

Abstract
Protein biomarkers have the potential to transform medicine as they are clinically used to diagnose diseases, stratify patients, and follow disease states. Even though a large number of potential biomarkers have been proposed over the past few years, almost none of them have been implemented so far in the clinic. One of the reasons for this limited success is the lack of technologies to validate proposed biomarker candidates in larger patient cohorts. This limitation could be alleviated by the use of antibody-independent validation methods such as selected reaction monitoring (SRM). Similar to measurements based on affinity reagents, SRM-based targeted mass spectrometry also requires the generation of definitive assays for each targeted analyte. Here, we present a library of SRM assays for 5568 N-glycosites enabling the multiplexed evaluation of clinically relevant N-glycoproteins as biomarker candidates. We demonstrate that this resource can be utilized to select SRM assay sets for cancer-associated N-glycoproteins for their subsequent multiplexed and consistent quantification in 120 human plasma samples. We show that N-glycoproteins spanning 5 orders of magnitude in abundance can be quantified and that previously reported abundance differences in various cancer types can be recapitulated. Together, the established N-glycoprotein SRMAtlas resource facilitates parallel, efficient, consistent, and sensitive evaluation of proposed biomarker candidates in large clinical sample cohorts.
Protein biomarkers measured in easily accessible samples are a critical element of personalized medicine, because detecting disease states at their early onset or following a response to treatment will undoubtedly enhance treatment efficacy and boost patient wellbeing. Protein biomarkers therefore have the potential to define diseases such as cancer at a molecular level, determine the best treatment by profiling a patient's disease, and assess the therapeutic efficacy over time (1–3). Over the last decade, long lists of biomarker candidates have been proposed from large scale proteomic and transcriptomic datasets. Additionally, recent extensive genomic efforts (4–9) and the integration of the obtained information at gene, transcript, and protein levels have led to an increased knowledge of molecular changes occurring during disease development. However, despite these advances in the biomarker candidate discovery, so far almost none of the proposed markers have been implemented in the clinic. Therefore, biomarker research efforts should shift the focus from the discovery of biomarker candidates to their systematic validation in suitable and well designed large clinical cohorts for evaluating their clinical value.
A major limitation for advancing biomarker research toward their validation in large clinical sample cohorts is the lack of quantitative assays for the multiplexed and consistent measurements of the proposed biomarker candidates (10). Proteins are mainly being measured by antibody-based methods, and there the scarcity of assays is particularly pronounced. High quality immunoassays relying on the availability of specific antibodies for protein quantification are only available for a small subset of the human proteome. Because the development of new affinity reagents is time-consuming and expensive (11), a larger coverage of the human proteome by immunoassays is not anticipated in the near future. Additionally, immunoassays are limited in their ability to quantify proteins in a multiplexed manner. However, for biomarker validation, the process in which large numbers of biomarker candidates are quantified in hundreds of patient samples to evaluate their clinical utility, multiplexed protein quantification is an important requirement (12, 13). Therefore, complementary novel technological platforms are critically needed to accelerate biomarker validation. The requirements for such a technology are high. Besides the capacity to consistently quantify multiple proteins simultaneously, it should reach limits of quantification (LOQ)1 in the low nanogram/ml level in plasma where tissue leakage products can be expected (14) and have a coefficient of variance below 15% as suggested by Addona et al. (15) to facilitate the detection of small abundance changes between multiple samples.
More recently, targeted mass spectrometric measurements via selected reaction monitoring (SRM) have been shown to fulfill the requirements for biomarker validation studies (11, 12, 15, 16). SRM supports the multiplexed (17–19), consistent (20), and sensitive quantification (16, 21–23) of hundreds of analytes in one measurement at coefficients of variance mostly below 20% (15). As for affinity reagent-based measurements, SRM requires the development of specific assays (24). A high quality SRM assay consists of the m/z and chromatographic retention time (RT) of the peptide precursor ion, as well as the m/z and relative intensities of the peptide fragment ion signals (24, 25). In contrast to affinity-based assays, SRM assays are easily portable between laboratories (15) and are generated faster and in a more cost-efficient manner. Essentially, once the peptide-specific coordinates are available for an SRM assay, it can be run on any triple quadrupole-based instrument and applied to any sample.
Here, we developed an SRM assay library for 2007 human and 1353 murine N-glycosylated proteins. Glycoproteins can be either N- or O-glycosylated. They represent a subproteome that is particularly relevant for clinical research, because these oligosaccharide-modified proteins are typically found either secreted by cells and tissues available for remote sensing in body fluids as potential biomarkers or at the cell surface as potential drug targets (26, 27). This is supported by the fact that the majority of the current clinically used biomarkers and drug targets are glycosylated (28). Furthermore, the enrichment of N-glycosites (i.e. deglycosylated peptides) from blood plasma in combination with SRM was shown to reach the required LOQ (23) and to facilitate the quantification of N-glycoproteins over a large concentration range reaching nanogram/ml levels in plasma (17). The sensitive and accurate quantification of N-glycoproteins by SRM therefore represents a promising biomarker validation strategy. For the development of the SRM assays, we used a high throughput approach based on the synthesis of equimolar synthetic peptides for 6279 N-glycosites. The synthetic peptides were employed as reference compounds to generate the corresponding fragment ion spectra and to extract the SRM assay coordinates (29). The final library consisting of SRM assays for 5568 N-glycosites is publicly available via the SRMAtlas (25). We show the utility of the developed SRM assay library for multiplexed quantification of selected cancer-associated N-glycoproteins in two independent cohorts of clinical specimens. The potential of the SRM assays was demonstrated by consistently quantifying 48 N-glycoproteins across 5 orders of magnitude in protein abundance and by recapitulating the abundance differences for 13 proteins in various cancer types that were previously discovered by antibody-based assays. Overall, with the N-glycoprotein SRMAtlas, we have generated a platform, which now solves the bottleneck of multiplexed, consistent, and sensitive quantification of potential biomarker candidates in suitably collected sample cohorts by using a complementary antibody-independent MS-based technology.
EXPERIMENTAL PROCEDURES
Protein and Peptide Selection
Peptide sequences were selected based on a combination of discovery-driven and PeptideAtlas experimental datasets collected from human and murine serum and plasma, various tissues, and cell lines and based on selected N-glycosylated peptides from the reference protein database. Peptides were selected based on the following criteria: fully tryptic peptides with no missed tryptic cleavage site; deamidated asparagines within the N-linked glycosylation motif (NX(S/T); N = asparagines, X = any amino acid except proline, and S/T = serine or threonine) that were identified with a high identification confidence (1% false discovery rate on the peptide level as determined by PeptideProphet (30) for the individual data sets); peptide length between 6 and 20 amino acids, and required hydrophobicity constraints for the peptide synthesis (as determined by JPT Peptide Technologies).
Additional N-glycosites were added from a reference protein database. Proteins that contain at least one of the following keywords, glycosylation, transmembrane, membrane, cell membrane, secreted, or extracellular, were extracted from the UniProt database (84). The selected proteins were digested in silico with trypsin cleaving at all lysine and arginine residues that are not followed by a proline residue. Next, the sequences were screened for conserved NX(S/T) motifs. Sequences without this motif were filtered out as non-N-glycopeptides. Furthermore, three additional criteria were applied to remove sequences that may fail to be detected in SRM studies. First, the molecular mass of the peptide must be between 400 and 4000 Da. Second, the length of the peptide sequence should be between 6 and 20 amino acids. Third, the peptide sequence should not contain methionine, which is labile to in vitro oxidation.
For the selection proteotypic peptides, peptides that are specific for one protein in the organism were preferentially selected, and the final list of peptides contained 4% nonproteotypic peptides (Supplemental Table 1).
Crude Peptide Library Generation
The final set of selected peptides was synthesized using the SPOT-synthesis technology (JPT Peptide Technologies) (31, 32). Synthesized peptides were lyophilized in 96-well plates with ∼50 nmol of unpurified peptide material per well. Crude peptides were resuspended in 20% acetonitrile, 0.1% formic acid, vortexed for 90 min, and sonicated for 15 min. Plates were kept frozen at −80°C until use. Pools of 96 peptides derived from one plate were prepared. All pools of target peptides taken together constitute the peptide library used for SRM assay generation. The peptide solvent was evaporated in vacuo, and the peptides were resolubilized in 2% acetonitrile, 0.1% formic acid and analyzed by LC-MS/MS on three different LC-MS instruments as described below. 200 fmol/μl of eight heavy isotope-labeled synthetic peptides (AAVYHHFISDGVR, HIQNIDIQHLAGK, GGQEHFAHLLILR, TEVSSNHVLIYLDK, TEHPFTVEEFVLPK, NQGNTWLTAFVLK, LVAYYTLIGASGQR, and TTNIQGINLLFSSR) with elution times spanning the whole gradient were added to each pool for the calculation of the indexed retention time (iRT) values (33).
SRM-triggered Peptide Fragmentation on the QTrap
Pools of peptides were analyzed in duplicate on a QTrap 4000 mass spectrometer equipped with a nano-electrospray ion source (AB Sciex) to generate full fragment ion spectra of the synthetic peptides as described previously (29). Chromatographic separations of peptides were performed by a Tempo nano-LC system (AB Sciex) coupled to a 15-cm fused silica emitter, 75 μm diameter (BGB Analytik) and packed with a Michrom Magic C18 AQ 5-μm resin (Michrom BioResources). Peptides were loaded onto the column from a cooled (4°C) Tempo autosampler (AB Sciex) and separated across a 60-min linear gradient of acetonitrile (5–35%) and water, containing 0.1% formic acid at a flow rate of 300 nl min−1. The mass spectrometer was operated in SRM mode, triggering acquisition of a full MS/MS spectrum upon detection of an SRM trace (threshold 400 ion counts). SRM acquisition was performed with Q1 and Q3 operated at unit resolution (0.7 m/z half-maximum peak width) with a dwell time of 10 ms for the ∼150–200 transitions per run resulting in a cycle time of 3.5–4 s. MS/MS spectra were acquired in enhanced product ion mode for the highest SRM transitions, using dynamic fill time, Q1 resolution low, scan speed 4000 Da s−1, and m/z range 250–1400. Collision energies (CEs) were calculated according to the formulas CE = 0.044 × m/zprecursor + 5.5 and CE = 0.051 × m/zprecursor + 0.55, for doubly and triply charged precursor ions, respectively. Declustering potential was set to 60.
Peptide Fragmentation on an Agilent 6520 Qtof
The peptide pools were analyzed in duplicate on an Agilent 6520 Qtof instrument (Agilent Technologies) in combination with the Agilent HPLC-Chip system (Agilent Technologies). The LC system consisted of an Agilent 1200 capillary pump, for loading the sample on the pre-column, and an Agilent 1200 nano pump, providing the gradient for the chromatographic separation. The peptides were enriched on a 160-nl enrichment column and separated over a 75-mm × 150-mm analytical column (C-18 SB-Zorbax material, 300A, 5-μm particle size), both embedded in the HPLC Chip. Two different LC-MS methods were used for the acquisition of the MS spectra. The first method was used to cover a large number of peptides using a longer gradient, and the second shorter gradient was applied to retarget peptides that were not identified in the first round. In the first LC-MS method, the samples were loaded using a flow of 3 μl/min of 3% (v/v) acetonitrile. The pre-column was further washed with 6 μl of 3% ACN in H2O. The peptides were separated using a flow rate of 250 nl/min and a gradient from 3% (v/v) to 25% (v/v) acetonitrile in 60 min. The gradient was followed by a step at a high acetonitrile concentration for column washing. Furthermore, the mass range for MS was set to 300–2000 m/z and 59–3000 m/z for MS/MS spectra. After each MS1 scan, the two most abundant precursor ions with a minimum count of 1000 ions were selected for MS2 fragmentation and dynamically excluded for 1 min. Total cycle time was ∼2 s. Singly charged precursors and precursors of unknown charge state were excluded from MS2. The collision energy applied was dependent on the m/z of the precursor ion and calculated with the formula CE = 3.0 x m/zprecursor/100 + 2. In the second LC-MS method, the peptides were separated on the analytical column using a flow of 300 nl/min and a gradient from 3% (v/v) to 50% (v/v) acetonitrile in 30 min. The gradient was followed by a step at high acetonitrile concentration for column washing. The mass range for MS was set to 400–1800 m/z and 150–2500 m/z for MS/MS spectra. All other parameters were set according to the first method.
Peptide Fragmentation on a Thermo LTQ Orbitrap XL
The LC-MS/MS analysis of the peptide pools was carried out on an Eksigent 1D-NanoLC-Ultra system (AB Sciex) connected to a Thermo LTQ Orbitrap XL mass spectrometer (Thermo Scientific) equipped with a standard nanoelectrospray source. The peptides were injected onto a 11-cm × 0.075-mm inner diameter column packed in-house with Michrom Magic C18 material (3-μm particle size and 200-Å pore size, Michrom BioResources). The separation was carried out using linear gradient 96% solvent A (0.15% formic acid, 2% acetonitrile) and 4% solvent B (0.15% formic acid, 98% acetonitrile) to 25% solvent B over 60 min at a flow rate of 0.3 μl/min.
Five MS/MS spectra were acquired in the linear ion trap per each FT-MS scan; the latter was acquired at 30,000 full widths at half-maximum resolution settings. One microscan was acquired per each MS/MS scan, and the repeat count was set to three to generate multiple MS/MS scans for each peptide ion. Charge state screening was employed, including all multiple charged ions for triggering MS/MS attempts and excluding all singly charged precursor ions, as well as ions for which no charge state could be determined. Only peptide ions exceeding a threshold of 150 ion counts were allowed to trigger MS/MS scans, followed by dynamic exclusion for 15 s. Inclusion lists for directed MS sequencing of peptide features were prepared containing the precursor ion masses in both the doubly and triply charged state of each peptide in a sample. The lists containing up to 1000 precursors per run with a mass range of 250 to 1600 m/z were used to trigger MS/MS attempts.
MS Database Search and Peptide Identification
Wiff-, RAW-, and d-files (derived from the different instrument platforms) were converted to mzML files using msconvert. X!tandem CYCLONE (2010.10.01.1) was used to search the mzML files against a synthetic peptide decoy database. The synthetic peptide database was generated by concatenating ordered synthetic peptides plus scrambled pseudo protein sequence, including the peptide sequences of the RT peptides. The pseudo protein database was composed of 300 protein entries, including 150 decoy entries. The following parameters were used for database searching: −2.0 to +4.0 precursor mass tolerances, tryptic digestion allowing up to two missed cleavages, carbamidomethyl cysteine as static modification, and oxidized methionine as variable modification. The X! TANDEM k-score version was applied for the database searches, which does not include the fragment ion tolerance option. C-terminal heavy isotope labeling of lysine and arginine (8.014199 Da for lysine and 10.008269 Da for arginine) was added as a variable modification to ensure the identification of the RT peptides. Database search results were validated using the Trans-Proteomics Pipeline (TPP Version 4.5 RAPTURE Revision 2, Seattle, Washington) including PeptideProphet, iProphet and ProteinProphet.
Two synthetic peptide PeptideAtlas builds were constructed, “Human Glyco Synthetic PeptideAtlas 2012-07” and “Mouse Glyco Synthetic PeptideAtlas 2012-07” for human and murine peptide identifications, respectively. We applied an iProphet probability ≥0.9 threshold to all data, which resulted in an individual peptide level false-discovery rate of 2% for Orbitrap, 0.45% for Qtof, and 0.6% for QTrap. The PeptideAtlas builds allow for browsing all peptide identifications via the PeptideAtlas webpage.
Spectral Library Generation for the SRMAtlas
SpectraST (Version 4.0, TPP Version 4.6) was used to generate the spectral libraries for SRMAtlas. We generated spectral libraries separately for each instrument and applied the same options to generate all the libraries. SpectraST options, -c_RWI (use raw intensity) -cP0.9 (iProphet probability), were used to generate the raw libraries, and -cJU (inclusion of all peptide ions in all the files), -cAC (generation of consensus spectrum of all replicate spectra of each peptide ion), -c_RWI (use raw intensity) were used to generate the consensus libraries. The consensus libraries for each instrument platform and each organism are available in Supplemental Data 1-6 and were uploaded in the SRMAtlas as the basis for the SRM assays.
Retention Time Extraction and Normalization
For the extraction of the RTs, instrument-specific SpectraST libraries were generated containing all raw-spectra identified on the different instrument platforms with an iProphet probability ≥0.9. An in-house developed script was used to perform the RT extraction, RT normalization between different instrument runs, and transformation to the iRT space. In the first step, the median RT for each peptide measured in one MS run was calculated by combining the RTs of all spectra acquired for the peptide (including all precursor ions) using a lower median estimator (median for odd n number of values; value at position n/2 − 1 for even number of values, whereby the values were ordered according to increasing values). The extracted median RTs for the RT peptides were subsequently used to align the RTs of each MS run. In the case that insufficient numbers of RT peptides were identified in an MS run, additional target N-glycosite peptides with known iRT values from one of the other instrument platforms were used for the alignment. After the alignment, the run-specific RTs were combined into a median RT for each peptide and each instrument platform, and the median RT was transformed to an iRT (33). In the final step, the instrument-specific iRTs were combined into an instrument platform-independent average peptide iRT. Therefore, the iRTs for each peptide were compared across the different instrument platforms. An iRT value was considered as outlier if it differed from the other two instrument platforms by more than 10 iRT units, but the difference between the other two instruments was below 10 iRT units. Outliers were removed only for peptides that were identified on all three instrument platforms. For each peptide, an average iRT and its standard deviation across the different instruments were calculated after the outlier removal. Peptides that are listed with an iRT but without standard deviation represent peptides that have been identified only on one instrument platform, and their iRT values have to be used cautiously as well as the iRTs that have a high standard deviation. The iRT values provided in the SRMAtlas represent an approximate value of all instrument platforms and allow monitoring the peptides of interest in a RT window of ±5% of the LC gradient length.
Functional Annotation of the Human N-Glycoproteins
Functional enrichment analysis was performed for the human N-glycoproteins covered by the N-glycoprotein SRMAtlas using the hypergeometric test implemented in the BiNGO plugin for Cytoscape (34). All biological processes were considered that had an adjusted p value by Benjamini-Hochberg of 1 × 10−23. The graph was generated using Cytoscape (35).
Quantification of Selected N-Glycosites in Human Sera from Healthy Blood Donors
The human serum samples for the verification and quantification experiments were from human single donations, provided by the St. Gallen Hospital. The Ethics Committee of the Kanton St. Gallen, Switzerland, approved all procedures involving human material, and all donors signed an informed consent. N-Glycosites of 32 human serum samples were isolated as described previously (17). A set of 49 SRM assays, targeting 48 glycoproteins, was selected from the generated SRM assay library. Isotopically labeled peptides of the target peptides were added into each sample at a concentration close to the expected concentration of the endogenous peptides (data not shown).
For each peptide, three transitions for the internal standard as well as the endogenous peptide were monitored in a scheduled SRM mode with a retention time window of 3 min and a cycle time fixed to 3.5 s. SRM analyses was performed on a 4000 QTrap (AB Sciex) equipped with a nano-electrospray ion source. Chromatographic separations of peptides were performed by a Tempo nano-LC system (AB Sciex) coupled to a 15-cm fused silica emitter, 75 μm in diameter (BGB Analytik), packed with a Magic C18 AQ 5-μm resin (Michrom BioResources). Peptides were loaded onto the column from a cooled (4°C) Tempo autosampler (AB Sciex) and separated in 35 min over a linear gradient of acetonitrile (5–35%) and water, containing 0.1% formic acid at a flow rate of 300 nl min−1. SRM acquisition was performed with Q1 and Q3 operated at unit resolution (0.7 m/z half-maximum peak width). Data analysis was done using the MultiQuantTM software (AB Sciex) as described elsewhere (17).
Quantification of Selected N-Glycosites in Human Plasma from Cancer Patients and Healthy Controls
The human plasma samples for the clinical evaluation of cancer-associated proteins were provided by University Hospital, Olomouc, Czech Republic. Patients signed an informed consent document. N-Glycosites of 120 human plasma samples were isolated as described above. A set of 22 SRM assays, targeting 15 glycoproteins, was selected from the generated SRM assay library. Isotopically labeled internal standard peptides were added for relative quantification of the target peptides. SRM data analysis was performed as described above. For each peptide, three transitions for the internal standard as well as the endogenous peptide were monitored in a scheduled SRM mode with a retention time window of 5 min and a cycle time fixed to 3 s. Significance analysis was performed within the statistical framework SRMstats (36).
RESULTS
Generation of the N-Glycoprotein SRMAtlas
For the selection of human and murine glycoproteins and their N-glycosites for the SRM assay generation, we focused on N-glycosites with empirical evidence. The primary sources for the selection of N-glycosites were large discovery-driven MS-based experiments in diverse human and murine tissues, cell lines, and plasma. In these experiments, N-glycosites were isolated with routinely used enrichment techniques (17, 27, 37–39) and identified by LC-MS/MS. This MS evidence-based set of N-glycosites was complemented with N-glycosites that were selected from the UniProt database. The final list of selected peptides included a total of 6279 N-glycosites (Supplemental Table 1). These N-glycosites correspond to 2142 human and 1440 murine glycoproteins (Supplemental Table 1) and cover 40% of the human and 30% of the murine proteins that are annotated in the UniProt database as glycoproteins. The selected proteins include 364 cancer-associated proteins (40) and 53 glycoproteins for which a clinical assay is available (Supplemental Table 1) (41).
To generate the N-glycosite SRM assays in an efficient way, we employed a recently described synthetic peptide strategy (29). The selected 6279 N-glycosite peptides were chemically synthesized, and equimolar peptides mixtures were generated for assay development. This strategy is devoid of a protein abundance bias, which occurs when assays are generated from fragment ion spectra from biological samples with a large protein dynamic range. Therefore, the high throughput synthetic approach employed here allows for the generation of SRM assays irrespective of the endogenous abundance of a protein.
To obtain the SRM assay coordinates, pools of synthetic peptides including retention time standards (33) were analyzed on the three most commonly used instrument platforms, a triple quadrupole-ion trap MS (QTrap), a quadrupole-time of flight MS (Qtof), and an Orbitrap-ion trap MS (Orbitrap). These measurements resulted in an SRM assay library composed of 5568 N-glycosite assays (Table I and Supplemental Table 1). RT peptide standards were used to calculate an iRT (33) for the SRM assays enabling the accurate RT estimation required for scheduled SRM measurements (Supplemental Table 2). Because it has been previously shown that different instrument platforms might result in different fragmentation patterns (29, 42), separate SRM assay libraries were generated for each instrument platform to provide high quality assays for all currently used targeted MS platforms.
Table I. Human and murine N-glycosite SRM assays included in the N-glycoprotein SRMAtlas.
| No. of proteins | No. of N-glycosites | |
|---|---|---|
| Human | 2007 | 4421 |
| Mouse | 1353 | 2402 |
| Total | 3360 | 5568 |
The generated SRM assays correspond to one or more N-glycosites per protein for 2007 human and 1353 mouse glycoproteins (Table I). Assays are available for glycoproteins involved in most cellular processes, but they are especially enriched for cell adhesion, glycoproteins involved in cell surface receptor-linked signaling, immune and inflammatory response, as well as system and anatomical structure development (Fig. 1). These cellular processes that are associated with the hallmarks of cancer development (43) can now be monitored by means of N-glycosite assays provided in the established SRMAtlas. The resource also includes assays for 48 proteins with already established clinical assays as well as currently used biomarkers in the clinic such as carcinoembryonic antigen, epidermal growth factor receptor, α-fetoprotein, or mesothelin (Supplemental Table 1).
Fig. 1.

Functional annotation of human N-glycoproteins covered by N-glycoprotein SRMAtlas. The graph shows highly enriched biological processes among the human N-glycoproteins (colored in red). The node size is according to the number of proteins corresponding to each biological process, and the intensity of the color is according to the p value.
The developed SRM assays are accessible via the SRMAtlas interface, where they can be queried and retrieved for specific N-glycosites and N-glycoproteins and directly plugged into any triple quadrupole-based instrument for measurements in samples of interest.
N-Glycosite SRM Assays Facilitate Multiplexed and Sensitive Quantification of N-Glycosites in Human Serum
Upon successful generation of the N-glycoprotein SRMAtlas, we set out to test the sensitivity and dynamic protein concentration range that can be achieved using N-glycosite enrichment in combination with the SRM assays in typical clinical samples. We selected human blood serum, a commonly sampled specimen readily available in sample biobanks that allows noninvasive testing. For this experiment, we selected a panel of 48 N-glycoproteins that are known to cover a large concentration range in the serum proteome. SRM assays for the N-glycosites as well as corresponding heavy isotope-coded internal standards (44) were multiplexed in a single SRM method and used to measure the N-glycosites in N-glycosite-enriched sera samples from 32 healthy blood donors. The obtained quantitative results show that the combination of N-glycosite enrichment and the subsequent SRM-based quantification using high quality SRM assays enabled the quantification of proteins in glycosite-enriched serum samples over a dynamic range of more than 5 orders of magnitude with an LOQ reaching low nanogram/ml concentrations (Fig. 2). This region of the serum proteome is especially interesting because it is the protein abundance range where clinically relevant tissue leakage products reside (10). Moreover, 47 of the 48 quantified N-glycoproteins were consistently quantified in all serum samples, which illustrates the high level of consistency and reproducibility achieved by targeted MS. Together, these results demonstrate that parallel and sensitive SRM measurements are now possible with the N-glycosite SRM assays retrieved from the newly established assay repository.
Fig. 2.

Quantification of N-glycoproteins in sera of healthy blood donors. 48 N-glycoproteins were quantified by SRM in sera obtained from 32 healthy blood donors. Each boxplot represents the concentrations of one protein measured across individual samples.
N-Glycosite SRM Assays Enable Consistent Quantitative Assessment of Clinically Relevant Proteins in Blood Plasma Samples of Patient Cohorts
To evaluate the established N-glycosite SRM assays in a clinical setting, we designed a proof-of-concept study to assess previously reported quantitative differences of disease-associated proteins in a larger clinical cohort of blood plasma samples. The clinical cohort was composed of three malignancy groups, i.e. colorectal cancer, lung cancer, and pancreatic cancer, and a control blood donor group. Each group consisted of 30 cases (120 cases in total). For the evaluation, we selected 15 N-linked glycoproteins that were previously assayed in clinical studies and frequently studied in the context of a variety of malignancies (Table II). This different selection criterion compared with the experiment testing the sensitivity and dynamic range of our approach resulted in different sets of proteins. The selected proteins are of high clinical impact and include markers such as tissue inhibitor of metalloproteinase 1 (TIMP1), CD44 antigen, serum paraoxonase/arylesterase 1 (PON1), members of the extracellular matrix (fibronectin and vitronectin), or members of the serpin family of serine proteinase inhibitors.
Table II. Selected cancer-associated proteins.
The proteins were selected based on their association with different cancer types, and references to the associations are provided in the table. The table provides information about the protein concentration in plasma and the reported and the measured trend in fold change of the selected proteins comparing cancer versus normal conditions (↑ = higher protein abundance in cancer specimens and ↓ = lower protein abundance in cancer specimens).
| Gene name | Description | Plasma concentrations (μg/ml) (40, 48, 49) | Reported trend cancer versus normal | Measured trend by SRM cancer versus normal | Refs. |
|---|---|---|---|---|---|
| AHSG | α-2-HS-glycoprotein (Fetuin-A) | 80 | ↓ | ↓ | 50 |
| APOB | Apolipoprotein B-100 | 720 | ↑ | ↑ | 51–53 |
| CD44 | Extracellular matrix receptor III) (hyaluronate receptor) | 223 | ↑ | - | 54–57 |
| CLU | Clusterin | 56 | ↓↑ | ↓ | 50, 58–62 |
| CP | Ceruloplasmin | 210 | ↑ | ↑ | 63–65 |
| F11 | Coagulation factor XI | 5 | ↑ | 66 | |
| FN1 | Fibronectin | 110 | ↑ | ↓ | 67–69 |
| HP | Haptoglobin | 880 | ↑ | ↑ | 50, 65, 70 |
| PON1 | Serum paraoxonase/aryl esterase 1 | 89 | ↓ | ↓ | 58, 71, 72 |
| SERPINA1 | α-1-Antitrypsin | 1100 | ↑ | ↑ | 73–75 |
| SERPINA3 | α-1-Antichymotrypsin | 450 | ↑ | ↑ | 73, 75 |
| SERPINA6 | Corticosteroid-binding globulin | 70 | ↑ | 76 | |
| SERPINA7 | Thyroxine-binding globulin | 19 | ↑ | ↑ | 77 |
| TIMP1 | Tissue inhibitor of metalloproteinases 1 | 95 | ↑ | ↑ | 78–80 |
| VTN | Vitronectin | 115 | 81–83 |
We then multiplexed and used the corresponding N-glycosite SRM assays to measure and quantify these proteins in N-glycosite-enriched blood plasma of the 120 subjects and to determine their abundance trends in the three malignancies. The generated clinical dataset showed that we are able to consistently quantify the selected proteins in almost all samples and that it is of a large enough size to perform statistical testing with high sensitivity (Fig. 3A). Nine of the quantified proteins showed a higher abundance, and four proteins showed a lower abundance in at least one malignancy group when compared with the healthy control group (Fig. 3B and Table II). The observed quantitative trends were uniform across different cancers suggesting that the selected proteins are directly or indirectly modulated in a variety of cancer types. This is in agreement with previously reported abundance trends of these cancer-associated proteins (Table II) and supports the use of SRM assays and the SRM platform for accurate quantification of protein biomarkers across more than a hundred clinical samples.
Fig. 3.
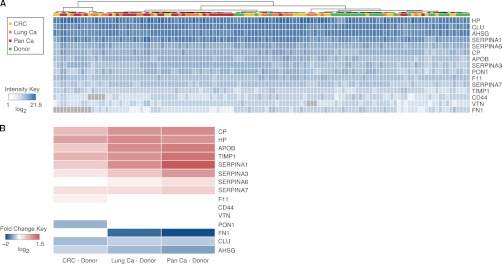
Quantitative abundance trends of cancer-associated proteins. A, 15 N-glycoproteins were quantified by SRM in plasma samples of 120 subjects. The clinical cohort was comprised of three malignancy groups (colorectal cancer (CRC), lung cancer (Lung Ca), and pancreatic cancer (Pan Ca), depicted in yellow, orange, and red, respectively) and a control donor group (depicted in green). Unsupervised clustering was applied on the quantitative data matrix. Higher abundance is depicted with higher intensity of blue, and gray color indicates missing values in the dataset. B, significant abundance changes between the distinct type of cancer (colorectal, lung, or pancreatic) and the controls (healthy blood donors) are depicted in red (as an increase in abundance in cancer) or in blue (as a decrease in abundance in cancer). White indicates no significant difference (at a cutoff of p ≤ 0.01, fold change ± 1.1).
Together, our results demonstrate the use of N-glycosite SRM assays for the parallel quantification of a set of clinical markers that are commonly assayed by immunoassays and provide a large collection of N-glycosite SRM assays for the discovery of novel markers for which assays were previously unavailable.
DISCUSSION
Despite the generation of extensive lists of biomarker candidates by large scale discovery-driven proteomic, transcriptomic, and genomic efforts in the last decade, almost none of the proposed biomarkers have been translated into the clinic so far. This is, to a large extent, due to the missing technological platform that allows the systematic evaluation of the clinical value of potential biomarkers in large patient cohorts in a multiplexed, consistent, and sensitive fashion. Here, we describe a platform based on SRM technology and provide an extensive collection of SRM assays that enable fast and parallel validation of biomarker candidates. The collection is focused on N-glycoproteins, because most of the currently used biomarkers are glycoproteins, and contains high quality and publicly accessible SRM assays for 2007 human and 1353 murine N-glycoproteins. The assays that were generated on three widely used mass spectrometric platforms can be queried and downloaded via the SRMAtlas webpage (25). These instrument-specific SRM assays account for potential variation in the fragmentation pattern of peptides measured on different instrument platforms, thereby providing high quality assays for all instrument platforms currently employed in targeted MS-based proteomics. This resource covers a large number of reported biomarker candidates and proteins involved in disease-related processes, which could not yet be tested for statistical significance across clinical specimens of larger cohorts, mainly due to the lack of a suitable biomarker validation strategy. The selection of N-glycosites for the SRMAtlas was mainly guided by available empirical evidence for the detectability of the peptides by MS. However, the automated high throughput approach applied for the SRM assay generation allows for fast and cost-efficient expansion of the resource to accommodate for whole human and murine N-glycoproteome.
The SRM assay coordinates for the SRMAtlas were extracted from fragment ion spectra derived from mixtures of synthetic peptides in equimolar amounts. In contrast to the extraction of the coordinates from the MS data of complex protein samples, the synthetic peptide approach is advantageous because it leads to the acquisition of high quality spectra for each peptide without sample-specific interferences and therefore ensures a high quality SRM assay. The N-glycoprotein SRMAtlas does not provide information about the LOQs and linear quantification ranges for the N-glycosites. These properties are dependent on sample preparation and instrument platform and should therefore be determined locally, preferentially using isotope-labeled internal standards before the validation of the proteins in large cohorts of clinical specimens.
To evaluate the performance of the established resource, we used a subset of SRM assays for the quantification of 48 N-glycoproteins across 32 blood serum samples. The glycoproteins were selected based on previous knowledge of their typical concentrations in human serum to examine the dynamic concentration range, which can be covered using the proposed SRM-based strategy. The results show that the combined approach of N-glycosite enrichment and the highly sensitive SRM measurements allow for consistent and parallel quantification of the selected proteins across all samples down to low nanogram/ml protein concentrations in serum. In a previous study, we developed a resource of SRM assays for cancer-associated proteins, which we screened in body fluids (20). The detectability of the cancer-associated proteins in plasma using SRM was limited to proteins with a higher or medium abundance in plasma (20). This result showed that fractionation or enrichment of proteins in body fluids prior to the SRM measurement is inevitable to detect and quantify the low abundance tissue leakage products, which are of high interest for biomarker research. The N-glycoprotein SRMAtlas provides SRM assays that in combination with the prior enrichment step overcome the limited sensitivity in detecting low abundance proteins in body fluids, allows the quantification of clinically relevant proteins, and therefore fulfills the sensitivity requirements for biomarker validation.
To demonstrate a direct application of the assays in a clinical setting, we selected a large cohort of 120 human plasma samples to evaluate protein abundance alterations in different cancer types. Targeted quantification of 15 previously reported cancer-associated N-glycoproteins in three malignancy groups and one control group substantially reproduced, in a single consistent measurement, the trends in abundance previously described in multiple studies. The obtained results demonstrate that the simultaneous profiling of biomarker candidates in a particular disease setting can be accomplished within complex clinical samples by using the SRM technology and the N-glycosite assays derived from the SRMAtlas. The previously reported cancer-associated proteins that have been studied in plasma comprise to a large extent plasma proteins of higher abundance underlining the need of new technologies, such as the one suggested in this study, that allow the consistent quantification of low abundance proteins in plasma for validating their clinical value systematically in large and well designed sample cohorts.
In conclusion, it is important to reiterate that the proposed strategy and resource enables the interrogation of “The roads less traveled” (45). To date, biomarker validation studies were limited by the availability of antibodies, which in turn implies that the existing quantitative assays were only representing a small minority of human proteins. This resulted in a bias for proteins being analyzed, for which assays were readily available and for which extensive knowledge has been accumulated (45). Therefore, we believe that the resource presented here is a turning point in plasma biomarker research (Fig. 4), because it represents a large collection of assays that can be used for an unbiased analysis of proteins that were previously not addressed. The N-glycoprotein SRMAtlas holds the promise to accelerate the systematic evaluation of biomarker candidates in a cost and time efficient screening mode across large cohorts of patient specimens. We expect that the suggested approach will close the gap between proposed biomarkers and clinical usage due to the possibility of a multiplexed hypothesis testing in larger clinical cohorts without requiring antibody development (Fig. 4). Currently, the SRM technology allows for parallel quantification of around 100 peptides, including their isotope-labeled counterparts in one measurement at high sensitivity. However, newly developed MS strategies based on data-independent acquisition and targeted extraction of peptide signals (46, 47) hold the promise of measuring all N-glycosites present in a sample in one MS run with only a 2–3-fold reduced LOQ compared with SRM (47). The N-glycoprotein SRMAtlas is also compatible with this newly developed MS acquisition mode. Therefore, we expect that biomarkers with clinical value will emerge from the widespread application of this unique collection of quantitative assays, and we envisage that the new bottleneck for the next phase in the biomarker pipeline (i.e. clinical validation) is going to be the availability of well annotated clinical cohorts of suitable quality and scale.
Fig. 4.

Impact of the N-glycoprotein SRMAtlas on the biomarker pipeline. The general biomarker pipeline consists of the generation of a candidate list and a lengthy and expensive development time of antibody-based assays for the validation of the candidates in clinical specimen like blood plasma. The N-glycoprotein SRMAtlas now accelerates this validation phase, because the assays for candidate quantification are publicly available. Additionally, in comparison with the gold-standard ELISA, the SRM technology facilitates multiplexed measurements of any set of candidates.
Supplementary Material
Acknowledgments
We acknowledge all contributors of the experimental N-glycosites. We thank Thomas Bock for help with MS measurements, Olga Schubert for helpful discussions, and Lucia Espona Pernas and Meena Choi for computational support. Author Contributions: R.H., S.S., R.O., R.L.M., B.W., and R.A. designed experiments; R.H., S.S., R.O., F.C., D.F.B., and U.K. performed the MS measurements for the SRMAtlas generation; R.S. performed the measurements in healthy blood donors; M.H. provided the patient plasma samples; S.S. performed the measurements in the patient plasma cohort; R.H., S.S., Z.S., and D.C. analyzed data; G.R. developed the script for the retention time extraction; D.C. and R.L.M. developed the platform for the accessibility of the data; J.C. selected the N-glycosites from UniProt; O.R. generated the methods for the MS measurements; R.H., S.S., B.W., and R.A. wrote the paper.
Footnotes
* This work was supported, in whole or in part, by American Recovery and Reinvestment Act funds through National Institutes of Health Grant R01 HG005805 (to R.L.M. and to Z.S., D.C., and U.K.), the EDRN program of the NCI (to R.A.), and in part by NIGMS Grant 2P50 GM076547 from Center for Systems Biology. This work was also supported by American Recovery and Reinvestment Act (ARRA) funds through grant number R01 HG005805 (to R.L.M.) the National Human Genome Research Institute; funding from the National Center of Competence in Research Neural Plasticity and Repair (to B.W.); the Swiss National Science Foundation Grant 31003A_135805 (to B.W.); SystemsX.ch/InfectX and SystemsX.ch/BIP (to B.W.); the Swiss National Science Foundation Grant 3100A0-107679 (to R.A.); the PRIME-XS Project funded by the European Union 7th Framework Programme Grant 262067 (to R.A.), the European Research Council Grant ERC-2008-AdG 233226 (to R.A.), funding from the Luxembourg Centre for Systems Biomedicine and the University of Luxembourg, from the National Science Foundation MRI Grant 0923536. U.K. was supported by a fellowship from the German Academic Exchange Service. Conflict of Interest: R.S. is CEO at ProteoMediX AG; O.R. is CSO at Biognosys AG, and R.O. is an employee of Biognosys AG. The other authors declare that they have no competing interests.
 This article contains Supplemental material.
This article contains Supplemental material.
1 The abbreviations used are:
- iRT
- indexed retention time
- LOQ
- limit of quantification
- RT
- retention time
- SRM
- selected reaction monitoring.
REFERENCES
- 1. Schröder F. H., Hugosson J., Roobol M. J., Tammela T. L., Ciatto S., Nelen V., Kwiatkowski M., Lujan M., Lilja H., Zappa M., Denis L. J., Recker F., Berenguer A., Määttänen L., Bangma C. H., Aus G., Villers A., Rebillard X., van der Kwast T., Blijenberg B. G., Moss S. M., de Koning H. J., Auvinen A., and ERSPC Investigators (2009) Screening and prostate-cancer mortality in a randomized European study. N. Engl. J. Med. 360, 1320–1328 [DOI] [PubMed] [Google Scholar]
- 2. Karapetis C. S., Khambata-Ford S., Jonker D. J., O'Callaghan C. J., Tu D., Tebbutt N. C., Simes R. J., Chalchal H., Shapiro J. D., Robitaille S., Price T. J., Shepherd L., Au H. J., Langer C., Moore M. J., Zalcberg J. R. (2008) K-ras mutations and benefit from cetuximab in advanced colorectal cancer. N. Engl. J. Med. 359, 1757–1765 [DOI] [PubMed] [Google Scholar]
- 3. Duffy M. J., van Dalen A., Haglund C., Hansson L., Holinski-Feder E., Klapdor R., Lamerz R., Peltomaki P., Sturgeon C., Topolcan O. (2007) Tumour markers in colorectal cancer: European Group on Tumour Markers (EGTM) guidelines for clinical use. Eur. J. Cancer 43, 1348–1360 [DOI] [PubMed] [Google Scholar]
- 4. Sjöblom T., Jones S., Wood L. D., Parsons D. W., Lin J., Barber T. D., Mandelker D., Leary R. J., Ptak J., Silliman N., Szabo S., Buckhaults P., Farrell C., Meeh P., Markowitz S. D., Willis J., Dawson D., Willson J. K., Gazdar A. F., Hartigan J., Wu L., Liu C., Parmigiani G., Park B. H., Bachman K. E., Papadopoulos N., Vogelstein B., Kinzler K. W., Velculescu V. E. (2006) The consensus coding sequences of human breast and colorectal cancers. Science 314, 268–274 [DOI] [PubMed] [Google Scholar]
- 5. Wood L. D., Parsons D. W., Jones S., Lin J., Sjöblom T., Leary R. J., Shen D., Boca S. M., Barber T., Ptak J., Silliman N., Szabo S., Dezso Z., Ustyanksky V., Nikolskaya T., Nikolsky Y., Karchin R., Wilson P. A., Kaminker J. S., Zhang Z., Croshaw R., Willis J., Dawson D., Shipitsin M., Willson J. K., Sukumar S., Polyak K., Park B. H., Pethiyagoda C. L., Pant P. V., Ballinger D. G., Sparks A. B., Hartigan J., Smith D. R., Suh E., Papadopoulos N., Buckhaults P., Markowitz S. D., Parmigiani G., Kinzler K. W., Velculescu V. E., Vogelstein B. (2007) The genomic landscapes of human breast and colorectal cancers. Science 318, 1108–1113 [DOI] [PubMed] [Google Scholar]
- 6. Berger M. F., Lawrence M. S., Demichelis F., Drier Y., Cibulskis K., Sivachenko A. Y., Sboner A., Esgueva R., Pflueger D., Sougnez C., Onofrio R., Carter S. L., Park K., Habegger L., Ambrogio L., Fennell T., Parkin M., Saksena G., Voet D., Ramos A. H., Pugh T. J., Wilkinson J., Fisher S., Winckler W., Mahan S., Ardlie K., Baldwin J., Simons J. W., Kitabayashi N., MacDonald T. Y., Kantoff P. W., Chin L., Gabriel S. B., Gerstein M. B., Golub T. R., Meyerson M., Tewari A., Lander E. S., Getz G., Rubin M. A., Garraway L. A. (2011) The genomic complexity of primary human prostate cancer. Nature 470, 214–220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Beroukhim R., Mermel C. H., Porter D., Wei G., Raychaudhuri S., Donovan J., Barretina J., Boehm J. S., Dobson J., Urashima M., Mc Henry K. T., Pinchback R. M., Ligon A. H., Cho Y. J., Haery L., Greulich H., Reich M., Winckler W., Lawrence M. S., Weir B. A., Tanaka K. E., Chiang D. Y., Bass A. J., Loo A., Hoffman C., Prensner J., Liefeld T., Gao Q., Yecies D., Signoretti S., Maher E., Kaye F. J., Sasaki H., Tepper J. E., Fletcher J. A., Tabernero J., Baselga J., Tsao M. S., Demichelis F., Rubin M. A., Janne P. A., Daly M. J., Nucera C., Levine R. L., Ebert B. L., Gabriel S., Rustgi A. K., Antonescu C. R., Ladanyi M., Letai A., Garraway L. A., Loda M., Beer D. G., True L. D., Okamoto A., Pomeroy S. L., Singer S., Golub T. R., Lander E. S., Getz G., Sellers W. R., Meyerson M. (2010) The landscape of somatic copy-number alteration across human cancers. Nature 463, 899–905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Cancer Genome Atlas Research Network (2011) Integrated genomic analyses of ovarian carcinoma. Nature 474, 609–615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Cancer Genome Atlas Network (2012) Comprehensive molecular characterization of human colon and rectal cancer. Nature 487, 330–337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Surinova S., Schiess R., Hüttenhain R., Cerciello F., Wollscheid B., Aebersold R. (2011) On the development of plasma protein biomarkers. J. Proteome Res. 10, 5–16 [DOI] [PubMed] [Google Scholar]
- 11. Whiteaker J. R., Lin C., Kennedy J., Hou L., Trute M., Sokal I., Yan P., Schoenherr R. M., Zhao L., Voytovich U. J., Kelly-Spratt K. S., Krasnoselsky A., Gafken P. R., Hogan J. M., Jones L. A., Wang P., Amon L., Chodosh L. A., Nelson P. S., McIntosh M. W., Kemp C. J., Paulovich A. G. (2011) A targeted proteomics-based pipeline for verification of biomarkers in plasma. Nat. Biotechnol. 29, 625–634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hüttenhain R., Malmström J., Picotti P., Aebersold R. (2009) Perspectives of targeted mass spectrometry for protein biomarker verification. Curr. Opin. Chem. Biol. 13, 518–525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Micheel G. M., Nass S. J., Omenn G. S., eds. (2012) Evolution of Translational Omics: Lessons Learned and the Path Forward, The National Academies Press, Washington D.C. [PubMed] [Google Scholar]
- 14. Anderson N. L., Anderson N. G. (2002) The human plasma proteome: history, character, and diagnostic prospects. Mol. Cell. Proteomics 1, 845–867 [DOI] [PubMed] [Google Scholar]
- 15. Addona T. A., Abbatiello S. E., Schilling B., Skates S. J., Mani D. R., Bunk D. M., Spiegelman C. H., Zimmerman L. J., Ham A. J., Keshishian H., Hall S. C., Allen S., Blackman R. K., Borchers C. H., Buck C., Cardasis H. L., Cusack M. P., Dodder N. G., Gibson B. W., Held J. M., Hiltke T., Jackson A., Johansen E. B., Kinsinger C. R., Li J., Mesri M., Neubert T. A., Niles R. K., Pulsipher T. C., Ransohoff D., Rodriguez H., Rudnick P. A., Smith D., Tabb D. L., Tegeler T. J., Variyath A. M., Vega-Montoto L. J., Wahlander A., Waldemarson S., Wang M., Whiteaker J. R., Zhao L., Anderson N. L., Fisher S. J., Liebler D. C., Paulovich A. G., Regnier F. E., Tempst P., Carr S. A. (2009) Multisite assessment of the precision and reproducibility of multiple reaction monitoring-based measurements of proteins in plasma. Nat. Biotechnol. 27, 633–641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Addona T. A., Shi X., Keshishian H., Mani D. R., Burgess M., Gillette M. A., Clauser K. R., Shen D., Lewis G. D., Farrell L. A., Fifer M. A., Sabatine M. S., Gerszten R. E., Carr S. A. (2011) A pipeline that integrates the discovery and verification of plasma protein biomarkers reveals candidate markers for cardiovascular disease. Nat. Biotechnol. 29, 635–643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Cima I., Schiess R., Wild P., Kaelin M., Schüffler P., Lange V., Picotti P., Ossola R., Templeton A., Schubert O., Fuchs T., Leippold T., Wyler S., Zehetner J., Jochum W., Buhmann J., Cerny T., Moch H., Gillessen S., Aebersold R., Krek W. (2011) Cancer genetics-guided discovery of serum biomarker signatures for diagnosis and prognosis of prostate cancer. Proc. Natl. Acad. Sci. U.S.A. 108, 3342–3347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Anderson L., Hunter C. L. (2006) Quantitative mass spectrometric multiple reaction monitoring assays for major plasma proteins. Mol. Cell. Proteomics 5, 573–588 [DOI] [PubMed] [Google Scholar]
- 19. Domanski D., Percy A. J., Yang J., Chambers A. G., Hill J. S., Freue G. V., Borchers C. H. (2012) MRM-based multiplexed quantitation of 67 putative cardiovascular disease biomarkers in human plasma. Proteomics 12, 1222–1243 [DOI] [PubMed] [Google Scholar]
- 20. Hüttenhain R., Soste M., Selevsek N., Röst H., Sethi A., Carapito C., Farrah T., Deutsch E. W., Kusebauch U., Moritz R. L., Nimèus-Malmström E., Rinner O., Aebersold R. (2012) Reproducible quantification of cancer-associated proteins in body fluids using targeted proteomics. Sci. Transl. Med. 4, 142ra194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Picotti P., Bodenmiller B., Mueller L. N., Domon B., Aebersold R. (2009) Full dynamic range proteome analysis of S. cerevisiae by targeted proteomics. Cell 138, 795–806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Keshishian H., Addona T., Burgess M., Kuhn E., Carr S. A. (2007) Quantitative, multiplexed assays for low abundance proteins in plasma by targeted mass spectrometry and stable isotope dilution. Mol. Cell. Proteomics 6, 2212–2229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Stahl-Zeng J., Lange V., Ossola R., Eckhardt K., Krek W., Aebersold R., Domon B. (2007) High sensitivity detection of plasma proteins by multiple reaction monitoring of N-glycosites. Mol. Cell. Proteomics 6, 1809–1817 [DOI] [PubMed] [Google Scholar]
- 24. Lange V., Picotti P., Domon B., Aebersold R. (2008) Selected reaction monitoring for quantitative proteomics: a tutorial. Mol. Syst. Biol. 4, 222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Picotti P., Lam H., Campbell D., Deutsch E. W., Mirzaei H., Ranish J., Domon B., Aebersold R. (2008) A database of mass spectrometric assays for the yeast proteome. Nat. Methods 5, 913–914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Roth J. (2002) Protein N-glycosylation along the secretory pathway: relationship to organelle topography and function, protein quality control, and cell interactions. Chem. Rev. 102, 285–303 [DOI] [PubMed] [Google Scholar]
- 27. Wollscheid B., Bausch-Fluck D., Henderson C., O'Brien R., Bibel M., Schiess R., Aebersold R., Watts J. D. (2009) Mass spectrometric identification and relative quantification of N-linked cell surface glycoproteins. Nat. Biotechnol. 27, 378–386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Schiess R., Wollscheid B., Aebersold R. (2009) Targeted proteomic strategy for clinical biomarker discovery. Mol. Oncol. 3, 33–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Picotti P., Rinner O., Stallmach R., Dautel F., Farrah T., Domon B., Wenschuh H., Aebersold R. (2010) High-throughput generation of selected reaction-monitoring assays for proteins and proteomes. Nat. Methods 7, 43–46 [DOI] [PubMed] [Google Scholar]
- 30. Keller A., Nesvizhskii A. I., Kolker E., Aebersold R. (2002) Empirical statistical model to estimate the accuracy of peptide identifications made by MS/MS and database search. Anal. Chem. 74, 5383–5392 [DOI] [PubMed] [Google Scholar]
- 31. Frank R. (2002) The SPOT-synthesis technique. Synthetic peptide arrays on membrane supports–principles and applications. J. Immunol. Methods 267, 13–26 [DOI] [PubMed] [Google Scholar]
- 32. Wenschuh H., Volkmer-Engert R., Schmidt M., Schulz M., Schneider-Mergener J., Reineke U. (2000) Coherent membrane supports for parallel microsynthesis and screening of bioactive peptides. Biopolymers 55, 188–206 [DOI] [PubMed] [Google Scholar]
- 33. Escher C., Reiter L., MacLean B., Ossola R., Herzog F., Chilton J., MacCoss M. J., Rinner O. (2012) Using iRT, a normalized retention time for more targeted measurement of peptides. Proteomics 12, 1111–1121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Maere S., Heymans K., Kuiper M. (2005) BiNGO: a Cytoscape plugin to assess overrepresentation of gene ontology categories in biological networks. Bioinformatics 21, 3448–3449 [DOI] [PubMed] [Google Scholar]
- 35. Shannon P. (2003) Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498–2504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Chang C.-Y., Picotti P., Hüttenhain R., Heinzelmann-Schwarz V., Jovanovic M., Aebersold R., Vitek O. (2012) Protein significance analysis in selected reaction monitoring (SRM) measurements. Mol. Cell. Proteomics 11, M111.014662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Zhang H., Li X. J., Martin D. B., Aebersold R. (2003) Identification and quantification of N-linked glycoproteins using hydrazide chemistry, stable isotope labeling, and mass spectrometry. Nat. Biotechnol. 21, 660–666 [DOI] [PubMed] [Google Scholar]
- 38. Hofmann A., Gerrits B., Schmidt A., Bock T., Bausch-Fluck D., Aebersold R., Wollscheid B. (2010) Proteomic cell surface phenotyping of differentiating acute myeloid leukemia cells. Blood 116, e26–e34 [DOI] [PubMed] [Google Scholar]
- 39. Zhang H., Loriaux P., Eng J., Campbell D., Keller A., Moss P., Bonneau R., Zhang N., Zhou Y., Wollscheid B., Cooke K., Yi E. C., Lee H., Peskind E. R., Zhang J., Smith R. D., Aebersold R. (2006) UniPep–a database for human N-linked glycosites: a resource for biomarker discovery. Genome Biol. 7, R73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Polanski M., Anderson N. L. (2007) A list of candidate cancer biomarkers for targeted proteomics. Biomark. Insights 1, 1–48 [PMC free article] [PubMed] [Google Scholar]
- 41. Anderson N. L. (2010) The clinical plasma proteome: A survey of clinical assays for proteins in plasma and serum. Clin. Chem. 56, 177–185 [DOI] [PubMed] [Google Scholar]
- 42. de Graaf E. L., Altelaar A. F., van Breukelen B., Mohammed S., Heck A. J. (2011) Improving SRM assay development: a global comparison between triple quadrupole, ion trap, and higher energy CID peptide fragmentation spectra. J. Proteome Res. 10, 4334–4341 [DOI] [PubMed] [Google Scholar]
- 43. Hanahan D., Weinberg R. (2011) Hallmarks of cancer: The next generation. Cell 144, 646–674 [DOI] [PubMed] [Google Scholar]
- 44. Gerber S. A., Rush J., Stemman O., Kirschner M. W., Gygi S. (2003) Absolute quantification of proteins and phosphoproteins from cell lysates by tandem MS. Proc. Natl. Acad. Sci. U.S.A. 100, 6940–6945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Edwards A. M., Isserlin R., Bader G. D., Frye S. V., Willson T. M., Yu F. H. (2011) Too many roads not taken. Nature 470, 163–165 [DOI] [PubMed] [Google Scholar]
- 46. Gillet L. C., Navarro P., Tate S., Rost H., Selevsek N., Reiter L., Bonner R., Aebersold R. (2012) Targeted data extraction of the MS/MS spectra generated by data-independent acquisition: a new concept for consistent and accurate proteome analysis. Mol. Cell. Proteomics 11, O111 016717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Liu Y., Hüttenhain R., Surinova S., Gillet L. C., Mouritsen J., Brunner R., Navarro P., Aebersold R. (2013) Quantitative measurements of N-linked glycoproteins in human plasma by SWATH-MS. Proteomics pmic. 201200417 [DOI] [PubMed] [Google Scholar]
- 48. Haab B. B., Geierstanger B. H., Michailidis G., Vitzthum F., Forrester S., Okon R., Saviranta P., Brinker A., Sorette M., Perlee L., Suresh S., Drwal G., Adkins J. N., Omenn G. S. (2005) Immunoassay and antibody microarray analysis of the HUPO plasma proteome project reference specimens: systematic variation between sample types and calibration of mass spectrometry data. Proteomics 5, 3278–3291 [DOI] [PubMed] [Google Scholar]
- 49. Hortin G. L., Sviridov D., Anderson N. L. (2008) High-abundance polypeptides of the human plasma proteome comprising the top 4 logs of polypeptide abundance. Clin. Chem. 54, 1608–1616 [DOI] [PubMed] [Google Scholar]
- 50. Dowling P., Clarke C., Hennessy K., Torralbo-Lopez B., Ballot J., Crown J., Kiernan I., O'Byrne K. J., Kennedy M. J., Lynch V., Clynes M. (2012) Analysis of acute-phase proteins, AHSG, C3, CLI, HP, and SAA, reveals distinctive expression patterns associated with breast, colorectal, and lung cancer. Int. J. Cancer 131, 911–923 [DOI] [PubMed] [Google Scholar]
- 51. Reddavide R., Misciagna G., Caruso M. G., Notarnicola M., Armentano R., Caruso M. L., Pirrelli M., Valentini A. M. (2011) Tissue expression of glycated apolipoprotein B in colorectal adenoma and cancer. Anticancer Res. 31, 555–559 [PubMed] [Google Scholar]
- 52. Ashur-Fabian O., Har-Zahav A., Shaish A., Wiener Amram H., Margalit O., Weizer-Stern O., Dominissini D., Harats D., Amariglio N., Rechavi G. (2010) apoB and apobec1, two genes key to lipid metabolism, are transcriptionally regulated by p53. Cell Cycle 9, 3761–3770 [PubMed] [Google Scholar]
- 53. Okuyama S., Marusawa H., Matsumoto T., Ueda Y., Matsumoto Y., Endo Y., Takai A., Chiba T. (2012) Excessive activity of apolipoprotein B mRNA editing enzyme catalytic polypeptide 2 (APOBEC2) contributes to liver and lung tumorigenesis. Int. J. Cancer 130, 1294–1301 [DOI] [PubMed] [Google Scholar]
- 54. Masson D., Denis M. G., Denis M., Blanchard D., Loirat M. J., Cassagnau E., Lustenberger P. (1999) Soluble CD44: quantification and molecular repartition in plasma of patients with colorectal cancer. Br. J. Cancer 80, 1995–2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Penno M. B., August J. T., Baylin S. B., Mabry M., Linnoila R. I., Lee V. S., Croteau D., Yang X. L., Rosada C. (1994) Expression of CD44 in human lung tumors. Cancer Res. 54, 1381–1387 [PubMed] [Google Scholar]
- 56. Galizia G., Gemei M., Del Vecchio L., Zamboli A., Di Noto R., Mirabelli P., Salvatore F., Castellano P., Orditura M., De Vita F., Pinto M., Pignatelli C., Lieto E. (2012) Combined CD133/CD44 expression as a prognostic indicator of disease-free survival in patients with colorectal cancer. Arch. Surg. 147, 18–24 [DOI] [PubMed] [Google Scholar]
- 57. Bohn O. L., Nasir I., Brufsky A., Tseng G. C., Bhargava R., MacManus K., Chivukula M. (2009) Biomarker profile in breast carcinomas presenting with bone metastasis. Int. J. Clin. Exp. Pathol. 3, 139–146 [PMC free article] [PubMed] [Google Scholar]
- 58. Okano T., Kondo T., Kakisaka T., Fujii K., Yamada M., Kato H., Nishimura T., Gemma A., Kudoh S., Hirohashi S. (2006) Plasma proteomics of lung cancer by a linkage of multidimensional liquid chromatography and two-dimensional difference gel electrophoresis. Proteomics 6, 3938–3948 [DOI] [PubMed] [Google Scholar]
- 59. Penno M. A., Klingler-Hoffmann M., Brazzatti J. A., Boussioutas A., Putoczki T., Ernst M., Hoffmann P. (2012) 2D-DIGE analysis of sera from transgenic mouse models reveals novel candidate protein biomarkers for human gastric cancer. J. Proteomics 77, 40–58 [DOI] [PubMed] [Google Scholar]
- 60. Jin J., Kim J. M., Hur Y. S., Cho W. P., Lee K. Y., Ahn S. I., Hong K. C., Park I. S. (2012) Clinical significance of clusterin expression in pancreatic adenocarcinoma. World J. Surg. Oncol. 10, 146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Rodríguez-Piñeiro A. M., García-Lorenzo A., Blanco-Prieto S., Alvarez-Chaver P., Rodríguez-Berrocal F. J., Cadena M. P., Martínez-Zorzano V. S. (2012) Secreted clusterin in colon tumor cell models and its potential as diagnostic marker for colorectal cancer. Cancer Invest. 30, 72–78 [DOI] [PubMed] [Google Scholar]
- 62. Hassan M. K., Watari H., Han Y., Mitamura T., Hosaka M., Wang L., Tanaka S., Sakuragi N. (2011) Clusterin is a potential molecular predictor for ovarian cancer patient's survival: targeting clusterin improves response to paclitaxel. J. Exp. Clin. Cancer Res. 30, 113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Yildirim A., Meral M., Kaynar H., Polat H., Ucar E. Y. (2007) Relationship between serum levels of some acute-phase proteins and stage of disease and performance status in patients with lung cancer. Med. Sci. Monit. 13, CR195–CR200 [PubMed] [Google Scholar]
- 64. Hanas J. S., Hocker J. R., Cheung J. Y., Larabee J. L., Lerner M. R., Lightfoot S. A., Morgan D. L., Denson K. D., Prejeant K. C., Gusev Y., Smith B. J., Hanas R. J., Postier R. G., Brackett D. J. (2008) Biomarker identification in human pancreatic cancer sera. Pancreas 36, 61–69 [DOI] [PubMed] [Google Scholar]
- 65. Walker C., Gray B. N. (1983) Acute-phase reactant proteins and carcinoembryonic antigen in cancer of the colon and rectum. Cancer 52, 150–154 [DOI] [PubMed] [Google Scholar]
- 66. Luque-García J. L., Martínez-Torrecuadrada J. L., Epifano C., Cañamero M., Babel I., Casal J. I. (2010) Differential protein expression on the cell surface of colorectal cancer cells associated to tumor metastasis. Proteomics 10, 940–952 [DOI] [PubMed] [Google Scholar]
- 67. Steffens S., Schrader A. J., Vetter G., Eggers H., Blasig H., Becker J., Kuczyk M. A., Serth J. (2012) Fibronectin 1 protein expression in clear cell renal cell carcinoma. Oncol. Lett. 3, 787–790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Ritzenthaler J. D., Han S., Roman J. (2008) Stimulation of lung carcinoma cell growth by fibronectin-integrin signalling. Mol. Biosyst. 4, 1160–1169 [DOI] [PubMed] [Google Scholar]
- 69. Saito N., Nishimura H., Kameoka S. (2008) Clinical significance of fibronectin expression in colorectal cancer. Mol. Med. Report. 1, 77–81 [PubMed] [Google Scholar]
- 70. Kim Y. W., Bae S. M., Kim I. W., Liu H. B., Bang H. J., Chaturvedi P. K., Battogtokh G., Lim H., Ahn W. S. (2012) Multiplexed bead-based immunoassay of four serum biomarkers for diagnosis of ovarian cancer. Oncol. Rep. 28, 585–591 [DOI] [PubMed] [Google Scholar]
- 71. Akcay M. N., Polat M. F., Yilmaz I., Akcay G. (2003) Serum paraoxonase levels in pancreatic cancer. Hepatogastroenterology 50, Suppl. 2, ccxxv–ccxxvii [PubMed] [Google Scholar]
- 72. Balci H., Genc H., Papila C., Can G., Papila B., Yanardag H., Uzun H. (2012) Serum lipid hydroperoxide levels and paraoxonase activity in patients with lung, breast, and colorectal cancer. J. Clin. Lab. Anal. 26, 155–160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Zelvyte I., Wallmark A., Piitulainen E., Westin U., Janciauskiene S. (2004) Increased plasma levels of serine proteinase inhibitors in lung cancer patients. Anticancer Res. 24, 241–247 [PubMed] [Google Scholar]
- 74. Orchekowski R., Hamelinck D., Li L., Gliwa E., vanBrocklin M., Marrero J. A., Vande Woude G. F., Feng Z., Brand R., Haab B. B. (2005) Antibody microarray profiling reveals individual and combined serum proteins associated with pancreatic cancer. Cancer Res. 65, 11193–11202 [DOI] [PubMed] [Google Scholar]
- 75. Kloth J. N., Gorter A., Fleuren G. J., Oosting J., Uljee S., ter Haar N., Dreef E. J., Kenter G. G., Jordanova E. S. (2008) Elevated expression of SerpinA1 and SerpinA3 in HLA-positive cervical carcinoma. J. Pathol. 215, 222–230 [DOI] [PubMed] [Google Scholar]
- 76. Wu J., Xie X., Liu Y., He J., Benitez R., Buckanovich R. J., Lubman D. M. (2012) Identification and confirmation of differentially expressed fucosylated glycoproteins in the serum of ovarian cancer patients using a lectin array and LC-MS/MS. J. Proteome Res. 11, 4541–4552 [DOI] [PubMed] [Google Scholar]
- 77. Kajita Y., Ishida M., Hachiya T., Miyazaki T., Yoshimura M., Ijichi H., Ochi Y. (1981) Clinical study on increased serum thyroxine-binding globulin in cancerous state. Endocrinol. Jpn. 28, 785–791 [DOI] [PubMed] [Google Scholar]
- 78. Pan S., Chen R., Crispin D. A., May D., Stevens T., McIntosh M. W., Bronner M. P., Ziogas A., Anton-Culver H., Brentnall T. A. (2011) Protein alterations associated with pancreatic cancer and chronic pancreatitis found in human plasma using global quantitative proteomics profiling. J. Proteome Res. 10, 2359–2376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Pan S., Chen R., Brand R. E., Hawley S., Tamura Y., Gafken P. R., Milless B. P., Goodlett D. R., Rush J., Brentnall T. A. (2012) Multiplex targeted proteomic assay for biomarker detection in plasma: a pancreatic cancer biomarker case study. J. Proteome Res. 11, 1937–1948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Holten-Andersen M. N., Christensen I. J., Nielsen H. J., Stephens R. W., Jensen V., Nielsen O. H., Sørensen S., Overgaard J., Lilja H., Harris A., Murphy G., Brünner N. (2002) Total levels of tissue inhibitor of metalloproteinases 1 in plasma yield high diagnostic sensitivity and specificity in patients with colon cancer. Clin. Cancer Res. 8, 156–164 [PubMed] [Google Scholar]
- 81. Ahn H. S., Shin Y. S., Park P. J., Kang K. N., Kim Y., Lee H. J., Yang H. K., Kim C. W. (2012) Serum biomarker panels for the diagnosis of gastric adenocarcinoma. Br. J. Cancer 106, 733–739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Ramani V. C., Haun R. S. (2008) Expression of kallikrein 7 diminishes pancreatic cancer cell adhesion to vitronectin and enhances urokinase-type plasminogen activator receptor shedding. Pancreas 37, 399–404 [DOI] [PubMed] [Google Scholar]
- 83. Kadowaki M., Sangai T., Nagashima T., Sakakibara M., Yoshitomi H., Takano S., Sogawa K., Umemura H., Fushimi K., Nakatani Y., Nomura F., Miyazaki M. (2011) Identification of vitronectin as a novel serum marker for early breast cancer detection using a new proteomic approach. J. Cancer Res. Clin. Oncol. 137, 1105–1115 [DOI] [PubMed] [Google Scholar]
- 84. UniProt Consortium (2008) The universal protein resource (UniProt). Nucleic Acids Res. 36, D190–D195 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.