

Abstract
Scalp-ear-nipple (SEN) syndrome is a rare, autosomal-dominant disorder characterized by cutis aplasia of the scalp; minor anomalies of the external ears, digits, and nails; and malformations of the breast. We used linkage analysis and exome sequencing of a multiplex family affected by SEN syndrome to identify potassium-channel tetramerization-domain-containing 1 (KCTD1) mutations that cause SEN syndrome. Evaluation of a total of ten families affected by SEN syndrome revealed KCTD1 missense mutations in each family tested. All of the mutations occurred in a KCTD1 region encoding a highly conserved bric-a-brac, tram track, and broad complex (BTB) domain that is required for transcriptional repressor activity. KCTD1 inhibits the transactivation of the transcription factor AP-2α (TFAP2A) via its BTB domain, and mutations in TFAP2A cause cutis aplasia in individuals with branchiooculofacial syndrome (BOFS), suggesting a potential overlap in the pathogenesis of SEN syndrome and BOFS. The identification of KCTD1 mutations in SEN syndrome reveals a role for this BTB-domain-containing transcriptional repressor during ectodermal development.
Main Text
Scalp-Ear-Nipple (SEN) syndrome (MIM 181270), or Finlay-Marks syndrome, is a rare autosomal-dominant disorder first described in 1978 by Finlay and Marks,1 and to date, fewer than 15 independent cases have been reported.2–12 SEN syndrome is characterized by aplasia cutis congenita (ACC) of the scalp, breast abnormalities that range from hypothelia or athelia to amastia, and minor anomalies of the external ears (Figure 1). Less frequent clinical characteristics include nail dystrophy, dental anomalies, cutaneous syndactyly of the digits, and renal malformations. The penetrance of SEN syndrome appears to be high, although it exhibits substantial variable expressivity within families. The phenotypic features of SEN syndrome overlap with those of several syndromes, perhaps most notably ulnar-mammary syndrome (MIM 181450) and ectrodactyly with ectodermal dysplasia and cleft lip/palate syndrome (MIM 129900). Nevertheless, despite screening of candidate genes (e.g., TBX3 [MIM 601621] and TP63 [MIM 603273]) in which mutations cause syndromes with similar phenotypic characteristics (data not shown), the genetic basis of SEN syndrome remains unknown.
Figure 1.
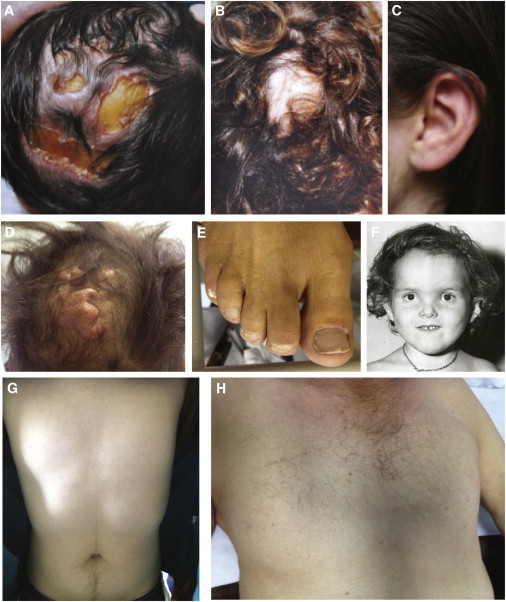
Characteristic Physical Findings in Individuals with SEN Syndrome
Clinical characteristics of SEN syndrome include cutis aplasia of the vertex of the scalp (A, B, and D), minor external ear defects, such as overfolded helix and anteverted ears (C and F), and aplasia of the nipples and sparse or absent secondary sexual hair (G and H). Less common findings include syndactyly and nail dysplasia (E) and craniofacial malformations such as telecanthus, a broad, flat nasal bridge, and frontal bossing (F). Case identifiers E:III-1 (A–C), F:II-1 (F), J:I-1 (D and H), and J:II-1 (E and G) correspond to those in Table 1 and Table S1, where a detailed description of the phenotype of each affected individual is provided.
To identify causative mutations for SEN syndrome, we performed a genome-wide linkage scan under a dominant model (f1 = 1; q = 0.0001) by using short tandem repeats in the ABI PRISM Linkage Mapping Set Version 2.5 MD10 on a large pedigree with 15 affected individuals spanning four generations previously reported by Edwards et al. (family A in Figure 2; Table 1 and Table S1, available online). This linkage-mapping set used 382 autosomal and 18 X-linked microsatellite markers to define an approximately 10 cM resolution human index map. A pairwise maximum LOD score (Zmax) of 2.94 was obtained with the marker D18S53 at a recombination fraction (Θ) of 0.001 (no other linkage peaks were found; Table S2). Haplotype analysis defined a ∼42.9 Mb critical interval containing ∼206 genes between D18S452 and D18S474. Without an obvious candidate gene within this interval to test, we next performed exome sequencing of the two most distantly related affected individuals in family A (Figure 2) and of the affected individuals from two additional unrelated families (families B and C in Figure 2). All studies were approved by the institutional review boards of the University of Washington and Seattle Children’s Hospital, and informed consent was obtained from participants or their parents.
Figure 2.
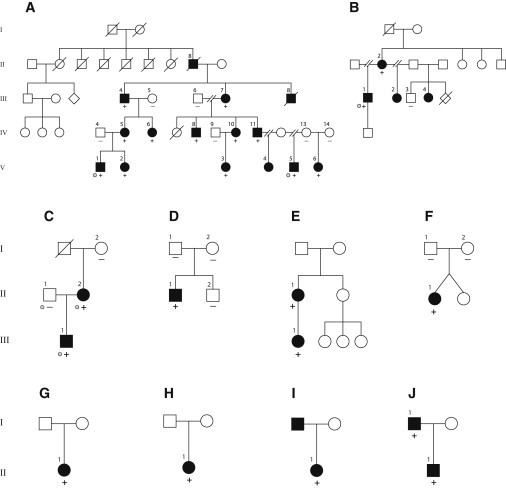
Pedigrees of Families A–J
Pedigrees illustrating the relationships and affected status of ten families affected by SEN syndrome. Case identifiers correspond to those in Table 1 and Table S1. Grey dots indicate individuals who underwent exome sequencing. Plus signs (+) indicate individuals in whom mutations were identified, and minus signs (–) indicate individuals in whom no mutation was found. Family A individuals with a plus or minus sign (except individuals IV-14 and V-6) were also included in the linkage analysis.
Table 1.
Mutations and Clinical Findings of Individuals with SEN Syndrome
| Case | Reference | Ancestry | Gender |
Mutation Information |
Clinical Findings |
||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| KCTD1 Exon | Nucleotide Change | GERP Score | Amino Acid Change | Predicted Effect | Cutis Aplasia | Ear Anomaly | Absent or Hypoplastic Nipples and/or Breast | ||||
| Family A | |||||||||||
| II-8 | Edwards et al.4 | European | male | 2 | c.89C>A | 5.68 | p.Ala30Glu | missense | + | + | + |
| III-4 | Edwards et al.4 | European | male | 2 | c.89C>A | 5.68 | p.Ala30Glu | missense | + | + | + |
| III-7 | Edwards et al.4 | European | female | 2 | c.89C>A | 5.68 | p.Ala30Glu | missense | + | + | + |
| III-8 | Edwards et al.4 | European | male | NT | NT | NT | NT | NT | + | + | − |
| IV-5 | Edwards et al.4 | European | female | 2 | c.89C>A | 5.68 | p.Ala30Glu | missense | + | + | − |
| IV-6 | Edwards et al.4 | European | female | 2 | c.89C>A | 5.68 | p.Ala30Glu | missense | + | + | − |
| IV-8 | Edwards et al.4 | European | male | 2 | c.89C>A | 5.68 | p.Ala30Glu | missense | + | + | + |
| IV-10 | Edwards et al.4 | European | female | 2 | c.89C>A | 5.68 | p.Ala30Glu | missense | + | + | + |
| IV-11 | Edwards et al.4 | European | male | 2 | c.89C>A | 5.68 | p.Ala30Glu | missense | + | + | + |
| V-1 | Edwards et al.4 | European | male | 2 | c.89C>A | 5.68 | p.Ala30Glu | missense | + | ND | ND |
| V-3 | Edwards et al.4 | European | female | 2 | c.89C>A | 5.68 | p.Ala30Glu | missense | + | + | + |
| V-4 | Edwards et al.4 | European | female | NT | NT | NT | NT | NT | + | + | + |
| V-5 | Edwards et al.4 | European | male | 2 | c.89C>A | 5.68 | p.Ala30Glu | missense | + | ND | ND |
| V-6 | Edwards et al.4 | European | female | 2 | c.89C>A | 5.68 | p.Ala30Glu | missense | + | + | + |
| Family B | |||||||||||
| II-2 | NA | European | female | 2 | c.92C>T | 5.68 | p.Pro31Leu | missense | + | + | + |
| III-1 | NA | European | male | 2 | c.92C>T | 5.68 | p.Pro31Leu | missense | + | + | + |
| III-4 | NA | European | female | NT | NT | NT | NT | NT | + | + | + |
| Family C | |||||||||||
| II-2 | NA | European | female | 2 | c.58C>T | 5.68 | p.Pro20Ser | missense | + | + | + |
| III-1 | NA | European | male | 2 | c.58C>T | 5.68 | p.Pro20Ser | missense | + | + | + |
| Family D | |||||||||||
| II-1 | Sobreira et al.9 | Brazilian | male | 2 | c.99C>A | 5.68 | p.His33Gln | missense | + | + | + |
| Family E | |||||||||||
| II-1 | NA | European | female | 2 | c.98A>C | 5.68 | p.His33Pro | missense | + | + | + |
| III-1 | NA | European | female | 2 | c.98A>C | 5.68 | p.His33Pro | missense | + | + | − |
| Family F | |||||||||||
| II-1 | NA | European | female | 3 | c.207C>A | 5.78 | p.Asp69Glu | missense | + | + | + |
| Family G | |||||||||||
| II-1 | Le Merrer et al.12 | North African | female | 2 | c.92C>G | 5.68 | p.Pro31Arg | missense | + | + | + |
| Family H | |||||||||||
| II-1 | Plessis et al.8 | European | female | 3 | c.221A>C | 5.78 | p.His74Pro | missense | + | + | + |
| Family I | |||||||||||
| I-1 | Picard et al.7 | European | male | NT | NT | NT | NT | NT | + | + | + |
| II-1 | Picard et al.7 | European | female | 3 | c.185G>A | 5.63 | p.Gly62Asp | missense | + | − | − |
| Family J | |||||||||||
| I-1 | NA | Brazilian | male | 2 | c.92C>A | 5.68 | p.Pro31His | missense | + | − | + |
| II-1 | NA | Brazilian | male | 2 | c.92C>A | 5.68 | p.Pro31His | missense | + | + | + |
This table depicts mutation information and associated clinical findings for individuals with SEN syndrome. Plus signs (+) indicate the presence of a finding, and minus signs (–) indicate the absence of a finding. GERP scores provide an estimate of conservation across species at a nucleotide site; a more positive score is associated with deleteriousness. An expanded phenotype table is provided in Table S1. The following abbreviations are used: GERP, Genomic Evolutionary Rate Profiling; ND, no data; NA, not applicable; and NT, not tested.
In brief, 1 μg of genomic DNA was subjected to a series of shotgun library construction steps, including fragmentation through acoustic sonication (Covaris), end polishing (NEBNext End Repair Module), A tailing (NEBNext dA-Tailing Module), and ligation of 8 bp barcoded sequencing adaptors (Enzymatics Ultrapure T4 Ligase). Prior to exome capture, the library was amplified via PCR (BioRad iProof). One microgram of barcoded shotgun library was hybridized for the capture of probes targeting 64 Mb of coding exons and miRNA (Roche Nimblegen SeqCap EZ Human Exome Library v.3.0) per the manufacturer’s protocol, and custom blockers complimentary to the full length of the flanking adaptor and barcodes were added. Enriched libraries were amplified via PCR before sequencing (BioRad iProof). Library quality was determined by the examination of molecular-weight distribution and sample concentration (Agilent Bioanalyzer). Pooled, barcoded libraries were sequenced via paired-end 50 bp reads with an 8 bp barcode read on Illumina HiSeq sequencers.
Demultiplexed BAM files were aligned to a human reference (UCSC Genome Browser hg19) with the Burrows-Wheeler Aligner.13 Read data from a flow-cell lane were treated independently for alignment and quality-control purposes in instances where the merging of data from multiple lanes was required. All aligned read data were subjected to: (1) removal of duplicate reads (Picard), (2) indel realignment with the GATK IndelRealigner, and (3) base-quality recalibration with GATK TableRecalibration. Variant detection and genotyping were performed with the UnifiedGenotyper tool from GATK (refv.1.529). Variant data for each sample were formatted (in variant call format [VCF]) as “raw” calls that contained individual genotype data for one or multiple samples. Low-quality and potentially false-positive sites (e.g., those with quality scores ≤ 50, allelic imbalance ≥ 0.75, long homopolymer runs [>3], and/or low quality by depth [<5]) were flagged with the filtration walker (GATK). Variant data were annotated with the SeattleSeq Annotation Server.
The region under the linkage peak was examined for rare and/or unique functional variation, including indels and missense, nonsense, and splice-site mutations, shared among affected individuals in family A. Variants found in dbSNP v.134 or in ∼1,530 exomes from a subset of the National Heart, Lung, and Blood Institute (NHLBI) Exome Sequencing Project (ESP) were excluded prior to analysis for all sequenced individuals. This approach identified two genes, KCTD1 (MIM 613420) and EPG5, with candidate missense variants. In the exome data from the two additional unrelated affected individuals with SEN syndrome, no candidate variants were identified in EPG5, but each individual had a different missense mutation in KCTD1 (Figure 3, Table 1, and Table S1). The presence of each variant found in KCTD1 (RefSeq accession number NM_001258221.1) was subsequently confirmed by Sanger sequencing.
Figure 3.
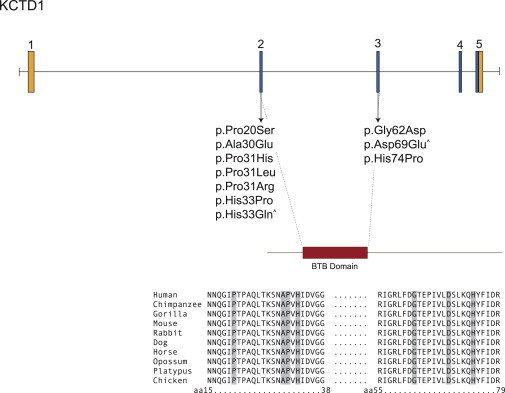
Genomic Structure and Allelic Spectrum of KCTD1 Mutations that Cause SEN Syndrome
KCTD1 is composed of five exons that encode UTRs (orange) and protein-coding-sequence domains (blue), including a BTB domain (red). Arrows indicate the locations of ten different mutations found in ten families affected by SEN syndrome. Carrots indicate mutations that were confirmed to be de novo. Deduced amino acid sequences of partial KCTD1 in multiple species are shown. Locations of amino acid residues affected by KCTD1 mutations are shaded.
To determine the extent to which KCTD1 mutations explain cases of SEN syndrome, we used Sanger sequencing to screen seven additional unrelated families (Figure 2) with a total of nine affected individuals available for sequencing. We identified missense mutations in KCTD1 in all seven families (Figure 3, Table 1, and Table S1). Collectively, between the three families used for discovery and the seven families used for validation studies, KCTD1 mutations were found in all ten kindreds tested. Accordingly, there is no evidence that SEN syndrome is genetically heterogeneous.
In total, ten unique missense KCTD1 mutations were identified in either exon two (n = 7) or exon three (n = 3) of KCTD1 (Figure 3, Table 1, and Table S1). None of the KCTD1 missense mutations identified in the subjects were found in >13,000 chromosomes sequenced as part of the NHLBI ESP. The clinical characteristics of individuals with different KCTD1 mutations were similar to one another (Table 1 and Table S1), despite the variable expressivity of SEN syndrome within families. Additionally, no heterozygous pathogenic KCTD1 variants were found in six tested families affected by dominantly inherited isolated ACC (MIM 107600).
KCTD1 is a member of a family of proteins that contain a potassium-channel tetramerization domain (KCTD).14,15 The amino acid sequence of KCTD1 is highly conserved, and humans and mice differ only by an 8 aa insertion immediately following the start codon; humans and rats differ only by a single amino acid residue.14 The N-terminal KCTD (T1) of KCTD1 overlaps with a bric-a-brac, tram track, and broad complex (BTB) domain (T1-type BTB domain), which is commonly found in transcriptional regulators that contain zinc-finger motifs and that mediate protein-protein interactions.16 The BTB domain of KCTD1 has been shown to mediate the transcriptional repressor activity of KCTD1 and to be required for the homomerization of KCTD1 proteins.14 Thus, alterations within the BTB domain of KCTD1 would be predicted to affect its transcriptional repressor activity.
From Drosophila to humans, highly conserved BTB domains are involved in dimerization, transcriptional repression, and nuclear localization. Crystollographic analysis of the BTB domain in human promyelocytic leukemia zinc finger (PLZF) identified a charged pocket formed by the apposition of two BTB monomers.17 Proper alignment of charge within this pocket is essential for BTB to act as a transcriptional repressor because altering the charge of critical residues (Asp35 and Arg49 in PLZF) in the BTB pocket results in partial or complete loss of repressor activity.
All of the KCTD mutations that cause SEN syndrome are missense mutations predicted to alter BTB-domain residues, including several residues that form the BTB pocket or that are adjacent to such residues. Specifically, in the crystal structure of the PLZF BTB domain, the floor of the pocket is formed by residues 33–35 whereas each wall is formed by residues 63, 64, and 66–68.17 Accordingly, we predict that KCTD1 mutations that cause SEN syndrome are likely to affect the transcriptional repressor activity of the BTB domain. Clustering of missense mutations within a structural hotspot and the absence of stops, frameshifts, and splice-site mutations could mean that the missense mutations we have identified cause loss of function via a dominant-negative mechanism. This would be consistent with the observation that KCTD1 forms homodimers and that some alterations of the PLZF pocket reduce transcriptional repression even though they do not disrupt dimerization. It remains to be determined whether substitutions of amino acids not directly contributing to the functional integrity of the BTB also cause SEN syndrome by disrupting transcriptional repression.
One protein family with which KCTD1 interacts is the AP-2α family of transcription factors. Specifically, KCTD1 interacts with TFAP2A, TFAP2B, and TFAP2C via its BTB domain and acts as a strong repressor of TFAP2A transcriptional activity.15 This is noteworthy because mutations in TFAP2A cause branchiooculofacial syndrome (BOFS [MIM 113620]), one of the features of which is branchial cutis aplasia.18 This observation suggests that the cutis aplasia observed in SEN syndrome might result from altered regulation of TFAP2A. Thus, the identified mutations within the region encoding the BTB domain of KCTD1 in SEN syndrome might affect its inhibitory activity on AP-2α transactivation during embryogenesis and result in developmental defects in skin formation. It is likely that the additional ectodermal abnormalities affecting the ears and nipples in SEN syndrome are a consequence of altered repressor activity of the KCTD1 BTB domain on other transcription factors involved in the formation of these ectodermally derived anatomic structures. This is not surprising given that proteins with a BTB domain have been shown to have multiple roles during embryogenesis and tissue homeostasis, including skin structure.19
In summary, we used linkage analysis and exome sequencing to discover that missense mutations in KCTD1 explain the cause of SEN syndrome in all ten families that we tested. Mutations in KCTD1 were clustered in the region that encodes a functional pocket of its BTB domain, the perturbation of which in other BTB-containing proteins causes loss of disruption of transcriptional activity. The findings reveal a function of the KCTD1 BTB domain during ectodermal development. Our findings also indicate that phenotypic overlap between SEN syndrome and BOFS might be due to a shared defect in the same biological process and suggest that similar clinical cases for which the gene has yet to be discovered might be due to variants in genes that have a similar role.
Acknowledgments
We thank the families for their participation and support; Tracy Dudding-Byth for clinical assistance; Kati Buckingham and Christa Poel for technical assistance; and Jennifer E. Below for discussion. Our work was supported in part by grants from the National Institutes of Health National Human Genome Research Institute (1U54HG006493 to M.B., D.N., and J.S.; 1RC2HG005608 to M.B., D.N., and J.S.; and 5RO1HG004316 to H.T.), the Life Sciences Discovery Fund (2065508 and 0905001), and the Washington Research Foundation.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
FASTX-Toolkit, http://hannonlab.cshl.edu/fastx_toolkit/
Human Genome Variation Society, http://www.hgvs.org/mutnomen/
Illumina HumanCytoSNP-12 DNA Analysis BeadChip Kit, http://www.illumina.com/products/humancytosnp_12_dna_analysis_beadchip_kits.ilmn
NHLBI Exome Sequencing Project (ESP) Exome Variant Server, http://evs.gs.washington.edu/EVS/
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org/
Picard, http://picard.sourceforge.net/
SAMtools, http://samtools.sourceforge.net/
SeattleSeq Annotation 137, http://snp.gs.washington.edu/SeattleSeqAnnotation137/
References
- 1.Finlay A.Y., Marks R. An hereditary syndrome of lumpy scalp, odd ears and rudimentary nipples. Br. J. Dermatol. 1978;99:423–430. doi: 10.1111/j.1365-2133.1978.tb06182.x. [DOI] [PubMed] [Google Scholar]
- 2.Al-Gazali L., Nath R., Iram D., Al Malik H. Hypotonia, developmental delay and features of scalp-ear-nipple syndrome in an inbred Arab family. Clin. Dysmorphol. 2007;16:105–107. doi: 10.1097/MCD.0b013e3280147217. [DOI] [PubMed] [Google Scholar]
- 3.Baris H., Tan W.H., Kimonis V.E. Hypothelia, syndactyly, and ear malformation—a variant of the scalp-ear-nipple syndrome?: Case report and review of the literature. Am. J. Med. Genet. A. 2005;134A:220–222. doi: 10.1002/ajmg.a.30612. [DOI] [PubMed] [Google Scholar]
- 4.Edwards M.J., McDonald D., Moore P., Rae J. Scalp-ear-nipple syndrome: additional manifestations. Am. J. Med. Genet. 1994;50:247–250. doi: 10.1002/ajmg.1320500307. [DOI] [PubMed] [Google Scholar]
- 5.Naik P., Kini P., Chopra D., Gupta Y. Finlay-Marks syndrome: report of two siblings and review of literature. Am. J. Med. Genet. A. 2012;158A:1696–1701. doi: 10.1002/ajmg.a.35389. [DOI] [PubMed] [Google Scholar]
- 6.Paik Y.S., Chang C.W. Stahl ear deformity associated with Finlay-Marks syndrome. Ear Nose Throat J. 2010;89:256–257. [PubMed] [Google Scholar]
- 7.Picard C., Couderc S., Skojaei T., Salomon R., de Lonlay P., Le Merrer M., Munnich A., Lyonnet S., Amiel J. Scalp-ear-nipple (Finlay-Marks) syndrome: a familial case with renal involvement. Clin. Genet. 1999;56:170–172. doi: 10.1034/j.1399-0004.1999.560216.x. [DOI] [PubMed] [Google Scholar]
- 8.Plessis G., Le Treust M., Le Merrer M. Scalp defect, absence of nipples, ear anomalies, renal hypoplasia: another case of Finlay-Marks syndrome. Clin. Genet. 1997;52:231–234. doi: 10.1111/j.1399-0004.1997.tb02553.x. [DOI] [PubMed] [Google Scholar]
- 9.Sobreira N.L., Brunoni D., Cernach M.C., Perez A.B. Finlay-Marks (SEN) syndrome: a sporadic case and the delineation of the syndrome. Am. J. Med. Genet. A. 2006;140:300–302. doi: 10.1002/ajmg.a.31063. [DOI] [PubMed] [Google Scholar]
- 10.Taniai H., Chen H., Ursin S. Finlay-Marks syndrome: another sporadic case and additional manifestations. Pediatr. Int. 2004;46:353–355. doi: 10.1111/j.1442-200x.2004.01905.x. [DOI] [PubMed] [Google Scholar]
- 11.Aase J.M., Wilroy S.R. The Finlay-Marks (SEN) syndrome: Report of a new case and review of the literature. Proceedings of the Greenwood Genetics Center. 1988;7:177–178. [Google Scholar]
- 12.Le Merrer M., Renier D., Briard M.L. Scalp defect, nipples absence and ears abnormalities: an other case of Finlay syndrome. Genet. Couns. 1991;2:233–236. [PubMed] [Google Scholar]
- 13.Li H., Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ding X.F., Luo C., Ren K.Q., Zhang J., Zhou J.L., Hu X., Liu R.S., Wang Y., Gao X., Zhang J. Characterization and expression of a human KCTD1 gene containing the BTB domain, which mediates transcriptional repression and homomeric interactions. DNA Cell Biol. 2008;27:257–265. doi: 10.1089/dna.2007.0662. [DOI] [PubMed] [Google Scholar]
- 15.Ding X., Luo C., Zhou J., Zhong Y., Hu X., Zhou F., Ren K., Gan L., He A., Zhu J. The interaction of KCTD1 with transcription factor AP-2alpha inhibits its transactivation. J. Cell. Biochem. 2009;106:285–295. doi: 10.1002/jcb.22002. [DOI] [PubMed] [Google Scholar]
- 16.Zollman S., Godt D., Privé G.G., Couderc J.L., Laski F.A. The BTB domain, found primarily in zinc finger proteins, defines an evolutionarily conserved family that includes several developmentally regulated genes in Drosophila. Proc. Natl. Acad. Sci. USA. 1994;91:10717–10721. doi: 10.1073/pnas.91.22.10717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ahmad K.F., Engel C.K., Privé G.G. Crystal structure of the BTB domain from PLZF. Proc. Natl. Acad. Sci. USA. 1998;95:12123–12128. doi: 10.1073/pnas.95.21.12123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Milunsky J.M., Maher T.A., Zhao G., Roberts A.E., Stalker H.J., Zori R.T., Burch M.N., Clemens M., Mulliken J.B., Smith R., Lin A.E. TFAP2A mutations result in branchio-oculo-facial syndrome. Am. J. Hum. Genet. 2008;82:1171–1177. doi: 10.1016/j.ajhg.2008.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gebhardt A., Kosan C., Herkert B., Möröy T., Lutz W., Eilers M., Elsässer H.P. Miz1 is required for hair follicle structure and hair morphogenesis. J. Cell Sci. 2007;120:2586–2593. doi: 10.1242/jcs.007104. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.