GraphPad prism. The initial velocity (v), measured in relative fluorescence units per unit time, was plotted versus the logarithm of substrate concentration. The model used to fit the data is a sigmoidal does-response curve with variable slope, and from this model all three kinetic constants were derived. The general equation of this model is Y = Bottom + (Top − Bottom)/(1 + 10((log EC50 − X) × Hill Slope)), where Y is the initial velocity, X is the logarithm of the substrate concentration, and Top, Bottom, EC50 (Km), and Hill Slope are free parameters fit to the data. A standard curve using pure AFC product was used to convert relative fluorescence units to units of concentration (μm). In determining kinetic constants for caspases, we observed that at saturating substrate concentrations, the enzyme exhibited decreasing activity as substrate concentration increased, most likely due to product inhibition. To correctly fit our data using nonlinear regression, data points exhibiting product inhibition were excluded.
To test our proposed kinetic model for wild-type caspase-1, enzyme and substrate were both varied using the above assay buffer of 50 mm HEPES (pH 8.0), 50 mm KCl, 200 mm NaCl, 10 mm DTT, 0.1% CHAPS. Caspase-1 was varied from 500 to 8.5 nm in serial 1.5-fold dilution steps, and substrate peptide Ac-WEHD-AFC (in 20% DMSO) was varied from 0.34 to 53.2 μm in a 1.4-fold dilution steps in 96-well format so that the final concentration of DMSO was <2% by volume. A linear region of the data within the first 2 min was selected for the steady-state activity measurement. Enzyme concentrations of 333 and 500 nm and substrate concentrations below 1.31 μm were excluded from analysis due to the fact that they consumed substrate too rapidly. To convert between relative fluorescence units and product concentration, a steady-state a curve was constructed using the relative fluorescence units at the highest enzyme concentration after substrate was fully consumed to internally correct for the effects of caspase-1 on the relative fluorescence.
Steady-state Kinetic Modeling
Chemical kinetic equations were derived from the scheme in Fig. 3C. These equations were solved assuming quasi-steady-state conditions (please see “steady-state kinetic model derivations” in the supplemental information for more details). The derivatives with respect to time of the concentration of species A, B, D, and E were set to 0. It was assumed that substrate concentrations at steady state were equal to their initial values; this common approximation was deemed appropriate as the data points used for fitting were under a linear product production per unit time phase. Enzyme mass conservation was used. To solve the equations, we made temporary approximations that were then updated over 10 iterations; these approximations used in our iterative analytic method gave results that were consistent with fully numerical solutions for the parameters reported here. Parameters were fit using the Matlab optimization toolkit using an objective function consisting of steady-state rates from the assay data and restraints for ratios of dimerization rates based on the AUC data, assuming the hemi-labeled dimerization affinity is close to the double-labeled dimerization affinity. Parameter optimization was performed in two stages. An initial unconstrained minimization (“fminsearch” function) was performed and then followed by a constrained nonlinear regression (“lsqnonlin”) with physically reasonable constraints; for example, to keep on-rates less than 1 × 109 s−1. The fitted parameters are given in Table 1.
FIGURE 3.
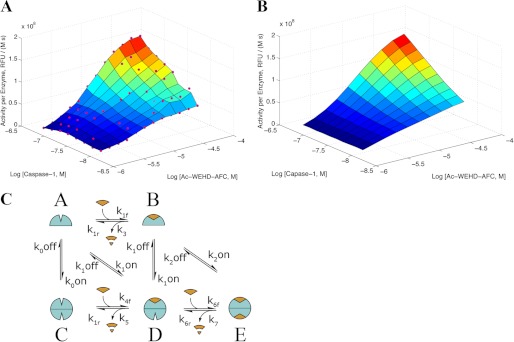
Steady-state kinetics for cleavage of WEHD-AFC by caspase-1. A, experimentally determined steady-state substrate cleavage kinetics show concentrations of caspase-1 from 8.7 to 150 nm and Ac-WEHD-fmk from 1.3 to 530 μm. RFU, relative fluorescence units. B, fitted steady-state cleavage rates from our kinetic model with parameters are shown below. C, shown is the steady-state kinetic model describing on- and off-states for caspase-1 (open and closed shapes, respectively) undergoing monomer to dimer transitions in the presence and absence of substrate (shown as a wedge) or product (cleaved wedge).
TABLE 1.
Parameters for caspase-1 kinetic steady-state model fit to assay data
| Parameter | Value | Units |
|---|---|---|
| k1r | 2.30 × 10−6 | s−1 |
| k0off | 5.22 | s−1 |
| k3 | 2.26 × 10−1 | s−1 |
| k1off | 2.43 × 101 | s−1 |
| k1f | 2.60 × 105 | m−1s−1 |
| k0on | 5.22 × 104 | m−1s−1 |
| k1on | 4.35 × 106 | m−1s−1 |
| k2off | 5.34 × 101 | s−1 |
| k2on | 1.06 × 107 | m−1s−1 |
| k4r | 1.91 | s−1 |
| K5 | 1.60 | s−1 |
| k4f | 1.53 × 102 | m−1s−1 |
| k6r | 2.44 × 101 | s−1 |
| K 7 | 1.74 | s−1 |
| K6f | 1.24 × 108 | m−1s−1 |
RESULTS
To examine how binding at the active site influences the monomer-dimer equilibrium constant in caspase-1, we employed AUC to determine the dimerization constant in the absence of substrate or inhibitor (Fig. 2). To prevent any autoproteolysis during the overnight AUC run, we made the active site C285A mutant. We also used the standard caspase-1 construct, which lacks the CARD domain; full-length capsase-1 is poorly expressed in E. coli and largely insoluble. The CARD domain is used to allow pro-caspase-1 to engage the inflammasome but is not believed to mediate dimerization because the CARD domain is topologically very far from the dimer interface (23). As shown in Fig. 2A, the dissociation constant (KD) calculated for the active site mutant of the CARD-less apo-caspase-1 (C285A) is 110 ± 1 μm. In sharp contrast, caspase-1 that is fully inhibited with the covalent inhibitor (z-VAD-fmk) forms a 20-fold tighter dimer complex, with a calculated KD of 5 ± 2 μm (Fig. 2B). These data show that active site occupancy dramatically increases the dimer affinity.
FIGURE 2.
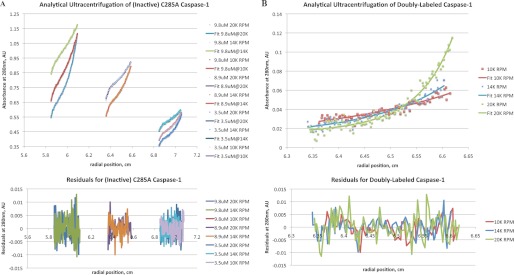
Solution measurements of the dimerization constants by AUC for caspase-1 yielded a KD of 109 μm for apocaspase-1 (90% confidence interval of 92–134 μm) and a KD of 5 μm for active site inhibitor-bound caspase-1 (90% confidence interval of 2.4–10 μm). Data points are shown as small colored squares, and fitted absorbance values are shown as colored lines. A, shown is analytical ultracentrifugation results of catalytically inactive C285A caspase-1 in the absence of ligands at 10,000, 14,000, and 20,000 rpm at three different final protein concentrations: 9.8, 8.9, and 3.5 μm. The curves are best fit to a monomer-dimer equilibrium having a KD value of 110 μm. AU, absorbance units. B, shown are analytical ultracentrifugation results of caspase-1 fully inhibited with the covalent active site inhibitor z-VAD-fmk at 10,000, 14,000, and 20,000 rpm at a final concentration of 1 μm.
To examine the dependence of enzyme activity on enzyme concentration and obtain data to fit a kinetic model, we evaluated the initial rates for hydrolysis of the Ac-WEHD-AFC substrate by changing both enzyme and substrate concentrations. We varied the enzyme concentration from 10 to 500 nm. Kinetic studies at enzyme concentrations >150 nm were challenging to interpret in the low range of substrate concentrations because the high enzyme concentrations depleted the substrate too quickly for accurate steady-state rates to be determined. Thus, data in this range were excluded from further analysis. As seen in Fig. 3A, the activity per enzyme subunit undergoes a dramatic rise as the enzyme concentration is increased. These data can be fit to the minimal kinetic model that describes two conformational on- and off-states and the monomer-to-dimer transitions (Fig. 3B). The model includes the possibilities that substrate can be hydrolyzed from the monomer or dimer states either when one site is occupied or when both sites are occupied. Using the dimerization affinities determined from AUC studies above, the kinetic constants were fitted to the experimentally determined steady-state activity per enzyme molecule (Table 1). This used a quasi-steady-state solution of the chemical kinetics equations (see “Experimental Procedures” and “steady-state kinetic model derivations” in the supplemental information for details). The model assumes that substrate (Ac-WEHD-AFC) provides the same enhancement in dimerization affinity as inhibitor (z-VAD-fmk) and that the dimerization affinity with one molecule of z-VAD-fmk bound is close to that with two molecules of z-VAD-fmk bound. Under these assumptions, we obtained an excellent fit to the measured steady-state activities. The measured dimerization constants (Fig. 2) are reasonably consistent with those calculated from the fit to the steady-state kinetic data in Fig. 3B (i.e. calculated KD values of 5 μm for caspase-1 with 2 Ac-WEHD-AFC bound, 5.6 μm for caspase-1 with 1 Ac-WEHD-AFC bound, and 100 μm with no Ac-WEHD-AFC bound).
We next wished to directly measure how binding at only one site affected the catalytic activity of caspase-1. To study this, we created a homogeneous preparation of hemi-labeled caspase-1 in which only one of the two active sites in the caspase-1 dimer was labeled with the covalent inhibitor, z-VAD-fmk. This was achieved by creating two tagged versions of caspase-1 in which the p20 subunit, containing the active site cysteine, was fused to either an N-terminal Strep tag or His6 tag (Fig. 4). We refolded each p20 in the presence of the p10 subunit and purified each. Control experiments showed that the tagged enzymes had virtually the same kinetic properties as the wild-type enzyme (supplemental Table 1). The His6-tagged caspase-1 was then fully labeled with the covalent active site inhibitor z-VAD-fmk. Complete labeling was confirmed by mass spectrometry and full inactivation of enzyme activity (supplemental Fig. 1). The labeled His6-tagged caspase-1 was denatured with guanidine-HCl in the presence of an excess of Strep-tagged caspase-1. The mixture was dialyzed and refolded, allowing scrambling of the p20 subunits and generation of hemi-labeled caspase-1 marked by the presence of both the His6 tag and Strep tag. The hemi-labeled dual-tagged enzyme could be purified away from the homo-tagged enzymes by a dual affinity column; first, a nickel column to recover any His6-tagged enzymes and then an avidin column to recover the His6/Strep-tagged enzyme. Mass spectrometry confirmed the dual-tagged caspase-1 contained a single z-VAD-fmk label on the His6-tagged p20 subunit (supplemental Fig. 1).
FIGURE 4.

A, the scheme show the preparation of the hemi-labeled caspase-1. Two separate p20 subunits were fused with either a His6 or Strep affinity tag and refolded with the p10 to generate the pure homodimers. The refolded His6 tag caspase-1 was fully inhibited with z-VAD-fmk, and complete labeling was confirmed by mass spectrometry. After gel filtration to remove the active site inhibitor, the enzyme was mixed in excess amounts of unlabeled Strep-tagged caspase-1. The proteins were denatured and refolded, allowing for scrambling of the subunits. The hemi-labeled species was isolated by affinity purification of the protein that bound to both the nickel and avidin column and confirmed by mass spectrometry (see supplemental Fig. 1). B, shown is a simplified kinetic scheme for the hemi-labeled enzyme assuming all activity comes from the dimer. C, shown is the fit of Km to the simplified kinetic scheme in panel B. The value Km is fit to the activity as a function of Ac-WEHD-AFC concentration at an enzyme concentration of 50 nm given the dimerization affinity from the AUC data and the chemical rate kchem from the wild-type model (k9). RU, relative units.
The kinetic constants for the labeled tagged variants were determined by Michaelis-Menten analysis and are shown in Table 2. When corrected for having only one active site available per dimer, the hemi-labeled enzyme shows roughly an 18-fold increase in kcat, a 2-fold increase in Km, and a resulting 9-fold increase in the catalytic efficiency (kcat/Km) compared with the unlabeled control. Thus, inhibitor binding at one site enhances catalysis both by stabilizing the dimer and propagating a conformational change in the unoccupied monomer that leads to enhanced catalytic efficiency. To compare the hemi-labeled experiments to our previous kinetic model on wild-type caspase-1, we solved a simplified kinetic model shown in Fig. 4B. For the parameters, we assumed a value of 5 μm for the dimerization KD in the presence of z-VAD-fmk based on the AUC data, and the maximal kcat calculated from parameter k7 in Fig. 3 and Table 1. Fixing these and fitting for Km from the steady-state kinetic data produced a Km value of 44 nm, which is in close agreement with the 30 nm value from the wild-type kinetic model (Km = (k6r + k7)/k6f). Note this value is different from the “effective” Km in Table 2, which does not account for the monomer-dimer equilibrium.
TABLE 2.
Hemi-labeling of caspase-1 dimer leads to enzyme activation
Kinetic constants for hybrid-tagged caspase-1 either with or without a single z-VAD-fmk on the His-tagged p20 subunit (see Fig. 4) are shown. The simple Michaelis-Menten fit here does not take into account the monomer-dimer equilibrium and thus yields a higher effective Km(eff) than in our more detailed model in Fig. 4, which provides the Km of the dimeric hemi-labeled enzyme. Standard errors are within 10% of reported values based on data collected in triplicate.
| Construct | Km(eff) | kcat | kcat/Km | Ratio |
|---|---|---|---|---|
| μm | s−1 | m−1·s−1 | kcat/Km | |
| Unlabeled caspase-1 control | 1.9 | 0.11 | 5.6 × 104 | 1 |
| Hemi-labeled caspase-1 | 3.8 | 1.93 | 5.1 × 105 | 9.1 |
If binding of inhibitor at one site enhances activity at the other site, it is possible that a direct titration of wild-type caspase-1 with z-VAD-fmk could activate at substoichiometric equivalents of inhibitor if hemi-labeled intermediates were generated. Indeed, titration of wild-type caspase-1 leads to systematic activation (up to 1.4-fold) that plateaus between 0.2 and 0.4 eq of inhibitor per active site. Further titration leads to a steep decline in activity, reaching 0 at about 1 eq of inhibitor per active site. The greater activity for the pure hemi-labeled over that obtained by substoichiometric titration (9-fold versus 1.4-fold) likely results from biased homo-labeling in the titration. That is, when one monomer gets labeled with z-VAD-fmk, this promotes dimerization and rapid labeling of the second site leading to a biased two-site labeling. We can calculate at any point in the titration the ratio of two-site versus hemi-labeled enzyme using a modified version of our kinetic model (Fig. 3) and assuming all inhibitor is irreversibly bound (Fig. 5B). Assuming the activities for apo- and hemi-labeled caspase-1 are those calculated in Table 1, we can predict the relative activity of inhibited wild-type caspase-1 compared with that of the uninhibited wild-type caspase-1. Using this simplified model, we obtained good quantitative agreement with the experimental data and are able to reproduce the “roller-coaster” curve that shows activation followed by inhibition at increasing inhibitor concentrations (Fig. 5). As a control, we examined the active site titration of the hemi-labeled enzyme and found virtually no increase in activity upon the addition of inhibitor (Fig. 5A). The fact that we observed such excellent agreement without adjusting parameters lends further support to these kinetic models and the parameter values shown in Table 1.
FIGURE 5.

Active site titration of wild-type caspase-1 (blue labels) or hemi-labeled hybrid caspase-1 (red labels). Each enzyme was titrated with increasing amounts of the active site-inhibitor z-VAD-fmk in substoichiometric increments up to 1.4 eq per active site. These were allowed to react to completion for 2 h at room temperature, and enzyme activity was measured as described under “Experimental Procedures.” A, shown is measured relative activity of wild-type and hemi-labeled caspase-1 and predicted relative activity of wild-type caspase-1 from the steady-state model. B, shown is a simplified steady-state model of inhibitor-assisted dimerization (assuming complete labeling with inhibitor), taking parameters from Table 1. C, shown is the predicted amount of hemi-labeled caspase-1 species formed during the active site titration using a quasi-steady-state solution to the model in Panel B.
In contrast to caspase-1, caspase-3 is a constitutive dimer and has a Hill coefficient of 1.0 (Table 3). We wished to test how hemi-labeling or spontaneous labeling of caspase-3 affected its kinetics. We generated the hemi-labeled construct for caspase-3 as we did for caspase-1 and determined the Michaelis-Menten constants (Table 3). As shown, once corrected for half of the active sites available in the dimer, the hemi-labeled caspase-3 shows only a 2.5-fold enhancement in kcat and a 2-fold increase in Km, resulting in only a 1.2-fold enhancement in kcat/Km. Next we evaluated how spontaneous titration of caspase-3 with z-VAD-fmk affects its kinetics (Fig. 6). As shown, titration with z-VAD-fmk caused a linear decrease in activity for both the wild-type caspase-3 as well as the hemi-labeled caspase-3. Thus, in sharp contrast to caspase-1, caspase-3 shows dramatically lower propensity for substrate- or inhibitor-induced activation. This correlates with caspase-3 existing as a constitutive dimer that exhibits little positive cooperativity.
TABLE 3.
Hemi-labeling of the caspase-3 dimer results in minor enzyme activation
Standard errors within 10% of reported values based on data collected in triplicate.
| Construct | Km(eff) | kcat | kcat/Km | Ratio |
|---|---|---|---|---|
| μm | s−1 | m−1·s−1 | kcat/Km | |
| Unlabeled caspase-3 control | 13 | 1.4 | 1.1 × 105 | 1 |
| Hemi-labeled caspase-3 | 28 | 3.7 | 1.3 × 105 | 1.2 |
FIGURE 6.

Active site titration of caspase-3 constructs under similar conditions as described in Fig. 5 for caspase-1.
DISCUSSION
The biophysical and enzymologic data presented strongly suggest that substrate and inhibitor binding drives dimerization of caspase-1. The hemi-label experiments show that binding at only one site is sufficient to promote dimer formation. Substoichiometric amounts of inhibitor can activate caspase-1 but not to the same level as the pure hemi-dimer. Together these data suggest that once substrate is bound at one site in the dimer, the active site of the other monomer is poised to bind and catalyze hydrolysis of a second substrate or react more rapidly with an active site inhibitor. Thus, caspase-1 appears cooperative in how substrate binding drives dimer formation and favors the active conformation at the other site in the dimer.
Does dimerization promote better catalytic activity or better substrate binding? The kinetic parameters derived from the steady-state data suggest that the apo dimer binds substrate slowly (k4f = 153 m−1s−1, k4r = 1.9 s−1), whereas the dimer with one substrate bound (“hemi-labeled”) binds substrate much more rapidly (k6f = 1.2 × 108 m−1s−1, k6r = 24.4 s−1). This large difference in rate constants suggests there is low flux from state C (apo dimer) to state D (dimer with substrate bound to one site). We note that our enzyme assay data do not provide much information about the on-rate for substrate binding to the apo dimer (k4f), as state C was lowly populated; a more accurate determination of this on-rate (k4f) would have required prohibitively large enzyme concentrations. Nonetheless, the data are consistent with binding of substrate to one site increasing the affinity for substrate at the second site on the dimer. In contrast, the catalytic rate for dimeric caspase-1 does not significantly depend on whether one or both sites are bound (k5 = 1.6 s−1 versus k7 = 1.74 s−1), indicating that cooperativity lies in substrate binding rather than in catalysis. These rates are roughly similar to kcat values for caspase-1 in the literature, albeit at lower enzyme concentrations (0.77 s−1 for Ac-WEHD-AFC at 10 nm caspase-1 (24) and 0.89 s−1 for Ac-YVAD-AMC at 1 nm caspase-1), from THP-1 cells, prepared using a column conjugated with reversible active site inhibitor, which according to our results would activate the enzyme and increase the population of the dimeric form (25). Thus, we suggest that in the absence of substrate or inhibitor, caspase-1 is in an inactive conformation whether in monomeric or dimeric form. This is consistent with the x-ray structure of apo-caspase-1 (11), which shows a dimeric enzyme in an inactive form that is close to the structure of dimeric pro-caspase-7 inactive zymogen. However, when substrate or inhibitor is bound to one subunit, the other subunit is more likely to be in the active conformation. Previous work on caspase-1 used mutational and structural studies to uncover a linear circuit of functional residues running between the two active sites through the allosteric site (8). Enzymatic activity is strongly affected by perturbations of this circuit, suggesting that the interactions are important for stabilizing the active conformation of caspase-1.
Caspase-1 and caspase-3 provide an interesting contrast in enzymatic properties, as they are respectively prototypical members of the inflammatory and executioner caspase families. Previous work demonstrated that whereas caspase-1 evidenced positive cooperativity, caspase-3 did not (8). In addition, a comparison of base-line enzymatic activity of wild-type enzyme shows that caspase-3 is a more active protease than caspase-1 (almost an order of magnitude greater kcat). We have now demonstrated that caspase-1 can be strongly activated by using the hemi-labeling technique to nearly the same catalytic efficiency as caspase-3. In contrast, caspase-3 shows only a slight degree of activation in the analogous hemi-labeled construct, likely because it exists predominantly as active dimer even in the apo-form.
Our measured dimerization affinities of 110 μm for apocaspase-1 (C285A) and 5 μm for inhibitor-bound caspase-1 are surprisingly similar to those of the initiator caspase-8. AUC measurements on caspase-8 showed that inhibitor-bound caspase-8 has a KD of 5 μm, whereas apocaspase-8 has a KD of 50 μm (26). In contrast, procaspase-3 (27) and caspase-6 (28) are constitutively dimmers in AUC experiments at micromolar concentrations of enzyme, whereas initiator caspase-9 (29) remains a monomer in solution in AUC experiments. Although caspase-6 monomer/dimer equilibrium is much tighter and, therefore, does not appear to be functionally regulated by ligand binding as in caspases-1 and -8, substrate binding changes the conformation of the caspase-6 dimer to that of canonical active caspases (30).
Taken together, these data suggest a model of caspase conformational states shown in Fig. 7. The free caspases transition from monomer to homodimer and equilibrate between an inactive and active conformation. The observations of lower intrinsic catalytic turnover, positive cooperativity, and hybrid activation all point to caspase-1 residing primarily in the inactive monomeric conformation. In addition, these data are consistent with previous studies using conformationally specific antibodies raised to the on- and off-forms of caspase-1, which also suggested that apocaspase-1 is in the off-state (31). In contrast, the higher intrinsic activity of caspase-3, lack of positive cooperativity, and inability to be activated by half-labeling all suggest that in the absence of ligand, caspase-3 is in a predominantly active conformation.
FIGURE 7.

Model of caspase conformational states. Caspases exist in a conformational equilibrium between active and inactive states. The inflammatory caspase-1 resides primarily in the inactive conformation (“off” state), and binding to the active site selects for the active conformation (“on” state), which can serve as a template for another monomer to bind and form an active dimer. The executioner caspase, caspase-3, resides primarily in the active conformation; binding in the active site has little additional effect on this equilibrium and enzyme activity.
Recent work by Denault et al. (40) produced engineered heterodimers of the executioner caspase-7, where only one of the subunits was rendered catalytically inactive by mutagenesis. Much like our result with caspase-3, they show that the resulting caspase-7 heterodimer had about half the activity of wild type. This suggests that the unaltered catalytic site maintained full activity and that the two catalytic domains in the caspase-7 dimer are equal and independent. Indeed, the structure of apo-caspase-7 looks very much like the inhibitor-bound structure (32), suggesting that very little active site reorganization occurs in response to substrate binding.
Previous reports have described a dominant-negative effect of a mutated caspase-1 construct. Friedlander et al. (33) created an active site caspase-1 mutant by knocking out the catalytic cysteine with a C285G mutation. This variant was then expressed in a transgenic mouse model under the control of a neuron-specific promoter. They were able to show that expression of this caspase-1 variant protected neurons from apoptosis after trophic factor withdrawal and reduced brain injury after ischemic events. A subsequent study showed that expression of the same caspase-1 dominant-negative mutant in a mouse model of Huntington disease delayed symptoms and prolonged survival (34). The molecular mechanism by which this caspase-1 mutant is able to exert a dominant-negative effect has not been elucidated but is consistent with our model, suggesting that caspase-1 is most active when both active sites are occupied by substrate.
We estimate the dimerization constant (KD) of caspase-1 in the presence of inhibitor or substrate to be ∼5 μm. Caspase concentrations vary in cells depending on the type but are generally in the range of 100 nm, which is well below the substrate-induced dimerization constant we measure for caspase-1. Pro-caspase-1 binds to inflammasomes using its CARD domain where it is both activated and concentrated to levels we propose that are well above the 5 μm dimerization constant. Thus, we suggest that both the concentration on inflammasomes and binding of substrate are regulatory events that drive dimerization and activation.
Although the structures of activated caspase-1 and -3 share much similarity, the biology they drive is quite different. Caspase-1 is primarily involved in processing of proinflammatory cytokines associated with the innate immune response, whereas caspase-3 is a major executioner for apoptosis. Recent proteomics studies show that there are large differences in the number and types of substrates cleaved by caspases 1 and 3 (35, 36). For example, the number of observed caspase-1 substrates cleaved during inflammatory signaling is on the order of about 30 (36), whereas there are 100s cleaved by caspase-3 in apoptosis (18, 19). The observation that the apocaspase-1 catalytic domain resides in an inactive conformation suggests that positive cooperativity may provide an additional selectivity filter for cleaving proinflammatory substrates only when they are concentrated in cells. It has been recently suggested that pro-IL-1β is concentrated at membranes in inflammasomes or in vesicles where it is cleaved by caspase-1 (37–39). These studies suggest that substrates may be sequestered with enzyme. Our observation of substrate-assisted dimerization suggests that the enzyme activity in vivo might be regulated by its concentration in the inflammasome. The inflammatory pathway, with few substrates cleaved in a specific cytokine signaling event, requires a protease under much tighter regulation. By contrast, in apoptotic signaling events, the number of reported caspase substrates is now >1000 (35). It is thus not surprising that caspase-3 is substantially more active than caspase-1 given the much larger number of substrates cleaved in apoptosis. Thus, the dimerization model provides an additional level of regulation through substrate activation.
Acknowledgments
We thank Scott Hansen and Danielle Canzio for assistance with the analytical ultracentrifugation experiments, and Justin Scheer for guidance in developing the tagged caspase-1 constructs. We also thank colleagues in the Wells laboratory, David Agard, William DeGrado, and Susan Miller at University of California, San Francisco and Luhua Lai at Peking University for useful discussions.
This work was supported, in whole or in part, by National Institutes of Health Grants R01-AI070292 (to J. A. W.) and GMO7618 (to D. D.). This work was also supported by a PhRMA Foundation fellowship and a National Science Foundation East Asia and Pacific Summer Institutes grant (to C. L. M., who is currently funded by National Institutes of Health Postdoctoral Fellowship F32-GM099197). This work was also supported by supercomputer time provided by the National Science Foundation Teragrid program at the Texas Advanced Supercomputing Center (Project TG-MCB090109 to M. P. J., a consultant for Schrodinger, Inc.).

This article contains supplemental Table 1, Fig. 1, and Equations 12–14.
- AUC
- analytical ultracentrifugation
- z-VAD-fmk
- benzyloxycarbonyl-Val-Ala-Asp-fluoromethylketone
- Ac-WEHD-AFC
- Ac-Trp-Glu-His-Asp-7-amino-4-trifluoromethylcoumarin.
REFERENCES
- 1. Changeux J. P., Edelstein S. J. (2005) Allosteric mechanisms of signal transduction. Science 308, 1424–1428 [DOI] [PubMed] [Google Scholar]
- 2. Kuriyan J., Eisenberg D. (2007) The origin of protein interactions and allostery in colocalization. Nature 450, 983–990 [DOI] [PubMed] [Google Scholar]
- 3. del Sol A., Tsai C. J., Ma B., Nussinov R. (2009) The origin of allosteric functional modulation. Multiple pre-existing pathways. Structure 17, 1042–1050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Brennan D. F., Dar A. C., Hertz N. T., Chao W. C., Burlingame A. L., Shokat K. M., Barford D. (2011) A Raf-induced allosteric transition of KSR stimulates phosphorylation of MEK. Nature 472, 366–369 [DOI] [PubMed] [Google Scholar]
- 5. Madsen N. B., Shechosky S. (1967) Allosteric properties of phosphorylase b. II. Comparison with a kinetic model. J. Biol. Chem. 242, 3301–3307 [PubMed] [Google Scholar]
- 6. Hauske P., Ottmann C., Meltzer M., Ehrmann M., Kaiser M. (2008) Allosteric regulation of proteases. Chembiochem 9, 2920–2928 [DOI] [PubMed] [Google Scholar]
- 7. Pop C., Salvesen G. S. (2009) Human caspases. Activation, specificity, and regulation. J. Biol. Chem. 284, 21777–21781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Datta D., Scheer J. M., Romanowski M. J., Wells J. A. (2008) An allosteric circuit in caspase-1. J. Mol. Biol. 381, 1157–1167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Black R. A., Kronheim S. R., Merriam J. E., March C. J., Hopp T. P. (1989) A pre-aspartate-specific protease from human leukocytes that cleaves pro-interleukin-1β. J. Biol. Chem. 264, 5323–5326 [PubMed] [Google Scholar]
- 10. Kostura M. J., Tocci M. J., Limjuco G., Chin J., Cameron P., Hillman A. G., Chartrain N. A., Schmidt J. A. (1989) Identification of a monocyte specific pre-interleukin 1β convertase activity. Proc. Natl. Acad. Sci. U.S.A. 86, 5227–5231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Cerretti D. P., Kozlosky C. J., Mosley B., Nelson N., Van Ness K., Greenstreet T. A., March C. J., Kronheim S. R., Druck T., Cannizzaro L. A. (1992) Molecular cloning of the interleukin-1β converting enzyme. Science 256, 97–100 [DOI] [PubMed] [Google Scholar]
- 12. Thornberry N. A., Bull H. G., Calaycay J. R., Chapman K. T., Howard A. D., Kostura M. J., Miller D. K., Molineaux S. M., Weidner J. R., Aunins J. (1992) A novel heterodimeric cysteine protease is required for interleukin-1 β processing in monocytes. Nature 356, 768–774 [DOI] [PubMed] [Google Scholar]
- 13. Wilson K. P., Black J. A., Thomson J. A., Kim E. E., Griffith J. P., Navia M. A., Murcko M. A., Chambers S. P., Aldape R. A., Raybuck S. A. (1994) Structure and mechanism of interleukin-1 β converting enzyme. Nature 370, 270–275 [DOI] [PubMed] [Google Scholar]
- 14. Talanian R. V., Dang L. C., Ferenz C. R., Hackett M. C., Mankovich J. A., Welch J. P., Wong W. W., Brady K. D. (1996) Stability and oligomeric equilibria of refolded interleukin-1β converting enzyme. J. Biol. Chem. 271, 21853–21858 [DOI] [PubMed] [Google Scholar]
- 15. Romanowski M. J., Scheer J. M., O'Brien T., McDowell R. S. (2004) Crystal structures of a ligand-free and malonate-bound human caspase-1. Implications for the mechanism of substrate binding. Structure 12, 1361–1371 [DOI] [PubMed] [Google Scholar]
- 16. Scheer J. M., Wells J. A., Romanowski M. J. (2005) Malonate-assisted purification of human caspases. Protein Expr. Purif. 41, 148–153 [DOI] [PubMed] [Google Scholar]
- 17. Schuck P. (2000) Size-distribution analysis of macromolecules by sedimentation velocity ultracentrifugation and lamm equation modeling. Biophys. J. 78, 1606–1619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Vistica J., Dam J., Balbo A., Yikilmaz E., Mariuzza R. A., Rouault T. A., Schuck P. (2004) Sedimentation equilibrium analysis of protein interactions with global implicit mass conservation constraints and systematic noise decomposition. Anal. Biochem. 326, 234–256 [DOI] [PubMed] [Google Scholar]
- 19. Stennicke H. R., Salvesen G. S. (1999) Caspases. Preparation and characterization. Methods 17, 313–319 [DOI] [PubMed] [Google Scholar]
- 20. Rano T. A., Timkey T., Peterson E. P., Rotonda J., Nicholson D. W., Becker J. W., Chapman K. T., Thornberry N. A. (1997) A combinatorial approach for determining protease specificities. Application to interleukin-1β converting enzyme (ICE). Chem. Biol. 4, 149–155 [DOI] [PubMed] [Google Scholar]
- 21. Talanian R. V., Quinlan C., Trautz S., Hackett M. C., Mankovich J. A., Banach D., Ghayur T., Brady K. D., Wong W. W. (1997) Substrate specificities of caspase family proteases. J. Biol. Chem. 272, 9677–9682 [DOI] [PubMed] [Google Scholar]
- 22. Thornberry N. A., Rano T. A., Peterson E. P., Rasper D. M., Timkey T., Garcia-Calvo M., Houtzager V. M., Nordstrom P. A., Roy S., Vaillancourt J. P., Chapman K. T., Nicholson D. W. (1997) A combinatorial approach defines specificities of members of the caspase family and granzyme B. Functional relationships established for key mediators of apoptosis. J. Biol. Chem. 272, 17907–17911 [DOI] [PubMed] [Google Scholar]
- 23. Bauernfeind F., Ablasser A., Bartok E., Kim S., Schmid-Burgk J., Cavlar T., Hornung V. (2011) Inflammasomes. Current understanding and open questions. Cell. Mol. Life Sci. 68, 765–783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Scheer J. M., Romanowski M. J., Wells J. A. (2006) A common allosteric site and mechanism in caspases. Proc. Natl. Acad. Sci. U.S.A. 103, 7595–7600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Thornberry N. A. (1994) Interleukin-1 β converting enzyme. Methods Enzymol. 244, 615–631 [DOI] [PubMed] [Google Scholar]
- 26. Donepudi M., Mac Sweeney A., Briand C., Grütter M. G. (2003) Insights into the regulatory mechanism for caspase-8 activation. Molecular Cell 11, 543–549 [DOI] [PubMed] [Google Scholar]
- 27. Bose K., Clark A. C. (2005) pH effects on the stability and dimerization of procaspase-3. Protein Science 14, 24–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Baumgartner R., Meder G., Briand C., Decock A., D'arcy A., Hassiepen U., Morse R., Renatus M. (2009) The crystal structure of caspase-6, a selective effector of axonal degeneration. Biochem. J. 423, 429–439 [DOI] [PubMed] [Google Scholar]
- 29. Shiozaki E. N., Chai J., Rigotti D. J., Riedl S. J., Li P., Srinivasula S. M., Alnemri E. S., Fairman R., Shi Y. (2003) Mechanism of XIAP-mediated inhibition of caspase-9. Mol. Cell 11, 519–527 [DOI] [PubMed] [Google Scholar]
- 30. Vaidya S., Velázquez-Delgado E. M., Abbruzzese G., Hardy J. A. (2011) Substrate-induced conformational changes occur in all cleaved forms of caspase-6. J. Mol. Biol. 406, 75–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Gao J., Sidhu S. S., Wells J. A. (2009) Two-state selection of conformation-specific antibodies. Proc. Natl. Acad. Sci. U.S.A. 106, 3071–3076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Chai J., Wu Q., Shiozaki E., Srinivasula S. M., Alnemri E. S., Shi Y. (2001) Crystal structure of a procaspase-7 zymogen. Mechanisms of activation and substrate binding. Cell 107, 399–407 [DOI] [PubMed] [Google Scholar]
- 33. Friedlander R. M., Gagliardini V., Hara H., Fink K. B., Li W., MacDonald G., Fishman M. C., Greenberg A. H., Moskowitz M. A., Yuan J. (1997) Expression of a dominant negative mutant of interleukin-1β-converting enzyme in transgenic mice prevents neuronal cell death induced by trophic factor withdrawal and ischemic brain injury. J. Exp. Med. 185, 933–940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ona V. O., Li M., Vonsattel J. P., Andrews L. J., Khan S. Q., Chung W. M., Frey A. S., Menon A. S., Li X. J., Stieg P. E., Yuan J., Penney J. B., Young A. B., Cha J. H., Friedlander R. M. (1999) Inhibition of caspase-1 slows disease progression in a mouse model of Huntington's disease. Nature 399, 263–267 [DOI] [PubMed] [Google Scholar]
- 35. Mahrus S., Trinidad J. C., Barkan D. T., Sali A., Burlingame A. L., Wells J. A. (2008) Global sequencing of proteolytic cleavage sites in apoptosis by specific labeling of protein N termini. Cell 134, 866–876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Agard N. J., Maltby D., Wells J. A. (2010) Inflammatory stimuli regulate caspase substrate profiles. Mol. Cell. Proteomics 9, 880–893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ferrari D., Pizzirani C., Adinolfi E., Lemoli R. M., Curti A., Idzko M., Panther E., Di Virgilio F. (2006) The P2X7 receptor. A key player in IL-1 processing and release. J. Immunol. 176, 3877–3883 [DOI] [PubMed] [Google Scholar]
- 38. Andrei C., Margiocco P., Poggi A., Lotti L. V., Torrisi M. R., Rubartelli A. (2004) Phospholipases C and A2 control lysosome-mediated IL-1 β secretion. Implications for inflammatory processes. Proc. Natl. Acad. Sci. U.S.A. 101, 9745–9750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. MacKenzie A., Wilson H. L., Kiss-Toth E., Dower S. K., North R. A., Surprenant A. (2001) Rapid secretion of interleukin-1β by microvesicle shedding. Immunity 15, 825–835 [DOI] [PubMed] [Google Scholar]
- 40. Denault J.-B., Bekes M., Scott F. L., Sexton K. M. B., Bogyo M., Salvesen G. S. (2006) Engineered hybrid dimers: tracking the activation pathway of caspase-7. Mol. Cell 23, 523–533 [DOI] [PubMed] [Google Scholar]