

Abstract
Recently, mutations in genes involved in the biosynthesis of the glycosylphosphatidylinositol (GPI) anchor have been identified in a new subclass of congenital disorders of glycosylation (CDGs) with a distinct spectrum of clinical features. To date, mutations have been identified in six genes (PIGA, PIGL, PIGM, PIGN, PIGO, and PIGV) encoding proteins in the GPI-anchor-synthesis pathway in individuals with severe neurological features, including seizures, muscular hypotonia, and intellectual disability. We developed a diagnostic gene panel for targeting all known genes encoding proteins in the GPI-anchor-synthesis pathway to screen individuals matching these features, and we detected three missense mutations in PGAP2, c.46C>T, c.380T>C, and c.479C>T, in two unrelated individuals with hyperphosphatasia with mental retardation syndrome (HPMRS). The mutations cosegregated in the investigated families. PGAP2 is involved in fatty-acid GPI-anchor remodeling, which occurs in the Golgi apparatus and is required for stable association between GPI-anchored proteins and the cell-surface membrane rafts. Transfection of the altered protein constructs, p.Arg16Trp (NP_001243169.1), p.Leu127Ser, and p.Thr160Ile, into PGAP2-null cells showed only partial restoration of GPI-anchored marker proteins, CD55 and CD59, on the cell surface. In this work, we show that an impairment of GPI-anchor remodeling also causes HPMRS and conclude that targeted sequencing of the genes encoding proteins in the GPI-anchor-synthesis pathway is an effective diagnostic approach for this subclass of CDGs.
Main Text
In the last 2 years, individuals with characteristic phenotypic features including severe neurological abnormalities were reported to have defects in the GPI-anchor-biosynthesis pathway, representing a new subclass of congenital disorders of glycosylation (CDGs).1 Mutations in PIGV (MIM 610274) and PIGO (MIM 614730) were shown to cause hyperphosphatasia with mental retardation syndrome (HPMRS [MIM 239300 and 214749]), which is also referred to as Mabry syndrome.2–7 Individuals with coloboma, congenital heart disease, ichthyosiform dermatosis, mental retardation, and ear anomalies syndrome (CHIME [MIM 280000]), also known as Zunich neuroectodermal syndrome, were reported to have mutations in PIGL (MIM 605947).8 A hypomorphic promoter mutation in PIGM (MIM 610273) causes portal venous thrombosis and absence seizures (MIM 610293).9 Germline mutations in PIGN (MIM 606097) and PIGA (MIM 311770) cause severe syndromes with multiple congenital anomalies, hypotonia, and seizures (MCAHS), now referred to as MCAHS1 (MIM 614080) and MCAHS2 (MIM 300868). Similar to other disorders of glycosylation, disorders caused by mutations interfering with the GPI-anchor pathway are characterized by a remarkable phenotypic diversity whereby the clinical impact seems to depend on the severity of the mutation.10 To date, all identified mutations are hypomorphic and no complete loss of function has been reported in any of these genes. Although distinct phenotypic features seem to be exclusive to single genes or are shared only by a subgroup, the phenotypic features of intellectual disability, seizures, and muscular hypotonia are present in a majority of the individuals described so far.
We therefore included 13 individuals with intellectual disability and elevated serum alkaline phosphatese (ALP) in a mutation screen of all genes encoding proteins in the GPI-anchor-biosynthesis pathway. In these individuals, mutations in PIGV had been excluded by Sanger sequencing. The Charité University Medicine ethics board approved this study, and we obtained informed consent from the responsible persons (parents) on behalf of all study participants. In this work, we report the molecular findings in two unrelated individuals (II-1 of family A and II-1 of family B in Figure 1) with the clinical diagnosis of HPMRS but without identifiable mutations in PIGV and PIGO (Table 1). We performed targeted capture sequencing in the affected individuals of families A and B. Family A is of Finnish origin, and family B is of Turkish origin. For targeted enrichment of exons of all known genes involved in GPI-anchor synthesis, we designed a customized SureSelect library (Agilent) comprising 1,202 different 120 bp oligonucleotide baits in total (see Supplemental Data, available online). Genomic DNA of both individuals was enriched for this target region according to the manufacturer’s protocol, and this was followed by single-read cluster generation on the Cluster Station (Illumina). The captured, purified, and clonally amplified library was then sequenced on an Illumina Genome Analyzer IIx and mapped to the human reference sequence GRCh37, resulting in a mean coverage of above 300-fold for all exons and more than 10-fold for >95% of the target region (see Figure S1). Variants were detected with SAMtools,12 annotated with ANNOVAR,13 and further analyzed in GeneTalk.14 In individual A, we detected a total of 30 single-nucleotide variants with respect to the reference sequence GRCh37, and these included 14 missense mutations, 9 of which were homozygous (Table S2). Three variants not listed in dbSNP135 coded for heterozygous missense mutations: one in PIGZ, c.214G>C (RefSeq accession number NM_025163.2) (p.Asp72His) (RefSeq NP_079439.2), and two in PGAP2, c.[46C>T];[479C>T] (RefSeq NM_001256240.1) (p.[Arg16Trp];[p.Thr160Ile]) (RefSeq NP_001243169.1). In individual B, we observed 32 variants, 17 synonymous and 15 homozygous (Table S3). Only one homozygous missense mutation in PGAP2 (c.380T>C [p.Leu127Ser] [RefSeq NM_001256240.1]) was not listed in dbSNP135.
Figure 1.
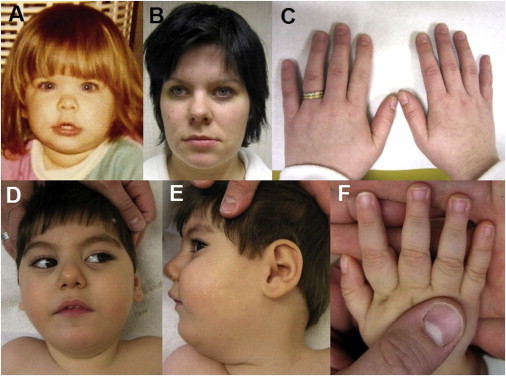
Phenotypic Features of HPMRS Associated with Mutations in PGAP2
(A and B) Face of individual A from family A at the ages of 3 (A) and 28 years (B).
(C) Normal-appearing fingernails of the affected individual in family A.
(D and E) Facial dysmorphism of the affected individual in family B at the age of 2 years includes wide palpebral fissures, a short nose with a broad nasal bridge, a tented upper lip, and a small jaw.
(F) Distal tapering of fingers and mild nail hypoplasia of the fifth digit of the affected individual in family B.
Table 1.
Summary of Clinical Findings in HPMRS-Affected Individuals Carrying PGAP2, PIGO and PIGV Mutations
| Features | Human Phenotype Ontology ID11 | Affected Individual in Family A | Affected Individual in Family B | Individuals with PIGO Mutations (n = 3) | Individuals with PIGV Mutations (n = 14)a |
|---|---|---|---|---|---|
| Sex | NA | female | male | females | 9 females and 5 males |
| Age at last assessment | NA | 28 years | 3.5 years | 20 months to 15 years | 7 months to 17 years |
| Origin | NA | Finnish | Turkish | European | German, Maroccan, Dutch, Polish, British, and European American |
| Height (SD) | NA | −0.9 | +0.6 | −1.4 to −4.2 | normal in 13/14 |
| Weight (SD) | NA | normal | −1.0 | +0.6 to −3.3 | normal in 13/14 |
| OFC (SD) | NA | normal | −4.5 | +0.7 to −5.5 | normal in 12/14 |
| Hyperphoshatasiab | HP:0003155 | + | + | 3/3 | 14/14 |
| Intellectual disabilityb | HP:0001263 | mild | + | 3/3 | 14/14 |
| Age at walking | NA | 18 months | no walking | delayed | delayed |
| Delayed speech and language development | HP:0000750 | − | + | 3/3 | 14/14 |
| Muscular hypotonia | HP:0001252 | − | + | 3/3 | 11/12 |
| Seizures | HP:0001250 | + | + | 1/3 | 9/12 |
| Apparent hypertelorism | HP:0000316 | − | + | 3/3 | + |
| Long palpebral fissures | HP:0000637 | − | + | 3/3 | + |
| Broad nasal bridge | HP: 0000431 | + | + | 3/3 | + |
| Broad nasal tip | HP:0000455 | − | + | 3/3 | + |
| Tented upper lip vermilion | HP:0010804 | + | + | 3/3 | + |
| Brachytelephalangy | HP:0009882 | normal appearing fingernails | short fifth fingernail | 3/3 | 14/14 |
| Anorectal abnormalities and/or constipation | HP:0002025 (anal stenosis) | − | + | 3/3 | 6/12 |
| Aganglionic megacolon | HP:0002251 | − | + | 1/3 | 2/14 |
| Heart defect | HP:0001631 | − | + | + | 1/14 |
| Cleft palate | HP:0000175 | − | + | 0/3 | 3/14 |
| Hearing impairment | HP:0000365 | − | + | 0/3 | 3/14 |
The following abbreviations are used: NA, not applicable; and OFC, occipitofrontal head circumference.
Not all features were documented in the reported individuals.
Consistent features.
All missense mutations (c.46C>T, c.380T>C, and c.479C>T) were analyzed for segregation in available family members (Figure 2). In family A, the mother is a carrier for c.46C>T. The healthy brother is a carrier of c.479C>T, allowing us to infer the same genotype for the father, who was not available for analysis. In family B, both parents and one healthy brother are carriers of c.380T>C, whereas one healthy brother has the wild-type sequence.
Figure 2.
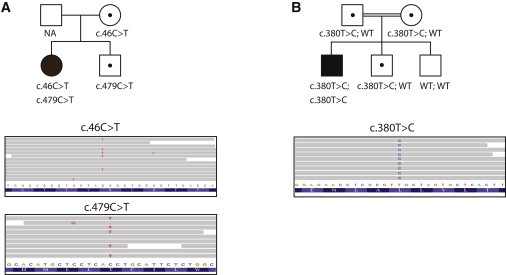
Identification and Segregation of the PGAP2 Mutations
Pedigrees showing segregation of the HPMRS phenotype with deleterious variants in PGAP2 in families A (A) and B (B). Circles represent females, squares represent males, filled symbols represent affected individuals, and dots within the symbols represent heterozygotes. Sequence reads show the mutation in short read alignments visualized in integrative genome viewer.
Individual II-1 of family A is the first child of nonconsanguineous Finnish parents. Her younger brother is healthy. There is a family history of febrile seizures and epilepsy, but not of intellectual disability. Her neonatal period was uneventful, and postnatal development was normal. She started to walk at the age of 18 months, and her initial speech development was normal. At that age, her facial dysmorphism was subtle in that she had only a broad nasal bridge and a tented upper lip (Figure 1A and Table 1). From the age of 8 months to the age of 2.5 years, she suffered from febrile seizures. At the age of 8 years, she began to have tonic-clonic seizures, which responded well to valproic acid. At the age of 22 years, her antiepileptic medication was discontinued and she showed no recurrence of seizures. A physical examination at 28 years revealed a height, weight, and head circumference within the normal range. There was no distinctive facial dysmorphism (Figure 1B). Her fingernails appeared to be normal (Figure 1C). A hand radiograph was not available. Individual II-1 of family A started at an ordinary school but has received special education since the age of 12 years, and she currently works in supported employment.
Her serum ALP activity was measured only once during childhood when she was 10 years old. This elevated value (3,470 U/l; the normal range for the corresponding age is 105–400 U/l) was interpreted as a laboratory mistake. When she was 28 years old, ALP was measured again. These values were repeatedly elevated (2,107–2,448 U/l; the normal range is 35–105 U/l).
Individual II-1 of family B is the third child of consanguineous parents of Turkish origin. The family history is unremarkable. Birth length and weight were normal, and the occipitofrontal head circumference (OFC) at birth was 33 cm (−2 SDs). After birth, physical examination revealed a median cleft palate, which was surgically corrected. Chronic constipation and acute illeus led to the diagnosis of Hirschsprung disease, which was histologically confirmed and surgically repaired. Examinations of this tissue or other tissues for intracellular inclusions were not performed. Echocardiography showed an atrial septal defect. Cranial computed tomography revealed hypoplasia of the corpus callosum.
The boy’s psychomotor development was severely delayed. At the age of 3.5 years, he was still not able to sit, stand, or walk. At the age of 2 years, he had no speech.
Since he was 7 months old, he has suffered from myoclonic and tonic-clonic seizures, which have responded well to anticonvulsants. Electroencephalography investigations indicated multifocal sharp waves. Brainstem auditory-evoked response demonstrated sensorineural hearing loss. Ophthalmologic examination gave normal results.
Physical examination of this 3.5-year-old male showed a height of 104 cm (+0.6 SD), a weight of 14 kg (−1 SD), marked secondary microcephaly and a head circumference of 45 cm (−4.5 SDs), scoliosis, and severe muscular hypotonia (Table 1). Facial dysmorphism included wide palpebral fissures and a wide mouth (Figures 1D and 1E). His fingers showed broad fingernails and a bilateral hypoplastic fifth fingernail (Figure 1F). ALP activity was elevated in repeated tests (2,022 U/l; the normal range is 120–320 U/l for the corresponding age). Conventional cytogenetic analysis gave normal results. Mutations and deletion of ZFHX1B were excluded for ruling out Mowat-Wilson syndrome (MIM 235730).
Thus, both affected individuals presented with intellectual disability, seizures of various degrees, and marked hyperphosphatasia (more than six times the age-adjusted upper limit of the normal range). In addition, mild shortness of fingernails was present in individual II-1 of family B (Table 1).
Whereas individual A, who harbors compound-heterozygous PGAP2 mutations, shows only mild manifestations regarding neurological involvement and physical features, individual B, who has the homozygous c.380T>C mutation, is severely affected by seizures, muscular hypotonia, and marked intellectual disability, as well as various malformations.
In comparison with the specific phenotypic pattern of all previously reported individuals with PIGV and PIGO mutations, the phenotype of individual A broadens the clinical range of HPMRS with the absence of syndrome-specific minor anomalies and malformations and only a mild degree of intellectual disability.
PGAP2 is a membrane protein mainly expressed in the Golgi and is required for reacylation of the lysoform intermediate GPI during fatty-acid remodeling.15 PGAP2 is hypothesized to play a role in the recruitment or recognition of fatty-acid donor substrate.15
All three identified PGAP2 alterations, p.Arg16Trp, Leu127Ser, and Thr160Ile, affect evolutionarily highly conserved amino acid residues (Figure 3) and are predicted to be deleterious by MutationTaster.16 We therefore hypothesized that they might impair the function of PGAP2 (Figure 3). We cloned human PGAP2 (RefSeq NM_00125640.1) from a cDNA library derived from Hep3B (a hepatoma cell line) cells, tagged with FLAG at the N terminus, and subcloned it into pME.17 Altered forms of PGAP2 were generated by site-directed mutagenesis. Altered and wild-type PGAP2 plasmids were transfected by electroporation into human-CD59-expressing PGAP2-deficient Chinese hamster ovary (CHO) cells that were derived from aerolysin-resistant clones from chemically mutagenized CHO cells as previously described.18 The protein levels of CD55 and CD59, both GPI-anchored proteins, at the cell surface were determined by cell staining with anti-FLAG and anti-hamster antibodies and analyzed by flow cytometry (BD FACSCanto II, BD Biosciences) with Flowjo software (Tommy Digital). In PGAP2-deficient cells, fatty-acid remodeling is terminated at the lysoform intermediate GPI as a result of a lack of PGAP2-dependent reacylation. The lysoform GPI-anchored proteins are transported to the cell surface, where they are cleaved by a phospholipase D, resulting in the release of GPI-anchored proteins lacking lipid moiety and a decrease in the cell-surface level of GPI-anchored proteins.15 After transfection, wild-type PGAP2 restored the levels of CD55 and CD59 at the cell surface more efficiently than did the p.Arg16Trp, p.Leu127Ser, and p.Thr160Ile altered forms (Figure 3). Of all three tested alterations, p.Arg16Trp reduced the levels of CD55 and CD59 to a lesser degree than did p.Leu127Ser or p.Thr160Ile. Although it is uncertain whether this result is relevant for the in vivo situation, it might suggest a less severe impairment of PGAP2 function and might correlate with the milder phenotype in individual II-1 of family A.
Figure 3.
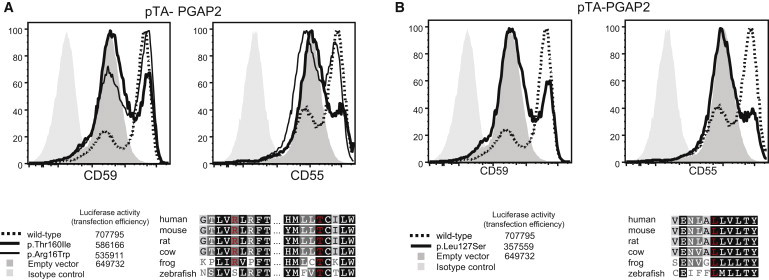
Reduced Activity of Altered Forms of PGAP2 in Restoring Surface Expression of GPI-Anchored Proteins after Transfection into PGAP2-Null Cell Lines
PGAP2-deficient CHO cells were transiently transfected with wild-type or altered forms (p.Arg16Trp, p.Thr160Ile [family A], and p.Leu127Ser [family B]) of pTA Flag-PGAP2 isoform 8 driven by a weak promoter. Restoration of the surface expression was assessed 2 days later by flow cytometry. p.Arg16Trp and p.Thr160Ile detected in family A and p.Leu127Ser detected in family B did not restore the surface expression of CD59 and CD55 as efficiently as the wild-type PGAP2. The reduction of surface protein levels associated with p.Arg16Trp was less severe. This correlates with a lower sequence conservation of this position and a milder phenotype in individual II-1of family A.
Elevated secretion of ALP, which is normally GPI anchored to the cell surface, into the serum leads to hyperphosphatasia. The biochemical mechanisms of hyperphosphatasia in PGAP2-deficient individuals described in this study and in PIGV- or PIGO-deficient individuals reported previously are distinct. In PGAP2-deficient cells, GPI-anchored proteins lacking the lipid moiety and having only the glycan moiety of GPI are released because of a defect in PGAP2-mediated reacylation during fatty-acid exchange in the Golgi and the subsequent cleavage by a phospholipase D after transport to the cell.15,19 In PIGV- or PIGO-deficient cells, the C-terminal GPI-attachment signal peptide of the GPI-anchored protein precursor tentatively acts as a membrane anchor in the endoplasmic reticulum and is cleaved by GPI transamidase but cannot be replaced by a GPI anchor because of a lack of mature GPI synthesis. This abnormality results in the release of soluble proteins completely lacking GPI moiety.20
In summary, we have identified a homozygous missense mutation in PGAP2 in an affected individual with the specific HPMRS phenotype and compound-heterozygous PGAP2 mutations causing a nonsyndromic intellectual-disability phenotype in a second individual. These findings suggest that the clinical range associated with PGAP2 mutations includes severe manifestations of HPMRS and nonsyndromic and mild intellectual disability. Recent data from exome-sequencing studies have shown that mutations of known genes associated with a specific syndrome diagnosis might also be identified in nonsyndromic intellectual disability. This suggests that present syndrome descriptions are strongly biased toward clinically recognizable phenotypes.21,22
Molecular and phenotypic characterization of more individuals with HPMRS will be required for determining whether there are any differences in the phenotypes caused by PIGV, PIGO, and PGAP2 mutations. The comprehensive sequence analysis of HPMRS cases, as well as intellectual-disability cases with a suspected GPI-anchor deficiency indicated by, for example, elevated serum ALP activity, will help to elucidate the phenotypic spectrum of mutations affecting this molecular pathway.
Acknowledgments
This work was supported by a grant from the Bundesministerium für Forschung und Technologie (0313911), by a Deutsche Forschungsgemeinschaft grant to P.M.K. (DFG KR 3985/1-1) and to S.M. (SFB 665), and by grants from the Ministry of Education, Culture, Sports, Science, and Technology and the Ministry of Health, Labour, and Welfare of Japan. We wish to thank all individuals involved in this study for their generous help.
Contributor Information
Peter N. Robinson, Email: peter.robinson@charite.de.
Denise Horn, Email: denise.horn@charite.de.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
1000 Genomes, http://www.1000genomes.org
Agilent eArray, https://earray.chem.agilent.com/earray/
Human Phenotype Ontology, http://www.human-phenotype-ontology.org
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org
NHLBI Exome Sequencing Project (ESP) Exome Variant Server, http://evs.gs.washington.edu/EVS/
GeneTalk, http://www.gene-talk.de
References
- 1.Jaeken J. Congenital disorders of glycosylation (CDG): it’s (nearly) all in it! J. Inherit. Metab. Dis. 2011;34:853–858. doi: 10.1007/s10545-011-9299-3. [DOI] [PubMed] [Google Scholar]
- 2.Horn D., Krawitz P., Mannhardt A., Korenke G.C., Meinecke P. Hyperphosphatasia-mental retardation syndrome due to PIGV mutations: expanded clinical spectrum. Am. J. Med. Genet. A. 2011;155A:1917–1922. doi: 10.1002/ajmg.a.34102. [DOI] [PubMed] [Google Scholar]
- 3.Krawitz P.M., Murakami Y., Hecht J., Krüger U., Holder S.E., Mortier G.R., Delle Chiaie B., De Baere E., Thompson M.D., Roscioli T. Mutations in PIGO, a member of the GPI-anchor-synthesis pathway, cause hyperphosphatasia with mental retardation. Am. J. Hum. Genet. 2012;91:146–151. doi: 10.1016/j.ajhg.2012.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Krawitz P.M., Schweiger M.R., Rödelsperger C., Marcelis C., Kölsch U., Meisel C., Stephani F., Kinoshita T., Murakami Y., Bauer S. Identity-by-descent filtering of exome sequence data identifies PIGV mutations in hyperphosphatasia mental retardation syndrome. Nat. Genet. 2010;42:827–829. doi: 10.1038/ng.653. [DOI] [PubMed] [Google Scholar]
- 5.Mabry C.C., Bautista A., Kirk R.F., Dubilier L.D., Braunstein H., Koepke J.A. Familial hyperphosphatase with mental retardation, seizures, and neurologic deficits. J. Pediatr. 1970;77:74–85. doi: 10.1016/s0022-3476(70)80047-6. [DOI] [PubMed] [Google Scholar]
- 6.Thompson M.D., Roscioli T., Marcelis C., Nezarati M.M., Stolte-Dijkstra I., Sharom F.J., Lu P., Phillips J.A., Sweeney E., Robinson P.N. Phenotypic variability in hyperphosphatasia with seizures and neurologic deficit (Mabry syndrome) Am. J. Med. Genet. A. 2012;158A:553–558. doi: 10.1002/ajmg.a.35202. [DOI] [PubMed] [Google Scholar]
- 7.Thompson M.D., Nezarati M.M., Gillessen-Kaesbach G., Meinecke P., Mendoza-Londono R., Mornet E., Brun-Heath I., Squarcioni C.P., Legeai-Mallet L., Munnich A., Cole D.E. Hyperphosphatasia with seizures, neurologic deficit, and characteristic facial features: Five new patients with Mabry syndrome. Am. J. Med. Genet. A. 2010;152A:1661–1669. doi: 10.1002/ajmg.a.33438. [DOI] [PubMed] [Google Scholar]
- 8.Ng B.G., Hackmann K., Jones M.A., Eroshkin A.M., He P., Wiliams R., Bhide S., Cantagrel V., Gleeson J.G., Paller A.S. Mutations in the glycosylphosphatidylinositol gene PIGL cause CHIME syndrome. Am. J. Hum. Genet. 2012;90:685–688. doi: 10.1016/j.ajhg.2012.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Almeida A.M., Murakami Y., Layton D.M., Hillmen P., Sellick G.S., Maeda Y., Richards S., Patterson S., Kotsianidis I., Mollica L. Hypomorphic promoter mutation in PIGM causes inherited glycosylphosphatidylinositol deficiency. Nat. Med. 2006;12:846–851. doi: 10.1038/nm1410. [DOI] [PubMed] [Google Scholar]
- 10.Freeze H.H. Genetic defects in the human glycome. Nat. Rev. Genet. 2006;7:537–551. doi: 10.1038/nrg1894. [DOI] [PubMed] [Google Scholar]
- 11.Robinson P.N., Köhler S., Bauer S., Seelow D., Horn D., Mundlos S. The Human Phenotype Ontology: a tool for annotating and analyzing human hereditary disease. Am. J. Hum. Genet. 2008;83:610–615. doi: 10.1016/j.ajhg.2008.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li H. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics. 2011;27:2987–2993. doi: 10.1093/bioinformatics/btr509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang K., Li M., Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38:e164. doi: 10.1093/nar/gkq603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kamphans T., Krawitz P.M. GeneTalk: an expert exchange platform for assessing rare sequence variants in personal genomes. Bioinformatics. 2012;28:2515–2516. doi: 10.1093/bioinformatics/bts462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tashima Y., Taguchi R., Murata C., Ashida H., Kinoshita T., Maeda Y. PGAP2 is essential for correct processing and stable expression of GPI-anchored proteins. Mol. Biol. Cell. 2006;17:1410–1420. doi: 10.1091/mbc.E05-11-1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schwarz J.M., Rödelsperger C., Schuelke M., Seelow D. MutationTaster evaluates disease-causing potential of sequence alterations. Nat. Methods. 2010;7:575–576. doi: 10.1038/nmeth0810-575. [DOI] [PubMed] [Google Scholar]
- 17.Takebe Y., Seiki M., Fujisawa J., Hoy P., Yokota K., Arai K., Yoshida M., Arai N. SR alpha promoter: an efficient and versatile mammalian cDNA expression system composed of the simian virus 40 early promoter and the R-U5 segment of human T-cell leukemia virus type 1 long terminal repeat. Mol. Cell. Biol. 1988;8:466–472. doi: 10.1128/mcb.8.1.466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hong Y., Ohishi K., Inoue N., Kang J.Y., Shime H., Horiguchi Y., van der Goot F.G., Sugimoto N., Kinoshita T. Requirement of N-glycan on GPI-anchored proteins for efficient binding of aerolysin but not Clostridium septicum alpha-toxin. EMBO J. 2002;21:5047–5056. doi: 10.1093/emboj/cdf508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Maeda Y., Tashima Y., Houjou T., Fujita M., Yoko-o T., Jigami Y., Taguchi R., Kinoshita T. Fatty acid remodeling of GPI-anchored proteins is required for their raft association. Mol. Biol. Cell. 2007;18:1497–1506. doi: 10.1091/mbc.E06-10-0885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Murakami Y., Kanzawa N., Saito K., Krawitz P.M., Mundlos S., Robinson P.N., Karadimitris A., Maeda Y., Kinoshita T. Mechanism for release of alkaline phosphatase caused by glycosylphosphatidylinositol deficiency in patients with hyperphosphatasia mental retardation syndrome. J. Biol. Chem. 2012;287:6318–6325. doi: 10.1074/jbc.M111.331090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.de Ligt J., Willemsen M.H., van Bon B.W., Kleefstra T., Yntema H.G., Kroes T., Vulto-van Silfhout A.T., Koolen D.A., de Vries P., Gilissen C. Diagnostic exome sequencing in persons with severe intellectual disability. N. Engl. J. Med. 2012;367:1921–1929. doi: 10.1056/NEJMoa1206524. [DOI] [PubMed] [Google Scholar]
- 22.Rauch A., Wieczorek D., Graf E., Wieland T., Endele S., Schwarzmayr T., Albrecht B., Bartholdi D., Beygo J., Di Donato N. Range of genetic mutations associated with severe non-syndromic sporadic intellectual disability: an exome sequencing study. Lancet. 2012;380:1674–1682. doi: 10.1016/S0140-6736(12)61480-9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.