

Abstract
PGAP2 encodes a protein involved in remodeling the glycosylphosphatidylinositol (GPI) anchor in the Golgi apparatus. After synthesis in the endoplasmic reticulum (ER), GPI anchors are transferred to the proteins and are remodeled while transported through the Golgi to the cell membrane. Germline mutations in six genes (PIGA, PIGL, PIGM, PIGV, PIGN, and PIGO) in the ER-located part of the GPI-anchor-biosynthesis pathway have been reported, and all are associated with phenotypes extending from malformation and lethality to severe intellectual disability, epilepsy, minor dysmorphisms, and elevated alkaline phosphatase (ALP). We performed autozygosity mapping and ultra-deep sequencing followed by stringent filtering and identified two homozygous PGAP2 alterations, p.Tyr99Cys and p.Arg177Pro, in seven offspring with nonspecific autosomal-recessive intellectual disability from two consanguineous families. Rescue experiments with the altered proteins in PGAP2-deficient Chinese hamster ovary cell lines showed less expression of cell-surface GPI-anchored proteins DAF and CD59 than of the wild-type protein, substantiating the pathogenicity of the identified alterations. Furthermore, we observed a full rescue when we used strong promoters before the mutant cDNAs, suggesting a hypomorphic effect of the mutations. We report on alterations in the Golgi-located part of the GPI-anchor-biosynthesis pathway and extend the phenotypic spectrum of the GPI-anchor deficiencies to isolated intellectual disability with elevated ALP. GPI-anchor deficiencies can be interpreted within the concept of a disease family, and we propose that the severity of the phenotype is dependent on the location of the altered protein in the biosynthesis chain.
Introduction
Posttranslational modification of proteins by the addition of glycosylphosphatidylinositol (GPI) anchors to the C termini is well conserved in eukaryotes. GPI anchors function as sorting signals for the transport of GPI-anchored proteins (GPI-APs) in the secretory and endocytic pathways, and they enable anchoring of the GPI-APs in the cell-membrane rafts.1 The GPI-anchor-biosynthesis pathway involves over 20 different gene products (Figure S1, available online). After synthesis, GPI anchors are transferred to the proteins in the endoplasmic reticulum (ER) and are further remodeled by the elimination of acylphosphate and ethanolaminephosphate chains. GPI-APs are then transported from the ER to the plasma membrane through the Golgi apparatus. In the Golgi apparatus, fatty-acid remodeling of the GPI anchor occurs; an unsaturated fatty acid is removed by PGAP3, and a saturated fatty acid is transferred back, most likely by the noncatalytic protein PGAP2 and presumably also by an uncharacterized acyltransferase given that PGAP2 itself does not resemble other acyltransferases (Figure S1).2,3
The GPI-anchor deficiencies represent a subgroup of congenital disorders of glycosylation.4 Germline mutations in six genes encoding proteins in the ER-located part of the GPI-anchor-biosynthesis pathway are known: those in PIGA (MIM 311770), PIGL (MIM 605947), PIGM (MIM 610273), PIGV (MIM 610274), PIGN (MIM 606097), and PIGO (MIM 614730) (Figure S1 and Table 1).5–8,10,11 Phenotypes of PIGA, PIGL, and PIGN mutations resulting in protein modification are associated with gross malformations and in some instances are even lethal.5,6,10 Mutations further downstream of the pathway in PIGV and PIGO lead to intellectual disability, hyperphosphatasia, epilepsy, and minor dysmorphisms.8,11 All mutations are recessive with residual protein function, and it seems that complete deficiency in GPI-anchor biosynthesis is incompatible with life.12 A single reported mutation in a regulatory element of PIGM leads to a different phenotype of portal and hepatic vein thrombosis and epilepsy.7
Table 1.
Mutations in Genes Involved in the GPI-Anchor-Biosynthesis Pathway
| Gene (RefSeq) | Protein Function | Disease | Phenotype | Mutation | Consequence | Origin | Reference |
|---|---|---|---|---|---|---|---|
| PIGA (NM_002641.3) | GPI-GlcNAc transferase | MCAHS2 (MIM 300868) |
multiple congenital anomalies involving cleft palate, neonatal seizures, CNS structural malformations, and other anomalies | c.[1234C>T];[1234C>T] | p.[Arg412∗];[Arg412∗] | unknown | Johnston et al.5 |
| PIGL (NM_004278.3) | GlcNAc-PI de-N-acetylase |
CHIME syndrome (MIM 280000) |
coloboma, congenital heart disease, ichthyosiform dermatosis, mental retardation, and ear anomalies | c.[274delC];[500T>C] c.[500T>C];[652C>T] c.[500T>C];[?] c.[427–1G>A];[500T>C] c.[500T>C]; deletion |
p.[Leu92Phefs∗15];[Leu167Pro] p.[Leu167Pro];[Gln218∗] p.[Leu167Pro];[?] p.[ex skip];[Leu167Pro] p.Leu167Pro |
unknown | Ng et al.6 |
| PIGM (NM_145167.2) | alpha1-4 mannosyl-transferase I | autosomal-recessive GPI-anchor deficiency (MIM 610293) |
portal and hepatic vein thrombosis in early childhood and seizures | c.[–270C>G];[–270C>G] | promoter | Middle Eastern, Turkish | Almeida et al.7 |
| PIGV (NM_017837.3) | alpha1-6 mannosyl-transferase II | HPMRS1 (MIM 239300) |
intellectual disability, hyperphosphatasia, distinct facial gestalt, seizures, brachytelephalangy | c.[1022C>A];[1022C>A] c.[1022C>A];[1154A>C] c.[766C>A];[766C>A] |
p.[Ala341Glu];[Ala341Glu] p.[Ala341Glu];[His385Pro] p.[Gln256Lys];[Gln256Lys] | unknown | Krawitz et al.8 |
| c.[467G>A];[1022C>A] c.[1022C>A];[1022C>A] |
p.[Cys156Tyr];[ Ala341Glu] p.[Ala341Glu];[ Ala341Glu] |
European | Horn et al.9 | ||||
| PIGN (NM_012327.5) | EtNP transferase I |
MCAHS1 (MIM 614080) |
multiple congenital anomalies, hypotonia, seizures | c.[2126G>A];[2126G>A] | p.[Arg709Gln];[Arg709Gln] | Israeli Arab | Maydan et al.,10 |
| PIGO (NM_032634.3) | EtNP transferase III |
HPMRS2 (MIM 614749) |
intellectual disability, hyperphosphatasia, distinct facial gestalt, seizures, brachytelephalangy | c.[2869C>T];[2361dup] c.[2869C>T];[3069+5G>A] |
p.[Leu957Phe];[Thr788Hisfs∗5] p.[Leu957Phe];[Val952Aspfs∗24] | European | Krawitz et al.,11 |
| PGAP2 (NM_001256240.1) | lyso-GPI-AP acyltransferase (noncatalytic) | MRT17 (MIM 614207) |
severe intellectual disability, hyperphosphatasia, absence seizures | c.[296A>G];[296A>G] c.[530G>C];[530G>C] |
p.[Tyr99Cys];[Tyr99Cys] p.[Arg177Pro];[Arg177Pro] |
Syrian Pakistani |
present study |
We recently reported genetic mapping in a large group of families affected by nonspecific autosomal-recessive intellectual disability (ARID) without clinical preselection. In two consanguineous families, we identified overlapping linkage to chromosomal region 11p15 (see Figure S2).13,14 Here, we report that each family harbors one homozygous PGAP2 (postglycosylphosphatidylinositol attachment to proteins factor 2) mutation encoding a protein involved in the remodeling of the GPI anchor in the Golgi apparatus. Functional analyses in PGAP2-deficient Chinese hamster ovary (CHO) cell lines showed reduced activity of the respective altered proteins and thus confirmed pathogenicity. Our results extend the phenotypic spectrum of the GPI-anchor deficiencies to isolated intellectual disability with elevated alkaline phosphatase (ALP). Our findings support the group of GPI-anchor deficiencies within the concept of a disease family as suggested by Brunner and van Driel,15 and we hypothesize that the severity of the phenotype of a GPI-anchor deficiency is dependent on the location of the altered protein in the synthesis pathway.
Subjects and Methods
Families and Clinical Phenotype of the Affected Members
This study was approved by the Institutional Research Ethics Committee of the National Institute for Biotechnology and Genetic Engineering, School of Biotechnology, Quaid-i-Azam University (Islamabad, Pakistan); by the National Committee on Health Research Ethics (Copenhagen, Denmark); and by the ethics committees of the University of Bonn and the University of Erlangen-Nuremberg (Germany). Informed consent was obtained from all examined persons or their guardians.
We examined family MR043 at their residence in northwestern Syria. The family consists of two branches, MR043a and MR043b, which have two affected girls and one affected girl, respectively (Figure 1 and Figure S2).13 The affected girls in MR043a were 6 (IV:1) and 4 (IV:2) years old at the time of examination. Pregnancy, delivery, and birth parameters of both girls were unremarkable. With estimated IQs below 35, both girls have severe intellectual disability. Their motor development is severely delayed, and they have pronounced muscular weakness and hypotonia. Both girls have strabismus, and neither of them has epilepsy. Sleep patterns of both girls are disordered. Metabolic screening (aminoacidopathies and fattyacidopathies) at the University Children’s Hospital in Damascus revealed no remarkable values. Serum ALP activity was elevated to 4,455 U/l in IV:1 and 4,375 U/l in VI-2 (the normal range is up to 837 U/l). Brain computed-tomography (CT) scans revealed atrophy and increased gyration in both girls. The elder girl (IV:1) showed signs of Dandy-Walker malformation (MIM 220200). At the time of examination, she was 101 cm tall (7 cm below the third percentile) and had a head circumference of 48 cm (1 cm below the fifth percentile). IV:2 was 100 cm tall (25th percentile) and had a head circumference of 47 cm (1 cm below the fifth percentile). Their healthy parents also had head circumferences below the fifth percentile (52 and 53 cm).
Figure 1.
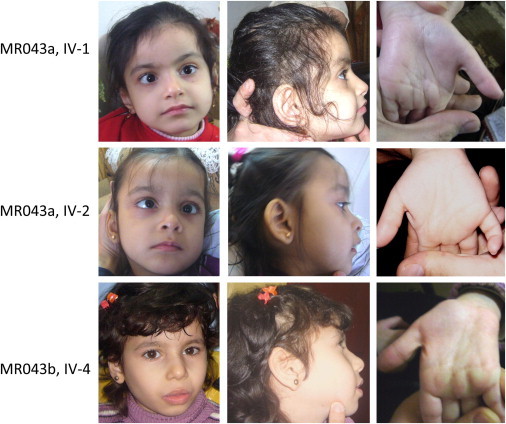
Affected Children of Family MR043
The three affected individuals with PGAP2 mutations; there are no major dysmorphisms or syndromic gestalt.
The affected girl, IV:4, in MR043b was 8.5 years old at the time of examination. During pregnancy, the mother noticed less fetal movement. Delivery and birth length were unremarkable, but severe muscular hypotonia was noted in the neonatal period. Her motor development was severely delayed: she sat at the age of 5 years and could not walk at the time of examination. She spoke single words like “mama” and “papa.” Since the age of 7 years, she has had absence epilepsy. Metabolic screening at the University Children’s Hospital in Damascus revealed no remarkable values. Serum ALP values were not available. A CT scan revealed brain atrophy. Muscle biopsy identified muscle atrophy. At the time of examination, she was 112 cm tall (6 cm below the third percentile) and had a head circumference of 50 cm (tenth percentile). Her siblings and parents had low normal head circumferences. She is a social and joyful girl who loves to play with her siblings and to move to music.
We examined family MR5 at their residence in the district of Faisalabad Punjab province, Pakistan (Figure S2).14 The prenatal, perinatal, and postnatal stages of all four affected members were uneventful. IQ assessments included verbal and motor abilities evaluated by the Slosson Intelligence Test16 and showed an average IQ score of 22. None of the affected members has epilepsy. The affected family members have head circumferences in the normal range. They are thin but do not show growth retardation. Dysmorphological, neurological, opthalmological, and otorhinolaryngological examinations were normal. Structural MRI revealed unremarkable results. Cytogenetic examination using a high-resolution G-banding technique showed normal karyotypes. Blood biochemistry of the liver, renal function, electrolyte levels, thyroid hormone profiles, hematology, chest X-rays, torch profiles, and echocardiography were, apart from anemia, unremarkable. There is no information about serum ALP values.
Genetic Mapping and Identification of Mutations
Parametric linkage analyses showed overlapping significant LOD scores between 0.2 and 6.0 Mb at 11p15.5–p15.4 (Figure S2).13,14 Because of the large number of genes in the candidate region, we performed next-generation sequencing (NGS). For the Pakistani family, MR5, a custom-designed DNA capture array for the region chr11: 1–6,000,000 (hg19) was purchased from NimbleGen (Roche NimbleGen, Madison, WI, USA). A size-fractionated genomic library was generated with DNA from individual V:6 and enriched for the target region according to the manufacture’s protocol. The captured DNA was sequenced with an Illumina Genome Analyzer II platform (Illumina, San Diego, CA, USA), and reads were aligned to the human reference sequence hg18 with the use of ELAND and Burrows-Wheeler Aligner software. The data were filtered for known variants on the basis of public databases (the National Heart, Lung, and Blood Institute [NHLBI] Exome Sequencing Project [ESP] Exome Variant Server and 1000 Genomes), and homozygous variants were selected as putative candidates. Finally, the variants were analyzed in silico with SIFT, PolyPhen-2, and MutationTaster.17–19 PCR and Sanger sequencing were done according to standard protocols for the exclusion of technical artifacts and for segregation testing.
DNA from individual IV:1 of family MR043 was enriched with the SureSelect Human All Exon 50M Kit (Agilent technologies, Santa Clara, CA, USA) and was paired-end sequenced on a SOLiD 5500 xl instrument (Life Sciences, Santa Clara, CA, USA). Image analysis and base calling were performed with SOLiD instrument control software with default parameters. Read alignment was performed with LifeScope 2.5 with default parameters and human genome assembly hg19 (GRCh37) as a reference. Single-nucleotide variants (SNVs) and small indels were detected with LifeScope, GATK 2, and SAMtools/BCFtools. After data were integrated from a variety of public databases, variant annotation was performed with Annovar.20 Additionally, variants were compared to an in-house database containing more than 200 sequenced exomes for the identification of further common variants not present in public databanks. Finally, the variants were analyzed in silico with SIFT, PolyPhen-2, and MutationTaster, and PCR and Sanger sequencing were done according to standard protocols for the exclusion of technical artifacts and for segregation testing.
Functional Analyses in CHO Cell Lines
The C84 PGAP2-deficient cell line and the pME vector systems are described by Tashima and colleagues.21 cDNA (RefSeq accession number NM_001256240) encoding wild-type PGAP2 isoform 8 was amplified from the cDNA library of human hepatoma cell line Hep3B and subcloned into a pME vector carrying a strong αSR promoter. pME vectors containing the mutant PGAP2 cDNAs were made by site-directed mutagenesis with Pfu-Taq DNA polymerase and DpnI endonuclease (Agilent Technologies, Horsholm, Denmark) according to standard protocols. Fragments encoding wild-type and altered PGAP2 were further ligated into the pTal-vector system with a medium-strength TK promoter and into the pTA-vector system with a weak TATA-box promoter. Wild-type and mutant vector constructs were Sanger sequenced for unwanted mutations before use in the C84 transfection experiments. PGAP2-deficient CHO cells (C84) were transiently transfected with all three promoters and either wild-type or altered PGAP2 by electroporation. Cells (5 × 106) were suspended in 0.4 ml of Opti-MEM and electroporated with 10 μg each of the plasmids at 260 V and 960 μF with a Gene Pulser (Bio-Rad, Hercules, CA, USA). Restoration of the surface expression of DAF and CD59 was assessed 2 days later by cell staining with mouse DAF (IA10) or CD59 (5H8) antibody followed by a PE-conjugated anti-mouse IgG antibody and then by flow-cytometry analysis (BD FACSCanto II, BD Biosciences, Franklin Lakes, NJ, USA) with Flowjo software (Tommy Digital, Tokyo, Japan). Levels of PGAP2 in cells were determined by immunoblotting. PGAP2-band intensities quantified with Image Gauge software were normalized for band intensities of endogenous GAPDH and for transfection efficiencies determined by luciferase activities in the transfected cells.
Lymphoblastoid Cell Lines and Flow Cytometry
Lymphoblastoid cell lines (LCLs) were cultured in RPMI 1640 (GIBCO, Life Technologies, Darmstadt, Germany) supplemented with 10% fetal calf serum (PAA Biotech, Cölbe, Germany) and other different supplements. Flow-cytometry analysis was performed with the BD FACSCanto II. For detection of relevant surface markers, the fluorochrome-conjugated antibodies DAF-PE and CD59-FITC (eBioscience, Frankfurt, Germany) were used. The mean fluorescent intensity (MFI) was used as an assessment criterion.
Results
NGS and the PGAP2 Mutations
NGS of IV:1 from the Syrian family, MR043, after whole-exome enrichment resulted in an average coverage of 153×. In the linkage region, 108 variants were nonsynonymous or at splice sites, and only two missense variants in PGAP2 and DNHD1 were not annotated, could be validated with Sanger sequencing, and segregated in the family. In silico analysis using MutationTaster, PolyPhen-2, and SIFT predicted that the variant in DNHD1 is tolerated but that variant c.296A>G (p.Tyr99Cys) (RefSeq NM_001256240.1) in PGAP2 is pathogenic (Table 2).
Table 2.
Overview of the SNVs Identified in the Linkage Region on Chromosome 11
| Family |
Number of Variants |
Position (hg19) | Nucleotide Change | Coverage | Gene | RefSeq Accession Number | Amino Acid Change | Conserved |
In Silico Analyses |
|||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Homozygous in Linkage Region | Coding | Nonsynonymous or at Splice Site | Not Annotated | PolyPhen-2 | SIFT | MutationTaster | ||||||||
| MR5 | 7,040a | 205 | 105 | 1 | chr11: 3,846,254 | c.530G>C | 108× | PGAP2 | NM_001256240.1 | p.Arg177Pro | yes | damaging (0.647) | tolerated (0.08) | disease causing (0.687) |
| MR043 | 398b | 190 | 108 | 2 | chr11: 3,845,243 | c.296A>G | 24× | PGAP2 | NM_001256240.1 | p.Tyr99Cys | yes | damaging (0.998) | deleterious (0) | disease causing (0.999) |
| chr11: 6,567,019 | c.4850A>G | 139× | DNHD1 | NM_144666.2 | p.Lys1617Arg | no | benign (0.021) | tolerated (0.26) | polymorphism (0.999) |
|||||
In family MR5, the genomic candidate region was sequenced.
In family MR043, exons and their boundaries were sequenced genome-wide after enrichment.
NGS of V:6 from the Pakistani family, MR5, after targeted enrichment of the whole genomic candidate region resulted in an average coverage of 142×. We identified 7,040 homozygous SNVs in the candidate region. One hundred and five variants were nonsynonymous or at splice sites, and only one variant, c.530G>C (p.Arg177Pro) (RefSeq NM_001256240.1) in PGAP2, could be verified by Sanger sequencing and segregated in the family (Table 2). In silico analysis using MutationTaster and PolyPhen-2 predicted that the variant in PGAP2 is pathogenic, whereas SIFT predicted the variant to be benign.
PGAP2 encodes a transmembrane Golgi protein that is involved in the modification of the lipid moiety of the GPI anchor before transport to the cell surface (Figure S1 and Figure 2). Human PGAP2 is represented by at least 16 different RNA transcript variants; eight of them encode different PGAP2 isoforms, and eight are noncoding RNA (NCBI gene ID 27315). PGAP2 isoform 8 (encoded by transcript variant 12) is 254 amino acids long and has more than 90% homology with other mammalian orthologs and 44%–57% homology with the zebrafish and Xenopus tropicalis orthologs (data not shown). The membrane prediction server TMHMM22 predicted that isoform 8 has five alpha-helix domains embedded in the Golgi membrane and that its N terminus is in the cytoplasm and its C terminus is in the Golgi lumen. It is supposed that isoform 8 is the biologically active form (T. Kinoshita, unpublished data). Substitutions p.Tyr99Cys and p.Arg177Pro are in the Golgi lumen between TM1 and TM2 and between TM3 and TM4, respectively (Figure 2).
Figure 2.
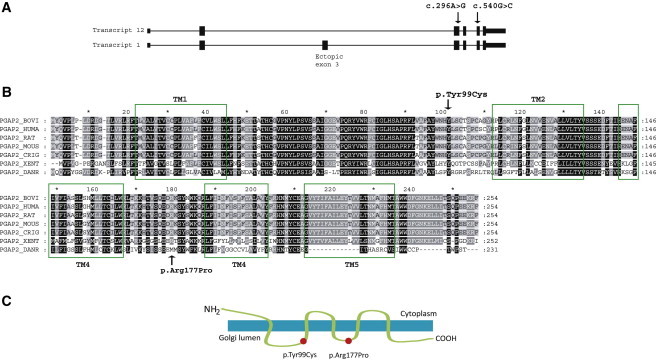
PGAP2 Exon-Intron Structure and PGAP2 Sequence and Structure
(A) The gene structure for the PGAP2 transcript variants 1 and 12. Isoform 1 (transcript variant 1) has a 61 aa insertion (Ala56 to Gly116) corresponding to an ectopic exon 3. It seems that transcript 12 (isoform 8) is the active one (Y. Murakami, unpublished data). Arrows denote the mutations.
(B) The alignment of the human PGAP2 isoform 8 shows homology to mammalian, zebrafish, and frog orthologous proteins. The TM1-5 alpha-helix regions (predicted by TMHMM22) are marked by rectangles, and the altered amino acids are denoted (RefSeq accession numbers NP_001243169.1 [Homo sapiens], NP_001092581.1 [Bos Taurus], NP_446347.1 [Rattus norvegicus], NP_663558.1 [Mus musculus], NP_001233740.1 [Cricetulus griseus], NP_001106477.1 [Xenopus tropicalis], and NP_001013562.1 [Danio rerio]).
(C) PGAP2 is a transmembrane protein, and the p.Tyr99Cys and p.Arg177Pro alterations are located in the Golgi lumen.
PGAP2-Deficient CHO Cells Demonstrate Reduced Activity for the Altered PGAP2 Proteins
PGAP2 was originally cloned by complementation in PGAP2-deficient mutant CHO cell lines expressing the marker proteins DAF (also called CD55) and CD59.21 We used the PGAP2-deficient CHO cell line C84 to analyze the functional consequences of the p.Tyr99Cys and p.Arg177Pro alterations. Because C84 cells express the GPI-anchored marker proteins DAF and CD59 on the cell surface at very low levels, they were used for expression studies after transient transfection of the wild-type and mutant PGAP2 cDNAs subcloned into the expression vectors. Expression of DAF or CD59 on the cell surface after transient transfection could be monitored by fluorescence-activated cell sorting (FACS) as a measurement of PGAP2 activity. We subcloned transcript variant 12 cDNAs encoding wild-type and altered isoform 8 into the mammalian expression vector under three different promoters: a strong SRα promoter, a medium-strength TK promoter, and a weak TATA promoter. When the strong promoter was used, the immunoblot was able to detect PGAP2 proteins and clearly demonstrated that the p.Tyr99Cys and p.Arg177Pro alterations and the wild-type PGAP2 were expressed at similar levels in the C84 cells (Figure 3A). Endogenous GAPDH expression was assessed for controlling loading efficiencies, and a luciferase expression plasmid was cotransfected for monitoring transfection efficiency. With the strong SRα promoter, the wild-type and the two mutant cDNAs fully restored the GPI-AP expression. However, with the medium TK promoter (Figure 3B) and the weak TATA box promoter constructs, the FACS analysis clearly showed that the wild-type constructs fully restored GPI-AP expression but that the mutant constructs had significantly decreased activity in restoring the surface expression (Figure 3B). These results demonstrate that the p.Tyr99Cys and p.Arg177Pro altered PGAP2 proteins have significantly low specific activity.
Figure 3.
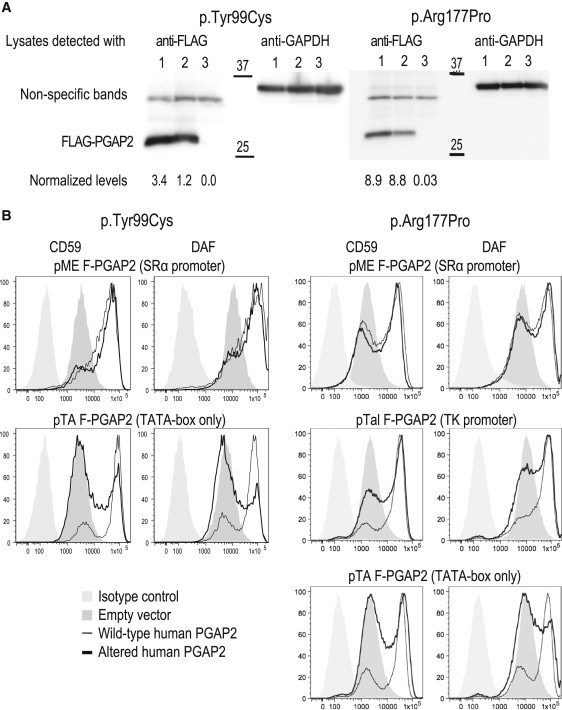
Results of Functional Analyses in CHO Cells
cDNAs encoding PGAP2 isoform 8 were subcloned into the vectors under promoters of different strengths and were expressed in PGAP2-deficient CHO cells.
(A) Immunoblot of cell lysates isolated with the strong SRα promoter after 2 days of expression showed similar levels of wild-type (lane 1) and altered (lane 2) PGAP2 proteins; an empty-vector construct did not express the protein (lane 3). The protein levels were normalized (levels are shown underneath the blot) with the intensities of GAPDH expression shown at the left part of the blots.
(B) Expression of the DAF and CD59 cell-surface proteins after transient transfection was monitored by FACS as a measurement of PGAP2 activities and is shown for the various promoter constructs. The left panel shows the p.Tyr99Cys alteration, and the right panel shows the p.Arg177Pro alteration. Under the strong SRα promoter, both altered proteins restore the cell-surface expression of DAF and CD59, suggesting a hypomorphic alteration. Expression under the weak promoters TK and TATA-box shows reduced activity of altered PGAP2 (see Results and Discussion for details).
FACS of LCLs with Altered PGAP2 Show Same DAF and CD59 Expression
It has been reported that mutations in genes involved in PGI-anchor biosynthesis lead to lower expression of GPI-anchored markers, such as DAF and CD59, in cells of an affected individual.6,7,10 We performed FACS with LCLs derived from three homozygous (−/−) and six heterozygous (+/−) healthy family members and from 15 healthy volunteers with a confirmed genotype (+/+). DAF and CD59 expression in LCLs derived from healthy donors showed high individual variances. The MFI for DAF and CD59 was 12,267 (6,522−22,105) and 31,040 (21,518–53,291), respectively. Cells from heterozygous or homozygous LCLs showed no significant difference in the distribution of DAF and CD59 expression (p > 0.2), although mean MFIs were seen to be somewhat lower in the homozygous group of individuals (11,326 and 30,018 for DAF and CD59, respectively, Figure S3).
Discussion
We investigated two unrelated consanguineous ARID-affected families sharing the overlapping loci MRT17 and MRT21.13,14 NGS identified in each family one homozygous missense variant in PGAP2 (Table 2). We considered both variants to be interesting candidate mutations because they are not annotated and affect conserved amino acids. Several prediction programs classified them as pathogenic. PGAP2 encodes a protein in the biosynthesis pathway of the GPI anchor;1 this protein probably has five transmembrane alpha-helix domains and is located in the Golgi apparatus (Figure 2). Changing the charged and strongly hydrophobic arginine to the nonpolar and weakly hydrophobic proline (p.Arg177Pro) and changing the hydrophobic tyrosine to the hydrophilic cysteine (p.Tyr99Cys) in the Golgi lumen suggest a change in the secondary and tertiary structures of the protein and might disturb protein folding. It is also tempting to speculate that the PGAP2 interaction with a yet unidentified putative acyltransferase is disturbed.
To substantiate our results, we performed in vitro functional analyses in CHO cell lines. For both mutations, we showed reduced expression activity of cell-surface proteins DAF and CD59 after transient transfection of C84 cells with mutant constructs. The CHO rescue experiments clearly demonstrated reduced activity for both PGAP2 alterations and restoration of GPI-AP expression by the wild-type protein (Figure 3). These functional experiments show reduced, but not eliminated, PGAP2 activity. A possible consequence of reduced PGAP2 activity might be interference with the export and anchoring of the immature GPI-APs in the cell-membrane rafts. If the immature GPI-APs without the second saturated fatty acid are transported to the cell surface, the stability of the integration into the cell membrane might be reduced as a result of only the fatty acyl chain and the consequence is release from the cell surface. In pulse-chase experiments, Tashima and colleagues showed that the lysoform GPI-APs in PGAP2-deficient CHO cells are transported to the cell surface and most likely outside of the rafts and are thereafter released to the medium.21 The decreased GPI-AP surface expression seen for the PGAP2 alterations in the C84 cells might be caused by the release of those proteins into the medium. The reduced, but not complete lack of, cell-surface-expressed GPI-APs suggests hypomorphic PGAP2 mutations. Results of FACS of LCLs from affected individuals showed no differences in the expression of the cell-surface markers DAF and CD59, which further supports a hypomorphic effect of the mutation.
Because mutations in PIGA, PIGO, and PIGV are associated with hyperphosphatasia,5,8,11 we retrospectively measured the ALP in the available affected members (IV:1 and IV:2 from family MR043) and found an elevation of more than five times its normal level. Thus, we extend the phenotypic spectrum of GPI-anchor deficiencies to isolated intellectual disability with elevated ALP.
We think that alterations in various members of the GPI-anchor pathway represent an overlapping disease family as discussed by Brunner and van Driel.15 In deficiencies of GPI-anchor proteins, we observe a relation between the location in the pathway of the deficient protein and the severity of the phenotype. Mutations in PGAP2 at the final step of GPI-anchor remodeling cause severe intellectual disability and elevated ALP, and mutations in PIGO and PIGV, upstream of PGAP2, are also associated with epilepsy, brachydactyly, and facial dysmorphism. Mutations in PIGA, PIGL, and PIGN, which encode proteins at the beginning and middle steps of the biosynthesis of the GPI anchor, have a more severe phenotype: malformations and even lethality. Furthermore, we observed that compared to wild-type cells, those with mutations in PIGL, PIGN, and PIGM showed reduced expression of GPI-APs (e.g., DAF, CD59, and CD24) on the cell surface;6,7,10 however, expression of DAF and CD59 did not show differences between cell lines from our affected individuals and cell lines with mutations in PIGO and PIGV (P.M. Krawitz, personal communication). It is thus possible that in this disease group there is a phenotypic gradient from severe at the beginning of the biosynthesis pathway to mild toward the end of the pathway; i.e., the remodeling steps of the GPI anchor. We cannot exclude that the phenotype is dependent on the kind of mutation, e.g., the regulating mutation in PIGM leads to a fully different phenotype. At this early phase of deciphering the phenotypic spectrum associated with the GPI-anchor-biosynthesis pathway, this hypothesis needs validation with larger numbers of well-characterized cases.
In conclusion, we have shown that hypomorphic mutations in PGAP2 lead to severe intellectual disability with elevated ALP and no obvious malformations. This represents a defect in the GPI-anchor-biosynthesis step that takes place in the Golgi and involves the remodeling of the anchors, and it expands the phenotypic spectrum of GPI-anchor deficiencies to isolated intellectual disability.
Acknowledgments
We are grateful to the families involved in this study for their participation. We thank Karen Friis Henriksen and Linda Boje Dalsgaard for their help in library generation, sequencing, and molecular biology analyses. We thank Kana Miyanagi for the excellent assistance in Chinese hamster cell transfection and fluorescence-activated cell sorting. We thank Bärbel Lippke and Margrieta Alblas from Bonn and Farah Radwan, Angelika Diem, Petra Rothe, Steffen Uebe, and Arif Ekici from Erlangen for assistance with lymphoblast cells lines, SNP array genotyping, and next-generation sequencing. This study was supported by the German Intellectual Disability Network through grants from the German Ministry of Research and Education to A.R. (01GS08160 and 01GR0804-4), by the Deutsche Forschungsgemeinschaft through a grant to R.A.J. (AB393/2-1), by the Detusches Austauschdienst DAAD through a grant to H.T., by the Lundbeck Foundation and the Danish National Research Foundation through grants to N.T., and by the Higher Education Commission in Pakistan and a research fellowship from European Molecular Biology Organization to S.u.R.
Contributor Information
Lars Hansen, Email: lah@sund.ku.dk.
Rami Abou Jamra, Email: rami.aboujamra@uk-erlangen.de.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
1000 Genomes, http://www.1000genomes.org/
Burrows-Wheeler Aligner, http://bio-bwa.sourceforge.net/
KEGG: Kyoto Encyclopedia of Genes and Genomes, http://www.genome.jp/kegg/
MutationTaster, http://www.mutationtaster.org/
NHLBI Exome Sequencing Project (ESP) Exome Variant Server, http://evs.gs.washington.edu/EVS/
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org
PolyPhen-2, http://genetics.bwh.harvard.edu/pph2/
SIFT, http://sift.jcvi.org/
TMHMM Server, http://www.cbs.dtu.dk/services/TMHMM/
UCSC Genome Browser, http://www.genome.ucsc.edu
References
- 1.Kinoshita T., Fujita M., Maeda Y. Biosynthesis, remodelling and functions of mammalian GPI-anchored proteins: recent progress. J. Biochem. 2008;144:287–294. doi: 10.1093/jb/mvn090. [DOI] [PubMed] [Google Scholar]
- 2.Fujita M., Kinoshita T. GPI-anchor remodeling: potential functions of GPI-anchors in intracellular trafficking and membrane dynamics. Biochim. Biophys. Acta. 2012;1821:1050–1058. doi: 10.1016/j.bbalip.2012.01.004. [DOI] [PubMed] [Google Scholar]
- 3.Maeda Y., Kinoshita T. Structural remodeling, trafficking and functions of glycosylphosphatidylinositol-anchored proteins. Prog. Lipid Res. 2011;50:411–424. doi: 10.1016/j.plipres.2011.05.002. [DOI] [PubMed] [Google Scholar]
- 4.Freeze H.H., Eklund E.A., Ng B.G., Patterson M.C. Neurology of inherited glycosylation disorders. Lancet Neurol. 2012;11:453–466. doi: 10.1016/S1474-4422(12)70040-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Johnston J.J., Gropman A.L., Sapp J.C., Teer J.K., Martin J.M., Liu C.F., Yuan X., Ye Z., Cheng L., Brodsky R.A., Biesecker L.G. The phenotype of a germline mutation in PIGA: the gene somatically mutated in paroxysmal nocturnal hemoglobinuria. Am. J. Hum. Genet. 2012;90:295–300. doi: 10.1016/j.ajhg.2011.11.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ng B.G., Hackmann K., Jones M.A., Eroshkin A.M., He P., Wiliams R., Bhide S., Cantagrel V., Gleeson J.G., Paller A.S. Mutations in the glycosylphosphatidylinositol gene PIGL cause CHIME syndrome. Am. J. Hum. Genet. 2012;90:685–688. doi: 10.1016/j.ajhg.2012.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Almeida A.M., Murakami Y., Layton D.M., Hillmen P., Sellick G.S., Maeda Y., Richards S., Patterson S., Kotsianidis I., Mollica L. Hypomorphic promoter mutation in PIGM causes inherited glycosylphosphatidylinositol deficiency. Nat. Med. 2006;12:846–851. doi: 10.1038/nm1410. [DOI] [PubMed] [Google Scholar]
- 8.Krawitz P.M., Schweiger M.R., Rödelsperger C., Marcelis C., Kölsch U., Meisel C., Stephani F., Kinoshita T., Murakami Y., Bauer S. Identity-by-descent filtering of exome sequence data identifies PIGV mutations in hyperphosphatasia mental retardation syndrome. Nat. Genet. 2010;42:827–829. doi: 10.1038/ng.653. [DOI] [PubMed] [Google Scholar]
- 9.Horn D., Krawitz P., Mannhardt A., Korenke G.C., Meinecke P. Hyperphosphatasia-mental retardation syndrome due to PIGV mutations: expanded clinical spectrum. Am. J. Med. Genet. A. 2011;155A:1917–1922. doi: 10.1002/ajmg.a.34102. [DOI] [PubMed] [Google Scholar]
- 10.Maydan G., Noyman I., Har-Zahav A., Neriah Z.B., Pasmanik-Chor M., Yeheskel A., Albin-Kaplanski A., Maya I., Magal N., Birk E. Multiple congenital anomalies-hypotonia-seizures syndrome is caused by a mutation in PIGN. J. Med. Genet. 2011;48:383–389. doi: 10.1136/jmg.2010.087114. [DOI] [PubMed] [Google Scholar]
- 11.Krawitz P.M., Murakami Y., Hecht J., Krüger U., Holder S.E., Mortier G.R., Delle Chiaie B., De Baere E., Thompson M.D., Roscioli T. Mutations in PIGO, a member of the GPI-anchor-synthesis pathway, cause hyperphosphatasia with mental retardation. Am. J. Hum. Genet. 2012;91:146–151. doi: 10.1016/j.ajhg.2012.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nozaki M., Ohishi K., Yamada N., Kinoshita T., Nagy A., Takeda J. Developmental abnormalities of glycosylphosphatidylinositol-anchor-deficient embryos revealed by Cre/loxP system. Lab. Invest. 1999;79:293–299. [PubMed] [Google Scholar]
- 13.Abou Jamra R., Wohlfart S., Zweier M., Uebe S., Priebe L., Ekici A., Giesebrecht S., Abboud A., Al Khateeb M.A., Fakher M. Homozygosity mapping in 64 Syrian consanguineous families with non-specific intellectual disability reveals 11 novel loci and high heterogeneity. Eur. J. Hum. Genet. 2011;19:1161–1166. doi: 10.1038/ejhg.2011.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rehman Su., Baig S.M., Eiberg H., Rehman Su., Ahmad I., Malik N.A., Tommerup N., Hansen L. Autozygosity mapping of a large consanguineous Pakistani family reveals a novel non-syndromic autosomal recessive mental retardation locus on 11p15-tel. Neurogenetics. 2011;12:247–251. doi: 10.1007/s10048-011-0286-5. [DOI] [PubMed] [Google Scholar]
- 15.Brunner H.G., van Driel M.A. From syndrome families to functional genomics. Nat. Rev. Genet. 2004;5:545–551. doi: 10.1038/nrg1383. [DOI] [PubMed] [Google Scholar]
- 16.Slosson R.L. Slosson Educational; East Aurora, NY: 1985. Slosson Intelligence Test for children and adults—revised. [Google Scholar]
- 17.Adzhubei I.A., Schmidt S., Peshkin L., Ramensky V.E., Gerasimova A., Bork P., Kondrashov A.S., Sunyaev S.R. A method and server for predicting damaging missense mutations. Nat. Methods. 2010;7:248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ng P.C., Henikoff S. SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003;31:3812–3814. doi: 10.1093/nar/gkg509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schwarz J.M., Rödelsperger C., Schuelke M., Seelow D. MutationTaster evaluates disease-causing potential of sequence alterations. Nat. Methods. 2010;7:575–576. doi: 10.1038/nmeth0810-575. [DOI] [PubMed] [Google Scholar]
- 20.Wang K., Li M., Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38:e164. doi: 10.1093/nar/gkq603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tashima Y., Taguchi R., Murata C., Ashida H., Kinoshita T., Maeda Y. PGAP2 is essential for correct processing and stable expression of GPI-anchored proteins. Mol. Biol. Cell. 2006;17:1410–1420. doi: 10.1091/mbc.E05-11-1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Krogh A., Larsson B., von Heijne G., Sonnhammer E.L. Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J. Mol. Biol. 2001;305:567–580. doi: 10.1006/jmbi.2000.4315. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.