

Abstract
Biochemical analysis and whole-exome sequencing identified mutations in the Golgi-localized UDP-galactose transporter SLC35A2 that define an undiagnosed X-linked congenital disorder of glycosylation (CDG) in three unrelated families. Each mutation reduced UDP-galactose transport, leading to galactose-deficient glycoproteins. Two affected males were somatic mosaics, suggesting that a wild-type SLC35A2 allele may be required for survival. In infancy, the commonly used biomarker transferrin showed abnormal glycosylation, but its appearance became normal later in childhood, without any corresponding clinical improvement. This may indicate selection against cells carrying the mutant allele. To detect other individuals with such mutations, we suggest transferrin testing in infancy. Here, we report somatic mosaicism in CDG, and our work stresses the importance of combining both genetic and biochemical diagnoses.
Main Text
Congenital disorders of glycosylation (CDGs) are a group of more than 60 mainly autosomal recessive disorders caused by impaired synthesis of glycoconjugates.1 Affected individuals often have profound neurological deficiencies, including intellectual disability, seizures, and a wide range of multiorgan symptoms.2 The majority of CDG defects involve the N-linked glycosylation pathway, and alterations can be detected by abnormal glycosylation of the convenient biomarker serum transferrin (Tf).3,4
All clinical samples were obtained with proper informed consent in accordance with the Sanford-Burnham Medical Research Institute’s institutional review board consent guidelines.
We identified an affected male (CDG-341) with an unusual Tf profile in comparison to a healthy control who showed loss of both galactose and sialic acid from multiple branches of complex type N-glycans (Figures 1A and 1B). Sanger sequencing of candidate genes encoding proteins in the glycosylation pathway identified hemizygosity for frameshift mutation c.15_91+48delinsA (p.Gly8Serfs∗9) in SLC35A2 (MIM 314375, RefSeq: NM_001042498.2) (Table 1, Figure S1A available online). This X-linked gene encodes the single known Golgi-localized UDP-galactose transporter. CDG-341 had a 46 XY karyotype, suggesting that he was a somatic mosaic, given that he carried both wild-type (WT) and mutant SLC35A2 alleles. This finding motivated us to examine exome sequencing (ES) data from 16 unrelated individuals with unclassified CDG and abnormal Tf profiles.
Figure 1.
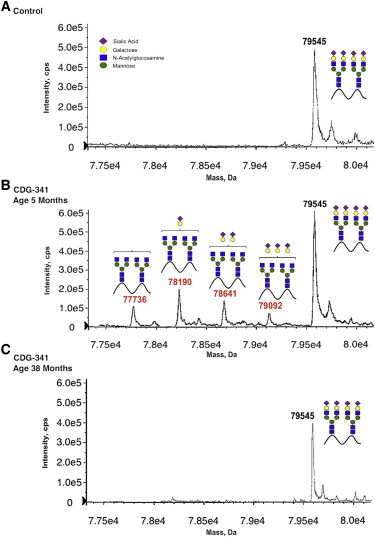
Electrospray Ionization Mass Spectrometry Analyses of Whole Serum Tf from the Control Sample and Individual CDG-341
(A) Serum Tf from an unaffected control.
(B) CDG-341 at 5 months of age showing loss of both galactose and sialic acid from multiple branches of complex type N-glycans.
(C) CDG-341 at 38 months of age showing that Tf glycosylation has become normal.
Table 1.
Clinical Presentation and SLC35A2 Mutations Identified in Three Families with CDG Type IIx
| CDG-341 | CDG-348 | CDG-352 | |
|---|---|---|---|
| Ancestry | European | European | European |
| Gender | Male | Female | Male |
| Current Age | 3 years | 3 years | 6 years |
| Inheritance | de novo | de novo | de novo |
| Nucleotide Change | chrX: 48,768,775_48,768,899 delinsT |
chrX: 48,768,911 C>T |
chrX: 48,762,195 C>T |
| cDNA Change | c.15_91+48 delinsA | c.3G>A | c.991G>A |
| Protein Change | p.Gly8Serfs∗9 | p.Met1? | p.Val331Ile |
| Likely Affect on Protein | Termination | Loss of initiator methionine | Improper positioning in membrane |
| Developmental Delay | Yes | Yes | Yes |
| Hypotonia | Yes | Yes | Yes |
| Seizures | Yes | Yes | No |
| Hypsarrhythmia | Yes | Yes | No |
| Hypertension | No | No | Yes |
| Dysmorphic features | No | Yes | Yes |
| Skeletal abnormalities | No | Shortened extremities | Shortened extremities |
| Renal | No | No | Acute nephrotic syndrome; requiring dialysis |
| Ocular | Nystagmus | Retinitis pigmentosa | Ocular flutter |
| Liver dysfunction | No | No | No |
| Coagulopathy | Yes | No | No |
| Gastrointestinal | No | Strictly G-tube feed | Gastro-esophageal reflux, esophagitis, duodenal perforation |
| Recurrent Infections | No | Yes | No |
| Microcephaly | No | Yes | Yes |
| Brain malformation | Small cerebellum | Thinning of corpus callosum with delayed myelinization | Cerebral hypoplasia/atrophy |
Two of these cases (CDG-348 and CDG-352) had Tf profiles similar to CDG-341 that showed loss of both galactose and sialic acid. One of these cases (CDG-348), a female, was heterozygous for a c.3G>A missense variant in the initiation codon (p.Met1?) that eliminated the normal start site (Table 1, Figure S1B). However, using our standard exome filtering pipeline, we did not find any mutations in SLC35A2 in a male individual, CDG-352, nor was he homozygous or compound heterozygous for rare variants in known CDG-causing genes. Motivated by the unique Tf pattern, we performed a manual review of all variants in CDG-352 to determine whether candidate variants had been excluded by one of the GATK filter flags. This review revealed a c.991G>A missense variant predicted to cause a deleterious substitution (p.Val331Ile) in the last transmembrane domain of SLC35A2 (Table 1, Figure S1C). This variant had been excluded in our standard pipeline because of a low genotype quality value of 16.99 (standard cutoff value ≥ 75). Furthermore, 9% of the SLC35A2 alleles identified in CDG-352 at the variant site were reference alleles, suggesting somatic mosaicism for p.Val331Ile. Sanger sequencing confirmed this observation (Figure S1C).
None of these mutations were present in the Single Nucleotide Polymorphism database, 1000 Genomes, or the NHLBI Exome Variant Server (EVS), the latter of which consists of SNVs found in >13,000 chromosomes from European Americans (n = 8600) and from African Americans (n = 4406). Sanger sequencing of the unaffected parents confirmed that all of these SLC35A2 mutations arose de novo (Table 1, Figures S1A–S1C). Collectively, these results were compelling evidence that mutations in SLC35A2 cause a CDG subtype with unique biochemical and genetic features.
Both affected males were hemizygous for their de novo mutation, but they also retained a functional allele, indicating somatic mosaicism for the mutant SLC35A2 allele. This observation suggests that the retention of a functional SLC35A2 allele might be required for survival and that deleterious alleles might be less tolerated in males than in females. To test this, we categorized each variant in the NHLBI Exome Sequencing Project (ESP)6900 data set as putatively deleterious if the variant was annotated by SeattleSeq134 as creating a stop loss or frameshift mutation or if the variant had a genomic evolutionary rate profiling score ≥ 3.0. To control for different numbers of males (N = 2,602) and females (N = 4,190) as well as different gene lengths, we analyzed the proportion of variants in a gene that were deleterious.
Consistent with this hypothesis, the proportion of SLC35A2 alleles predicted to be deleterious was both higher in females (0.41) in comparison to males (0.25) and ranked higher among all genes on the X chromosome in females (358, 713th) in comparison to males (256, 704th) in the NHLBI ESP data set (Figure S2).
To test whether each of these variants in SLC35A2 altered galactosylation, we analyzed glycans from primary fibroblasts and whole serum of each individual. Loss of SLC35A2 severely limits Golgi-localized pools of UDP-galactose, resulting in truncated β-GlcNAc-terminated N-glycans and α-GalNAc-terminated O-glycans. These changes predict increased binding to the lectins GSII (Griffonia simplicifolia) for terminal β-GlcNAc on N-glycans and to VVA (Vicia villosa) for terminal α-GalNAc on O-glycans.5–7 Flow cytometry analysis with fluorescently tagged lectins proves that all affected individual fibroblast lines fit the predictions, indicating incomplete galactosylation, which leads to incomplete sialylation. (Figure 2)
Figure 2.
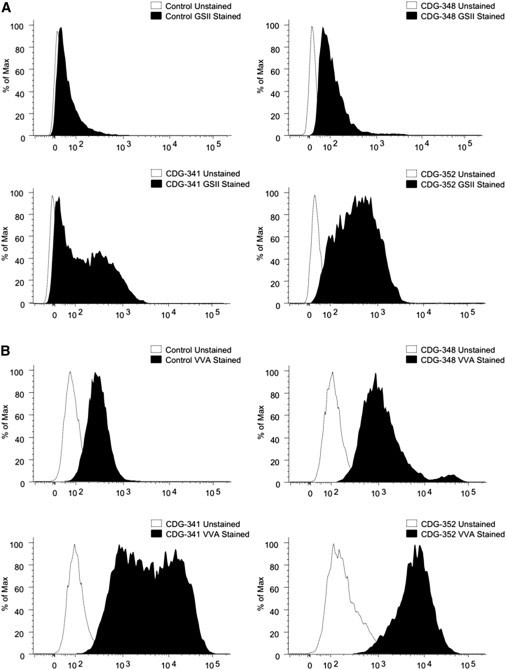
Cell-Surface Analysis of Deficient Galactosylation and Increased Terminal β-GlcNAc and α-GalNAc
(A) Flow cytometry analysis of Alexa-647-conjugated GSII (Griffonia simplicifolia) lectin in control and individual fibroblasts showing increased lectin binding in all three individuals. The parallel control is representative of three different lines that were tested.
(B) Flow cytometry analysis of fluorescein-labeled VVA (Vicia Villosa) lectin in control and individual fibroblasts also showing increased binding in all three individuals. The parallel control is representative of three different lines that were tested.
We directly measured the transport of UDP [6-3H]-galactose into the Golgi using streptolysin-O-permeabilized fibroblasts to preserve intact Golgi and SLC35A2 transporter activity.8 Despite having a mixed population of both WT and mutant cells, all three individuals had considerably reduced UDP-galactose transport when compared to control cells (Figure 3).
Figure 3.
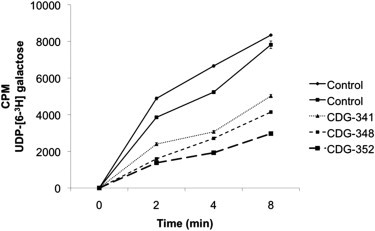
SLC35A2 Transport Activity in Fibroblasts from Both Control and Affected Individuals
Golgi transport of UDP [6-3H]-galactose in streptolysin-O permeabilized fibroblasts from controls and affected individuals. Each data point was done in duplicate, and error bars were calculated by SD.
UDP [6-3H]-galactose transport assays with a 90% pure population of CDG-341-GSII-sorted mutant fibroblasts showed a further 20% decrease in activity and not its elimination (data not shown). This supports the presence of an alternate UDP-galactose transporter that accounts for the activity.
Analysis of N-glycans from whole serum by MALDI-TOF mass spectrometry also showed increased levels of hypogalactosylated glycans, particularly biantennary species (Figure S3). Given that cells from CDG-348 and CDG-341 must have either a normal allele with complete galactosylation or a functionally null allele with no galactosylation at all, the presence of a substantial amount of partially galactosylated glycans on serum glycoproteins further suggests the possible existence of a previously unrecognized UDP-gal transporter.
Other studies suggest that a normal UDP-GlcNAc transporter, SLC35A3 [MIM 605632], can also improve galactosylation in the UDP-galactose-deficient CHO cell line LEC8. Furthermore, many putative nucleotide sugar transporters are incompletely characterized.9
The clinical phenotypes and severity of biochemical defects of these individuals varied substantially (Table 1). Nevertheless, at 5–7 months of age, all individuals had similar galactosylation-deficient Tf profiles; but a sample obtained from CDG-348 at 3 years of age showed a nearly normal pattern without clinical improvement (Figure S4). Samples taken at an older age in both CDG-352 (5 years of age) and CDG-341 (3 years of age) (Figure 1C) were also completely normal. This normalization in all three individuals suggests that SLC35A2-CDG cases could have a limited abnormal Tf diagnostic window, and, without that biochemical lead, standard exome analysis would have discounted the complex results from these somatic mosaics. Accordingly, individuals suspected of having a CDG should be tested for abnormal Tf as early in life as possible. In each individual, it is very likely that Tf testing would have been normal had it been delayed, making diagnosis of a CDG much more challenging.
One explanation for the normalized Tf profiles is that hepatocytes carrying the mutant allele are selected against during infancy. In support of this, we found that GSII-enriched fibroblasts (CDG-341) containing the mutant allele grew ∼25% slower than cells with a WT allele (data not shown). Moreover, DNA prepared from peripheral blood leukocytes had nearly undetectable levels of the mutant allele (data not shown). Again, this suggests possible selection against abnormal cells.
We tried to improve galactosylation in each fibroblast line by providing 150–300 uM galactose for between 2–4 days, but we saw no significant improvement in GSII binding.
Our study highlights the importance of combining biochemical data and detailed knowledge of glycosylation pathways to narrow the candidate genes from ES. We suggest that this approach will help solve the other glycosylation disorders, even if Tf was not analyzed or was uninformative. Heightened awareness of glycobiology and the accelerating discovery of new glycosylation disorders by ES will enhance both basic science and medical practice.10,11
Acknowledgments
We thank the families for their participation and support. Our work is supported by the Rocket Fund, the National Institutes of Health (NIH) (R01DK55615 to H.H.F.), and is also supported in part by grants from the NIH and the National Human Genome Research Institute (1U54HG006493 to M.J.B., D.A.N., and J.S.). We thank Stephanie Grunewald and Margaret J. McMillin for their contributions.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
1000 Genomes Project, http://www.1000genomes.org/
NHLBI Exome Sequencing Project (ESP) Exome Variant Server,http://evs.gs.washington.edu/EVS
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org/
The Single Nucleotide Polymorphism database (dbSNP), http://www.ncbi.nlm.nih.gov/SNP/
References
- 1.Hennet T. Diseases of glycosylation beyond classical congenital disorders of glycosylation. Biochim. Biophys. Acta. 2012;1820:1306–1317. doi: 10.1016/j.bbagen.2012.02.001. [DOI] [PubMed] [Google Scholar]
- 2.Freeze H.H., Eklund E.A., Ng B.G., Patterson M.C. Neurology of inherited glycosylation disorders. Lancet Neurol. 2012;11:453–466. doi: 10.1016/S1474-4422(12)70040-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lacey J.M., Bergen H.R., Magera M.J., Naylor S., O’Brien J.F. Rapid determination of transferrin isoforms by immunoaffinity liquid chromatography and electrospray mass spectrometry. Clin. Chem. 2001;47:513–518. [PubMed] [Google Scholar]
- 4.Freeze H.H. Genetic defects in the human glycome. Nat. Rev. Genet. 2006;7:537–551. doi: 10.1038/nrg1894. [DOI] [PubMed] [Google Scholar]
- 5.Maszczak-Seneczko D., Olczak T., Wunderlich L., Olczak M. Comparative analysis of involvement of UGT1 and UGT2 splice variants of UDP-galactose transporter in glycosylation of macromolecules in MDCK and CHO cell lines. Glycoconj. J. 2011;28:481–492. doi: 10.1007/s10719-011-9348-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Deutscher S.L., Hirschberg C.B. Mechanism of galactosylation in the Golgi apparatus. A Chinese hamster ovary cell mutant deficient in translocation of UDP-galactose across Golgi vesicle membranes. J. Biol. Chem. 1986;261:96–100. [PubMed] [Google Scholar]
- 7.Miura N., Ishida N., Hoshino M., Yamauchi M., Hara T., Ayusawa D., Kawakita M. Human UDP-galactose translocator: molecular cloning of a complementary DNA that complements the genetic defect of a mutant cell line deficient in UDP-galactose translocator. J. Biochem. 1996;120:236–241. doi: 10.1093/oxfordjournals.jbchem.a021404. [DOI] [PubMed] [Google Scholar]
- 8.Wu X., Steet R.A., Bohorov O., Bakker J., Newell J., Krieger M., Spaapen L., Kornfeld S., Freeze H.H. Mutation of the COG complex subunit gene COG7 causes a lethal congenital disorder. Nat. Med. 2004;10:518–523. doi: 10.1038/nm1041. [DOI] [PubMed] [Google Scholar]
- 9.Maszczak-Seneczko D., Sosicka P., Majkowski M., Olczak T., Olczak M. UDP-N-acetylglucosamine transporter and UDP-galactose transporter form heterologous complexes in the Golgi membrane. FEBS Lett. 2012;586:4082–4087. doi: 10.1016/j.febslet.2012.10.016. [DOI] [PubMed] [Google Scholar]
- 10.National Research Council . The National Academies Press; Washington, D.C.: 2012. Transforming Glycoscience: A Roadmap for the Future. [PubMed] [Google Scholar]
- 11.Freeze H.H. Understanding Human Glycosylation Disorders: Biochemistry Leads the Charge. J. Biol. Chem. 2013 doi: 10.1074/jbc.R112.429274. http://www.jbc.org/cgi/doi/10.1074/jbc.R112.429274 Epub. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.