

Abstract
Characterizing protein GalNAc-type O-glycosylation has long been a major challenge, and as a result, our understanding of this glycoproteome is particularly poor. Recently, we presented a novel strategy for high throughput identification of O-GalNAc glycosites using zinc finger nuclease gene-engineered “SimpleCell” lines producing homogeneous truncated O-glycosylation. Total lysates of cells were trypsinized and subjected to lectin affinity chromatography enrichment, followed by identification of GalNAc O-glycopeptides by nLC-MS/MS, with electron transfer dissociation employed to specify sites of O-glycosylation. Here, we demonstrate a substantial improvement in the SimpleCell strategy by including an additional stage of lectin affinity chromatography on secreted glycoproteins from culture media (secretome) and by incorporating pre-fractionation of affinity-enriched glycopeptides via IEF before nLC-MS/MS. We applied these improvements to three human SimpleCells studied previously, and each yielded a substantial increase in the number of O-glycoproteins and O-glycosites identified. We found that analysis of the secretome was an important independent factor for increasing identifications, suggesting that further substantial improvements can also be sought through analysis of subcellular organelle fractions. In addition, we uncovered a substantial nonoverlapping set of O-glycoproteins and O-glycosites using an alternative protease digestion (chymotrypsin). In total, the improvements led to identification of 259 glycoproteins, of which 152 (59%) were novel compared with our previous strategy using the same three cell lines. With respect to individual glycosites, we identified a total of 856 sites, of which 508 (59%) were novel compared with our previous strategy; this includes four new identifications of O-GalNAc attached to tyrosine. Furthermore, we uncovered ∼220 O-glycosites wherein the peptides were clearly identified, but the glycosites could not be unambiguously assigned to specific positions. The improved strategy should greatly facilitate high throughput characterization of the human GalNAc-type O-glycoproteome as well as be applicable to analysis of other O-glycoproteomes.
Post-translational modifications (PTMs)1 provide for a nearly limitless expansion of the structural space of all the proteins of an organism and, by extension, add manifold layers of potential complexity to the organism's interactome. Of all known PTMs, protein glycosylation is arguably the most abundant and certainly the most diverse, and genes involved in glycosylation occupy at a minimum 1% of mammalian genomes (1). Among the major classes of complex protein glycosylation, “mucin-type” (GalNAc-type) O-linked glycosylation, initiated by attachment of an α-GalNAc residue to Ser, Thr, or Tyr residues, is one of the most abundant and complex protein modification pathways. At the same time, this type of protein glycosylation is the least understood, especially with respect to sites of glycosylation and specific biological functions (2, 3). As a prime example, the possibility of attachment of GalNAc to Tyr residues has only recently been discovered (4, 5). Currently, site-specific O-glycosylation as a regulator of protein function is a dynamic area of investigation, with the most recently discovered function being co-regulation of proprotein convertase processing of proteins (6–9). A major challenge in the field today is to fully identify the GalNAc O-glycoproteome to uncover site-specific biological functions of this abundant PTM.
Compared with other types of protein glycosylation, GalNAc-type O-glycosylation is unique in that multiple (up to 20) distinct isoenzymes, the polypeptide GalNAc transferases, direct positions of O-glycan attachment to proteins (2), which provides for temporal and spatial regulation of the O-glycoproteome not found in other types of protein glycosylation. The GalNAc-type O-glycoproteome of a given cell is dependent on protein expression in general and in particular the expression of a subset of the 20 GalNAc-T isoenzymes that initiate O-glycosylation on proteins (2). Moreover, it appears that the subcellular topology of the O-glycosylation apparatus may be altered in cancer cells with partial relocation of GalNAc-Ts to the ER (10), which could affect the O-glycoproteome quantitatively and possibly even qualitatively by competition with ER-located O-mannosylation, for example (3). The O-glycoproteome is therefore likely cell-specific and, in addition, dynamic, as at least some GalNAc-T isoforms are regulated in response to external stimuli (6, 11).
With respect to analysis, LC-MS-based O-glycoproteomic studies are muddied by natural glycan structural heterogeneity, which complicates enrichment and chromatographic strategies; in addition, glycosylation in itself hinders the more commonly used ion trap/Orbitrap CID- or HCD-MS/MS peptide sequencing approaches, because in the former mode fragmentation is dominated by sugar losses, and in the latter mode abundant peptide sequence ions are observable, but usually at the cost of virtually complete elimination of the glycan and with it the information about where the glycan was located. Furthermore, even HCD-MS/MS begins to lose its usefulness when the number of saccharide residues exceeds ∼4–5 (whether in a single glycan or spread out among multiple sites), reverting again to domination by sugar losses and lack of peptide fragmentation at reasonable collision energies (5, 12). This situation is alleviated partly by the use of electron capture dissociation/ETD-MS/MS, which provides peptide sequence ions with substantial retention of the glycan, making its site known in a relatively uncomplicated way. However, even electron capture dissociation/ETD-MS/MS has its limitations, with heavy glycosylation and/or a lack of abundant precursors with required multiple charge states (e.g. +3, +4, and +5) being two typical reasons for failure to observe sufficient fragmentation for site-specific sequence analysis (5, 12, 13). The limitations can become acute when there are many adjacent Ser and Thr residues with potential for glycosylation and many Pro residues interrupting the continuity of c/z fragmentation, both situations being frequent in O-glycosylated peptides. A further problem occurs in mucins, many of which are resistant to proteolytic cleavage, yielding O-glycopeptides of prohibitively high molecular weight.
A key tool of glycoproteome analysis is selective enrichment of glycoproteins and/or glycopeptides to increase sensitivity. Two useful strategies have been developed that exploit properties of sialic acid as follows: (i) its affinity for titanium dioxide, as demonstrated by Larsen et al. (14), and (ii) its extreme sensitivity to oxidation and weak acid hydrolysis, thus enabling a reliable hydrazide bead capture and release protocol (15). The disadvantages of both these methods are that on the one hand they specifically require the presence of sialylated glycans, and on the other hand they are nonspecific for either N-linked or several of the O-linked types of glycans that can be sialylated. More general glycan oxidative methods have also been introduced (16, 17), but despite a longer history, these have so far been demonstrated in practice only for N-glycoproteomics. One alternative is to employ lectin weak affinity chromatography (LWAC) to the proteolytic digest (12, 18, 19), which can be used to considerable advantage where glycosylation has been simplified to a degree of homogeneity and the lectin is sufficiently specific to exclude unwanted glyco-types (5). LWAC can be performed in two stages, at both the O-glycoprotein and O-glycopeptide level (9, 20), with considerable advantage in overall enrichment.
Another challenge specific to the field of O-glycoproteomics is that there are no reliable consensus sequence motifs, which allow prediction of O-glycosites and O-glycoproteins. The most widely used prediction server, NetOGlyc 3.1 (21), identified only 38% of the sites identified in our first screen of O-glycosites in three SimpleCells (5). This is likely because the algorithm is trained on a selective set of known O-glycoproteins, but probably it is also due to the complex regulation of sites by up to 20 distinct GalNAc-T enzymes with different substrate specificities (2).
As suggested above, the major problems with heterogeneity in O-glycan structures can be effectively attenuated by artificially reducing the complexity of attached glycans. One approach utilized is the exo-glycosidase treatment prior to LC-MS/MS analysis (12, 13). We have recently introduced a genetic engineering approach to transform the entire GalNAc-type O-glycoproteome of a cell into homogeneous simple truncated O-glycans with GalNAc (Tn antigen) and/or NeuAcα2–6GalNAc (STn antigen) structures (5). This is achieved by zinc finger nuclease-mediated knockout of either the core-extending glycosyltransferase (T-synthase) or its essential chaperone (cosmc) (5). This so-called SimpleCell strategy, like the exo-glycosidase strategy, sacrifices knowledge about glycan complexity and site-specific glycan heterogeneity for a straightforward path to wide scale, high throughput access to the O-glycoproteome, determination of which proteins are O-glycosylated, and where the O-glycan sites are. Because of the need for efficient cloning and propagation of gene-targeted cells, SimpleCells are based on immortalized cancer cell lines derived from different organs. Whereas the use of cancer-derived cell lines carries some limitations as to the value of detailed analysis of specific O-glycoproteins and subtle changes in the O-glycoproteome in given cells, the strategy is highly useful to begin to assess the ultimate boundaries of the human O-glycoproteome. We envision that the SimpleCell O-glycoproteome strategy will serve as a foundation for more detailed studies of the glycosylation status of individual proteins in natural systems; in addition, it is anticipated that extending the library of potential O-glycosylation sites will eventually enable new strategies for high throughput structure-specific O-glycosite analysis. Because achieving these foundational goals will be well served by maximizing the discovery of previously unknown O-glycosites and novel O-glycoproteins, we have explored a number of modifications to our original analytical methodology (5) to increase O-glycoproteome coverage. These include the use of isoelectric focusing (IEF), which has been suggested as an effective orthogonal prefractionation method for peptides in conventional proteomics (22, 23), and of an alternate protease, a strategy offered as a way to increase coverage by cleaving at peptide residues not reached by trypsin or by leaving more intact stretches where trypsin leaves only short peptides (24–27).
Here, we demonstrate that orthogonal prefractionation IEF and alternative proteolysis with chymotrypsin, neither of which have been applied previously to mass O-glycoproteomics, are effective means for increasing the ultimate depth of our analytical strategy, thereby expanding the GalNAc-type O-glycoproteome. Furthermore, using two-stage Vicia villosa agglutinin (VVA) LWAC protocol (9), we demonstrate that analysis of the secretomes of SimpleCell lines provides a major source of new identifications. The results described here produced a dramatic expansion of the human O-glycoproteome using just three cell lines, more than doubling the total number of detected O-glycopeptides, -glycosites, and -glycoproteins obtained in our previous investigation (5), which provides a promising strategy for gaining more insight into the O-glycoproteome.
MATERIALS AND METHODS
Zinc Finger Nuclease Gene Targeting
The three SimpleCell lines, originally chosen to represent different human cellular origins, K562 (chronic myelogenous leukemia), COLO-205 (colonic adenocarcinoma), and Capan-1 (pancreatic adenocarcinoma) were generated by zinc finger nuclease targeting of cosmc and characterized as described previously (5).
Cell Culture and LWAC Isolation of Tn O-Glycopeptides from Total Cell Lysates
Cell culture and VVA LWAC isolation of O-glycopeptides were performed as described previously (5). In brief, the cells were cultured in 10% fetal bovine serum and seeded in two T175 (175 cm2) flasks; the media were changed the following day, and the cells were then cultured for 48–72 h to 90% confluence. Cell pellets were obtained by removing media and washing with PBS, followed by scraping of the cells in PBS. The media were reserved for processing as described below, and the cell pellets were lysed in RapiGest (Waters) and sonicated. The cleared lysate was reduced, alkylated with iodoacetamide, and then digested overnight with trypsin or chymotrypsin (20 μg), followed by a 2-h digest with additional (5 μg) protease. The digest was then treated with TFA, purified by C18 Sep-Pak chromatography (Waters), and desialylated with neuraminidase. Finally, the digest was fractionated on a 2.6-m long VVA-agarose column (Vector Laboratories) as described previously (5).
Two-stage LWAC Enrichment of O-Glycopeptides from Culture Medium
Isolation of secreted O-glycoproteins was carried out as described previously (9), with some additional details noted herein. In brief, a sample of SimpleCell culture supernatant (∼100 ml), reserved as noted above, was cleared by centrifugation and dialyzed. The dialysis retentate was neuraminidase-treated (10 units of Clostridium perfringens neuramidase type VI (Sigma)) and loaded onto a short column of 0.8 ml of VVA-agarose (Vector Laboratories) to enrich the glycoproteins prior to digestion. After a column wash, the glycoproteins were eluted two times with 2 ml of 0.2 m GalNAc (COLO-205 and Capan-1) or four times with 1 ml of 0.15 m GalNAc followed by two times with 1.5 ml of 0.4 m GalNAc (K562). The eluate was then dialyzed against ammonium bicarbonate and lyophilized. The lyophilized sample was re-dissolved in ammonium bicarbonate, reduced, alkylated, and proteolyzed, and the digest was subjected to LWAC on the long VVA column as described above for the cell lysate sample.
SDS-PAGE and Western Blot
Fractions from secretome purification were loaded and separated on NuPage BisTris 4–12% gels (Invitrogen). Electrophoresis was performed in MES running buffer at 200 V for 35 min. The gel was stained with InstantBlue (Invitrogen) for protein visualization or blotted onto nitrocellulose membranes. Membranes were blocked in 1% (w/v) polyvinylpyrrolidone in 50 mm Tris-HCl, pH 7.4, 150 mm NaCl for 1 h and stained with Helix pomatia lectin (1 μg/ml) in blocking buffer supplemented with 0.05% Tween 20 and 1 mm CaCl2/MgCl2/MnCl2/ZnCl2 overnight at 4 °C. The membrane was washed and incubated for 40 min with streptavidin-alkaline phosphatase conjugate (Dako, 1:4000 dilution) and washed and developed with 5-bromo-4-chloro-3-indolyl phosphate/nitro blue tetrazolium ready to use reagent (Kem-En-Tec).
Peptide Isoelectric Focusing
For a preliminary evaluation of the potential advantage of using IEF (22, 23) for O-glycopeptide fractionation, tryptic digests of cell lysates from the Capan-1 SimpleCell line were subjected to VVA-LWAC enrichment, typically collecting 6–8 sequential fractions; after desalting by SepPak cartridges (Waters), LWAC fractions were screened by preliminary LC-MS of an aliquot of each for glycopeptide content (see below), and those most enriched in glycopeptides were pooled together, dried by vacuum centrifugation, reconstituted in IPG rehydration buffer, and submitted to IEF fractionation. Isoelectric focusing was performed by a 3100 OFFGEL fractionator (Agilent) using OFFGEL low resolution kit, pH 3–10 (Agilent), according to the manufacturer's instructions. Typically, 12 fractions were collected and desalted by StageTips (Empore 3 M) and submitted to LC-MS and HCD/ETD-MS/MS as described below.
Liquid Chromatography-Mass Spectrometry
O-Glycopeptide-enriched samples were analyzed using a system composed of an EASY-nLC II (Thermo Fisher Scientific) interfaced via a nanospray Flex ion source to an LTQ-Orbitrap XL hybrid spectrometer (Thermo Fisher Scientific), equipped for both HCD- and ETD-MS2 modes, enabling peptide sequence analysis without and with retention of glycan site-specific fragments, respectively.
In general each sample was split and analyzed in two separate runs (see “Results”). The conditions of LC analysis were essentially as described previously (5), except that the nLC was operated in a single analytical column set up using PicoFrit Emitters (New Objectives, 75 μm inner diameter) packed in-house with Reprosil-Pure-AQ C18 phase (Dr. Maisch, 3-μm particle size, ∼19-cm column length), with the flow rate lowered to 200 nl min−1 to compensate for the increased column length compared with the previous setup. In addition, the water/acetonitrile gradient elution program was altered somewhat; samples dissolved in 0.1% formic acid were injected onto the column and eluted in a gradient from 2 to 20% acetonitrile in either 65 or 95 min and from 20 to 80% acetonitrile in 10 min, followed by isocratic elution at 80% acetonitrile for 15 min (total elution time 90 or 120 min, respectively, chosen depending on complexity of the sample). The nanospray Flex ion source was operated at a spray voltage of 2.0 kV and heated capillary at 250 °C.
A data-dependent mass spectral acquisition routine, HCD triggering of subsequent ETD scan, was used for most runs. Briefly, a precursor MS1 scan (m/z 350–1700) of intact peptides was acquired in the Orbitrap at a nominal resolution setting of 30,000, followed by Orbitrap HCD-MS2 (m/z 100–2000, nominal resolution 15,000) of the three most abundant multiply charged precursors above 5000 counts in the MS1 spectrum (“top three” method). The appearance of a HexNAc fragment at m/z 204.086 (in practice a ± m/z 0.15 window was used) in the HCD-MS2 spectrum triggered a subsequent ETD-MS2 from the same precursor (m/z 100–2000, nominal resolution 15,000) (5). Activation times were 30 and 100–200 ms for HCD and ETD fragmentation, respectively; isolation width was 4 mass units, and usually 1 microscan was collected for each spectrum. Automatic gain control targets were 100,000 ions for Orbitrap MS1 and 10,000 for MS2 scans, and the automatic gain control for fluoranthene ion used for ETD was 300,000. Supplemental activation (20%) of the charge-reduced species was used in the ETD analysis to improve fragmentation. Dynamic exclusion for 30 s was used to prevent repeated analysis of the same components. Polysiloxane ion at m/z 445.12003 were used as a lock mass in all runs.
In cases where preliminary screening of fractions for glycopeptide enrichment was carried out prior to IEF, the triggered ETD-MS2 step was omitted, and HCD-MS2 (m/z 100–2,000) of the five most abundant multiply charged precursors was acquired (“top five method”). These HCD-MS2 spectra were simply screened for the appearance of the HexNAc fragment at m/z 204.086.
Data Analysis
The raw data were processed, in a manner similar to previous publications (5, 9), using Proteome Discoverer 1.2 software (PD 1.2; Thermo Fisher Scientific) and searched against the human-specific UniProt KB/SwissProt-reviewed database downloaded on July 8, 2010, containing 20,212 entries. In addition, another 205 common contaminants such as keratin, bovine serum albumin, and trypsin were included in the search. HCD and ETD data were searched using the SEQUEST node in PD 1.2 (algorithm version 28 build 60; node version 1.13); the ZCore node in PD 1.2 (algorithm version 1.0 build 60; node version 1.11) was also used for searching ETD data. In all cases, the precursor mass tolerance was set to 10 ppm and fragment ion mass tolerance to 50 millimass units. Carbamidomethylation on cysteine residues was used as a fixed modification; methionine oxidation and HexNAc attachment to serine and threonine were used as variable modifications; a maximum of 12 variable modifications were allowed per peptide. As a further pre-processing procedure, all HCD data showing the presence of fragment ions at m/z 204.08 were extracted into a single .mgf file, and the exact mass of 1–4× HexNAc units was subtracted from the corresponding precursor ion mass, generating four distinct files, each identified by a file name containing the number of HexNAc units subtracted. For this purpose, a script written in Microsoft Visual Basic 6.5 was used. Subtraction of more than 4× HexNAc units was not considered, because under the conditions of analysis, sugar losses so dominated the HCD spectra that peptide sequence-specific fragment ions were no longer produced in useful abundance. In these cases, it was often sufficient to obtain both peptide sequence and O-glycosite information from ETD-MS2 spectra alone. The pre-processed data files were submitted to a SEQUEST data base search under the same conditions mentioned above, again considering HexNAc attachment.
All spectra were separately searched against non-decoy and decoy databases to allow calculation of the false discovery rate. Spectra were grouped using a precursor tolerance of 15 ppm and a time window of 0.25 min. Afterward, a false discovery rate of 5% (high to-medium) was applied, and the resulting list was filtered to include only peptides with glycosylation as a modification. This resulted in a preliminary list of glycoproteins identified by at least one unique glycopeptide. Based on this preliminary list, all candidate-matched glycopeptides associated with each protein were validated by manual inspection. In cases where assigned ETD spectra were initially absent, a search of low confidence level assignments was used to find additional candidate spectra. In the next step, in addition to the highest confidence matches, we also considered glycopeptides with a low confidence level related to identified proteins from the preliminary list. The criteria used for acceptance versus rejection were as follows: (i) a rejection of HCD spectra not exhibiting the m/z 204/186 fragment pair; (ii) a good distribution of fragment ions over peptide sequence in HCD and ETD spectra; (iii) the clear presence of confirming ETD fragments on both sides of an assigned glycosylation site (allowing for interruption by adjacent Pro residues) or one or more fragments confirming site or clustered sites closest to the C or N terminus; (iv) the rejection of ETD site assignments based on one or more crucial fragments that have been assigned a charge state ≥2 in the absence of confirming isotope peaks or in obvious cases of charge state misassignment; and (v) consistent peptide sequence assignments between HCD- and ETD-MS2 spectra acquired from the identical precursor, where both are available.
In cases where the spectral data are sufficient to identify a definite peptide sequence bearing a specific number of HexNAc residues, but insufficient to yield some or all of their precise locations, we use the designation “ambiguous site assignment” (this covers site ambiguities on all or part of a peptide). In general, this situation occurred where a quality HCD-MS2 spectrum was acquired, but the ETD-MS2 was either missing or exhibited insufficient fragmentation to confirm all O-glycosite locations. An exception to this was made when the number of HexNAc residues detected was found to agree exactly with the number of potential O-glycosites in the sequence; then the sites were considered to be specified. It should be noted that neither SEQUEST nor ZCore provides any statistical measure of the reliability of site assignments.
To search efficiently for GalNAc O-glycosylation of tyrosine, a separate processing run on Proteome Discoverer was carried out on the data set with HexNAc allowed on Tyr in addition to Ser/Thr as one of the 12 variable modifications. High scoring hits were accepted after careful validation of ETD-MS2 spectra to make sure that sufficient fragments were clearly observable above noise to unambiguously define the sequence and HexNAc position. To identify cases where peptides from bovine proteins might contaminate the data set, a text string search of all proposed human peptide sequences was carried out against the bovine UniProt database (UniProtKB, reviewed, 6879 canonical sequences, September 2012 release), and any peptide sequences found to be 100% identical between the two species were put in a separate category (see under “Results”).
The overall workflow is summarized in Fig. 1. Lists of all identified proteins, peptide sequences, and detected O-glycosites are compiled in supplemental Tables 1 and 2.
Fig. 1.
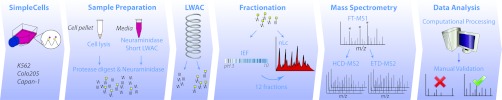
Outline of modified workflow. SimpleCell pellet is subjected to lysis, followed by protease and neuraminidase digestion; this is followed by O-glycopeptide enrichment by LWAC on a long column of immobilized VVA. In parallel, culture medium is treated with neuraminidase and then subjected to LWAC on a short column of VVA; this is followed by protease and neuraminidase digestion and LWAC enrichment on a long VVA column as for the cell lysate. After a pre-screening nLC-MS run to determine which LWAC fractions contained the most O-glycopeptides, these were divided into portions, with one part of each submitted directly to nLC-MS and the other parts pooled and submitted to IEF fractionation. IEF fractions (12) were then submitted to nLC-MS. Following computational processing of nLC-MS data as described, all spectral identifications were validated by inspection.
RESULTS
Isoelectric Focusing
The first question we sought to answer was whether the advantages of IEF separation (measured by increased identification of O-glycoprotein/glycosites) would sufficiently compensate for overall sample losses resulting from the extra fractionation and purification steps, which were estimated to be up to ∼40%. Each VVA-LWAC fraction from a Capan-1 SimpleCell lysate was divided into two equal parts, and each part was analyzed by two different protocols, one interpolating IEF before nLC-MS and the other submitting fractions directly to nLC-MS (workflow, Fig. 1). Comparison of the observed results revealed that despite the use of another step of fractionation and an additional round of purification, the incorporation of IEF fractionation gave an increase in identifications by a factor of 2 for proteins and more than 2.5 for glycosites (Fig. 2). Based on this result, IEF fractionation was used in all further analyses.
Fig. 2.

Venn diagrams showing uniqueness and overlap between data sets obtained using workflows incorporating LC/MS only versus IEF-LC/MS, with respect to number of O-glycoproteins (left) and O-glycosites (right) identified in equal portions of a Capan-1 SimpleCell lysate.
To further improve the yield of glycopeptides from IEF, we tested splitting each LWAC fraction into 10 and 90% portions, where 10% of each fraction was submitted to nLC-MS with a “top-five” HCD-MS2 protocol as a preliminary screening to determine which fractions had significant amounts of glycopeptides. This was accomplished by monitoring the appearance of the diagnostic oxonium fragment for HexNAc at m/z 204.08. Following this, the remaining 90% of the fractions most enriched for glycopeptides (typically 2 fractions of 1 ml each) was pooled together and fractionated via IEF, and the fractions from IEF were submitted to nLC-MS with the top three HCD/ETD-MS2 protocol. This “10–90” experimental scheme allowed elimination of fractions containing little or no glycopeptides from further processing, and it was applied to all subsequent analyses of the three SimpleCells as follows: Capan-1, COLO-205, and K562.
Secreted Glycoproteins
To employ normal cell growth media containing bovine serum, we developed a two-stage lectin affinity chromatography procedure that included an additional LWAC step to enrich for secreted O-glycoproteins in the experimental workflow already established (Fig. 1). The additional LWAC step not only facilitated enrichment of the glycoproteins but also removed the majority of highly abundant serum proteins, including BSA (Fig. 3A). For the K562 secretome, the short VVA column was more extensively eluted with GalNAc; however, subsequent Western blotting with Tn-binding lectin H. pomatia showed that the majority of glycoproteins were eluted within the first fraction (Fig. 3A). The concentrated glycoproteins were digested and fractionated with the long LWAC column by the same protocol as employed for cell lysates. This was followed by a preliminary nLC-MS screening, and the LWAC fractions exhibiting the most glycopeptides were combined into a single pool, further fractionated via IEF, and submitted to nLC-MS with the top three HCD/ETD-MS2 protocol as described above. By employing a comparison via text string search, a few cases were found where the human and bovine peptide sequences were 100% identical; these have been pointed out by a footnote in the supplemental Tables as potential false-positives, although the likelihood that the bovine sequences would also have truncated Tn type glycans as detected is rather low.
Fig. 3.

A, monitoring of fractions from K562 secretome preparation, visualized by Coomassie and lectin blot staining (left and right panels, respectively). Conditioned medium was dialyzed (lane 1) and neuraminidase-treated (lane 2). The medium was then filtered and diluted with LAC A buffer (lane 3) and passed twice over a short VVA column collecting a sample from the first (lane 4) and second flow-through (lane 5). The column was subsequently washed (lane 6) and eluted four times with 1 ml of 0.15 m GalNAc (lanes 7–10) and two times with 1.5 ml of 0.4 m GalNAc (lanes 11 and 12). B, Venn diagrams showing uniqueness and overlap between O-glycosite data sets obtained from secretomes versus cell lysates of the three SimpleCell lines studied, presented individually and as cumulative (nonredundant) totals.
Although we assumed some degree of overlap would be observed between total lysate and secretome analysis, the level of unique information, with respect to both glycoproteins and glycosites, found in the secretome exceeded our expectations. On average, the number of nonredundant identified glycosites in Capan-1 was distributed as 27% unique for the cell lysate, 34% unique for the secretome, and only 39% common for both (see Fig. 3B). In comparison, the overlap between two independent runs of Capan-1 cell lysate was 76%, with only 11 and 13% unique to each, respectively (as illustrated in supplemental Fig. 1). A similar picture was observed for COLO-205 (31% unique for cell lysate and 45% unique for secretome) (Fig. 3B) and K562 (32% unique for cell lysate and 37% for secretome) cell lines (Fig. 3B). We then asked whether the potential overlap between lysate and secretome would be different when comparing different cell lines; therefore, the combined O-glycosite data obtained from the secretomes of all three cell lines were compared with those data from the corresponding combined cell lysates (Fig. 3B). In this case, the degree of unique versus overlap was not substantially different, showing that the secretomes indeed added many novel glycosites not attributable simply to random statistics and run-to-run reproducibility. For example, lipolysis-stimulated lipoprotein receptor was newly identified in this study as a GalNAc-O-glycoprotein, and even though stimulated lipoprotein receptor was found in lysates of Capan-1 and COLO-205 as well as in secretomes of all three cell lines, the secretomes contributed to the identification of almost all the O-glycosites observed. In addition, peptides found exclusively in the secretomes of COLO-205 and K562 pointed to several new O-glycosites not previously identified in studies of amyloid β A4 precursor (APP) (see “Discussion”) (4, 28, 29).
In general, the Capan-1 secretome appeared to be the most significant contributor in this study; for example, many new sites were identified in the nontandem repeat region of MUC16, compared with our previous study, and a major portion of these were contributed by the Capan-1 secretome. A newly identified protein in this study, the proteoglycan versican (VCAN), was also found in Capan-1, and it was possible to identify twice as many O-glycosites on this protein in the secretome than the cell lysate. Capan-1 and COLO-205 contributed 10 new O-glycosites to the previously identified proteoglycan agrin (Fig. 4, A and B, depicting the HCD and ETD mass spectra identifying the doubly glycosylated peptide 1288APHPSHTSQPVAK1300 (peptides with the same sequence and 1–2 occupied sites were also detected)), but only three of these were identified in both cell lysates and secretomes, although all 10 were identified in the Capan-1 secretome.
Fig. 4.
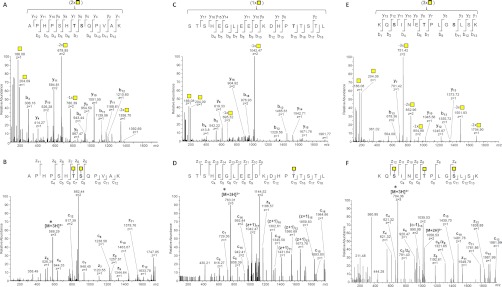
ESI-Orbitrap-MS2 spectra of selected O-glycopeptides identified in this study. HCD-MS2 (A) and ETD-MS2 (B) of O-glycopeptide from agrin (residues 1288–1300; precursor m/z 626.3097, z = 3+) data consistent only with glycosylation of Thr-1294 and Ser-1295; HCD-MS2 (C) and ETD-MS2 (D) of O-glycopeptide from CD44 (residues 514–532; precursor m/z 763.0129, z = 3+) data consistent only with glycosylation of Thr-528; HCD-MS2 (E) and ETD-MS2 (F) of O-glycopeptide from GalNAc-T5 (residues 290–303; precursor m/z 704.3594, z = 3+) data consistent only with glycosylation of Ser-292, Thr-296, and Ser-300. Precursors selected are indicated by an asterisk in the ETD-MS2 spectra. Deduced glycosylated residues are denoted by Consortium for Functional Glycomics standard yellow square in the peptide sequence heading each panel.
Chymotrypsin Versus Trypsin
Finally, we wished to probe whether use of another protease would substantially improve O-glycosite identification and especially if this would result in identification of more classical mucins with tandem repeat sequences largely devoid of Arg/Lys residues. For this purpose, proteins from total cell lysate of the Capan-1 SimpleCell line were digested by chymotrypsin instead of trypsin and enriched using our newly established workflow protocols, including IEF fractionation prior to LC-MS analysis for glycosite identification. Although we have compared a single run of chymotrypsin data containing 91 defined glycosites from 42 glycoproteins versus a triplicate set of Capan-1 data obtained using trypsin (2× cell lysate and 1× secretome) containing 538 glycosites from 134 glycoproteins, a minimal overlap of O-glycosites was observed between them, calculated as only 26% (see supplemental Fig. 2). On the glycoprotein level, the overlap was observed as 36%. Although the absolute numbers obtained from chymotrypsin digestion were approximately three times less than for trypsin, chymotrypsin not only allowed identification of more O-glycosites on already identified proteins, it also allowed characterization of 27 additional proteins as O-glycoproteins (supplemental Fig. 2). Even if one includes the data set from the other two cell lines (COLO-205 and K562, both cell lysate and secretome) the comparison still shows that the single analysis using chymotrypsin provides a substantial number of unique glycoproteins/glycosites (25/65 in this case) (data not shown).
Interestingly, the use of chymotrypsin did not in this case provide further identifications of sites in classical mucins besides the already identified MUC16 and made only a minor contribution of new O-glycosites in the nontandem repeat region of MUC16. However, it contributed eight peptides from CD44 bearing 20 O-glycosites, of which 13 were not found on any overlapping tryptic peptides from this or the previous study (see Fig. 4, C and D, showing HCD and ETD mass spectra identifying the singly glycosylated chymotryptic peptide 514STSHEGLEEDKDHPTTSTL532 from CD44; peptides with the same sequence and 2–4 occupied sites were also detected). In addition, chymotrypsin clearly contributed a heptaglycosylated mucin-like fragment uniquely identifying LAMP2 and two unique peptides contributing eight additional sites in the stem region of SDC1.
O-Glycosylation of Tyrosine
Because GalNAc O-glycosylation of tyrosine residues has been reported as a novel modification on glycoprotein APP (4) and NUCB2 (5), the possibility of this modification was routinely considered. To search for the Tyr modification efficiently, a separate processing run on Proteome Discoverer was carried out on the complete data set with HexNAc on Tyr included as one of the allowed variable modifications. This search, besides identifying the same O-glycopeptide from NUCB2, yielded four additional identifications. Interestingly, unlike the previously published sequences, all four contained one or more additional O-glycosylations of Ser and Thr residues. These formed a rather diverse set of sequences from CD44 antigen, nucleobindin 1 (NUCB1), extracellular matrix protein 1 (ECM1), and proline-rich acidic protein 1 (PRAP1). Such instances could easily be missed or misinterpreted in a search focused only on classical Ser/Thr sites, and in fact three out of four peptide sequences were already on the list of Ser/Thr-linked O-glycopeptides.
The CD44 case was particularly complex, observed as a 24-residue peptide 545RDPNHSEGSTTLLEGYTSHYPHTK568 with seven potential Ser/Thr sites but bearing eight HexNAc residues; nevertheless, sufficient spectral coverage was available to define the 8th site as one of the two available Tyr residues (Tyr-560), which is contiguous with two other occupied Ser/Thr residues (Fig. 5A). The NUCB1 peptide 32GAPNKEETPATESPDTGLYYHR53 also contains both glycosylated and unglycosylated Ser/Thr residues, as well as a pair of contiguous Tyr residues, of which one is clearly glycosylated (Tyr-50) (Fig. 5B). Fragmentation in the ETD-MS2 spectrum characterizing the ECM1 peptide 32QLRPEHFQEVGYAAPPSPPLSR53 is somewhat sparse, due at least in part to the densely Pro-rich region near the C terminus; nevertheless, there is sufficient fragmentation around Tyr-43 to show it is glycosylated (Fig. 5C), even though support for assignment of the second O-GalNAc to Ser-48 rather than Ser-52 is provided by only one weak c fragment. The PRAP1 peptide 102VLSPEPDHDSLYHPPPEEDQGEERPR127 was not identified in the first round of data processing, although it has two Ser residues, of which one is glycosylated (Ser-104); the ETD-MS2 spectrum for this peptide clearly shows glycosylation at Tyr-113 and not Ser-111 (Fig. 5D). The four corresponding HCD-MS2 spectra linked to these ETD-MS2 spectra are reproduced in supplemental Fig. 3, A–D, although they were not essential for characterizing the O-glycopeptides. Interestingly, a glycoform of the PRAP1 peptide with the occupied sites at Ser-104 and Ser-111, but not Tyr-113, was also observed in the same run but at a different retention time (supplemental Fig. 4, A and B). Also observed was a glycoform having only a single site at Ser-104, characterized by associated ETD and HCD spectra from the same precursor (supplemental Figs. 4C and 5, respectively). These data serve to strengthen the identification of the peptide, while at the same time exhibiting fragments clearly differentiating the glycosite assignments.
Fig. 5.
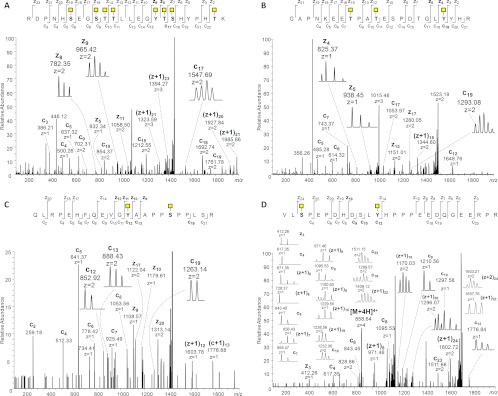
ESI-ETD-Orbitrap-MS2 spectra of selected tyrosine O-glycopeptides identified in this study. ETD-MS2 spectra of O-glycopeptides from CD44 (residues 545–568; precursor m/z 871.1893, z = 5+) are consistent with glycosylation of Tyr-560 along with Ser-653, Thr-654, Thr-655, Thr-661, Ser-662, Thr-667 (A); NUCB1 (residues 32–53; precursor m/z 761.3487, z = 4+) is consistent with glycosylation of Tyr-50 along with Thr-39 and Thr-42 (B); ECM1 (residues 32–53; precursor m/z 721.3652, z = 4+) is consistent with glycosylation of Tyr-43 along with Ser-48 (C); and PRAP1 (residues 102–127; precursor m/z 858.6449, z = 4+) is consistent with glycosylation of Tyr-113 along with Ser-104 (D).
Cumulative Data
Cumulative data collected from our previous study (5), triplicate runs of Capan-1 (two cell lysates and one secretome) from duplicate runs of COLO-205 (one cell lysate and one secretome), and K562 (one cell lysate and one secretome), all obtained by using trypsin as the proteolytic enzyme and from a single run of Capan-1 (1 cell lysate) using chymotrypsin for the digestion, are presented in supplemental Tables 1 and 2. In total, using these modified workflows incorporating additional protocols, we identified 259 O-glycoproteins, of which 107 were reported in our previous work (Fig. 6A) (5). In terms of O-glycosite identification, the work in this study contributed 508 newly defined and more than 220 new sites that are ambiguously defined within all or part of the peptide (Fig. 6B). Thus, by counting the new data along with the 115 unique and 233 common O-glycosites from our previous work (5), the total number of reported O-glycosites is more than 1000 (Fig. 6B), a number that is unprecedented in the field of GalNAc-O-glycoproteomics. Comparing our results with those compiled in the UniProt database, in which 242 human O-GalNAc sites are reported in toto, we found only 30 sites in common. Because mass spectrometry alone cannot distinguish between GalNAc and GlcNAc (for example), it has to be acknowledged that some of the sites detected could be of the O-GlcNAc type (30). Likely candidates are proteins with a clearly nuclear origin, and the six possible cases identified in our data set are pointed out by the footnote in the supplemental tables; however, we have established previously that the likelihood of a more significant proportion being of the O-GlcNAc type is low (see under “Discussion”) (5, 9).
Fig. 6.

Venn diagrams showing uniqueness and overlap between data sets obtained from three SimpleCell lines studied with respect to identified O-glycoproteins (A) and O-glycosites (B). Cell line totals are presented individually versus each other (top) and as cumulative totals compared with data obtained previously by Steentoft et al. (5) (bottom).
Site Occupancy
Histograms showing distribution of defined O-glycosites per protein and total O-glycosites (unambiguously plus ambiguously assigned) per peptide are plotted in Fig. 7, A and B, respectively. In addition to 31 and 15% of all identified proteins exhibiting 1 and 2 glycosites, respectively, we also observed a cluster of proteins with a high degree of glycosylation. Interestingly, although the high degree of glycosylation is expected for, e.g. the MUC16 protein, the detection of 28 glycosites for GalNAc-T5 (e.g. 290KQSINETPLGSLSK303, with three O-glycosites, Fig. 4, E and F), a member of the family of enzymes that initiate GalNAc glycosylation of proteins, was surprising. Previously (5), we have remarked on the characterization of new O-glycosites within glycoproteins bearing heavily O-glycosylated stretches of peptide, such as dystroglycan. Large O-glycoproteins that we have newly observed or greatly expanded O-glycosite coverage in this study include versican, agrin, and CD44, all being important multifunctional proteoglycans.
Fig. 7.

Analysis of cumulative results to date (this paper and Steentoft et al. (5)) with respect to O-glycosite distributions. Histograms show O-glycosite distributions by number of Tn identified per number of proteins (A) and per number of glycopeptides (B).
DISCUSSION
We previously developed the SimpleCell O-glycoproteomic strategy and implemented this in a pilot study on three human cancer cell lines (5). Although MS cannot be used to distinguish between isobaric/isomeric residues, as mentioned above, other criteria are used to establish that we are, with few exceptions, observing O-GalNAc rather than O-GlcNAc modification. These criteria include the following: the high specificity of VVA for GalNAc; the presence of signal peptide sequences and other ontological evidence showing that the vast majority of proteins identified have been processed through the secretory pathway; site locations in extracellular, as opposed to cytosolic, domains; O-glycoproteomic characterization of untransformed K562 cells showing many of the same O-glycosites as in the K562 SimpleCells but with Hex-O-HexNAc or other complex glycans, confirming that these are true Gal-O-GalNAc-type sites (5), not O-GlcNAc, which is not observed to be further modified (30). We have identified in these studies a small number of proteins of likely nuclear origin, and these can be expected to exhibit O-GlcNAcylation, and are listed as such in the supplemental tables.
We are currently in the process of expanding this strategy to a larger panel of human cell lines derived from the major organs to obtain a first view of the global GalNAc-type O-glycoproteome. Here, we reported on a series of improvements to the original analytical workflow, which all provided a substantial increase in identification of O-glycoproteins and glycosites. We tested and implemented these strategies on the same three human SimpleCell lines used previously (5), and we demonstrated positive effects of incorporating the following additional steps: (i) IEF for an added dimension of sample fractionation; (ii) inclusion of an additional source of biological material, e.g. extraction of secreted glycoproteins from the cell culture media (“secretome”); and (iii) use of a second proteolytic enzyme, e.g. chymotrypsin. In addition, we introduced a scheme for splitting, pre-screening, and pooling of LWAC-enriched O-glycopeptide fractions prior to IEF to improve sample usage.
Isoelectric Focusing
Although it has been debated that IEF could result in significant sample loss without enhanced identifications, we found that incorporation of IEF fractionation yielded an increase in identifications by a factor of 2 for proteins and more than 2.5 for glycosites. The advantage of employing IEF pre-fractionation would thus appear to outweigh the potential sample loss and extra effort involved. Moreover, careful inspection of data from the 12 IEF fractions used in this study showed that the sample complexity is high in each fraction, and use of either a faster and more sensitive mass spectrometer or alternative pre-fractionation methods could significantly further extend the depth of glycosite/glycoprotein identification.
Secretome Versus Total Cell Lysate
Although a total lysate of cells should in principle yield access to the entire proteome of a cell, the relative abundance in subcellular fractions and/or secretions will vary tremendously. We chose to address this initially by testing the contribution that analysis of the secretome could provide. To this end, we developed an initial VVA lectin capture step for secreted O-glycoproteins (Fig. 1). A general problem with secretome analysis is the abundant proteins derived from serum in regular cell culture media, and although it is possible to use serum/protein-free media for growth of many cell lines, it is often desirable to avoid this due to marked changes in growth and biological parameters. Enrichment of secreted O-glycoproteins from SimpleCells was efficiently achieved, and almost all of the bovine glycoproteins were eliminated because they do not carry truncated O-glycans reactive with VVA. Those few cases where a peptide was 100% identical to a bovine as well as human sequence are noted in supplemental Tables 1 and 2. We recently applied the secretome strategy to define the contribution of one polypeptide GalNAc-T to the O-glycoproteome in one SimpleCell line (human liver HepG2), and we demonstrated that it was possible to identify several O-glycoproteins that were glycosylated only when this GalNAc-T isoform was expressed (9). The strategy used involved analysis of the differential O-glycoproteomes of isogenic HepG2 SimpleCells with and without zinc finger nuclease knockout of the GalNAc-T2 isoform.
Chymotrypsin Versus Trypsin
Finally, we wished to probe whether use of a protease other than trypsin would contribute significant additional information, as has already been established for general and phosphoproteomics (24–27). Hypothetically, we should be able to expand O-glycosite identification to areas of O-glycoproteins that cannot be observed simply by replicate analysis. We found that although the absolute numbers obtained from chymotrypsin digestion were approximately three times less than for trypsin, the use of this enzyme opened significant O-glycosylation space apparently not observable using trypsin alone. In this perspective, we intend to evaluate the use of other proteolytic enzymes (e.g. Glu-C, Asp-N, Lys-N, etc.) for SimpleCell O-glycoproteome analysis and optimize our workflow again potentially to use them in one run.
Discussion of Selected O-Glycoproteins
Amyloid-β A4 is central to the etiology of Alzheimer disease, and the recent understanding that this is an O-glycoprotein with multiple O-glycans is intriguing. In our previous study, we identified amyloid β A4 peptides containing one and two missed cleavage sites, together covering the sequence 649GLTTRPGSGLTNIKTEEISEVK670 (numbering from the canonical amyloid protein precursor isoform APP770) having glycosylation of up to all four Thr residues. In this study, we also found these peptides and identified glycoforms bearing up to five of the six available sites. This region of APP has been identified in two previous MS-based studies, as recombinant human isoform APP695 secreted from CHO cells (28), and in the form of short APP/Aβ glycopeptides immunoprecipitated from human cerebrospinal fluid (4). In neither of these studies were more than three sites within the 649–670 segment unambiguously identified. Our results indicate the possibility that both Thr-663 and Ser-667 can be modified, of particular interest considering that the 649–670 peptide is just upstream of the β-secretase cleavage site (Met-671 to Asp-672), which releases soluble Aβ from the membrane-bound C-terminal peptide (processing further downstream by α-, γ-, and θ-secretases is also possible). In a recent study, Kitazume et al. (29) concluded that there should be no O-glycosylation beyond Thr-652; the possibility that further O-glycosylation along this peptide is likely and that this could have an effect on β-secretase processing was not envisioned.
Importantly, in this study we have now identified seven more glycosites distributed over four additional peptides, at either Thr-352 or Thr-353 and at Thr-366, Thr-367, Ser-370, Thr-371, Thr-381, and Thr-600; of these, Kitazume et al. (29) identified Thr-353, and Perdivara et al. (28) identified Thr-366 and Thr-367 in a peptide with a different N-terminal sequence from the APP695 isoform. We thus show that APP is a significantly more O-glycosylated protein than previously suspected.
Agrin
Altogether we have now identified in SimpleCells seven peptides from agrin; it is especially well represented in Capan-1, both cell lysate and secretome, as now shown in this study. The human agrin full-length transcript codes for a 2045-amino acid multidomain protein with a mass upwards of 200 kDa not including post-translational processing, which attaches heavy glycosylation that can double or even triple the apparent molecular weight. The mature protein has been characterized as a proteoglycan containing O-linked heparan sulfate chains along with N-linked glycans, GalNAc- and Fuc-O-linked glycosylation (31, 32). Multiple splice forms exist, and a plethora of signaling and structural organizing functions have been attributed to agrin, including a well established central role in the formation and maintenance of the neuromuscular junction (33). Most importantly for this study, it contains two Ser/Thr/Pro-rich mucin-like domains in linkage regions on either side of a single sea urchin sperm protein domain. However, the actual sites and structures of O-glycans on human agrin were previously unknown. Karlsson and McGuckin (34) observed O-Fuc and heparan sulfate-type glycans released from agrin from ovarian cancer ascites, but did not find mucin-type O-glycans. Including an additional peptide observed previously but not here (5), six of the seven detected peptides cover the entire “mucin-like” sequence (residues 1247–1321) between the sea urchin sperm protein domain (residues 1130–1252) and the first of several EGF-like domains (residues 1329–1367). In this sequence, all of the 20 potential O-GalNAc sites were detected. It has been proposed that O-glycans with characteristic antigenic structures in the mucin domains of rat agrin play an important role in modulating its binding to glycans on muscle cell proteins such as α-dystroglycan, activation of muscle-specific receptor tyrosine kinase MuSK, and stimulation of AChR clustering (31). It was hypothesized that the modulation of these intermolecular interactions is effected through intramolecular interactions of the O-glycans on the agrin mucin domains. Our study provides the first direct structural evidence for the GalNAc-O-glycosylation in one of these regions, and further studies should probe the functional importance of O-glycans on agrin.
CD44
CD44 is an important, well studied, and multifunctional molecule associated with cell-cell and cell-matrix interactions mediated through interactions with hyaluronans and other glycosaminoglycans, as well as being implicated in a variety of signaling processes, inflammatory response, development, cartilage deposition, early stages of carcinogenic transformation, and metastasis. It is known to possess both N- and O-glycans and, in forms that have been characterized as being heavily decorated with sulfated, sialylated, Lewis antigen determinants, an important interaction partner of E- and L-selectins and other carbohydrate-binding proteins (35–37). No careful structural studies have been performed on either type of glycan; however, although the N-glycosites are more or less known, almost nothing has been explored with respect to O-glycosite number, location, density, contribution to molecular variation, or intermolecular interactions. Using the sialoglycopeptide trapping approach, Halim et al. (38) detected a urinary glycopeptide mapping to CD44 showing simple O-glycosylation on either Thr-637 or Thr-638 (Net-O-Glyc predicts Thr-638). Overall, we detected a total of 22 O-glycopeptides representing a minimum of 39 occupied O-glycosites, which represents 15 new sites or a 39% increase compared with our previous study.
GalNAc-T5 (GALNT5)
Our SimpleCell glycoproteomic strategy has in particular provided insight into ER and Golgi membrane O-glycoproteins, including many type II transmembrane glycosyltransferases. One example of these is the polypeptide GalNAc-T5 isoform, which is remarkable among Golgi enzymes by having an ∼500-amino acid-long stem region (2, 39). We find that the stem region is extensively O-glycosylated with some 28 O-glycosites detected distributed throughout the stem region. We have detected O-glycosylation of the stem regions in many other type II glycosyltransferases (5, 9) but with fewer sites, consistent with their much shorter stem regions (typically 30–60 amino acids). It is likely that O-glycosylation of these enzymes is involved in protection from proteolytic cleavage and shedding of the catalytic C-terminal domains, and in some cases this may be a regulated event as recently discussed (2).
O-Glycosylation of Tyrosine
An additional result of this study has been the expansion of the list of O-glycoproteins identified having the recently discovered GalNAc O-glycosylation of Tyr residues in APP (4) and NUCB2 (5). Because this data subset is still extremely limited, it is premature to speculate too much with respect to pattern or functional significance. It does appear that the identified proteins are diverse and found in all three cell types studied here, as well as being found in both secretomes and cell lysates (and, in the case of APP, in cerebrospinal fluid (4)). Interestingly, glycosylation of Tyr-681 in APP, close to the α-secretase cleavage site (or proposed alternative γ-secretase site (4)) identified in cerebrospinal fluids, was not identified in any of the three SimpleCell lines despite the expanded number of O-glycosites that our strategy revealed. All of the Tyr-glycosylated proteins identified herein are consistent with ER-Golgi processing, found either membrane-bound or secreted (e.g. ECM1, found only in the secretomes; CD44 and PRAP1, found in both lysates and secretomes), or Golgi resident (e.g. NUCB2, found in cell lysates).
Summary and Outlook
We have shown that, in conjunction with the now established SimpleCell genetic engineering strategy for truncating O-glycans, IEF and alternative proteolytic enzymes are useful tools for further expanding the O-glycoproteome. Following incorporation of IEF into the sample handling strategy, we also found that secretome analysis yielded a major nonoverlapping increase in the depth and breadth of O-glycosite/O-glycoprotein detection. The key parameter contributing to identification of novel O-glycosites in these studies was reduced sample complexity, and this suggests the likelihood that future studies, including subcellular fractionation, could produce a further enhancement of O-glycoproteome coverage. Confirming previous observations, we found that computational approaches, such as NetOGlyc, are not adequate in their current forms to be of much help in O-glycosite prediction except for mucins and mucin-like domains with typical clusters of PTS residues. Regardless, our first steps to uncover the human O-glycoproteome suggest that more isolated O-glycosites are far more numerous, so far even dominant, than previously thought. In this respect, the term mucin-type O-glycosylation somewhat misrepresents the broader situation with this class of PTM residues. Moreover, the GalNAc O-glycoproteome is far larger than ever conceived. Our current protocol has not yet yielded any classical mucin domains, which could be due either to the selection of cell lines or to limitations of the digestion. This will need to be further investigated. In any case, more cell types, representing a wider cross-section of human organ systems, need to be investigated. Moreover, to gain insight into the regulation and functions of site-specific O-glycosylation, it will be necessary to expand analysis of glycoproteomes to isogenic SimpleCell lines with differential expression of the many GalNAc-T isoforms with the aim to identify nonredundant functions of individual isoforms as we have recently shown in a pilot study (9). The improvements established in this study should greatly aid in this bigger goal.
Toward this goal, our enabling strategy for direct detection of glycosites in high throughput mode will allow faster and broader characterization of the human O-glycoproteome, feeding investigations into functions of GalNAc-T isoforms, as well as improving O-glycosite prediction and identifying new biomarker candidates. These improvements, with a few additional modifications, should also be applicable to studies of other forms of O-glycosylation, such as O-mannosylation, O-fucosylation, and O-xylosylation.
Supplementary Material
Footnotes
* This work was supported by Kirsten og Freddy Johansen Fonden, A. P. Møller og Hustru Chastine Mc-Kinney Møllers Fond til Almene Formaal, The Carlsberg Foundation, The Novo Nordisk Foundation, The Alfred Benzon Foundation, The Danish Research Councils, and Program of Excellence from the University of Copenhagen. The Copenhagen Center for Glycomics is supported by Danish National Research Foundation (DNRF107).
 This article contains supplemental material.
This article contains supplemental material.
1 The abbreviations used are:
- PTM
- post-translational modification
- ETD
- electron transfer dissociation
- GalNAc-T
- polypeptide GalNAc transferase
- HCD
- higher energy collisional dissociation
- HexNAc
- N-acetylhexosamine
- IEF
- isoelectric focusing
- LWAC
- lectin weak affinity chromatography
- Tn
- GalNAcα-O-Ser/Thr
- VVA
- V. villosa agglutinin
- BisTris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol
- ER
- endoplasmic reticulum
- APP
- amyloid β A4 precursor.
REFERENCES
- 1. Lowe J. B., Marth J. D. (2003) A genetic approach to mammalian glycan function. Annu. Rev. Biochem. 72, 643–691 [DOI] [PubMed] [Google Scholar]
- 2. Bennett E. P., Mandel U., Clausen H., Gerken T. A., Fritz T. A., Tabak L. A. (2012) Control of mucin-type O-glycosylation: A classification of the polypeptide GalNAc-transferase gene family. Glycobiology 22, 736–756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Gill D. J., Clausen H., Bard F. (2011) Location, location, location: new insights into O-GalNAc protein glycosylation. Trends Cell Biol. 21, 149–158 [DOI] [PubMed] [Google Scholar]
- 4. Halim A., Brinkmalm G., Rüetschi U., Westman-Brinkmalm A., Portelius E., Zetterberg H., Blennow K., Larson G., Nilsson J. (2011) Site-specific characterization of threonine, serine, and tyrosine glycosylations of amyloid precursor protein/amyloid β-peptides in human cerebrospinal fluid. Proc. Natl. Acad. Sci. U.S.A. 108, 11848–11853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Steentoft C., Vakhrushev S. Y., Vester-Christensen M. B., Schjoldager K. T., Kong Y., Bennett E. P., Mandel U., Wandall H., Levery S. B., Clausen H. (2011) Mining the O-glycoproteome using zinc-finger nuclease-glycoengineered SimpleCell lines. Nat. Methods 8, 977–982 [DOI] [PubMed] [Google Scholar]
- 6. Kato K., Jeanneau C., Tarp M. A., Benet-Pagès A., Lorenz-Depiereux B., Bennett E. P., Mandel U., Strom T. M., Clausen H. (2006) Polypeptide GalNAc-transferase T3 and familial tumoral calcinosis. Secretion of fibroblast growth factor 23 requires O-glycosylation. J. Biol. Chem. 281, 18370–18377 [DOI] [PubMed] [Google Scholar]
- 7. Schjoldager K. T., Vester-Christensen M. B., Bennett E. P., Levery S. B., Schwientek T., Yin W., Blixt O., Clausen H. (2010) O-Glycosylation modulates proprotein convertase activation of angiopoietin-like protein 3: possible role of polypeptide GalNAc-transferase-2 in regulation of concentrations of plasma lipids. J. Biol. Chem. 285, 36293–36303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Schjoldager K. T., Vester-Christensen M. B., Goth C. K., Petersen T. N., Brunak S., Bennett E. P., Levery S. B., Clausen H. (2011) A systematic study of site-specific GalNAc-type O-glycosylation modulating proprotein convertase processing. J. Biol. Chem. 286, 40122–40132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Schjoldager K. T., Vakhrushev S. Y., Kong Y., Steentoft C., Nudelman A. S., Pedersen N. B., Wandall H. H., Mandel U., Bennett E. P., Levery S. B., Clausen H. (2012) Probing isoform-specific functions of polypeptide GalNAc-transferases using zinc finger nuclease glycoengineered SimpleCells. Proc. Natl. Acad. Sci. U.S.A. 109, 9893–9898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Gill D. J., Chia J., Senewiratne J., Bard F. (2010) Regulation of O-glycosylation through Golgi-to-ER relocation of initiation enzymes. J. Cell Biol. 189, 843–858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Frishberg Y., Ito N., Rinat C., Yamazaki Y., Feinstein S., Urakawa I., Navon-Elkan P., Becker-Cohen R., Yamashita T., Araya K., Igarashi T., Fujita T., Fukumoto S. (2007) Hyperostosis-hyperphosphatemia syndrome: a congenital disorder of O-glycosylation associated with augmented processing of fibroblast growth factor 23. J. Bone Miner. Res. 22, 235–242 [DOI] [PubMed] [Google Scholar]
- 12. Darula Z., Medzihradszky K. F. (2009) Affinity enrichment and characterization of mucin core-1 type glycopeptides from bovine serum. Mol. Cell. Proteomics 8, 2515–2526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Darula Z., Chalkley R. J., Baker P., Burlingame A. L., Medzihradszky K. F. (2010) Mass spectrometric analysis, automated identification, and complete annotation of O-linked glycopeptides. Eur. J. Mass Spectrom. 16, 421–428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Larsen M. R., Jensen S. S., Jakobsen L. A., Heegaard N. H. (2007) Exploring the sialiome using titanium dioxide chromatography and mass spectrometry. Mol. Cell. Proteomics 6, 1778–1787 [DOI] [PubMed] [Google Scholar]
- 15. Nilsson J., Rüetschi U., Halim A., Hesse C., Carlsohn E., Brinkmalm G., Larson G. (2009) Enrichment of glycopeptides for glycan structure and attachment site identification. Nat. Methods 6, 809–811 [DOI] [PubMed] [Google Scholar]
- 16. Zhang H., Li X. J., Martin D. B., Aebersold R. (2003) Identification and quantification of N-linked glycoproteins using hydrazide chemistry, stable isotope labeling, and mass spectrometry. Nat. Biotechnol. 21, 660–666 [DOI] [PubMed] [Google Scholar]
- 17. Sun B., Ranish J. A., Utleg A. G., White J. T., Yan X., Lin B., Hood L. (2007) Shotgun glycopeptide capture approach coupled with mass spectrometry for comprehensive glycoproteomics. Mol. Cell. Proteomics 6, 141–149 [DOI] [PubMed] [Google Scholar]
- 18. Chalkley R. J., Thalhammer A., Schoepfer R., Burlingame A. L. (2009) Identification of protein O-GlcNAcylation sites using electron transfer dissociation mass spectrometry on native peptides. Proc. Natl. Acad. Sci. U.S.A. 106, 8894–8899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Trinidad J. C., Barkan D. T., Gulledge B. F., Thalhammer A., Sali A., Schoepfer R., Burlingame A. L. (2012) Global identification and characterization of both O-GlcNAcylation and phosphorylation at the murine synapse. Mol. Cell. Proteomics 11, 215–229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Darula Z., Sherman J., Medzihradszky K. F. (2012) How to dig deeper? Improved enrichment methods for mucin core-1 type glycopeptides. Mol. Cell. Proteomics 11, O111.016774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Julenius K., Mølgaard A., Gupta R., Brunak S. (2005) Prediction, conservation analysis, and structural characterization of mammalian mucin-type O-glycosylation sites. Glycobiology 15, 153–164 [DOI] [PubMed] [Google Scholar]
- 22. Krijgsveld J., Gauci S., Dormeyer W., Heck A. J. (2006) In-gel isoelectric focusing of peptides as a tool for improved protein identification. J. Proteome Res. 5, 1721–1730 [DOI] [PubMed] [Google Scholar]
- 23. Hubner N. C., Ren S., Mann M. (2008) Peptide separation with immobilized pI strips is an attractive alternative to in-gel protein digestion for proteome analysis. Proteomics 8, 4862–4872 [DOI] [PubMed] [Google Scholar]
- 24. Choudhary G., Wu S. L., Shieh P., Hancock W. S. (2003) Multiple enzymatic digestion for enhanced sequence coverage of proteins in complex proteomic mixtures using capillary LC with ion trap MS/MS. J. Proteome Res. 2, 59–67 [DOI] [PubMed] [Google Scholar]
- 25. Swaney D. L., Wenger C. D., Coon J. J. (2010) Value of using multiple proteases for large-scale mass spectrometry-based proteomics. J. Proteome Res. 9, 1323–1329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zhou H., Hou W., Lambert J. P., Figeys D. (2010) New ammunition for the proteomic reactor: strong anion exchange beads and multiple enzymes enhance protein identification and sequence coverage. Anal. Bioanal. Chem. 397, 3421–3430 [DOI] [PubMed] [Google Scholar]
- 27. Casado-Vela J., Ruiz E. J., Nebreda A. R., Casal J. I. (2007) A combination of neutral loss and targeted product ion scanning with two enzymatic digestions facilitates the comprehensive mapping of phosphorylation sites. Proteomics 7, 2522–2529 [DOI] [PubMed] [Google Scholar]
- 28. Perdivara I., Petrovich R., Allinquant B., Alliquant B., Deterding L. J., Tomer K. B., Przybylski M. (2009) Elucidation of O-glycosylation structures of the β-amyloid precursor protein by liquid chromatography-mass spectrometry using electron transfer dissociation and collision-induced dissociation. J. Proteome Res. 8, 631–642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kitazume S., Tachida Y., Kato M., Yamaguchi Y., Honda T., Hashimoto Y., Wada Y., Saito T., Iwata N., Saido T., Taniguchi N. (2010) Brain endothelial cells produce amyloid β from amyloid precursor protein 770 and preferentially secrete the O-glycosylated form. J. Biol. Chem. 285, 40097–40103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hart G. W., Slawson C., Ramirez-Correa G., Lagerlof O. (2011) Cross-talk between O-GlcNAcylation and phosphorylation: roles in signaling, transcription, and chronic disease. Annu. Rev. Biochem. 80, 825–858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Xia B., Martin P. T. (2002) Modulation of agrin binding and activity by the CT and related carbohydrate antigens. Mol. Cell. Neurosci. 19, 539–551 [DOI] [PubMed] [Google Scholar]
- 32. Kim M. L., Chandrasekharan K., Glass M., Shi S., Stahl M. C., Kaspar B., Stanley P., Martin P. T. (2008) O-Fucosylation of muscle agrin determines its ability to cluster acetylcholine receptors. Mol. Cell. Neurosci. 39, 452–464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Bezakova G., Ruegg M. A. (2003) New insights into the roles of agrin. Nat. Rev. Mol. Cell Biol. 4, 295–308 [DOI] [PubMed] [Google Scholar]
- 34. Karlsson N. G., McGuckin M. A. (2012) O-Linked glycome and proteome of high molecular mass proteins in human ovarian cancer ascites: identification of sulfation, disialic acid, and O-linked fucose. Glycobiology 22, 918–929 [DOI] [PubMed] [Google Scholar]
- 35. Dimitroff C. J., Lee J. Y., Fuhlbrigge R. C., Sackstein R. (2000) A distinct glycoform of CD44 is an L-selectin ligand on human hematopoietic cells. Proc. Natl. Acad. Sci. U.S.A. 97, 13841–13846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Dimitroff C. J., Lee J. Y., Rafii S., Fuhlbrigge R. C., Sackstein R. (2001) CD44 is a major E-selectin ligand on human hematopoietic progenitor cells. J. Cell Biol. 153, 1277–1286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Sackstein R., Dimitroff C. J. (2000) A hematopoietic cell L-selectin ligand that is distinct from PSGL-1 and displays N-glycan-dependent binding activity. Blood 96, 2765–2774 [PubMed] [Google Scholar]
- 38. Halim A., Nilsson J., Ruetschi U., Hesse C., Larson G. (2012) Human urinary glycoproteomics; attachment site-specific analysis of N- and O-linked glycosylations by CID and ECD. Mol. Cell. Proteomics 11, M111.013649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ten Hagen K. G., Hagen F. K., Balys M. M., Beres T. M., Van Wuyckhuyse B., Tabak L. A. (1998) Cloning and expression of a novel, tissue specifically expressed member of the UDP-GalNAc:polypeptide N-acetylgalactosaminyltransferase family. J. Biol. Chem. 273, 27749–27754 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.