

Abstract
Adams-Oliver syndrome (AOS) is a rare, autosomal-dominant or -recessive disorder characterized primarily by aplasia cutis congenita and terminal transverse limb defects. Recently, we demonstrated that homozygous mutations in DOCK6 cause an autosomal-recessive form of AOS. In this study, we sought to determine the contribution of DOCK6 mutations to the etiology of AOS in several consanguineous families. In two of the five families studied, we identified two homozygous truncating mutations (a splice-site mutation and a frameshift duplication). DOCK6 sequencing revealed no mutation in the remaining three families, consistent with their autozygosity mapping and linkage-analysis results, which revealed a single candidate locus in 3p14.1 on three different haplotype backgrounds in the three families. Indeed, exome sequencing in one family revealed one missense mutation in EOGT (C3orf64), and subsequent targeted sequencing of this gene revealed a homozygous missense mutation and a homozygous frameshift deletion mutation in the other two families. EOGT encodes EGF-domain-specific O-linked N-acetylglucosamine (O-GlcNAc) transferase, which is involved in the O-GlcNAcylation (attachment of O-GlcNAc to serine and threonine residues) of a subset of extracellular EGF-domain-containing proteins. It has a documented role in epithelial-cell-matrix interactions in Drosophila, in which deficiency of its ortholog causes wing blistering. Our findings highlight a developmental role of O-GlcNAcylation in humans and expand the genetic heterogeneity of autosomal-recessive AOS.
Main Text
Adams-Oliver syndrome (AOS [MIM 100300]) is a rare congenital disorder characterized by aplasia cutis congenita (ACC) and terminal transverse limb defects (TTLDs). Additional abnormalities in other organs (e.g., the heart, brain, and eye) might also be present with variable expressivity.1–3 Although AOS was originally described as an autosomal-dominant entity, subsequent reports on consanguineous families have suggested that autosomal-recessive inheritance is also possible.4,5 Indeed, the recent revelation that dominant mutations in ARHGAP31 (MIM 610911) and recessive mutations in DOCK6 (MIM 614194), both of which encode proteins involved in the regulation of two important actin cytoskeleton regulators, RAC1 and CDC42, confirms that notion.6,7 The heterogeneity of autosomal-dominant AOS was demonstrated by the recent identification of dominant mutations in RBPJ (MIM 147183), encoding the primary transcriptional regulator for NOTCH signaling, in some AOS individuals.8 It remains unclear how RAC1- and CDC42-mediated cytoskeletal dysregulation and perturbed NOTCH signaling converge on the pathogenesis of AOS, which has historically been proposed to entail vascular disruption, given that the latter can theoretically explain the major findings of AOS. It also remains unknown whether recessive AOS is genetically heterogeneous and, if it is, how the additional locus (or loci) might add to our understanding of the pathogenesis of this developmental disorder. In view of the power that consanguineous pedigrees have in revealing autosomal-recessive disease loci,9 we set out in this study to conduct genetic analysis of a number of families with one or more AOS-affected individuals to explore the genetics of the recessive form of the disease.
Seven affected individuals from five families were enrolled under a protocol approved by the institutional review board at King Faisal Specialist Hospital and Research Center (research advisory council 2080006) after signing written informed consent. Blood samples were collected from the affected individuals, their parents, and, when available, their unaffected siblings. The five families are consanguineous; Table 1 summarizes their clinical features, and a few representative images are shown in Figure 1. Autozygome analysis was performed on all affected individuals with the Axiom SNP platform (Affymetrix, Santa Clara, CA, USA) and was followed by autoSNPa genome-wide determination of runs of homozygosity (ROHs) as surrogates of autozygosity as described before.9
Table 1.
Summary of Clinical Findings of AOS Individuals
|
Family |
|||||||
|---|---|---|---|---|---|---|---|
|
AOS_F1 |
AOS_F2 |
AOS_F3 |
AOS_F4 |
AOS_F5 |
|||
| IV:4 | IV:1 | IV:4 | IV:2 | V:1 | IV:4 | IV:5 | |
| Homozygous mutation | DOCK6a c.2520dupT | DOCK6a c.4107–1G>C | EOGT c.620G>C | EOGT c.1074delA | EOGT c.1130G>A | EOGT c.1130G>A | EOGT c.1130G>A |
| Age | 12 months | 2 years | 35 days | 32 months | 3 years | 10 years | 12 years |
| Gender | male | female | female | male | female | female | male |
| Cutis aplasia | occipital scalp and abdomen | + | parietal scalp defect 11 × 15 cm in diameter | + (5 cm scalp defect) | a small bony defect (5 cm) in the skull vault involves both parietal bones and is covered with hairless scalp skin | a small midline bony defect (1 cm) in the skull vault is covered with sparsely haired scalp skin | a large midline skull defect (5 cm) involves both parietal bones and is covered with hairless scalp skin |
| Terminal transverse limb defects | tapering fingers and hypoplastic distal phalanges of the hands and feet | + (hypoplastic terminal phalanges) | distal transverse defect, dysplastic right big toe, and hypoplastic nails in the other right toes | bilateral hypoplasia in third toes and toenails (r > l) | left foot: absent distal phalanges, absent middle phalanges of third and fourth toes, soft tissue syndactyly, and hypoplastic nails | left foot: absent terminal phalanges of all toes. | symmetrical in both feet; absent terminal phalanges of all toes and severe dysplastic nails |
| right foot: absent nails of first, second, third, and fourth toes | dysplastic nails in all toes | ||||||
| right foot: hypoplastic second and third phalanges in second, third, and fourth toes and absent terminal phalanx of fifth toe | |||||||
| Congenital heart disease | aortic valve dysplasia | ND | ASD-II | muscular VSD and PDA (resolved) | no | no | no |
| Microphthalmia | no | hypoplastic disc | no | no | no | no | no |
| Other | depressed nasal bridge, bulbous nasal tip, single palmar creases, dilation of cerebral ventricles, and periventricular calcifications | intracranial periventricular calcification, pachygyria (especially posteriorly), and uncontrolled seizures | normal neuroimaging | left temporal and occipital lobe infarcts (presumed prenatal origin), umbilical hernia, and speech and fine motor delays | cutis marmorata, six café au lait patches (less than 0.5 cm) on abdomen and chest, and small umbilical hernia | ND | ND |
| sister with dilation of cerebral ventricles, periventricular calcifications, aplasia cutis congenita, gastroschisis, convulsions, and distal malformed fingers and toes | |||||||
The following abbreviations are used: ND, not determined; ASD-II, atrial septal defect type II; VSD, ventricular septal defect; and PDA, patent ductus arteriosus.
RefSeq accession number NM_020812.3.
Figure 1.
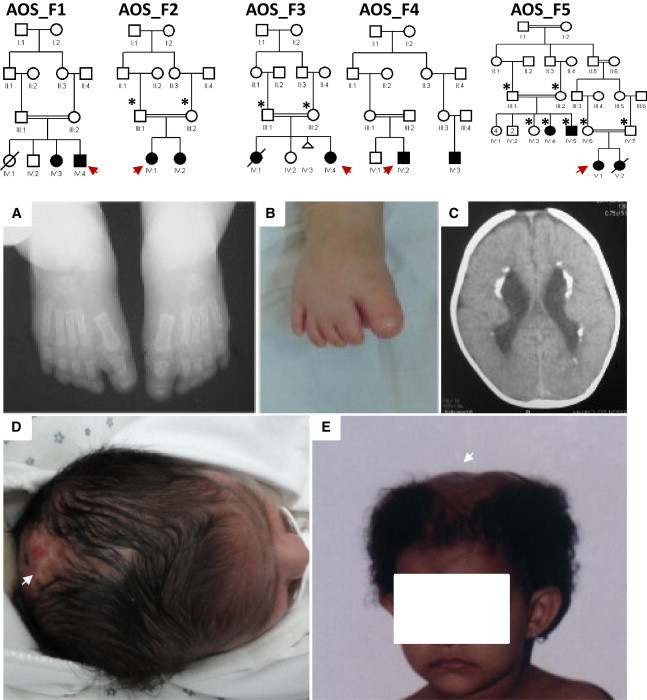
Identification of Five Consanguineous AOS-Affected Families
The index individual is indicated with an arrow in each pedigree, and asterisks denote individuals whose DNA was available for analysis. Please note that the degree of consanguinity between III:1 and III:2 in AOS_F4 is uncertain.
(A–E) Representative clinical images of the index individuals from families AOS_F2, AOS_F3, and AOS_F5. Clinical photographs of AOS_F2 IV:1 show the hypoplastic terminal phalanges (A and B) and periventricular calcifications, thick cortex, and posterior pachygyria (C). Clinical photographs show the cutis aplasia in AOS_F3 IV:4 (D) and AOS_F5 V:1 (E).
The index individuals in families AOS_F1 (IV:4) and AOS_F2 (IV:1) were each found to harbor an ROH that overlaps with DOCK6. Subsequent direct sequencing revealed that AOS_F1 IV:4 harbors a 1 bp duplication that results in a frameshift and the introduction of a premature stop codon (c.2520dupT [p.Arg841Serfs∗6]; RefSeq accession number NM_020812.3) (Figure S1A, available online). Similarly, sequencing of DOCK6 in AOS_F2 IV:1 revealed a homozygous splice-acceptor-site mutation (c.4107-1G>C) in DOCK6. RT-PCR using specific DOCK6 cDNA primers confirmed that the mutation replaces the consensus acceptor site by a cryptic site in the downstream exon (exon 33), leads to the loss of 7 bp from exon 33, and thus results in a frameshift and the introduction of a premature stop codon (p.Thr1370Metfs∗19) (Figures S1B and S1C).
None of the affected individuals in the remaining three families—AOS_F3 (IV:4), AOS_F4 (IV:2), and AOS_F5 (V:1, IV:4, and IV:5)—had an ROH overlapping with DOCK6, and, consistent with that finding, sequencing of this gene revealed no mutation. On the other hand, the autozygome of these individuals overlapped on a previously unreported 2,744,933 bp locus corresponding to the genomic region chr3: 66,612,406–69,357,338 (GRCh37/hg19) (Figure 2A). This locus was confirmed by linkage analysis, which revealed a single peak (LOD of ∼3.7) corresponding to the same critical ROH highlighted by autozygome analysis (Figure 2B). Exome sequencing of AOS_F3 IV:4 and subsequent application of the filtration scheme that we described previously (after filtering out all variants listed in dbSNP, we considered only those that were homozygous, coding or splicing, predicted to be pathogenic in silico, and absent in 230 Saudi exomes), revealed five missense variants, two of which turned out to be sequencing artifacts. However, we prioritized the candidacy of the missense variant (c.620G>C [p.Trp207Ser]; RefSeq NM_173654.1; ENST00000383701.3) in EOGT (C3orf64) because it was the only one among the three variants to fall within the critical locus. Indeed, Sanger sequencing confirmed this homozygous mutation in AOS_F3 IV:4, and her parents were found to be carriers (Figure 2C). We then fully sequenced EOGT in AOS_F4 IV:2 and AOS_F5 members V:1, IV:4, and IV:5. We identified a 1 bp homozygous deletion creating a frameshift and premature stop codon (c.1074delA [p.Gly359Aspfs∗28]) in AOS_F4 IV:2 and a homozygous missense mutation (c.1130G>A [p.Arg377Gln]) in AOS_F5 individuals V:1, IV:4, and IV:5. These variants were absent in 230 Saudi exomes, the 1000 Genomes Project, and the National Heart, Lung, and Blood Institute (NHLBI) Exome Variant Server. Furthermore, we fully sequenced EOGT in 100 normal Arabs (all affected individuals are Arabs) in an attempt to address the possibility that this gene accumulates pathogenic variants that are tolerated in normal individuals, but we identified no such variants. In addition, the two missense variants affect two absolutely conserved residues down to C. elegans, and both SIFT and PolyPhen predict them to be highly pathogenic (Figure S2). These data strongly suggest that each of the three alleles in EOGT is disease causing in their respective families in the homozygous state.
Figure 2.

Identification of an AOS-Associated Locus on Chromosome 3
(A) AutoSNPa output for chromosome 3 reveals an ROH (boxed in red) exclusively shared among AOS_F3 IV:4, AOS_F4 IV:2, and AOS_F5 V:1, IV:4, and IV:5.
(B) Combined genome-wide linkage analysis of the three families revealed a single maximal peak with a LOD score of ∼3.7 on chromosome 3.
(C) Upper panel: diagram of EOGT (red color denotes coding exons, yellow denotes UTRs, and triangles denote mutation sites). Lower panel: sequence chromatograms of the three mutations in EOGT (control tracing is shown for comparison) show the two missense variants in AOS_F3 IV:4 and AOS_F5 V:1 (mutation sites are denoted by asterisks, and the 1 bp deletion in AOS_F4 IV:2 is denoted by a red line). Please note that the NCBI contains only an alternatively spliced version (RefSeq NM_173654.1), according to which the nomenclature of the three mutations will be as follows: c.620G>C (p.Trp207Ser) (unchanged), c.832–791delA (instead of c.1074delA [p.Gly359Aspfs∗28]), and c.878G>A (p.Arg293Gln) (instead of c.1130G>A [p.Arg377Gln]).
The addition of O-linked N-acetylglucosamine (O-GlcNAc) moieties is a posttranslational modification that influences the stability of a number of proteins and regulates their role in various cellular mechanisms, including cytokinesis, protein trafficking, epigenetic and transcriptional regulation, and intracellular signaling in response to environmental stimuli, such as nutrition and stress.10–13 This glycosylation reaction is catalyzed by the enzyme O-linked N-acetylglucosamine transferase (OGT) but is readily reversed by another enzyme, N-acetyl-β-glucosaminidase (OGA), in order to maintain the dynamic nature of this modification, which is key to its role in regulating the above-mentioned cellular processes.14–16 In humans, the attachment of O-GlcNAc to serine and threonine residues as a form of posttranslational protein modification (O-GlcNAcylation) has been implicated in the pathogenesis of diabetes, Alzheimer disease (MIM 104300), and cancer, but no developmental role has been assigned to this posttranslational modification to date.17–20 OGT is the only known enzyme to effect O-GlcNAcylation, and it targets intracellular proteins.21 However, it has recently been shown that another enzyme, named EOGT (ER resident O-GlcNAc transferase), is capable of O-GlcNAcylating secreted extracellular proteins in Drosophila and mammals.22,23
In Drosophila, the O-GlcNAcylation of the extracellular EGF domains of two membrane proteins (Dumpy and Notch) was demonstrated.22,24,25 Remarkably, loss of Eogt in developing wing discs of Drosophila caused wing blistering, a well-documented phenotypic readout of the dysregulation of cell-cell adhesion and cell-matrix interaction.22,26 Although loss of Eogt was associated with demonstrable loss of O-GlcNAcylation of Notch, the activity of Notch was unaltered, and it was concluded that the observed defect in the interaction of epithelial cells with the apical extracellular matrix was mediated by Dumpy instead.22 More recently, the same target motif (a Thr residue located between the fifth and sixth conserved cysteines of the folded EGF-like domains, or the CXXGXS/TGXXC motif) in Drosophila Notch and Dumpy was shown to be modified by O-GlcNAcylation in murine NOTCH1 in a reaction catalyzed by the murine ortholog EOGT.23 Furthermore, wing blistering in Eogt-knockout Drosophila was rescued by the murine ortholog, confirming the conserved functional and developmental role of this enzyme between Drosophila and mammals.23 In addition, Eogt has been found to be expressed in the presomitic mesoderm and a number of adult mouse tissues (including the heart, lung, liver, spleen, and skeletal muscles) derived from the mesoderm.23,27
In order to determine the developmental expression pattern of Eogt, we performed whole-mount in situ hybridization on mouse embryos of various stages of development. Expression of Eogt at embryonic day (E) 10.5 was observed in the growing edge of the limb buds (Figure 3A). At E11.5, Eogt mRNA was enriched in the apical ectodermal ridge of the limbs (Figure 3B). By E12.5, the expression of Eogt assumed a digit-condensation pattern in the four limbs (Figures 3C–3C2). Lack of comparable staining with the corresponding sense probes confirmed specificity of the observed signals (see Figure 3D for a representative example).
Figure 3.

Whole-Mount In Situ Hybridization of Eogt during Mouse Embryonic Development
(A) An E10.5 mouse embryo shows expression in the growing edge of the limb bud.
(B) An E11.5 mouse embryo shows expression in the apical ectodermal ridge of the limbs.
(C) An E12.5 embryo shows the digit-condensation expression of Eogt mRNA (triangles) in the limbs.
(C1 and C2) Close-up views of the expression in the digits of the hindlimbs (C1) and forelimbs (C2) of the same E12.5 embryo as in (C).
(D) Sense control is shown for comparison in an E12.5 embryo. Eogt probes correspond to the area spanning nucleotides 91–816 and 830–1,537 of the coding region (RefSeq NM_175313.4).
The above data suggest that EOGT deficiency in humans causes AOS through its effect on cell-cell or cell-matrix interaction, processes that are known to be perturbed by the actin cytoskeletal defects observed in individuals with ARHGAP31 and DOCK6 mutations, thus providing tantalizing evidence of a potential mechanistic overlap that explains the phenotypic overlap. Another potential overlap between EOGT-related AOS and that caused by RBPJ mutations is that RBPJ is assumed (but not proven) to modulate NOTCH signaling and that EOGT is known to glycosylate NOTCH1 in mammals. The observation of intact Notch signaling in the fruit fly that lacks Eogt might not necessarily hold true in mammals. Unfortunately, we have no cells from any of the individuals with EOGT mutations to experimentally test their cytoskeleton and their NOTCH signaling activity. Of note, very recent data suggest that the motif recognized by EOGT is shared among a large number of human proteins, many of which reside in the extracellular matrix, so it is possible that the pathogenesis of EOGT-related AOS might involve one or more of these substrate proteins.28
In conclusion, we present evidence of genetic heterogeneity of autosomal-recessive AOS and implicate EOGT-mutation-causing defects in extracellular-protein O-GlcNAc modification in its pathogenesis. Our finding of an individual with gastroschisis and molecularly confirmed AOS calls for future investigation of the role of abnormal vascular development in the pathogenesis of this disease in light of the expanding repertoire of molecular targets.
Acknowledgments
We thank the affected individuals and their families for their enthusiastic participation. We are grateful to Mais Hashem and Niema Ibrahim for their help as clinical research coordinators. We also thank the Genotyping and Sequencing Core Facilities at King Faisal Specialist Hospital and Research Center for their technical help. This work was supported by King Abdulaziz City for Science and Technology grant 09-MED941-20 (to F.S.A.) and a Dubai Harvard Foundation for Medical Research Collaborative Research Center grant (to F.S.A.).
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
NHLBI Exome Sequencing Project (ESP) Exome Variant Server, http://evs.gs.washington.edu/EVS/
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org/
PolyPhen, www.genetics.bwh.harvard.edu/pph2/
SIFT, www.sift.jcvi.org/
UCSC Genome Browser, http://genome.ucsc.edu
References
- 1.Adams F.H., Oliver C.P. Hereditary deformities in man due to arrested development. J. Hered. 1945;36:3–7. [Google Scholar]
- 2.Whitley C.B., Gorlin R.J. Adams-Oliver syndrome revisited. Am. J. Med. Genet. 1991;40:319–326. doi: 10.1002/ajmg.1320400315. [DOI] [PubMed] [Google Scholar]
- 3.Snape K.M., Ruddy D., Zenker M., Wuyts W., Whiteford M., Johnson D., Lam W., Trembath R.C. The spectra of clinical phenotypes in aplasia cutis congenita and terminal transverse limb defects. Am. J. Med. Genet. A. 2009;149A:1860–1881. doi: 10.1002/ajmg.a.32708. [DOI] [PubMed] [Google Scholar]
- 4.Temtamy S.A., Aglan M.S., Ashour A.M., Zaki M.S. Adams-Oliver syndrome: further evidence of an autosomal recessive variant. Clin. Dysmorphol. 2007;16:141–149. doi: 10.1097/MCD.0b013e3280f9df22. [DOI] [PubMed] [Google Scholar]
- 5.Temtamy S.A., McKusick V.A. Alan R. Liss; New York: 1978. The Genetics of Hand Malformations. [PubMed] [Google Scholar]
- 6.Southgate L., Machado R.D., Snape K.M., Primeau M., Dafou D., Ruddy D.M., Branney P.A., Fisher M., Lee G.J., Simpson M.A. Gain-of-function mutations of ARHGAP31, a Cdc42/Rac1 GTPase regulator, cause syndromic cutis aplasia and limb anomalies. Am. J. Hum. Genet. 2011;88:574–585. doi: 10.1016/j.ajhg.2011.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shaheen R., Faqeih E., Sunker A., Morsy H., Al-Sheddi T., Shamseldin H.E., Adly N., Hashem M., Alkuraya F.S. Recessive mutations in DOCK6, encoding the guanidine nucleotide exchange factor DOCK6, lead to abnormal actin cytoskeleton organization and Adams-Oliver syndrome. Am. J. Hum. Genet. 2011;89:328–333. doi: 10.1016/j.ajhg.2011.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hassed S.J., Wiley G.B., Wang S., Lee J.Y., Li S., Xu W., Zhao Z.J., Mulvihill J.J., Robertson J., Warner J., Gaffney P.M. RBPJ mutations identified in two families affected by Adams-Oliver syndrome. Am. J. Hum. Genet. 2012;91:391–395. doi: 10.1016/j.ajhg.2012.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Alkuraya F.S. Discovery of rare homozygous mutations from studies of consanguineous pedigrees. Curr. Protoc. Hum. Genet. 2012;Chapter 6 doi: 10.1002/0471142905.hg0612s75. Unit 6.12. [DOI] [PubMed] [Google Scholar]
- 10.Butkinaree C., Park K., Hart G.W. O-linked beta-N-acetylglucosamine (O-GlcNAc): Extensive crosstalk with phosphorylation to regulate signaling and transcription in response to nutrients and stress. Biochim. Biophys. Acta. 2010;1800:96–106. doi: 10.1016/j.bbagen.2009.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang Z., Udeshi N.D., Slawson C., Compton P.D., Sakabe K., Cheung W.D., Shabanowitz J., Hunt D.F., Hart G.W. Extensive crosstalk between O-GlcNAcylation and phosphorylation regulates cytokinesis. Sci. Signal. 2010;3:ra2. doi: 10.1126/scisignal.2000526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hart G.W., Slawson C., Ramirez-Correa G., Lagerlof O. Cross talk between O-GlcNAcylation and phosphorylation: roles in signaling, transcription, and chronic disease. Annu. Rev. Biochem. 2011;80:825–858. doi: 10.1146/annurev-biochem-060608-102511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fujiki R., Chikanishi T., Hashiba W., Ito H., Takada I., Roeder R.G., Kitagawa H., Kato S. GlcNAcylation of a histone methyltransferase in retinoic-acid-induced granulopoiesis. Nature. 2009;459:455–459. doi: 10.1038/nature07954. [DOI] [PubMed] [Google Scholar]
- 14.Haltiwanger R.S., Holt G.D., Hart G.W. Enzymatic addition of O-GlcNAc to nuclear and cytoplasmic proteins. Identification of a uridine diphospho-N-acetylglucosamine:peptide beta-N-acetylglucosaminyltransferase. J. Biol. Chem. 1990;265:2563–2568. [PubMed] [Google Scholar]
- 15.Dong D.L., Hart G.W. Purification and characterization of an O-GlcNAc selective N-acetyl-beta-D-glucosaminidase from rat spleen cytosol. J. Biol. Chem. 1994;269:19321–19330. [PubMed] [Google Scholar]
- 16.Slawson C., Housley M.P., Hart G.W. O-GlcNAc cycling: how a single sugar post-translational modification is changing the way we think about signaling networks. J. Cell. Biochem. 2006;97:71–83. doi: 10.1002/jcb.20676. [DOI] [PubMed] [Google Scholar]
- 17.Dentin R., Hedrick S., Xie J., Yates J., 3rd, Montminy M. Hepatic glucose sensing via the CREB coactivator CRTC2. Science. 2008;319:1402–1405. doi: 10.1126/science.1151363. [DOI] [PubMed] [Google Scholar]
- 18.Slawson C., Hart G.W. O-GlcNAc signalling: implications for cancer cell biology. Nat. Rev. Cancer. 2011;11:678–684. doi: 10.1038/nrc3114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yuzwa S.A., Shan X., Macauley M.S., Clark T., Skorobogatko Y., Vosseller K., Vocadlo D.J. Increasing O-GlcNAc slows neurodegeneration and stabilizes tau against aggregation. Nat. Chem. Biol. 2012;8:393–399. doi: 10.1038/nchembio.797. [DOI] [PubMed] [Google Scholar]
- 20.Yuzwa S.A., Macauley M.S., Heinonen J.E., Shan X., Dennis R.J., He Y., Whitworth G.E., Stubbs K.A., McEachern E.J., Davies G.J., Vocadlo D.J. A potent mechanism-inspired O-GlcNAcase inhibitor that blocks phosphorylation of tau in vivo. Nat. Chem. Biol. 2008;4:483–490. doi: 10.1038/nchembio.96. [DOI] [PubMed] [Google Scholar]
- 21.Kearse K.P., Hart G.W. Topology of O-linked N-acetylglucosamine in murine lymphocytes. Arch. Biochem. Biophys. 1991;290:543–548. doi: 10.1016/0003-9861(91)90579-8. [DOI] [PubMed] [Google Scholar]
- 22.Sakaidani Y., Nomura T., Matsuura A., Ito M., Suzuki E., Murakami K., Nadano D., Matsuda T., Furukawa K., Okajima T. O-linked-N-acetylglucosamine on extracellular protein domains mediates epithelial cell-matrix interactions. Nat. Commun. 2011;2:583. doi: 10.1038/ncomms1591. [DOI] [PubMed] [Google Scholar]
- 23.Sakaidani Y., Ichiyanagi N., Saito C., Nomura T., Ito M., Nishio Y., Nadano D., Matsuda T., Furukawa K., Okajima T. O-linked-N-acetylglucosamine modification of mammalian Notch receptors by an atypical O-GlcNAc transferase Eogt1. Biochem. Biophys. Res. Commun. 2012;419:14–19. doi: 10.1016/j.bbrc.2012.01.098. [DOI] [PubMed] [Google Scholar]
- 24.Matsuura A., Ito M., Sakaidani Y., Kondo T., Murakami K., Furukawa K., Nadano D., Matsuda T., Okajima T. O-linked N-acetylglucosamine is present on the extracellular domain of notch receptors. J. Biol. Chem. 2008;283:35486–35495. doi: 10.1074/jbc.M806202200. [DOI] [PubMed] [Google Scholar]
- 25.Wilkin M.B., Becker M.N., Mulvey D., Phan I., Chao A., Cooper K., Chung H.J., Campbell I.D., Baron M., MacIntyre R. Drosophila dumpy is a gigantic extracellular protein required to maintain tension at epidermal-cuticle attachment sites. Curr. Biol. 2000;10:559–567. doi: 10.1016/s0960-9822(00)00482-6. [DOI] [PubMed] [Google Scholar]
- 26.Prout M., Damania Z., Soong J., Fristrom D., Fristrom J.W. Autosomal mutations affecting adhesion between wing surfaces in Drosophila melanogaster. Genetics. 1997;146:275–285. doi: 10.1093/genetics/146.1.275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sewell W., Sparrow D.B., Smith A.J., Gonzalez D.M., Rappaport E.F., Dunwoodie S.L., Kusumi K. Cyclical expression of the Notch/Wnt regulator Nrarp requires modulation by Dll3 in somitogenesis. Dev. Biol. 2009;329:400–409. doi: 10.1016/j.ydbio.2009.02.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Alfaro J.F., Gong C.X., Monroe M.E., Aldrich J.T., Clauss T.R., Purvine S.O., Wang Z., Camp D.G., 2nd, Shabanowitz J., Stanley P. Tandem mass spectrometry identifies many mouse brain O-GlcNAcylated proteins including EGF domain-specific O-GlcNAc transferase targets. Proc. Natl. Acad. Sci. USA. 2012;109:7280–7285. doi: 10.1073/pnas.1200425109. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.