

Abstract
The genetic causes of premature ovarian failure (POF) are highly heterogeneous, and causative mutations have been identified in more than ten genes so far. In two families affected by POF accompanied by hearing loss (together, these symptoms compose Perrault syndrome), exome sequencing revealed mutations in LARS2, encoding mitochondrial leucyl-tRNA synthetase: homozygous c.1565C>A (p.Thr522Asn) in a consanguineous Palestinian family and compound heterozygous c.1077delT and c.1886C>T (p.Thr629Met) in a nonconsanguineous Slovenian family. LARS2 c.1077delT leads to a frameshift at codon 360 of the 901 residue protein. LARS2 p.Thr522Asn occurs in the LARS2 catalytic domain at a site conserved from bacteria through mammals. LARS2 p.Thr629Met occurs in the LARS2 leucine-specific domain, which is adjacent to a catalytic loop critical in all species but for which primary sequence is not well conserved. A recently developed method of detecting remote homologies revealed threonine at this site in consensus sequences derived from multiple-species alignments seeded by human and E. coli residues at this region. Yeast complementation indicated that LARS2 c.1077delT is nonfunctional and that LARS2 p.Thr522Asn is partially functional. LARS2 p.Thr629Met was functional in this assay but might be insufficient as a heterozygote with the fully nonfunctional LARS2 c.1077delT allele. A known C. elegans strain with the protein-truncating alteration LARS-2 p.Trp247Ter was confirmed to be sterile. After HARS2, LARS2 is the second gene encoding mitochondrial tRNA synthetase to be found to harbor mutations leading to Perrault syndrome, further supporting a critical role for mitochondria in the maintenance of ovarian function and hearing.
Main Text
Premature ovarian failure (POF) is a major cause of infertility in young women and is characterized by primary or secondary amenorrhea and elevated levels of gonadotropins long before the natural age of menopause. Severe cases can involve ovarian dysgenesis. Nongenetic causes of POF include autoimmune disorders, viral infection, radiation, or chemotherapy. Genetic causes are highly heterogeneous and include both isolated (nonsyndromic) POF and syndromic forms. Mutations that cause POF have so far been identified in more than ten genes,1–12 but most cases are still unresolved. Perrault syndrome (MIM 233400) is characterized by POF in females and progressive hearing loss in both females and males. Mutations in HSD17B4 (encoding 17-beta hydroxysteroid dehydrogenase 4; MIM 601860), HARS2 (encoding mitochondrial histidyl-tRNA synthetase; MIM 600783), and CLPP (encoding mitochondrial ATP-dependent chambered protease; MIM 601119) are responsible for POF in the context of Perrault syndrome.9,10,12 Similarly to other cases of POF, the majority of cases of Perrault syndrome remain unresolved.
Gene discovery for POF is challenging for a number of reasons: its causes are extremely heterogeneous, infertility limits the size of informative families, and the genes that harbor causal mutations include tRNA-synthetase-encoding genes, among the oldest in evolution. Such extreme antiquity can lead to conserved protein architecture that includes domains both with and without apparent conserved primary sequence, rendering the interpretation of missense mutations even more difficult than usual. In an effort to address this problem, we augmented functional analysis by applying a recently developed protein-sequence-alignment method that generates consensus profiles from deep evolutionary roots.13,14
Genomic DNA samples from unrelated probands with POF and hearing loss and from their unaffected parents were evaluated in our laboratory by exome sequencing according to previously published methods.15 The study was approved by the human subjects committees of the institutional review boards of Shaare Zedek Medical Center (with approval from the Israel National Ethics Committee), Ljubljana University Medical Center, and the University of Washington. The present analysis focuses on two families affected by candidate mutations in the same gene (Figure 1). Family 1 is consanguineous and of Palestinian ancestry. At 17 years old, the proband presented with primary amenorrhea and postmenopausal levels of follicle-stimulating hormone (FSH; 76.9 IU/l) and luteinizing hormone (LH; 30.3 IU/l). Her uterus was prepubertal in size, and her ovaries were not visualized on abdominal ultrasound. The family 1 proband and her two brothers were diagnosed with sensorineural hearing loss at 3–5 years of age. The brothers’ hearing loss is severe at lower frequencies and less severe at higher frequencies, resulting in unusual upsloping audiograms. The profile of hearing loss in the proband is unique in our experience with Perrault-syndrome-affected families. When she was 8 years old, hearing loss in her right ear was mild to moderate in mid frequencies and was mild in her left ear. When she was 17 years old, hearing loss in her right ear was severe at lower frequencies and less severe at higher frequencies, resulting in an upsloping audiogram, whereas hearing loss remained mild in her left ear. She does not use a hearing aid. Family 2 is nonconsanguineous and of Slovenian ancestry. The proband, an only child, presented with POF and severe hearing loss. She had apparently normal menarche at age 13 years and regular menses until age 18 years, but she had no cycles between ages 19 and 30 years, her present age. Her FSH levels at age 30 years were high (101 IU/l). Both probands had 46XX karyotypes, normal neurological function, and normal intelligence. In both families, family structure is consistent with autosomal-recessive inheritance. In family 2, family structure is also consistent with X-linked recessive inheritance or a dominant de novo mutation.
Figure 1.
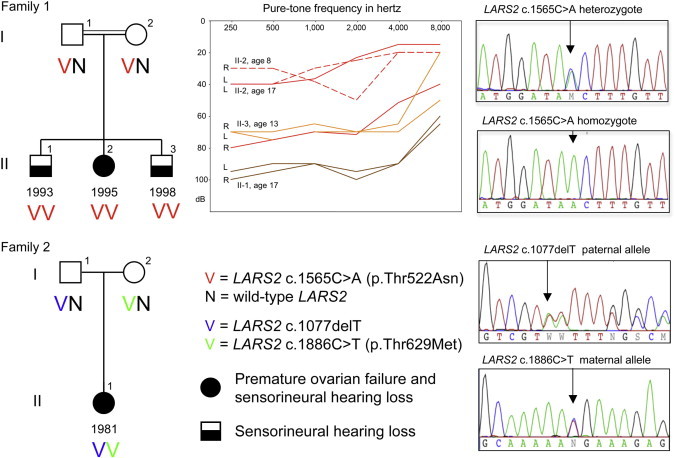
LARS2 Mutations in Families Affected by POF and Hearing Loss
Family 1 is a consanguineous family of Palestinian ancestry. Daughter II-2 has POF, and all three children have sensorineural hearing loss. Pure-tone audiometry measurements demonstrate hearing loss in II-1 (brown line), II-2 (dashed red line [age 8 years] and solid red line [age 17 years]), and II-3 (orange line). Abbreviations are as follows: R, right ear; and L, left ear. The parents are heterozygous and all three children are homozygous for LARS2 c.1565C>A (p.Thr522Asn). Family 2 is a nonconsanguineous family of Slovenian ancestry. Daughter II-1 has POF and severe hearing loss. She is compound heterozygous for paternal allele LARS2 c.1077delT (p.Ile360PhefsTer15) and maternal allele LARS2 c.1886C>T (p.Thr629Met). Mutations validated by Sanger sequencing were first identified by whole-exome sequencing.
Exome sequencing was carried out on paired-end libraries constructed from genomic DNA extracted from blood and sonicated to approximately 200 bp.15 After quality control, the libraries were hybridized to biotinylated DNA oligonucleotide baits from the SeqCap EZ Human Exome Library v.2.0 (Roche NimbleGen), purified by streptavidin-bound magnetic beads, amplified, and sequenced on a HiSeq 2000 instrument (Illumina). The exome design covers 44 Mb of the human genome, corresponding to the exons and flanking intronic regions of ∼300,000 coding exons, 700 miRNAs from miRBase v.14, and 300 noncoding RNAs. Four barcoded samples were multiplexed per lane, generating median coverage of 120-fold; 93% of targeted reads had >10-fold coverage. DNA variants were classified by predicted function: nonsense mutations, frameshift mutations, variants within 1 bp of a splice site, and putatively damaging missense variants (defined as predicted to be damaging by PolyPhen-2 and/or SIFT online tools).
Given that family 1 is consanguineous, we filtered variants of this family’s proband to identify homozygous damaging mutations. Only one mutation fulfilled this criterion: c.1565C>A (p.Thr522Asn) (RefSeq accession number NM_015340.3) at chr3: 45,537,808C>A (hg19) in LARS2 (MIM 604544), encoding mitochondrial leucyl-tRNA synthetase. The variant is predicted to be damaging by PolyPhen-2 (HumDiv score 1.000, HumVar score 1.000) and SIFT (p = 0.00). The variant lies in a 44 Mb region of homozygosity, the largest homozygous region in the proband’s genome, at 3p25.1–p21.1 between approximately 9.7 Mb and 53.7 Mb. PCR amplification and Sanger sequencing indicated that the proband and both her affected brothers are homozygous and their parents are heterozygous for the variant (Figure 1). We filtered variants of the proband of family 2 to identify homozygous, compound heterozygous, or de novo damaging mutations. No potentially damaging homozygous or de novo mutations were detected. The proband was compound heterozygous for one pair of variants both predicted to be damaging: LARS2 c.1077delT (p.Ile360PhefsTer15) (at chr3: 45,527,241) from her father and LARS2 c.1886C>T (p.Thr629Met) (at chr3: 45,557,610) from her mother. LARS2 p.Thr629Met is predicted to be damaging by PolyPhen-2 HumDiv (score 0.999) and SIFT (p = 0.00) and to be possibly damaging by PolyPhen-2 HumVar (score 0.791). None of the three LARS2 mutations were found in 239 Palestinian controls, in 362 Slovenian controls, or in 6,500 controls of European American or African American ancestries with variants released on the National Heart, Lung, and Blood Institute (NHLBI) Exome Variant Server.
LARS2 encodes a 903 aa mitochondrial leucyl-tRNA synthetase (Figure 2A).16 Aminoacyl-tRNA synthetases are evolutionarily conserved enzymes that attach specific amino acids to the 3′ ends of their cognate tRNAs.17 Aminoacyl-tRNA-synthetase activity is required in the cytoplasm and in mitochondria for the translation of nuclearly and mitochondrially encoded genes, respectively. In mammals, most cytoplasmic and mitochondrial tRNA synthetases are encoded by different genes. LARS2 c.1077delT in family 2 is predicted to yield a 373 aa truncated protein with 14 novel amino acids at the C terminus. Parts of the catalytic and editing domains, the leucine-specific domain, and the anticodon-binding domain would be deleted, and it is thus unlikely that a functional protein would be produced. In family 1, the site of the mutant residue, LARS2 p.Thr522, is highly conserved among prokaryotic and eukaryotic species (Figure 2B). In contrast, in family 2, the mutant residue, LARS2 p.Thr629, is at a site conserved in mammals but is poorly conserved in more distantly related species.
Figure 2.
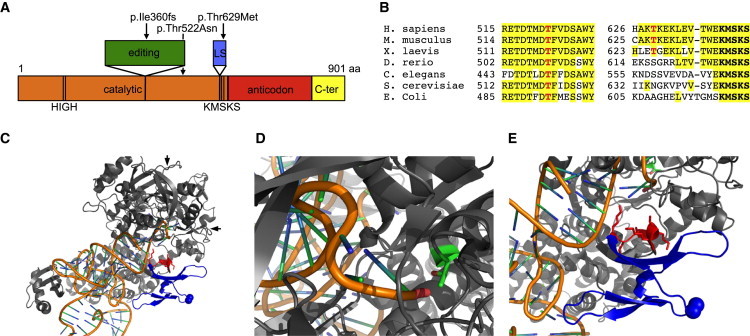
Domain Architecture and Conservation of LARS2 and Structural Analysis of the LARS2 E. coli Ortholog
(A) The domain architecture of LARS2 includes the catalytic domain, the editing domain, the leucine-specific (LS) domain, the anticodon-recognition domain, and the C-terminal (C-ter) domain. HIGH and KMSKS are catalytically important motifs.
(B) Protein sequence alignments of LARS2 orthologs show the regions surrounding altered residues p.Thr522 and p.Thr629 (indicated in red).
(C) Structure of E. coli LeuRS-tRNALeu in the aminoacylation conformation (Protein Data Bank [PDB] 4AQ7). The tRNA backbone is orange, and the bases are shown as blue and green sticks. LeuRS is gray, the leucine-specific domain is blue, and the KMSKS catalytic loop is red. LeuRS p.Thr492 (analogous to human LARS2 p.Thr522) is in green at the intersection point indicated by the two arrows. LeuRS p.Ala508 (analogous to human LARS2 p.Thr629) is indicated by spheres.
(D) A close view of LeuRS p.Thr492 shows that it is in very close proximity to the tRNA 3′ end, to which leucine will be covalently attached. Substitution at this position might shift the position of the tRNA and reduce aminoacylation efficiency.
(E) A close view shows that the KMSKS loop is in close proximity to the tRNA 3′ end and adjacent to the leucine-specific domain. Substitution at the position of LeuRS p.Ala508 with a nonpolar methionine could disrupt the structure and/or position of the leucine-specific domain and thus shift the position of the KMSKS loop and reduce catalytic efficiency.
The three-dimensional structures of the LARS2 E. coli ortholog, LeuRS, reveal possible consequences of the missense mutations in the human families. Two structures of the E. coli enzyme were informative: LeuRS in the aminoacylation conformation in complex with the 3′ end of tRNALeu(UAA) and a nonhydrolyzable analog of the leucyl-adenylate (Protein Data Bank [PDB] 4AQ7) (Figure 2C); and LeuRS in the editing conformation in complex with the 3′ end of tRNALeu(UAA) (PDB 4ARC).18 In family 1, LARS2 p.Thr522 (equivalent to LeuRS p.Thr492), is located in the highly conserved catalytic domain (Figure 2D). The residue sits at the N-terminal end of an α-helix that forms part of a pocket into which the 3′ end of the tRNA strand binds, and the hydroxyl group of the threonine side chain points directly at the terminal phosphate group of the tRNA strand. A substitution at this position could change the position of the 3′ end of the tRNA, causing a reduction in aminoacylation efficiency. In family 2, the mutant missense residue, LARS2 p.Thr629 (equivalent to LeuRS p.Ala508), occurs near the C-terminal end of the poorly conserved leucine-specific domain, just N-terminal to the catalytically important and highly conserved KMSKS loop (Figure 2E).19 In E. coli LeuRS, the KMSKS loop moves as an integral part of the leucine-specific domain during the aminoacylation and proofreading catalytic cycle,18 suggesting that the structure of the leucine-specific domain is important, despite lack of conservation of the primary sequence.
The conserved position of the leucine-specific domain in eukaryotic mitochondrial and bacterial leucyl-tRNA synthetases suggests possible underlying homology and functional conservation. To search for homology, we applied a method that uses iterative comparisons of hidden Markov models to detect remote homology and to build alignments of highly diverse sequences.13,14 As queries, we used 49 residue and 59 residue sequences, including the leucine-specific domains of human LARS2 and E. coli LeuRS, respectively. The two domains are bounded by conserved residues QG at the N-terminal end and KMSKS at the C-terminal end. Each query sequence was aligned to all possible sequences from bacterial, fungal, and protist kingdoms for which any similarity could be detected by the HHblits algorithm (Figure S1, available online). Each set of alignments then yielded a consensus sequence bounded by QG and KMSKS (Figure 3). Despite the fact that human and E. coli sequences differ throughout this domain, the consensus sequences generated by the two deep alignments are identical—KATGEPVV—at the region immediately surrounding LARS2 p.Thr629. Furthermore, methionine is never seen in the position of p.Thr629 in any species (Figure S1). The shared consensus sequences at the region surrounding p.Thr629 suggest that there is indeed underlying homology among species in this region of the protein, that there is likely to be functional and possibly structural conservation, and that threonine at this site is conserved. The consensus region forms a highly hydrophilic and surface-exposed loop attached to a β strand that leads directly to the KMSKS catalytic loop. Substitution of the small threonine side chain with a longer, more aliphatic methionine side chain could affect the loop’s ability to take on an exposed position in the structure.
Figure 3.
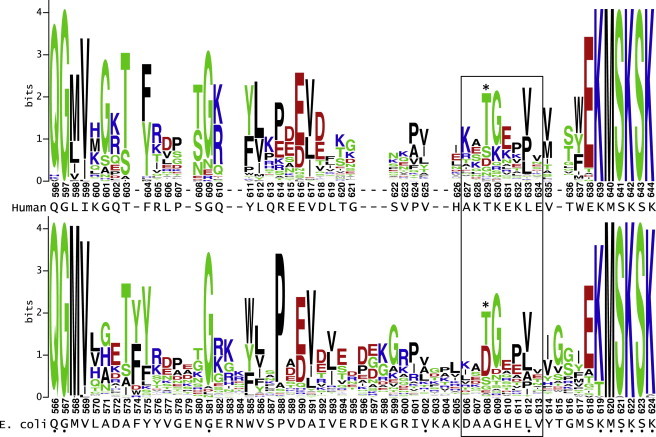
Consensus Sequences of LARS2 Leucine-Specific Domains from Alignments to Human and E. coli Sequences
Despite the dissimilarity in the sequences of human LARS2 and E. coli LeuRS leucine-specific domains, the consensus sequences for alignments to the human (upper) and E. coli (lower) leucine-specific domains are identical in the boxed region surrounding human LARS2 p.Thr629 (indicated by asterisk). Alignments were generated with the HHblits algorithm and displayed with WebLogo. Human and E. coli query sequences are shown below each respective logo. Dots below indicate positions of identity between human and E. coli sequences.
We investigated the functional effects of the LARS2 mutations in families 1 and 2 by using a yeast complementation assay. We determined the ability of wild-type LARS2 and all LARS2 mutant alleles to complement the mitochondrial dysfunction caused by deletion of the Saccharomyces cerevisiae LARS2 ortholog, NAM2. We generated a NAM2 deletion strain (nam2Δ::KAN), which also carried a mutation in the adenine biosynthesis gene ADE2. S. cerevisiae ade2 mutant cells produce a red pigment, but only in the presence of functional mitochondria.20 Cells of the ade2 nam2Δ::KAN strain, expressing EGFP as a negative control, are thus white (Figure 4A). Expression of wild-type human LARS2 or yeast NAM2 restored production of red pigment, indicating rescue of mitochondrial function. LARS2 c.1077delT did not rescue pigment production. Production of red pigment was partially rescued by LARS2 p.Thr522Asn and fully rescued by LARS2 p.Thr629Met. These results indicate that LARS2 c.1077delT is nonfunctional and that the activity of LARS2 p.Thr522Asn is reduced relative to wild-type LARS2. In contrast, when expressed as the sole source of mitochondrial-leucyl-tRNA-synthetase activity, as in this assay, LARS2 p.Thr629Met provided enough activity to support wild-type production of red pigment. We speculate that in the proband of family 2, the compound heterozygous combination of LARS2 p.Thr629Met and LARS2 c.1077delT would yield reduced total LARS2 activity and would thus lead to inadequate mitochondrial function in the ovary and inner ear.
Figure 4.
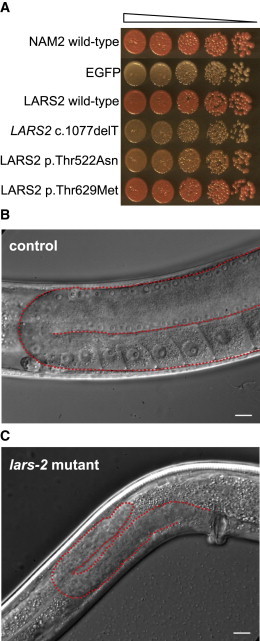
Analysis of Mitochondrial-Leucyl-tRNA-Synthetase Alterations in Yeast and C. elegans
(A) Yeast strain ade2 nam2Δ::KAN was transformed with integrating vectors expressing yeast wild-type mitochondrial leucyl-tRNA synthetase NAM2, negative control EGFP, wild-type LARS2, and each of the three LARS2 alterations. Cells were serially diluted 5-fold, spotted on complete media containing 2% glucose, and incubated for 2 days at 30°C. Red color indicates complementation of the nam2Δ::KAN mutation by the integrated allele.
(B and C) Adult C. elegans (of the strain lars-2(mg312);unc-29(e1072)) homozygous for the protein-truncating alteration LARS-2 p.Trp247Ter (C) are smaller and have smaller gonads (outlined in red) than do control unc-29(e1072) animals with wild-type LARS-2 function (B). The absence of oocytes in the lars-2 mutant strain indicates that germline development is arrested.
To investigate the effects of reduced activity of mitochondrial leucyl-tRNA synthetase on a multicellular organism, we examined a C. elegans strain with alteration LARS-2 p.Trp247Ter, which truncates mitochondrial leucyl-tRNA synthetase.21 This strain, lars-2(mg312);unc-29(e1072), had been described as small, slow growing, sterile, and unable to produce differentiated germ cells. We confirmed that worms homozygous for the lars-2 mutation were completely sterile—compared to the wild-type controls (n > 30), which produced 200–250 progeny per animal, they produced no progeny at all. Germ cell development was arrested such that oocytes were never observed (Figures 4B and 4C).
It is a clinical challenge to identify Perrault syndrome, and hence ovarian failure, among prepubertal girls who present with hearing loss. This challenge is particularly marked given the variability of hearing loss among affected children in families afflicted with Perrault syndrome.10,12 In the families reported here and in some affected persons in the family harboring HARS2 mutations, audiograms have a distinctive upward-sloping shape. Such audiograms offer clues to a possible Perrault phenotype in affected girls. More generally, families affected by recessively inherited, genetically unexplained hearing loss, detected only in prepubertal girls or in boys, might represent new cases of Perrault syndrome.
The identification of two Perrault-syndrome-affected families harboring different LARS2 mutations, evolutionary evidence, and model-organism results together provide strong evidence that mutations in LARS2 cause POF and hearing loss in these families. The 19 mitochondrial tRNA synthetases perform analogous roles in translating the 13 proteins encoded by the mitochondrial genome. Nonetheless, mutations in genes encoding these mitochondrial tRNA synthetases and their cognate tRNAs result in disease phenotypes that are highly variable among genes and, for many of the mitochondrial tRNAs, among alleles of the same gene.17,22–27 Mutations in the genes encoding eight mitochondrial tRNA synthetases other than LARS2 and HARS2 cause various multisystemic disorders involving nervous-system dysfunction, cardiomyopathy, exercise intolerance, anemia, and kidney failure.17,22–26 The mechanisms for such phenotypic diversity are not clear. Cells and tissues might vary in vulnerability to the effects of loss of function of specific tRNA synthetases as a result of cell-type-specific variation in gene expression or in the availability of cofactors required for optimal enzyme activity. In addition, the extreme genetic and allelic heterogeneity might indicate that the components of the mitochondrial translation machinery engage in complex interactions that are not revealed by their canonical activities. For example, in addition to its tRNA-synthetase activity, mitochondrial leucyl-tRNA synthetase in yeast is involved in the splicing of transcripts for mitochondrially encoded proteins.28
The identification of mutations in a fourth gene as responsible for Perrault syndrome underscores the genetic heterogeneity of this phenotype. In our small series of 15 families affected by clinically defined Perrault syndrome, four families’ phenotypes are explained by mutations is HARS2, HSD17B4, and LARS2, seven families have wild-type sequences of these genes and CLPP, and four families are presently being evaluated. Mutations in additional genes associated with Perrault syndrome remain to be discovered.
In mouse models, genetic changes that cause perturbations in mitochondrial protein translation have been shown to lead to hearing loss as a result of tissue-specific apoptosis.29,30 Given the role of apoptosis in ovarian development,31 accelerated or inappropriately timed apoptosis might explain both cardinal features of Perrault syndrome. The observation that mutations in a second gene encoding a mitochondrial tRNA synthetase lead to this phenotype suggests that mutations in genes involved in mitochondrial tRNA processing are candidates for yet unresolved cases and further supports a role for mitochondria in the maintenance of both ovarian function and hearing.
Acknowledgments
We are grateful to Sergey Ovchinnikov for advice on the analysis of protein homology, to Stephen Cusack (European Molecular Biology Laboratory, University of Grenoble, Centre National de la Recherche Scientifique) for providing structural coordinates prior to publication, to Karen Avraham and Zippora Brownstein for technical advice, and to Anne Thornton for technical assistance. This work was supported by National Institutes of Health National Institute on Deafness and Other Communication Disorders grant R01DC005641 (to M.C.K.) and a grant from the US Agency for International Development program for Middle East Research Cooperation (TA-MOU-10-M30-021 to E.L.L.).
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
Homology detection by iterative HMM-HMM comparison (HHblits), http://toolkit.tuebingen.mpg.de/hhblits
Mamit-tRNA: Compilation of mammalian mitochondrial tRNA genes, http://mamit-tRNA.u-strasbg.fr/
NHLBI Exome Sequencing Project (ESP) Exome Variant Server, http://evs.gs.washington.edu/EVS/
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org/
PolyPhen-2, www.genetics.bwh.harvard.edu/pph2/
RSCB Protein Data Bank, http://www.pdb.org/
UCSC Genome Browser, http://genome.ucsc.edu
WebLogo, http://weblogo.berkeley.edu/logo.cgi
References
- 1.Aittomäki K., Lucena J.L.D., Pakarinen P., Sistonen P., Tapanainen J., Gromoll J., Kaskikari R., Sankila E.-M., Lehväslaiho H., Engel A.R. Mutation in the follicle-stimulating hormone receptor gene causes hereditary hypergonadotropic ovarian failure. Cell. 1995;82:959–968. doi: 10.1016/0092-8674(95)90275-9. [DOI] [PubMed] [Google Scholar]
- 2.Bione S., Sala C., Manzini C., Arrigo G., Zuffardi O., Banfi S., Borsani G., Jonveaux P., Philippe C., Zuccotti M. A human homologue of the Drosophila melanogaster diaphanous gene is disrupted in a patient with premature ovarian failure: evidence for conserved function in oogenesis and implications for human sterility. Am. J. Hum. Genet. 1998;62:533–541. doi: 10.1086/301761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Harris S.E., Chand A.L., Winship I.M., Gersak K., Aittomäki K., Shelling A.N. Identification of novel mutations in FOXL2 associated with premature ovarian failure. Mol. Hum. Reprod. 2002;8:729–733. doi: 10.1093/molehr/8.8.729. [DOI] [PubMed] [Google Scholar]
- 4.Di Pasquale E., Beck-Peccoz P., Persani L. Hypergonadotropic ovarian failure associated with an inherited mutation of human bone morphogenetic protein-15 (BMP15) gene. Am. J. Hum. Genet. 2004;75:106–111. doi: 10.1086/422103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lacombe A., Lee H., Zahed L., Choucair M., Muller J.M., Nelson S.F., Salameh W., Vilain E. Disruption of POF1B binding to nonmuscle actin filaments is associated with premature ovarian failure. Am. J. Hum. Genet. 2006;79:113–119. doi: 10.1086/505406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Qin Y., Choi Y., Zhao H., Simpson J.L., Chen Z.-J., Rajkovic A. NOBOX homeobox mutation causes premature ovarian failure. Am. J. Hum. Genet. 2007;81:576–581. doi: 10.1086/519496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhao H., Chen Z.-J., Qin Y., Shi Y., Wang S., Choi Y., Simpson J.L., Rajkovic A. Transcription factor FIGLA is mutated in patients with premature ovarian failure. Am. J. Hum. Genet. 2008;82:1342–1348. doi: 10.1016/j.ajhg.2008.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lourenço D., Brauner R., Lin L., De Perdigo A., Weryha G., Muresan M., Boudjenah R., Guerra-Junior G., Maciel-Guerra A.T., Achermann J.C. Mutations in NR5A1 associated with ovarian insufficiency. N. Engl. J. Med. 2009;360:1200–1210. doi: 10.1056/NEJMoa0806228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pierce S.B., Walsh T., Chisholm K.M., Lee M.K., Thornton A.M., Fiumara A., Opitz J.M., Levy-Lahad E., Klevit R.E., King M.-C. Mutations in the DBP-deficiency protein HSD17B4 cause ovarian dysgenesis, hearing loss, and ataxia of Perrault Syndrome. Am. J. Hum. Genet. 2010;87:282–288. doi: 10.1016/j.ajhg.2010.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pierce S.B., Chisholm K.M., Lynch E.D., Lee M.K., Walsh T., Opitz J.M., Li W., Klevit R.E., King M.-C. Mutations in mitochondrial histidyl tRNA synthetase HARS2 cause ovarian dysgenesis and sensorineural hearing loss of Perrault syndrome. Proc. Natl. Acad. Sci. USA. 2011;108:6543–6548. doi: 10.1073/pnas.1103471108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zangen D., Kaufman Y., Zeligson S., Perlberg S., Fridman H., Kanaan M., Abdulhadi-Atwan M., Abu Libdeh A., Gussow A., Kisslov I. XX ovarian dysgenesis is caused by a PSMC3IP/HOP2 mutation that abolishes coactivation of estrogen-driven transcription. Am. J. Hum. Genet. 2011;89:572–579. doi: 10.1016/j.ajhg.2011.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jenkinson E.M., Rehman A.U., Walsh T., Clayton-Smith J., Lee K., Morell R.J., Drummond M.C., Khan S.N., Naeem M.A., Rauf B. Perrault syndrome is caused by recessive mutations in CLPP, encoding a mitochondrial ATP-dependent chambered protease. Am. J. Hum. Genet. 2013;92 doi: 10.1016/j.ajhg.2013.02.013. Published online March 28, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Remmert M., Biegert A., Hauser A., Söding J. HHblits: lightning-fast iterative protein sequence searching by HMM-HMM alignment. Nat. Methods. 2012;9:173–175. doi: 10.1038/nmeth.1818. [DOI] [PubMed] [Google Scholar]
- 14.Angermüller C., Biegert A., Söding J. Discriminative modelling of context-specific amino acid substitution probabilities. Bioinformatics. 2012;28:3240–3247. doi: 10.1093/bioinformatics/bts622. [DOI] [PubMed] [Google Scholar]
- 15.Walsh T., Shahin H., Elkan-Miller T., Lee M.K., Thornton A.M., Roeb W., Abu Rayyan A., Loulus S., Avraham K.B., King M.-C., Kanaan M. Whole exome sequencing and homozygosity mapping identify mutation in the cell polarity protein GPSM2 as the cause of nonsyndromic hearing loss DFNB82. Am. J. Hum. Genet. 2010;87:90–94. doi: 10.1016/j.ajhg.2010.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bullard J.M., Cai Y.C., Spremulli L.L. Expression and characterization of the human mitochondrial leucyl-tRNA synthetase. Biochim. Biophys. Acta. 2000;1490:245–258. doi: 10.1016/s0167-4781(99)00240-7. [DOI] [PubMed] [Google Scholar]
- 17.Antonellis A., Green E.D. The role of aminoacyl-tRNA synthetases in genetic diseases. Annu. Rev. Genomics Hum. Genet. 2008;9:87–107. doi: 10.1146/annurev.genom.9.081307.164204. [DOI] [PubMed] [Google Scholar]
- 18.Palencia A., Crépin T., Vu M.T., Lincecum T.L., Jr., Martinis S.A., Cusack S. Structural dynamics of the aminoacylation and proofreading functional cycle of bacterial leucyl-tRNA synthetase. Nat. Struct. Mol. Biol. 2012;19:677–684. doi: 10.1038/nsmb.2317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cusack S., Yaremchuk A., Tukalo M. The 2 A crystal structure of leucyl-tRNA synthetase and its complex with a leucyl-adenylate analogue. EMBO J. 2000;19:2351–2361. doi: 10.1093/emboj/19.10.2351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jones B.A., Fangman W.L. Mitochondrial DNA maintenance in yeast requires a protein containing a region related to the GTP-binding domain of dynamin. Genes Dev. 1992;6:380–389. doi: 10.1101/gad.6.3.380. [DOI] [PubMed] [Google Scholar]
- 21.Lee S.S., Lee R.Y., Fraser A.G., Kamath R.S., Ahringer J., Ruvkun G. A systematic RNAi screen identifies a critical role for mitochondria in C. elegans longevity. Nat. Genet. 2003;33:40–48. doi: 10.1038/ng1056. [DOI] [PubMed] [Google Scholar]
- 22.Suzuki T., Nagao A., Suzuki T. Human mitochondrial tRNAs: biogenesis, function, structural aspects, and diseases. Annu. Rev. Genet. 2011;45:299–329. doi: 10.1146/annurev-genet-110410-132531. [DOI] [PubMed] [Google Scholar]
- 23.Steenweg M.E., Ghezzi D., Haack T., Abbink T.E., Martinelli D., van Berkel C.G., Bley A., Diogo L., Grillo E., Te Water Naudé J. Leukoencephalopathy with thalamus and brainstem involvement and high lactate ‘LTBL’ caused by EARS2 mutations. Brain. 2012;135:1387–1394. doi: 10.1093/brain/aws070. [DOI] [PubMed] [Google Scholar]
- 24.Elo J.M., Yadavalli S.S., Euro L., Isohanni P., Götz A., Carroll C.J., Valanne L., Alkuraya F.S., Uusimaa J., Paetau A. Mitochondrial phenylalanyl-tRNA synthetase mutations underlie fatal infantile Alpers encephalopathy. Hum. Mol. Genet. 2012;21:4521–4529. doi: 10.1093/hmg/dds294. [DOI] [PubMed] [Google Scholar]
- 25.Shamseldin H.E., Alshammari M., Al-Sheddi T., Salih M.A., Alkhalidi H., Kentab A., Repetto G.M., Hashem M., Alkuraya F.S. Genomic analysis of mitochondrial diseases in a consanguineous population reveals novel candidate disease genes. J. Med. Genet. 2012;49:234–241. doi: 10.1136/jmedgenet-2012-100836. [DOI] [PubMed] [Google Scholar]
- 26.Bayat V., Thiffault I., Jaiswal M., Tétreault M., Donti T., Sasarman F., Bernard G., Demers-Lamarche J., Dicaire M.J., Mathieu J. Mutations in the mitochondrial methionyl-tRNA synthetase cause a neurodegenerative phenotype in flies and a recessive ataxia (ARSAL) in humans. PLoS Biol. 2012;10:e1001288. doi: 10.1371/journal.pbio.1001288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pütz J., Dupuis B., Sissler M., Florentz C. Mamit-tRNA, a database of mammalian mitochondrial tRNA primary and secondary structures. RNA. 2007;13:1184–1190. doi: 10.1261/rna.588407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Herbert C.J., Labouesse M., Dujardin G., Slonimski P.P. The NAM2 proteins from S. cerevisiae and S. douglasii are mitochondrial leucyl-tRNA synthetases, and are involved in mRNA splicing. EMBO J. 1988;7:473–483. doi: 10.1002/j.1460-2075.1988.tb02835.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Raimundo N., Song L., Shutt T.E., McKay S.E., Cotney J., Guan M.X., Gilliland T.C., Hohuan D., Santos-Sacchi J., Shadel G.S. Mitochondrial stress engages E2F1 apoptotic signaling to cause deafness. Cell. 2012;148:716–726. doi: 10.1016/j.cell.2011.12.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Someya S., Yamasoba T., Kujoth G.C., Pugh T.D., Weindruch R., Tanokura M., Prolla T.A. The role of mtDNA mutations in the pathogenesis of age-related hearing loss in mice carrying a mutator DNA polymerase gamma. Neurobiol. Aging. 2008;29:1080–1092. doi: 10.1016/j.neurobiolaging.2007.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Boumela I., Assou S., Aouacheria A., Haouzi D., Dechaud H., De Vos J., Handyside A., Hamamah S. Involvement of BCL2 family members in the regulation of human oocyte and early embryo survival and death: gene expression and beyond. Reproduction. 2011;141:549–561. doi: 10.1530/REP-10-0504. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.