

Abstract
Perrault syndrome is a genetically and clinically heterogeneous autosomal-recessive condition characterized by sensorineural hearing loss and ovarian failure. By a combination of linkage analysis, homozygosity mapping, and exome sequencing in three families, we identified mutations in CLPP as the likely cause of this phenotype. In each family, affected individuals were homozygous for a different pathogenic CLPP allele: c.433A>C (p.Thr145Pro), c.440G>C (p.Cys147Ser), or an experimentally demonstrated splice-donor-site mutation, c.270+4A>G. CLPP, a component of a mitochondrial ATP-dependent proteolytic complex, is a highly conserved endopeptidase encoded by CLPP and forms an element of the evolutionarily ancient mitochondrial unfolded-protein response (UPRmt) stress signaling pathway. Crystal-structure modeling suggests that both substitutions would alter the structure of the CLPP barrel chamber that captures unfolded proteins and exposes them to proteolysis. Together with the previous identification of mutations in HARS2, encoding mitochondrial histidyl-tRNA synthetase, mutations in CLPP expose dysfunction of mitochondrial protein homeostasis as a cause of Perrault syndrome.
Main Text
More than 400 syndromic forms of deafness have been defined.1 The specific genes causing some of these conditions have been identified, providing important insights into the molecular pathways and structures impaired in sensorineural hearing loss (SNHL).2 Perrault syndrome (MIM 233400) is an autosomal-recessive disorder characterized by SNHL and premature ovarian failure (POF) secondary to ovarian dysgenesis.3 It is clinically and genetically heterogeneous.4,5 A spectrum of additional clinical features, including cerebellar ataxia, learning disability, and peripheral neuropathy, have been described in some affected individuals.5–7
Mutations in two genes, HSD17B4 (MIM 601860), encoding D-bifunctional protein (DBP), and HARS2 (MIM 600783), encoding mitochondrial histidyl-tRNA synthetase, have been identified as underlying causes of Perrault syndrome,8,9 but these genes do not account for all cases of this heterogeneous condition.5,8,9 In our studies of hereditary deafness and Perrault syndrome, written informed consent was obtained from all participants after approval from the University of Manchester (reference 06138) and National Health Service ethics committees (approval 06/Q1406/52), the University of Washington (protocol 33468), the Combined Neuroscience Institutional Review Board (protocol IRB OH93-DC-0016) at the National Institutes of Health, and the institutional review boards at the National Centre of Excellence in Molecular Biology, the University of the Punjab (Lahore, Pakistan), the Quaid-I-Azam University (Islamabad, Pakistan), and the Baylor College of Medicine and Affiliated Hospitals (protocol H-17566).
Family PDF1 is a consanguineous Pakistani family living in the United Kingdom (Figure 1).5 All three affected sisters have profound congenital SNHL (>90 decibels hearing level [dBHL] at all test frequencies [Figure S1, available online]). The youngest affected sibling (II-6) was evaluated for delayed puberty at 15 years of age. Pelvic ultrasonography revealed streak ovaries and a hormone profile consistent with hypergonadotropic hypogonadism (Table 1). Subsequent hormone profiles in her elder siblings (II-2 and II-4) with hearing loss revealed elevated gonadotropin levels consistent with incipient POF. The menstrual cycles of both sisters had become erratic with infrequent menses. Of note, the oldest sister (II-2) had given birth to two healthy sons, demonstrating significant prior ovarian reserve, despite the fact that only one of her ovaries was detectable on pelvic ultrasonography when she was 22 years old. All three affected siblings were prescribed estrogen replacement therapy for the prevention of osteoporosis. In addition to having SNHL and ovarian failure, the three affected siblings have epilepsy, short stature (less than the third percentile), microcephaly (less than the third percentile), and moderate learning difficulties. Phenotypic features also include truncal and cerebellar ataxia with signs of lower-limb spasticity. An MRI brain scan of the eldest affected sibling (II-2) showed abnormally high signal intensity in the deep white matter and corticospinal tract (data not shown).
Figure 1.
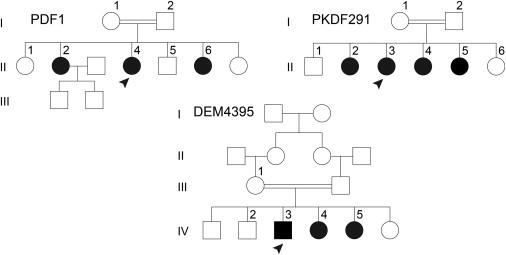
Pedigrees of the Three Families—PDF1, PKDF291, and DEM4395—Affected by Homozygous CLPP Mutations
Numbers are assigned only to individuals whose DNA was available for this study. Arrowheads denote individuals whose genomic DNA was subjected to exome sequencing. Parents of the six siblings in family PKDF291 have the same great-great grandparents.10 A double horizontal line denotes a consanguineous union. Family PDF1 was ascertained because of profound hearing loss in three sisters with subsequent POF. Families PKDF291 and DEM4395 first came to attention because of profound hearing loss in the affected family members. In family PKDF291, POF was revealed by subsequent evaluation of the affected sisters. In family DEM4395, no hormonal evaluation was possible.
Table 1.
Hormone Profiles of Individuals from Families PDF1 and PKDF291
| Family | Individual (Age at Assessment) | FSH (Normal Range) | LH (Normal Range) | Estradiol (Normal Range) |
|---|---|---|---|---|
| PDF1 | II-2 (22 years) | 41.5 (2–14 IU/l) | 64.6 (2–14 IU/l) | 777 (70–1,480 pmol/l) |
| II-4 (21 years) | 24.6 (2–14 IU/l) | 17.7 (2–14 IU/l) | 281 (70–1,480 pmol/l) | |
| II-6 (15 years) | 45 (2–14 IU/l) | 104 (2–14 IU/l) | 89 (70–1,480 pmol/l) | |
| PKDF291 | II-2 (15 years) | 111 (2.8–11.1 IU/l) | 29.4 (0–11.6 IU/l) | <20 (ND–160 pg/ml) |
| II-3 (20 years) | 104 (2.8–11.1 IU/l) | 49.8 (0–11.6 IU/l) | <20 (ND–160 pg/ml) | |
| II-4 (23 years) | 81 (2.8–11.1 IU/l) | 26 (0–11.6 IU/l) | <20 (ND–160 pg/ml) | |
| II-5 (25 years) | 101 (2.8–11.1 IU/l) | 31 (0–11.6 IU/l) | <20 (ND–160 pg/ml) | |
| II-6 (18 years) (unaffected) | 3.28 (1.2–9 IU/l) | 4.85 (0–14.7 IU/l) | 185 (27–246 pg/ml) |
The following abbreviations are used: FSH, follicle stimulating hormone; LH, luteinizing hormone; and ND, not determined.
Genome-wide homozygosity mapping of the three affected sisters and an unaffected sibling from family PDF1 was performed with an Affymetrix Human Array 6.0, as previously described,11 and was analyzed with AutoSNPa software.12 The only >1 Mb homozygosity region shared by the three affected sisters, but not by their unaffected sister, was chr19: 5,765,869–16,392,163 (reference human genome sequence GRCh37, UCSC hg19), flanked by SNPs rs4366824 and rs3852916, in the region 19p13.3–p13.11 (Figure S2). This 10.63 Mb region includes approximately 300 annotated genes.
A second large unrelated consanguineous Pakistani family, PKDF291 (Figure 1 of Rehman et al.10), affected by severe to profound congenital SNHL was also found to display linkage to chromosomal region 19p13. Linkage analysis, which was performed with 388 microsatellite markers, defined a 4.17 Mb SNHL locus with a maximum multipoint LOD score of 3.35 for the marker D19S391, located in this region.10 This locus, designated DFNB81, encompasses 104 genes. GIPC3 (MIM 608792), which is located adjacent to the DFNB81 interval, was Sanger sequenced, and no mutations in the affected individuals of family PKD291 were identified.10 Further evaluation of the phenotypic features revealed primary amenorrhea and hormone profiles indicative of hypergonadotropic hypogonadism in all four affected female siblings (Table 1). The three elder affected female siblings (PKDF291 II-3, II-4, and II-5) each had a rudimentary uterus and small ovaries on pelvic ultrasonography, whereas the youngest affected sibling (II-2) had a small uterus and normal-sized ovaries at 15 years of age. The unaffected sibling (II-6) had normal imaging of her uterus and ovaries. There was no evidence of learning disability, microcephaly, short stature, epilepsy, or neurological deficit in this family.
In a third consanguineous Pakistani family, DEM4395 (Figure 1), a 25-year-old male (IV-3; Figure S1) and two of his sisters were found to have profound congenital SNHL (>90 dBHL at 250–8,000 Hz). Both affected female siblings, IV-4 and IV-5, were reported to have normal menstrual cycles at ages 28 and 22 years, respectively, although formal evaluation of hormone profiles was not possible. No additional medical problems were self-reported by this family.
DNA samples from five individuals in family DEM4395 were genotyped at the Center for Inherited Disease Research with the use of the Illumina Linkage Panel 12. In chromosomal region 19p13, a 15.64 Mb homozygous locus flanked by 19pter and rs1273522 and with a maximum multipoint LOD score of 2.53 was observed. GIPC3 was included in the region of homozygosity in family DEM4395, and Sanger sequencing of GIPC3 coding exons revealed no mutations.
Exome sequencing was undertaken for affected individuals II-4 from family PDF1, II-3 from family PKDF291, and IV-3 from family DEM4395 (Table S1). For families PDF1 and DEM4395, exome sequencing was independently carried out in the King lab and in the Department of Genome Sciences sequencing core, respectively, at the University of Washington with the use of previously reported methods,13,14 whereas family PKDF291 was exome sequenced on an Applied Biosystems SOLiD 5500 platform in the National Institute on Deafness and Other Communication Disorders sequencing core.15 All high-quality reads were mapped to the reference human genome sequence (GRCh37, UCSC hg19).
The mapped loci in chromosomal region 19p13 overlapped in the three families, and homozygous variants absent from the National Heart, Lung, and Blood Institute (NHLBI) Exome Variant Server (ESP6500) were identified in only one gene, CLPP (MIM 601119), in the three families. The three variants were confirmed by PCR amplification from genomic DNA and subsequent Sanger sequence analysis (Figure 2). In family PDF1, a missense variant, c.433A>C (RefSeq accession number NM_006012) (p.Thr145Pro [RefSeq NP_006003]), was identified at chr19: 6,364,528. In family PKDF291, another missense variant, c.440G>C (p.Cys147Ser), was identified at chr19: 6,364,535. Both variants cosegregated with the phenotype and were absent in 193 (for c.433A>C [p.Thr145Pro]) and 483 (for c.440G>C [p.Cys147Ser]) ethnically matched controls. Evidence of pathogenicity was supported by the high conservation of both substituted residues (Figure 2C) and structural defects predicted from the analysis of the primary amino acid sequence. SIFT, MutationTaster, and PolyPhen-2 predicted both substitutions to be damaging. Sanger DNA sequencing of all coding exons of CLPP in 20 other Perrault-syndrome-affected families from the University of Washington8,9 and University of Manchester5 cohorts did not reveal other families with mutations.
Figure 2.
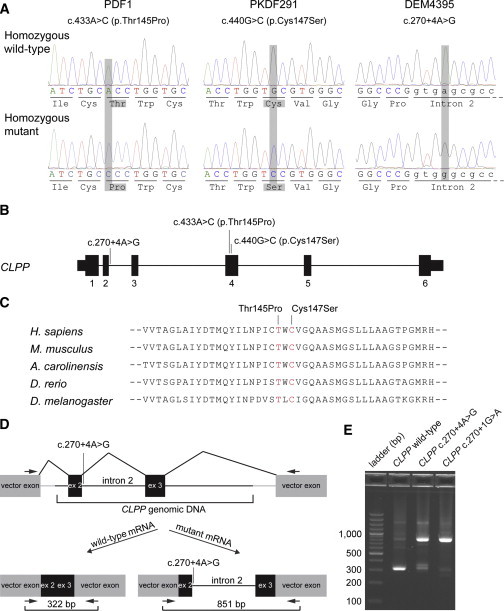
Identification of CLPP Mutations in Families PDF1, PKDF291, and DEM4395
(A) Chromatograms obtained from a wild-type control and homozygous affected individuals from three families. Mutations are highlighted in gray.
(B) Gene structure of CLPP and the location of the three mutations identified in this study. Thin bars represent 5′ and 3′ UTRs, and thick bars represent exons. The horizontal lines joining exons represent intronic sequence.
(C) A ClustalW alignment of CLPP orthologs in five animal species from human to Drosophila shows conservation of residues 145 and 147, which correspond to the substitutions identified in families PDF1 and PKDF291, respectively.
(D) Schematic presentation of the exon-splicing-assay vector used in COS-7 cells for the evaluation of the predicted splice-site mutation. CLPP exon 2, either with a wild-type donor site or a mutant donor splice site (c.270+4A>G or control c.270+1G>A), was cloned into the pSPL3 expression vector (gray). The CLPP sequence also included intron 2, exon 3, and 660 bp of flanking intronic sequences (black). Horizontal arrows indicate locations of vector-specific primers used for PCR amplification of cDNA containing the CLPP sequence.
(E) Splicing-assay products were separated by size on a 2% agarose gel for the wild-type allele, the DEM4395 c.270+4A>G mutant allele, and the c.270+1G>A control mutation. The 851 bp band is from CLPP transcripts that include intron 2. Sequencing of the two ∼300–350 bp gel-purified bands demonstrates that the c.270+4A>G mutant allele results in some wild-type splicing between CLPP exons 2 and 3. Additional splicing assay data in Table S2 support this conclusion.
A homozygous CLPP variant, c.270+4A>G at chr19: 6,361,955, was identified in the three affected individuals of family DEM4395. This variant was absent in 386 ethnically matched controls. MutationTaster predicted it to abolish the splice donor site of CLPP exon 2. Because CLPP mRNA could not be amplified from saliva samples of either affected individuals or controls (Oragene RNA, DNA Genotek), the effect of CLPP c.270+4A>G on splicing was tested experimentally in COS-7 cells (Figures 2D and 2E and Table S2). Splicing assays compared the wild-type allele, the CLPP c.270+4A>G mutant allele of family DEM4395, and CLPP c.270+1G>A, a donor-site control mutation expected to ablate exon 2 splicing to exon 3. For the wild-type sequence, 62% (23/37) of cloned transcripts had canonical splicing of exons 2 and 3, 30% (11/37) of clones retained intron 2, and 8% (3/37) of clones had other aberrant transcripts. For control CLPP c.270+1G>A, all (39/39) clones retained intron 2. The CLPP c.270+4A>G allele was evaluated in four experiments. From all four experiments combined, 84% (41/49) of clones from CLPP c.270+4A>G transcripts retained intron 2, 4% (2/49) of clones spliced at a cryptic donor site (CLPP c.255_256G>T), 10% (5/49) of clones had aberrant splicing that excluded either exon 2 or exon 3 and were most likely artifacts, and 2% (1/49) of clones had wild-type splicing. Wild-type splicing was independently confirmed in additional experiments. Nested PCR splicing products (of approximately 300–350 bp) detected on an agarose gel indicated possible wild-type splicing for the c.270+4A>G allele, and we confirmed this by gel purification, cloning, and Sanger sequencing (Figure 2E). Intron 2 includes stop codons in all reading frames. If the reading frame of exon 2 is retained, then translation termination occurs 38 codons after wild-type codon 90. We conclude from these data that, as predicted, the c.270+4A>G mutant allele of CLPP does not fully ablate donor splice-site function but rather weakens it.
Human CLPP (EC 3.4.21.92; UniProtKB Q16740) is the caseinolytic peptidase, proteolytic subunit homolog of E. coli ClpP. The protein is highly conserved at the primary amino acid sequence level through the quaternary structure level among all prokaryotes and eukaryotes.16–19 CLPP is an endopeptidase component of a mitochondrial ATP-dependent proteolytic machine.16 Degradation of proteins by CLPP occurs when an unfolded polypeptide chain is translocated into its interior chamber. The active unit of CLPP is a barrel-shaped tetradecamer, which results from the face-to-face association of two heptameric rings.18–20 This barrel creates the large central cavity in which proteolysis occurs. Substrate peptides enter the chamber via an axial pore and are exposed to 14 active sites within the barrel. In order to allow proteolysis of larger substrates, the CLPP tetradecamer binds the hexameric protein CLPX (caseinolytic peptidase X). CLPX is an AAA+ protein family member (i.e., an ATPase associated with a wide variety of cellular activities). CLPX recognizes and unfolds specific protein substrates. The newly unfolded peptides are extruded through the central pore of its hexameric ring into the CLPP proteolytic chamber.21
Multiple crystal structures and electron-microscopy density maps have been determined for CLPP. These structures can be used for predicting the consequences of amino acid substitutions in the protein. Crystal structures include a 2.1-Å structure of human CLPP and several bacterial ClpP orthologs.18,19,22–31 Each CLPP monomer consists of a globular “head” domain and an extended “handle” domain (Figure 3A). The catalytic triad, Ser153 (97)-His178 (122)-Asp227 (171), is formed in the cleft between these two domains. The parenthetical numbers are the residues after removal of 56 amino acids composing the N-terminal mitochondrial targeting sequence (MTS); the MTS is cleaved after translocation of CLPP to the mitochondrion, yielding the mature polypeptide. The head region, in which the missense substitutions of families PDF1 and PKDF291 are found, consists of two perpendicular β sheets associated with six α helices (Figure 3B). The two affected residues lie within the β-3 strand of the first β sheet and contribute to backbone hydrogen bonding of the β sheet. The side chain of residue Thr145 (89) can also form a hydrogen bond with residue Leu159 (103) of the α4 helix, which contains the catalytic residue Ser153 (97). Substitution of Thr145 (89) with proline (p.Thr145Pro) removes the possibility of hydrogen-bond formation to Leu159 (103) and Met166 (110). Indeed, the β sheet properties of proline are likely to cause significant disruption of the structure in this region. The consequences of the replacement of Cys147 (91) with serine (p.Cys147Ser) are less clear, but one result would be reduction in hydrophobicity of the side chain.
Figure 3.
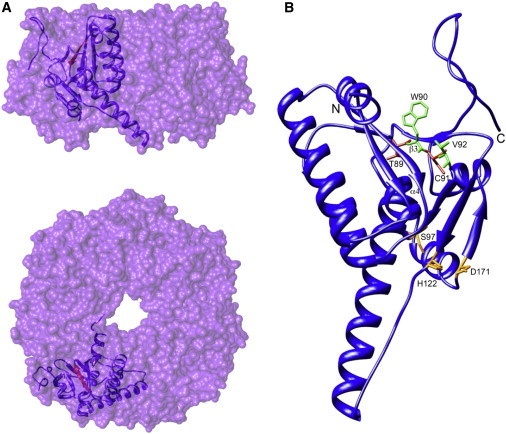
Location of Substitutions within the Crystal Structure of Human CLPP
(A and B) Surface representations show the side (A) and top (B) views of a single heptameric ring of CLPP subunits (Protein Data Bank 1tg6). The ribbon representation shows a single monomer within the ring in each case. The β strand affected by the two substitutions is highlighted in red.
(B) A ribbon representation shows the position of substitutions at the base of the CLPP hydrophobic pocket. Substitutions are shown in red, and adjacent hydrophobic residues known to be important in CLPX binding are shown in green. The catalytic triad is highlighted in orange. The single-letter codes for amino acid residues and numbers shown here are after cleavage of the mitochondrial targeting sequence (MTS), which removes the N-terminal 56 residues. Hence, the residue labeled T89 is equivalent to T145 in the unprocessed polypeptide. This is for consistency with studies relating to the structure of CLPP. The poorly resolved N-terminal region (residues 1–17) was omitted for clarity.
The affected β3 strand lies at the base of a deep hydrophobic cleft, which is important in mediating the interaction with the CLPX ATPase in the CLPXP holoenzyme.22,26 The two residues, Trp146 (90) and Val148 (92), which neighbor the Perrault syndrome substitutions, have been shown to play a role in binding to the macrolactone core of acyldepsipeptides ADEP1 and ADEP2 in B. subtilis.29 ADEPs are activators of bacterial ClpP, and the interaction between ClpP and ADEPs has been proposed to mimic the interaction between ClpP and the Ile-Gly-Phe/Lys loops of the Clp ATPases.28 A change in the position of key residues within the hydrophobic pocket might therefore affect the docking of CLPX onto CLPP, thus reducing the ability of the CLPXP complex to perform targeted degradation of substrates. The p.Thr145Pro substitution is expected to cause a more dramatic structural change than is p.Ser147Cys, and this might explain the more severe phenotype of this family.
CLPP localization was evaluated specifically for human and mouse ovaries and for mouse organ of Corti (Figure 4). In the initial descriptions of Perrault syndrome, ovarian dysgenesis resulting in primary ovarian failure is described.3 Therefore, human fetal ovarian tissue was evaluated for accumulation of CLPP during follicular development. At approximately 18 weeks of gestation (20 mm foot length), CLPP localized to germ cells of developing tissue. CLPP was not detected in the stromal layers. Staining was particularly evident around the nuclei of germ cells, consistent with the localization of mitochondria (Figure 4A).32 In mice, Clpp was widely expressed in adult and fetal tissues (Figure S3). In the adult mouse ovary, CLPP was localized in granulosa cells and oocytes (Figures 4C and 4D), whereas in the immature organ of Corti at postnatal day 3 (Figures 4E and 4F), CLPP localized predominantly in supporting cells. At this age, a weak CLPP signal was detected in adjacent sensory hair cells despite their abundance of mitochondria, as evidenced by strong cytochrome c signal by immunolocalization (Figure 4F) and previously published transmission-electron-microscopy images.33 The functional relevance of apparently restricted localization of CLPP mainly in germ cells of the developing ovary, granulosa cells and oocytes of the mature ovary, and supporting cells of the organ of Corti is not known. Perhaps higher levels of CLPP in these cell types might indicate a specific need for proteolysis of particular unfolded or misfolded proteins to preclude damage.
Figure 4.
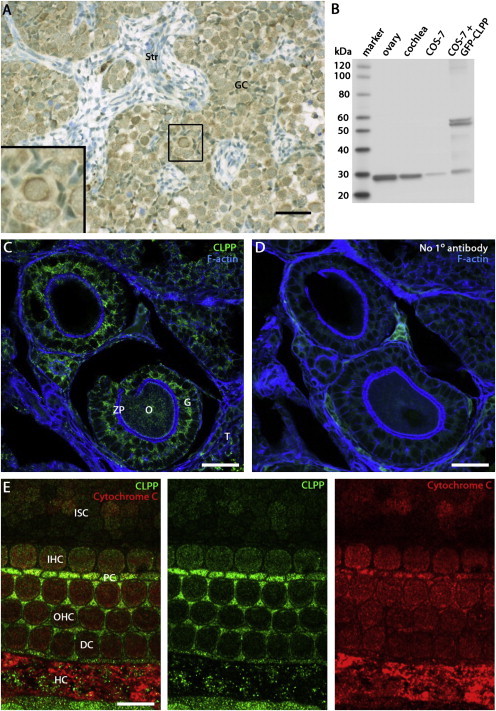
Immunolocalization of CLPP in Human Fetal Ovary, Adult Mouse Ovary, and P3 Mouse Organ of Corti
(A) Immunodetection (brown staining) of CLPP in germ cells of human fetal ovary (approximately 18 weeks of gestation; 20 mm foot length). Staining was particularly evident around nuclei of germ cells, consistent with the localization of mitochondria. Staining was not evident in the stromal layers. Human fetal tissue was collected with ethical approval under the Codes of Practice of the UK Human Tissue Authority and staged by foot length. For bright-field studies, endogenous peroxidase was quenched by incubation with 30% hydrogen peroxide and antigen retrieval was undertaken by heating at 95°C for 5 minutes in sodium citrate pH 6. Rabbit CLPP antibody (Sigma-Aldrich HPA010649) was used as the primary antibody, and unconjugated goat anti-rabbit IgG (Vector Labs AI-1000) was used as the secondary antibody. A system of streptavidin, horseradish peroxidase, and diaminobenzidine (Vector Labs) was used for generating a brown signal. Scale bars represent 50 μm.
(B) Immunoblot analysis for the validation of the rabbit monoclonal CLPP antibody (AbCam ab124822). When used at a 1:2,000 dilution in ECL Prime blocking reagent (GE RPN418V), this rabbit monoclonal antibody recognizes a predicted 26 kDa CLPP protein in 4 μg of a postnatal day (P) 25 mouse ovary lysate, 4 μg of a P3 mouse cochlea lysate, and 0.5 μg of COS-7 cell lysate. COS-7 cells were also transfected with pAcGFP-N2-CLPP and expressed GFP-tagged CLPP proteins of 57 kDa and 51 kDa. The latter protein presumably lacks the predicted 6 kDa MTS. Endogenous 26 kDa CLPP was also detected in these COS-7 cells.
(C and D) Immunohistochemistry of an adult mouse ovary reveals CLPP expression in granulosa cells (G) and oocytes (O). CLPP staining is absent in the zona pellucida (ZP) and theca (T). Fixed-frozen sections (Zyagen MF-406) were labeled with a rabbit monoclonal CLPP antibody as described below and counterstained with a 1:100 dilution of phalloidin-Atto 390 (Sigma 50556). Scale bars represent 30 μm.
(E) An optical section of a P3 mouse organ of Corti at the level of the cuticular plate reveals that CLPP (green) is more abundant in Deiter’s cells (DCs), inner pillar cells (PCs), and Hensen’s cells (HCs) than in inner sulcus cells (ISCs), inner hair cells (IHCs), and outer hair cells (OHCs). CLPP colocalized with the mitochondrial protein cytochrome c (red). After dissection from the temporal bone, cochleae were fixed in 4% paraformaldehyde. Tissues were finely dissected, permeabilized in PBS containing 0.5% Triton X-100, and incubated overnight in 1:100 dilutions of rabbit CLPP antibody and mouse cytochrome c antibody (BD Biosciences 556433) in 5% goat serum and 2% BSA blocking solution. Primary antibodies were detected with 1:400 dilutions of Alexa Fluor 488 donkey anti-rabbit IgG (Invitrogen 21206) and Alexa Fluor 568 goat anti-mouse IgG (Invitrogen 11004) in blocking solution. Scale bars represent 10 μm.
Our data strongly suggest that biallelic recessive mutations in CLPP result in Perrault syndrome. The phenotypic variability within and between members of the three families affected by CLPP mutations is striking. In family PDF1, in addition to comprising POF and SNHL, the phenotype includes progressive spastic paraplegia, growth restriction, learning disability, and nonspecific white-matter changes on brain imaging. In contrast, in family PKDF291, the phenotype includes only POF and SNHL. In family DEM4395, all affected individuals have SNHL, but formal evaluation of hormone levels could not be carried out in affected females.
The severe phenotype of family PDF1 might be due to their CLPP allele and/or to additional homozygous missense mutations. In the critical autozygous region in 19p13, two additional variants in PCP2 (MIM 602454) and GTF2F1 (MIM 189968) were identified in family PDF1. The c.392C>G (RefSeq NM_174895) (p.Pro131Arg [RefSeq NP_777555]) mutation in PCP2 was predicted to be damaging by MutationTaster, PolyPhen-2, and SIFT. The other variant, c.1328G>T (RefSeq NM_002096) (p.Gly443Val [RefSeq NP_002087]) in GTF2F1, was predicted to be damaging by MutationTaster and PolyPhen-2 but “tolerated” by SIFT. Both variants were absent in a panel of ethnically matched controls (n = 193) and affect residues conserved in mouse (PCP2) and zebrafish (GTF2F1). Variants in the protein-coding regions of these genes were not present in the affected individuals from families PKDF291 and DEM4395 and had not been previously associated with inherited disorders. GTF2F1 encodes the Rap74 subunit of human general transcription factor IIF (TFIIF), which is an initiator of transcription and acts by recruiting RNA polymerase II to the initiation complex.34 PCP2 encodes Purkinje cell protein 2, which is expressed exclusively in Purkinje cells and the retina35 and might have a role in Purkinje cell development or regulation.36 Pcp2-knockout mice have mild cerebellar hypoplasia, and null female mice are affected by anxiety.37,38
Evidence that the CLPP mutation is entirely responsible for the severe phenotype of family PDF1 can be drawn from other genes associated with spastic paraplegia. Recessive mutations in SPG7 cause spastic paraplegia.39 SPG7 encodes paraplegin, which, like CLPP, is part of an ATP-dependent proteolytic complex that degrades misfolded proteins and regulates ribosome assembly in the mitochondrial inner membrane.40 In addition, expression profiling of lymphocytes and fibroblasts from an individual with dominant spastic paraplegia type 13 (SPG13) due to a mutation in HSPD1, encoding a mitochondrial heat-shock protein, revealed that this individual had lower levels of CLPP message and protein than did controls.41 Furthermore, homozygous mutations in AFG3L2, which forms a heterooligomeric complex with paraplegin, cause an early-onset spastic-ataxia-neuropathy phenotype.42 It is thus possible that accumulation of misfolded proteins, but not those degraded by dysfunctional CLPP, is consistent with progressive spastic paraplegia. It is notable that this phenotype is only present in the individuals with the p.Thr145Pro missense variant, which by protein modeling is predicted to have a more significant effect on CLPP function. Sanger sequencing of the coding exons of CLPP in 20 individuals with spastic paraplegia consistent with autosomal-recessive inheritance and no mutation in known spastic paraplegia genes43 identified no pathogenic mutations (data not shown). This is not surprising given the significant genetic heterogeneity associated with spastic paraplegia.
Severe to profound prelingual SNHL was observed in all affected individuals of the three families reported here. The severity of hearing loss in the individuals with CLPP mutations contrasts with that described in individuals with Perrault syndrome due to HARS2 mutations or to HSD17B4 mutations. In the individuals with HARS2 mutations, the SNHL was progressive in all five affected siblings but varied strikingly in age of onset and severity;9 in the individuals with HSD17B4 mutations, there was variable severe progressive SNHL.8 The profound hearing loss in our cases with CLPP mutations might reflect an ascertainment bias in that severely affected individuals are more likely to be brought to the attention of clinicians. It is possible that other alleles of CLPP might contribute to milder degrees of hearing loss. Mutations in CLPP might be relevant to nonsyndromic hearing loss in males because in the absence of an affected female sibling, the diagnosis of Perrault syndrome would not be considered. The infertility associated with Perrault syndrome might be either primary or secondary.8,9 Therefore, there might be hearing-loss-affected females for whom a diagnosis of Perrault syndrome has not been suspected because they have had temporarily normal menstrual cycles.
The previous identification of HARS2 mutations indicated the role of mitochondrial dysfunction in the pathogenesis of some forms of Perrault syndrome.9 HARS2 encodes mitochondrial histidyl-tRNA synthetase, which catalyzes the covalent linkage of histidine to its cognate tRNA and is required in the mitochondria for protein translation. CLPP is required for protein degradation in the mitochondria, which also indicates an important role for mitochondrial protein homeostasis in disease pathogenesis. Genes encoding other members of interlinking pathways (Figure S4)44 involved in the mitochondrial unfolded-protein response (UPRmt) are attractive candidates for other as-yet-undefined causes of Perrault syndrome.
Acknowledgments
We thank the families for their participation in the study. We thank Inna Belyantseva, Dennis Drayna, and Andrew J. Griffith for their critique of this manuscript. This study was supported by the Infertility Research Trust; by the Manchester University Biomedical Research Centre; by the Wellcome Trust (N.A.H. is a Senior Fellow in Clinical Science); by National Institutes of Health (NIH) grants and contracts R01 DC005641, R01 DC011651, R01 DC003594, N01 HG065403, and U54 HG006493; by the Higher Education Commission and the Ministry of Science and Technology of Pakistan; and by the International Center for Genetic Engineering and Biotechnology, Trieste, Italy (CRP/PAK08-01 contract 08/009 to Sh.R). Genotyping of family DEM4395 was performed at the Center for Inherited Disease Research, which is funded through the NIH to The Johns Hopkins University (contract number N01-HG-65403). N.B. was supported by National Heart, Lung, and Blood Institute (NIH) intramural funds (HL004232) to James Sellers. Work at the National Institute on Deafness and Other Communication Disorders (NIH) was supported by intramural funds (DC000039-15) to T.B.F. The authors also acknowledge GeneMANIA, the development of which was funded by Genome Canada through the Ontario Genomics Institute (2007-OGI-TD-05) and which is now funded by the Ontario Ministry of Research and Innovation.
Contributor Information
Thomas B. Friedman, Email: friedman@nidcd.nih.gov.
William G. Newman, Email: william.newman@manchester.ac.uk.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
GeneMANIA, http://genemania.org/
MutationTaster, http://www.mutationtaster.org/
NHLBI Exome Sequencing Project (ESP) Exome Variant Server, http://evs.gs.washington.edu/EVS/
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org
PolyPhen-2, http://genetics.bwh.harvard.edu/pph2/
Primer3, http://frodo.wi.mit.edu/
Primer-BLAST, http://www.ncbi.nlm.nih.gov/tools/primer-blast/
UCSC Genome Browser, http://genome.ucsc.edu/index.html
References
- 1.Guest S.S., Evans C.D., Winter R.M. The Online London Dysmorphology Database. Genet. Med. 1999;1:207–212. doi: 10.1097/00125817-199907000-00007. [DOI] [PubMed] [Google Scholar]
- 2.Lenz D.R., Avraham K.B. Hereditary hearing loss: from human mutation to mechanism. Hear. Res. 2011;281:3–10. doi: 10.1016/j.heares.2011.05.021. [DOI] [PubMed] [Google Scholar]
- 3.Perrault M., Klotz B., Housset E. Deux cas de syndrome de Turner avec surdi-mutite dans une meme fratrie. Bull. Mem. Soc. Med. Hop. Paris. 1951;16:79–84. [PubMed] [Google Scholar]
- 4.Fiumara A., Sorge G., Toscano A., Parano E., Pavone L., Opitz J.M. Perrault syndrome: evidence for progressive nervous system involvement. Am. J. Med. Genet. A. 2004;128A:246–249. doi: 10.1002/ajmg.a.20616. [DOI] [PubMed] [Google Scholar]
- 5.Jenkinson E.M., Clayton-Smith J., Mehta S., Bennett C., Reardon W., Green A., Pearce S.H., De Michele G., Conway G.S., Cilliers D. Perrault syndrome: further evidence for genetic heterogeneity. J. Neurol. 2012;259:974–976. doi: 10.1007/s00415-011-6285-5. [DOI] [PubMed] [Google Scholar]
- 6.Linssen W.H., Van den Bent M.J., Brunner H.G., Poels P.J. Deafness, sensory neuropathy, and ovarian dysgenesis: a new syndrome or a broader spectrum of Perrault syndrome? Am. J. Med. Genet. 1994;51:81–82. doi: 10.1002/ajmg.1320510117. [DOI] [PubMed] [Google Scholar]
- 7.Gottschalk M.E., Coker S.B., Fox L.A. Neurologic anomalies of Perrault syndrome. Am. J. Med. Genet. 1996;65:274–276. doi: 10.1002/(SICI)1096-8628(19961111)65:4<274::AID-AJMG5>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 8.Pierce S.B., Walsh T., Chisholm K.M., Lee M.K., Thornton A.M., Fiumara A., Opitz J.M., Levy-Lahad E., Klevit R.E., King M.C. Mutations in the DBP-deficiency protein HSD17B4 cause ovarian dysgenesis, hearing loss, and ataxia of Perrault Syndrome. Am. J. Hum. Genet. 2010;87:282–288. doi: 10.1016/j.ajhg.2010.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pierce S.B., Chisholm K.M., Lynch E.D., Lee M.K., Walsh T., Opitz J.M., Li W., Klevit R.E., King M.C. Mutations in mitochondrial histidyl tRNA synthetase HARS2 cause ovarian dysgenesis and sensorineural hearing loss of Perrault syndrome. Proc. Natl. Acad. Sci. USA. 2011;108:6543–6548. doi: 10.1073/pnas.1103471108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rehman A.U., Gul K., Morell R.J., Lee K., Ahmed Z.M., Riazuddin S., Ali R.A., Shahzad M., Jaleel A.U., Andrade P.B. Mutations of GIPC3 cause nonsyndromic hearing loss DFNB72 but not DFNB81 that also maps to chromosome 19p. Hum. Genet. 2011;130:759–765. doi: 10.1007/s00439-011-1018-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Daly S.B., Urquhart J.E., Hilton E., McKenzie E.A., Kammerer R.A., Lewis M., Kerr B., Stuart H., Donnai D., Long D.A. Mutations in HPSE2 cause urofacial syndrome. Am. J. Hum. Genet. 2010;86:963–969. doi: 10.1016/j.ajhg.2010.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Carr I.M., Flintoff K.J., Taylor G.R., Markham A.F., Bonthron D.T. Interactive visual analysis of SNP data for rapid autozygosity mapping in consanguineous families. Hum. Mutat. 2006;27:1041–1046. doi: 10.1002/humu.20383. [DOI] [PubMed] [Google Scholar]
- 13.Ng S.B., Bigham A.W., Buckingham K.J., Hannibal M.C., McMillin M.J., Gildersleeve H.I., Beck A.E., Tabor H.K., Cooper G.M., Mefford H.C. Exome sequencing identifies MLL2 mutations as a cause of Kabuki syndrome. Nat. Genet. 2010;42:790–793. doi: 10.1038/ng.646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Walsh T., Shahin H., Elkan-Miller T., Lee M.K., Thornton A.M., Roeb W., Abu Rayyan A., Loulus S., Avraham K.B., King M.-C., Kanaan M. Whole exome sequencing and homozygosity mapping identify mutation in the cell polarity protein GPSM2 as the cause of nonsyndromic hearing loss DFNB82. Am. J. Hum. Genet. 2010;87:90–94. doi: 10.1016/j.ajhg.2010.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.McKernan K.J., Peckham H.E., Costa G.L., McLaughlin S.F., Fu Y., Tsung E.F., Clouser C.R., Duncan C., Ichikawa J.K., Lee C.C. Sequence and structural variation in a human genome uncovered by short-read, massively parallel ligation sequencing using two-base encoding. Genome Res. 2009;19:1527–1541. doi: 10.1101/gr.091868.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bross P., Andresen B.S., Knudsen I., Kruse T.A., Gregersen N. Human ClpP protease: cDNA sequence, tissue-specific expression and chromosomal assignment of the gene. FEBS Lett. 1995;377:249–252. doi: 10.1016/0014-5793(95)01353-9. [DOI] [PubMed] [Google Scholar]
- 17.Yu A.Y., Houry W.A. ClpP: a distinctive family of cylindrical energy-dependent serine proteases. FEBS Lett. 2007;581:3749–3757. doi: 10.1016/j.febslet.2007.04.076. [DOI] [PubMed] [Google Scholar]
- 18.Kang S.G., Maurizi M.R., Thompson M., Mueser T., Ahvazi B. Crystallography and mutagenesis point to an essential role for the N-terminus of human mitochondrial ClpP. J. Struct. Biol. 2004;148:338–352. doi: 10.1016/j.jsb.2004.07.004. [DOI] [PubMed] [Google Scholar]
- 19.Wang J., Hartling J.A., Flanagan J.M. The structure of ClpP at 2.3 A resolution suggests a model for ATP-dependent proteolysis. Cell. 1997;91:447–456. doi: 10.1016/s0092-8674(00)80431-6. [DOI] [PubMed] [Google Scholar]
- 20.Flanagan J.M., Wall J.S., Capel M.S., Schneider D.K., Shanklin J. Scanning transmission electron microscopy and small-angle scattering provide evidence that native Escherichia coli ClpP is a tetradecamer with an axial pore. Biochemistry. 1995;34:10910–10917. doi: 10.1021/bi00034a025. [DOI] [PubMed] [Google Scholar]
- 21.Wojtkowiak D., Georgopoulos C., Zylicz M. Isolation and characterization of ClpX, a new ATP-dependent specificity component of the Clp protease of Escherichia coli. J. Biol. Chem. 1993;268:22609–22617. [PubMed] [Google Scholar]
- 22.Kim Y.I., Levchenko I., Fraczkowska K., Woodruff R.V., Sauer R.T., Baker T.A. Molecular determinants of complex formation between Clp/Hsp100 ATPases and the ClpP peptidase. Nat. Struct. Biol. 2001;8:230–233. doi: 10.1038/84967. [DOI] [PubMed] [Google Scholar]
- 23.Szyk A., Maurizi M.R. Crystal structure at 1.9A of E. coli ClpP with a peptide covalently bound at the active site. J. Struct. Biol. 2006;156:165–174. doi: 10.1016/j.jsb.2006.03.013. [DOI] [PubMed] [Google Scholar]
- 24.Bewley M.C., Graziano V., Griffin K., Flanagan J.M. The asymmetry in the mature amino-terminus of ClpP facilitates a local symmetry match in ClpAP and ClpXP complexes. J. Struct. Biol. 2006;153:113–128. doi: 10.1016/j.jsb.2005.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ingvarsson H., Maté M.J., Högbom M., Portnoï D., Benaroudj N., Alzari P.M., Ortiz-Lombardía M., Unge T. Insights into the inter-ring plasticity of caseinolytic proteases from the X-ray structure of Mycobacterium tuberculosis ClpP1. Acta Crystallogr. D Biol. Crystallogr. 2007;63:249–259. doi: 10.1107/S0907444906050530. [DOI] [PubMed] [Google Scholar]
- 26.Effantin G., Maurizi M.R., Steven A.C. Binding of the ClpA unfoldase opens the axial gate of ClpP peptidase. J. Biol. Chem. 2010;285:14834–14840. doi: 10.1074/jbc.M109.090498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kim D.Y., Kim K.K. The structural basis for the activation and peptide recognition of bacterial ClpP. J. Mol. Biol. 2008;379:760–771. doi: 10.1016/j.jmb.2008.04.036. [DOI] [PubMed] [Google Scholar]
- 28.Li D.H., Chung Y.S., Gloyd M., Joseph E., Ghirlando R., Wright G.D., Cheng Y.Q., Maurizi M.R., Guarné A., Ortega J. Acyldepsipeptide antibiotics induce the formation of a structured axial channel in ClpP: A model for the ClpX/ClpA-bound state of ClpP. Chem. Biol. 2010;17:959–969. doi: 10.1016/j.chembiol.2010.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lee B.G., Park E.Y., Lee K.E., Jeon H., Sung K.H., Paulsen H., Rübsamen-Schaeff H., Brötz-Oesterhelt H., Song H.K. Structures of ClpP in complex with acyldepsipeptide antibiotics reveal its activation mechanism. Nat. Struct. Mol. Biol. 2010;17:471–478. doi: 10.1038/nsmb.1787. [DOI] [PubMed] [Google Scholar]
- 30.Lee B.G., Kim M.K., Song H.K. Structural insights into the conformational diversity of ClpP from Bacillus subtilis. Mol. Cells. 2011;32:589–595. doi: 10.1007/s10059-011-0197-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gersch M., List A., Groll M., Sieber S.A. Insights into structural network responsible for oligomerization and activity of bacterial virulence regulator caseinolytic protease P (ClpP) protein. J. Biol. Chem. 2012;287:9484–9494. doi: 10.1074/jbc.M111.336222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Motta P.M., Nottola S.A., Makabe S., Heyn R. Mitochondrial morphology in human fetal and adult female germ cells. Hum. Reprod. 2000;15(Suppl 2):129–147. doi: 10.1093/humrep/15.suppl_2.129. [DOI] [PubMed] [Google Scholar]
- 33.Weaver S.P., Schweitzer L. Development of gerbil outer hair cells after the onset of cochlear function: an ultrastructural study. Hear. Res. 1994;72:44–52. doi: 10.1016/0378-5955(94)90204-6. [DOI] [PubMed] [Google Scholar]
- 34.Flores O., Lu H., Killeen M., Greenblatt J., Burton Z.F., Reinberg D. The small subunit of transcription factor IIF recruits RNA polymerase II into the preinitiation complex. Proc. Natl. Acad. Sci. USA. 1991;88:9999–10003. doi: 10.1073/pnas.88.22.9999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Willard F.S., McCudden C.R., Siderovski D.P. G-protein alpha subunit interaction and guanine nucleotide dissociation inhibitor activity of the dual GoLoco motif protein PCP-2 (Purkinje cell protein-2) Cell. Signal. 2006;18:1226–1234. doi: 10.1016/j.cellsig.2005.10.003. [DOI] [PubMed] [Google Scholar]
- 36.Zhang X., Zhang H., Oberdick J. Conservation of the developmentally regulated dendritic localization of a Purkinje cell-specific mRNA that encodes a G-protein modulator: comparison of rodent and human Pcp2(L7) gene structure and expression. Brain Res. Mol. Brain Res. 2002;105:1–10. doi: 10.1016/s0169-328x(02)00379-0. [DOI] [PubMed] [Google Scholar]
- 37.Iscru E., Serinagaoglu Y., Schilling K., Tian J., Bowers-Kidder S.L., Zhang R., Morgan J.I., DeVries A.C., Nelson R.J., Zhu M.X., Oberdick J. Sensorimotor enhancement in mouse mutants lacking the Purkinje cell-specific Gi/o modulator, Pcp2(L7) Mol. Cell. Neurosci. 2009;40:62–75. doi: 10.1016/j.mcn.2008.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Walton J.C., Schilling K., Nelson R.J., Oberdick J. Sex-dependent behavioral functions of the Purkinje cell-specific Gαi/o binding protein, Pcp2(L7) Cerebellum. 2012;11:982–1001. doi: 10.1007/s12311-012-0368-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Casari G., De Fusco M., Ciarmatori S., Zeviani M., Mora M., Fernandez P., De Michele G., Filla A., Cocozza S., Marconi R. Spastic paraplegia and OXPHOS impairment caused by mutations in paraplegin, a nuclear-encoded mitochondrial metalloprotease. Cell. 1998;93:973–983. doi: 10.1016/s0092-8674(00)81203-9. [DOI] [PubMed] [Google Scholar]
- 40.Koppen M., Metodiev M.D., Casari G., Rugarli E.I., Langer T. Variable and tissue-specific subunit composition of mitochondrial m-AAA protease complexes linked to hereditary spastic paraplegia. Mol. Cell. Biol. 2007;27:758–767. doi: 10.1128/MCB.01470-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hansen J., Corydon T.J., Palmfeldt J., Dürr A., Fontaine B., Nielsen M.N., Christensen J.H., Gregersen N., Bross P. Decreased expression of the mitochondrial matrix proteases Lon and ClpP in cells from a patient with hereditary spastic paraplegia (SPG13) Neuroscience. 2008;153:474–482. doi: 10.1016/j.neuroscience.2008.01.070. [DOI] [PubMed] [Google Scholar]
- 42.Pierson T.M., Adams D., Bonn F., Martinelli P., Cherukuri P.F., Teer J.K., Hansen N.F., Cruz P., Mullikin J.C., for the NISC Comparative Sequencing Program. Blakesley R.W. Whole-exome sequencing identifies homozygous AFG3L2 mutations in a spastic ataxia-neuropathy syndrome linked to mitochondrial m-AAA proteases. PLoS Genet. 2011;7:e1002325. doi: 10.1371/journal.pgen.1002325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cleeter M., Houlden H., Simons P., Al-Shawi R., Stevanin G., Durr A., Hsuan J., Warner T.T. Screening for mutations in the phosphatidylinositol 4-kinase 2-alpha gene in autosomal recessive hereditary spastic paraplegia. Amyotroph. Lateral Scler. 2011;12:148–149. doi: 10.3109/17482968.2010.543689. [DOI] [PubMed] [Google Scholar]
- 44.Mostafavi S., Ray D., Warde-Farley D., Grouios C., Morris Q. GeneMANIA: a real-time multiple association network integration algorithm for predicting gene function. Genome Biol. 2008;9(Suppl 1):S4. doi: 10.1186/gb-2008-9-s1-s4. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.