

Abstract
The immunoglobulin heavy-chain locus (IGH) encodes variable (IGHV), diversity (IGHD), joining (IGHJ), and constant (IGHC) genes and is responsible for antibody heavy-chain biosynthesis, which is vital to the adaptive immune response. Programmed V-(D)-J somatic rearrangement and the complex duplicated nature of the locus have impeded attempts to reconcile its genomic organization based on traditional B-lymphocyte derived genetic material. As a result, sequence descriptions of germline variation within IGHV are lacking, haplotype inference using traditional linkage disequilibrium methods has been difficult, and the human genome reference assembly is missing several expressed IGHV genes. By using a hydatidiform mole BAC clone resource, we present the most complete haplotype of IGHV, IGHD, and IGHJ gene regions derived from a single chromosome, representing an alternate assembly of ∼1 Mbp of high-quality finished sequence. From this we add 101 kbp of previously uncharacterized sequence, including functional IGHV genes, and characterize four large germline copy-number variants (CNVs). In addition to this germline reference, we identify and characterize eight CNV-containing haplotypes from a panel of nine diploid genomes of diverse ethnic origin, discovering previously unmapped IGHV genes and an additional 121 kbp of insertion sequence. We genotype four of these CNVs by using PCR in 425 individuals from nine human populations. We find that all four are highly polymorphic and show considerable evidence of stratification (Fst = 0.3–0.5), with the greatest differences observed between African and Asian populations. These CNVs exhibit weak linkage disequilibrium with SNPs from two commercial arrays in most of the populations tested.
Introduction
Structural variants, including deletions, insertions, and duplications that result in changes in gene copy number (copy-number variants, CNVs), are common features of the human genome and are a significant source of interindividual sequence variation.1–5 This form of variation has been implicated in a broad spectrum of human phenotypes, including adaptive traits,6 developmental and neurological disorders,7–9 and infectious and autoimmune diseases.10–12 Despite the impressively large number of CNVs identified, many regions of the genome remain poorly characterized with respect to alternate CNV-containing haplotypes, as illustrated by recent resequencing efforts that have identified a substantial portion of novel sequence not found in the current human genome reference assembly (GRCh37).1–3,13 Particularly in regions characterized by segmental duplication, missing or incomplete sequence data hampers the ability to accurately tag such variation by using neighboring single nucleotide polymorphisms (SNPs).14 As a result, genes within these complex regions have not been fully investigated as part of routine disease-association studies, highlighting the need for directed sequencing to discover alternate haplotypes, particularly in genomic regions encompassing genes with potential biomedical relevance.
The human immunoglobulin heavy chain (IGH) locus is essential to the biosynthesis of functional heavy chains of antibodies—primary components of the adaptive immune system.15 Despite the importance of IGH, our understanding of locus-wide genetic variation at the nucleotide level is largely incomplete.16 The IGH locus spans approximately 1 megabase (Mb) of chromosome 14 (14q32.33) and consists of an estimated 123–129 variable (V [MIM147070]) genes (comprising 38–46 functional, 4–5 open reading frame [ORF], and 79–81 pseudogenes) that are located upstream of 27 diversity (D [MIM 146910]) genes (23 functional and 4 ORF), 9 joining (J [MIM 147010]) genes (6 functional and nonfunctional genes 3 pseudogenes), and 5–11 constant (C) genes (5–9 functional, 0–1 ORF, and 0–1 pseudogene)15,17–21 (IMGT Repertoire). Each group of IGH genes (IGHV, IGHD, IGHJ, and IGHC) is the result of duplication and divergence, making the IGH locus one of the most segmentally duplicated regions of the human genome. Due to several well-characterized multigene deletions in the IGHC locus, healthy individuals can vary in IGHC copy number from 5 to 11 genes; deletions of functional IGHC genes result in the absence of corresponding subclasses.15,17–21 These CNVs, which were the first described in the human IGH locus, occur within hot spots of recombination involving highly homologous sequences.17 Allelic polymorphisms within IGHC genes also contribute to haplotype diversity, as demonstrated by the characterization of IGHC allotypes and alleles.22 In the IGHV gene cluster, targeted studies have also indicated that both allelic variation15 and CNVs contribute to extreme haplotype diversity.23–27 Many genome-wide studies of structural variation, however, have excluded the locus due to the difficulty of distinguishing between structural events that occur as the result of V-(D)-J somatic rearrangements in B-lymphocyte derived DNA and genuine germline variation.28 Because the majority of IGHV haplotypes harboring CNVs have not been sequenced, the direct impacts of haplotype diversity on IGHV gene expression and function remain largely unexplored.
Given the role of antibodies in the immune system, immunoglobulin loci are attractive candidates for human disease, and several links between the IGH locus and disease have been reported;29–33 yet, few of these associations have been replicated or fine mapped. Disease links to IGH polymorphisms might be expected to be common, especially for autoimmune and infectious diseases, but whether the apparent scarcity of such associations represents a genuine absence or results from the difficulty of effectively assaying genomic variation at this locus remains an open question. The fact that only a single genome-wide association study (GWAS) has identified an association in IGH32 suggests a potential disconnect between known IGH haplotype diversity and current high-throughput genotyping tools. For example, the IGHV, IGHD, and IGHJ loci have only been sequenced and fully assembled once, and this sequence, which stands as the current human reference assembly, is a mosaic of large-insert clones from three libraries.20 Furthermore, this sequence is missing at least 11 functional and ORF IGHV genes, and of those IGHV genes represented, at least 16 are known or suspected to vary in copy number.16 Thus, given both the complexity of the locus and the lack of genomic data, it is unclear whether GWAS and high-throughput techniques based on next-generation sequencing fully capture genetic variation in IGH.
In order to identify and characterize additional IGH genomic haplotypes, we undertook a project to generate a high-quality alternate sequence assembly of the IGH locus by using large-insert bacterial artificial chromosome (BAC) clones from a haploid hydatidiform mole library (CHORI-17). Hydatidiform moles result from the fertilization of an enucleated egg followed by the doubling of paternal germline material. The CHORI-17 hydatidiform mole BAC library (CH17) genome is composed of only one haplotype, and as a result, any variation observed in assemblies can be attributed to paralogous sequence variation rather than allelic variation. This resource is, therefore, ideal for generating reliable haploid reference sequences within complicated regions of the genome associated with segmental duplication.34 Moreover, this tissue has not been subjected to V-(D)-J somatic rearrangement and thus should, in principle, harbor an unadulterated locus representative of the germline. In this study, we generate the most complete haploid sequence assembly of the IGHV, IGHD, and IGHJ loci from the CH17 library and also conduct resequencing from additional large-insert fosmid clones from diverse ethnicities. We use these data to map and annotate missing genes, discover large-scale CNVs, and characterize the frequencies of a subset of these polymorphisms in the human population. This sequence resource provides a comprehensive set of alternate IGH assemblies, a means to explore germline genetic variation, and a substrate for more effective disease-association studies.
Material and Methods
Sequencing of IGHV, IGHD, and IGHJ Loci from the CHORI-17 BAC Library
Based on BAC-end read mapping to the GRCh37 reference genome, nine clones were selected from CH17, representing a tiling path across IGHV, IGHD, and IGHJ gene loci, with the exception of a 21 kbp region at the most telomeric end of IGHV, for which no CH17 clones were identified based on analysis of BAC DNA fingerprinting and clone-end read mapping data. All sequenced BAC clones are listed in Table S1 available online. The CH17 BAC library was constructed from a complete hydatidiform mole at BACPAC Resources by Drs. Mikhail Nefedov & Pieter J. de Jong by using the cell line CHM1htert created by Dr. Urvashi Surti (BACPAC Resources Center). Clones were subjected to traditional shotgun capillary-based dideoxy sequencing at ≥8X coverage. Initial assemblies were constructed on a per clone basis and finished to high quality (phred quality ≥30); each base was covered by at least two shotgun clones, unless finished by PCR, and efforts to resolve issues related to repeat and segmental duplication structure were made. All individual clone assemblies were validated by restriction digest. IGH BAC clones were then assembled into a larger contig spanning the length of the IGHV, IGHD, and IGHJ loci. Contig assembly statistics are shown in Table S2. Based on sequence quality scores, sequences generated from individual BAC clones of this haplotype are estimated to have error rates between 1/100,000 and 1/10,000 bases. Alignment data from overlapping BAC clones in the tiling path suggest that this error rate is likely to be much lower for the majority of the locus, because we discovered 0 bp discrepancies in 279,483 junction bp from contig alignments of the six most centromeric BAC clones in the tiling path (Table S2). We did, however, observe minor sequence assembly discrepancies resulting from two simple tandem repeats occurring within the overlap junction of two BAC clones in the telomeric region of the IGHV locus (CH17-212P11 and CH17-314I7). It is important to note that the sequence identity within the overlap junction of these two BAC clones outside of the simple tandem repeat sequences was 100%, and the minor discrepancies identified in this junction did not impact analyses of CNVs or IGHV gene allelic variation characterized from CH17.
Mapping, Identification, and Sequencing of Discordant Fosmids in IGH
We downloaded clone names and mapping positions for all discordant fosmid clones (predicted to harbor structural variants based on clone-end read mapping1,2) from nine human fosmid libraries mapping to the IGH locus (GRCh37 coordinates, chr14:105,928,955-107,289,540) from the UCSC genome browser. These data were based on the mapping of approximately one million fosmid end-sequence reads per library/genome to the GRCh37 human reference by using previously described methods.1 Among the discordant fosmid clones, only those that mapped to the IGHV locus were considered for sequencing. In some cases, existing fosmids available in GenBank were also used to directly characterize CNVs or inform additional sequencing in particular regions to complete specific insertion haplotypes greater than 40 kbp; in three cases, two or more overlapping fosmid clones were used to build contiguous assemblies in order to fully represent CNV-containing haplotypes. Clones spanning one or more of any of the IGHV, IGHD, IGHJ, or IGHC gene genomic regions were assumed to represent V-(D)-J somatic rearrangements, because these events are known to occur in B lymphocyte cell lines; ten clones with one end mapping to an IGHV gene and the other end mapping to IGHC or IGHJ were sequenced and analyzed to validate this assumption. Selected clones were sequenced at ≥8X coverage by Sanger and assembled and finished to high quality by using methods described above for CH17 BAC clones when possible. Completed assemblies were submitted to GenBank (Table S1).
Identification of Genes and Regulatory Regions from Sanger-Sequenced BAC and Fosmid Clones
For IGHV gene identification and positioning, we analyzed complete sequences from each of the 9 BAC and 55 fosmid clones individually. Sequences of all functional and ORF IGHV, IGHD, and IGHJ genes were downloaded from IMGT/GENE-DB (GENE-DB35 at IMGT®, the international ImMunoGeneTics information system®,36–39 and IMGT Alignments of alleles) and aligned to BAC and fosmid sequences using BLAST.40 Once positioned, sequences at each gene locus were extracted, and alleles were determined using IMGT/V-QUEST.41,42 For the identification of regulatory elements, we extracted sequences of IGHV genes and flanking regions identified in the CH17 haplotype, as well as those IGHV genes identified in completed fosmids for which regulatory elements had not been previously characterized; these sequences were aligned with homologous IGHV gene sequences from GRCh37 and visually scanned for previously identified motifs.20
Analyses of IGHV Gene Copy Number Variation Identified in BAC and Fosmid Clones
Based on methods described previously,2,43 clones were compared to the human genome reference assembly using the program Miropeats.44 The breakpoints of identified variants were analyzed by aligning extracted sequences that spanned the predicted variant-associated breakpoints from GRCh37 and the BAC or fosmid clone sequences being compared. To identify any putative repeat sequences flanking or spanning CNV breakpoints, intrasequence alignments of sequence generated from the haplotype in which the event was suspected to have occurred were carried out using BLAST.45 Alignments of identified repeats were then generated by using Needle46 to calculate pairwise sequence similarities. Multisequence alignments of event-associated repeats spanning the breakpoints from GRCh37 and variant haplotypes were generated using Clustal W47 and visualized in SeqMan Pro (DNASTAR Lasergene, Madison, WI, USA), in most cases allowing for the determination of the region in which crossovers associated with each CNV occurred.
Given the identification of several diverse CNVs located between IGHV4-28 to IGHV4-34, putative variant breakpoints in this region were characterized by constructing a large multisequence alignment containing all portions of the event-mediating ∼25 kbp segmental duplication blocks annotated from every BAC and fosmid clone spanning the region (Figure S1). Stretches of sequence between segmental duplications exhibiting the highest sequence similarity were then identified to determine the most likely segmental duplications mediating each event, generally allowing for the delineation of regions harboring event breakpoints. Gene synteny based on allelic descriptions at genes in these haplotypes was also considered.
PCR Genotyping of CNVs in 1000 Genomes Project Population Samples
PCR assays were designed for four of the CNVs identified from the fosmid and BAC clones. Where possible, primers were designed to generate products spanning identified variant breakpoints to allow for allele-specific amplification of either the reference or BAC/fosmid alleles. For the insertion containing IGHV1-69D, IGHV2-70D, IGHV1-f, and IGHV3-h genes, TaqMan copy number assay primers and probes were designed per manufacturer instructions by using primer express software (ABI). Additional primers targeting unique sequence near IGHV1-f were also designed to test for the presence of this haplotype by using standard PCR. PCR primers and probes are listed in Table S3. PCR primers were first validated by using BAC or fosmid clone DNA from which the variants were identified, as well as a selected panel of individuals from the 1000 Genomes (1KG) Project, including those individuals used to construct fosmid libraries analyzed in this study. Validated PCR assays were subsequently genotyped in a total of 425 unrelated 1KG individuals from each of 9 geographic populations: Han Chinese (CHB, n = 45), Japanese (JPT, n = 46), Finnish (FIN, n = 48), British (GBR, n = 48), Iberian (IBS, n = 48), Toscani (TSI, n = 48), Yoruba (YRI, n = 48), Luhya (LWK, n = 48), and Maasai (MKK, n = 46) (Table S4). The use of human subjects was approved by the Human Subjects Review Committees of the University of Washington. In addition, PCR assays were used to screen DNA from four nonhuman primate species (Pan troglodytes, n = 5; Gorilla gorilla, n = 5; Pongo pygmaeus, n = 2; Pongo abelii, n = 3). Products of CNV breakpoint allele-specific PCR amplifications were visualized on agarose gels for genotyping; PCR products produced from nonhuman primates were sequenced and aligned to characterized CNV-containing BAC or fosmid clones to confirm the presence of the correct amplification products. Copy-number estimates for each individual, using the IGHV1-69 and IGHV2-70 duplication assay, were analyzed using ΔΔCt. TaqMan copy number assay estimates were used to infer the frequency of the one-copy or two-copy genotypes, and these were compared to the IGHV1-f insertion assay results. PLINK was used to assess allele frequencies for genotyped polymorphisms,45 and pairwise Fst was used to assess population differentiation for each of the genotyped loci. Genotypes for SNPs found on the Affy6.0 and Illumina Omni 1 Quad arrays were downloaded from the 1KG data sets48 for the 319 individuals that overlapped with those genotyped above (Table S4). Linkage disequilibrium (LD) estimates between alleles at these SNPs and alleles at each of the structurally variant loci genotyped above were assessed using r2 in PLINK,45 considering all SNP genotypes within the IGH locus (GRCh37 coordinates, chr14:105,928,955–107,289,540).
Results
Sequencing and Assembly of the IGHV, IGHD, and IGHJ Loci from the CH17 BAC Library
We sequenced a complete haplotype of the IGHV, IGHD, and IGHJ loci (14q32.33) by selecting CH17 hydatidiform mole BAC clones whose end-sequences specifically mapped to the IGH locus. High-quality capillary-based Sanger shotgun sequence was obtained for each of the IGH BAC clones, and overlapping clones were aligned to create a contiguous assembly encompassing the IGHV, IGHD, and IGHJ genes. The resulting IGH haplotype consists of 1,073 kbp of sequence spanning IGHJ6 to 49 kbp upstream of IGHV3-74. The most telomeric 5′ end of the locus (∼21 kbp based on GRCh37), including the previously described IGHV gene, IGHV7-81, and four IGHV pseudogenes,20 was not included in the CH17 assembly, as no BAC clones were identified from the library in this region. Despite this small gap, the CH17 IGH haplotype represents the most complete sequence spanning the IGHV, IGHD, and IGHJ loci generated from a single chromosome. Accession numbers and sequences for CH17 BAC clones analyzed in this study have been deposited in GenBank (Table S1), and the AGP tiling path for our assembly is described in Table S2.
Identification of IGHV, IGHD, and IGHJ Genes and Alleles from the CH17 Sanger Assembly
Based on mapping of known IGH genes available in IMGT/GENE-DB35 and IMGT Alignments of alleles at IMGT®,36–39 the CH17 haplotype includes 47 IGHV genes (44 functional and 3 ORF), 27 IGHD genes (23 functional and four ORF), and 6 functional IGHJ genes. Alleles at these loci were annotated by comparing their sequences to IMGT® (Figure 1, Table S5). Previously uncharacterized alleles were identified in the CH17 haplotype for five single-copy IGHV genes (IGHV4-28∗07, IGHV3-20∗02, IGHV1-18∗04, IGHV3-13∗05, and IGHV3-11∗06); in addition, we described an allele at the duplicated gene IGHV3-64D∗06. These alleles were approved by the WHO/IUIS/IMGT Nomenclature Committee.49,50 In each case, allelic variants were represented as SNPs in dbSNP135 and in the 1KG data sets48 (Table S5). The remaining IGHV alleles from the CH17 haplotype were listed in IMGT Alignments of alleles51 and in IMGT/GENE-DB,35 as were all alleles at IGHD and IGHJ gene loci.
Figure 1.
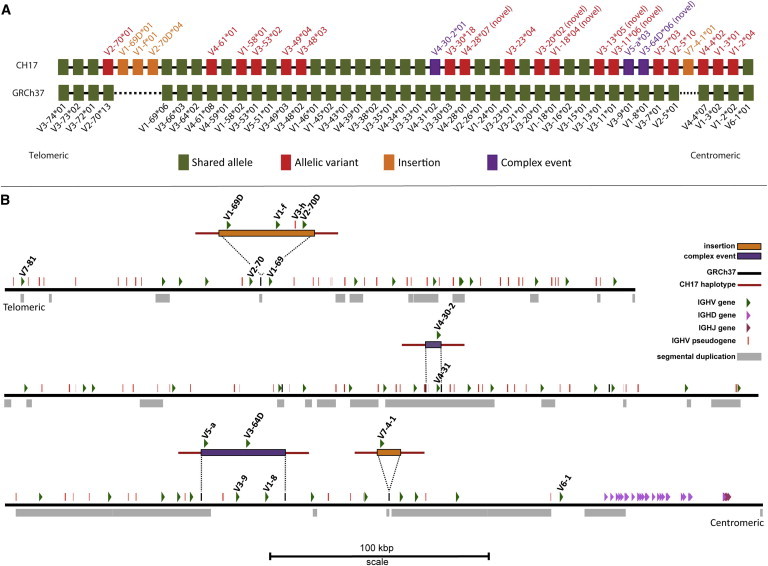
Schematic Comparisons of IGHV Haplotype between CH17 and the Human Reference Genome Assembly (GRCh37)
(A) Functional and ORF IGHV genes annotated from each reference are depicted by filled boxes with corresponding IGHV gene locus and allele identifiers located above and below the haplotypes.
(B) The positions of two insertions and two complex events characterized from the CH17 haplotype are shown mapped to the GRCh37 IGH human reference assembly sequence (black line; chr14:105,928,955-107,289,540). The locus is presented in the same orientation as that depicted by IMGT. Functional and ORF IGHV, IGHD, and IGHJ genes (not to scale) and IGHV pseudogenes are shown (to scale); the names of IGHV genes involved in the characterized structural variants are indicated. Segmental duplications82,83 downloaded from the UCSC genome browser are shown below GRCh37.
A direct comparison of this CH17 assembly to that of the human genome reference revealed the presence of four large CNVs involving ten functional IGHV genes, including two insertions and two complex insertion/deletion events (Figure 1; Table 1; Figure S2). We refer to complex events as those for which a haplotype harboring a CNV requires more than two breakpoints to reconcile the variant with the human reference genome and is therefore not a simple insertion, deletion, or inversion (e.g., the IGHV5-a and IGHV3-64D haplotype versus the IGHV3-9 and IGHV1-8 haplotype). Importantly, a complex event can be best described in the context of a haplotype other than the human reference sequence; for example, although the complex event in CH17 involving IGHV4-30-2 is significantly different from GRCh37 with respect to nucleotide similarity, based on sequence analysis (Figures S1 and S8) this event was most likely mediated by an alternate insertion haplotype described from fosmid clones in this study (see below).
Table 1.
CNVs Identified from BAC and Fosmid Clones
| Individual | Population | CNV Type | IGHV Genes Included in CNVa | GRCh37 Outer-Start (Breakpoint)b | GRCh37 Outer-End (Breakpoint)b | Event Size (∼kbp) | Accessions/Clones |
|---|---|---|---|---|---|---|---|
| CH17 | nd | Insertion | V1-69D, V1-f, V3-h, V2-70D (gain) | 107174927 | 107174941 | 46.6 | AC245023, AC245094 |
| CH17 | nd | Complex event | V4-30-2 (gain) V4-31 (loss) | 106804332 | 106810878 | 6.5c/48.8d | AC245166 |
| CH17 | nd | Complex event | V5-a, V3-64D (gain) V3-9, V1-8 (loss) | 106531320 | 106569343 | 38c/37.7e | AC245085, AC247036 |
| CH17 | nd | Insertion | V7-4-1 (gain) | 106483362 | 106484225 | 9.5 | AC244226, AC245085, AC247036 |
| NA12156 | CEPH | Deletion | V4-39, V3-38 (loss) | 106866357 | 106899042 | 32.7 | AC244497 |
| NA15510 and NA19240 | nd and Yoruba | Insertion | V1-c, V3-d, V3-43D, V4-b (gain) | 106877146 | 106877535 | 61.1 | AC241995, AC234225; AC233755, KC162926f |
| NA18555 (haplotype A) | Han Chinese | Complex event | V3-30-5, V4-30-4, V3-30-3, V4-30-2 (gain) | 106804332 | 106804333 | 49.2 | KC162924, AC231260, AC244456, KC162925 |
| NA18555 (haplotype B) | Han Chinese | Deletion | V4-31, V3-30 (loss) | 106786254 | 106811213 | 24.9c/73.9d | AC244464 |
| NA18507 | Yoruban | Complex eventg | V4-30-4, V3-30-3 (gain) V3-30 (loss) | 106784242 | nd | 25.2 | AC244411 |
| NA18502 | Yoruban | Complex eventg | V3-30-5 (gain) V3-33, V4-31 (loss) | nd | 106820685 | 24.7 | AC245243 |
| NA18956 and NA12156 | Japanese and CEPH | Duplication | V3-23D (gain) | 106716650 | 106727861 | 10.8 | AC244473, AC206018; AC244492 |
| NA19240 and NA12878 | Yoruban and CEPH | Insertion | V7-4-1 (gain) | 106483362 | 106484225 | 9.5 | AC241513; AC245090 |
nd, not defined;
Genes either lost or gained in the context of GRCh37 as part of CNV-containing haplotypes are noted.
Coordinates delineate regions in which event breakpoints are predicted to have occurred.
Event size with respect to GRCh37.
Event size with respect to NA18555 haplotype A (see Figure 4).
Event size with respect to CH17.
Clones from these two individuals were used to build a composite haplotype.
Partially characterized.
We determine that the CH17 haplotype harbors 101 kbp of sequence not represented in GRCh37. With respect to gene copy number differences, the CH17 IGHV haplotype differs from GRCh37 by four CNVs that involve ten IGHV genes (seven gains and three losses; Figure 1; Figures S2 and S3A). Of the 47 IGHV genes identified in the CH17 haplotype and the 43 identified in the human genome reference assembly (excluding IGHV7-81), 40 were shared (Figure S3B). Allelic differences were observed at 18 of these 40 IGHV genes, 14 of which involved amino acid changes between the two haplotypes (Figure 1; Table S5). In contrast to the IGHV locus, we did not observe any allelic differences at IGHD and IGHJ loci between the CH17 haplotype and GRCh37 (Table S5).
We also annotated 5′ and 3′ regulatory regions of the IGHV genes in CH17, including the 3′ recombination signal (RS) sequences, which are necessary for proper somatic rearrangement of IGHV genes to partially rearranged IGHD-J15 (Table S6). For genes that occur in identified CNVs, we provided a complete description of their regulatory regions (e.g., IGHV7-4-1). Regulatory sequence motifs for all CH17 haplotype IGHV genes were also compared to those reported in Matsuda et al.20 No variation was observed in the characterized regulatory sequences for IGHV genes shared between the CH17 haplotype and the GRCh37 assembly, even for genes at which we identified different alleles. However, for the IGHV3-64D duplicate gene that occurred as part of a complex event in CH17 (Figure 1), the RS nonamer differed from that described for IGHV3-64∗0220 by two nucleotides (Table S6). This sequence matched with 100% identity to RS nonamers from other IGHV genes.
Discovery and Sequence Characterization of Additional CNV Haplotypes in IGHV
We further explored CNVs in the IGHV locus by complete sequencing of large-insert fosmid clones mapping to the region. The clones were part of a fosmid end-sequence mapping project previously developed to characterize structural variation in the human genome.1–3 The primary libraries used here were constructed from nine individuals (two YRI, two JPT, two CHB, two individuals of European descent (CEPH), and one individual of unknown ethnicity). We identified a total of 191 discordant clones mapping to the IGH region from the nine libraries (47 insertion and 144 deletion clones). Of the 144 clones suspected to harbor deletions, 86 spanned IGHV, IGHD, IGHJ, and/or IGHC gene loci, based on end-read mapping positions, and were presumed to represent somatic V-(D)-J rearrangements. These are expected because the DNA used for fosmid library construction was isolated from Epstein-Barr virus (EBV) transformed B cell lines that undergo somatic rearrangements at immunoglobulin loci. To validate this assumption, we sequenced ten fosmid clones with suspected somatic rearrangements (Table S8)—an example of one of these fosmids in comparison to GRCh37 is shown in Figure S5. In addition, 23 deletion clones and 3 insertion clones mapped within the IGHC gene locus only and were excluded from further analysis; however, it is worth noting that IGHC-containing CNVs have been described previously.17–19,21,22,52,53 In addition to this set of clones, fully sequenced fosmids from a previous genome-wide data set/analysis43 based on a second set of libraries (eight individuals, from the same ethnicities) were also searched for possible CNV-containing clones in IGHV.
In total, we analyzed complete inserts of 17 discordant fosmid clones mapping to the IGHV locus, characterizing 8 CNV haplotypes (Table 1; Figure 2; Figure S4; an additional nine fosmids were also analyzed and found to contain either partial or no structural variation, Table S1). The corresponding CNVs from the aforementioned 17 clones involve copy number changes in 13 functional and ORF genes (not including those that overlapped with CH17), two of which (IGHV3-43D and IGHV3-38) had not been associated with CNVs prior to this study. In addition to that characterized above in CH17, we generated an additional 121 kbp of insertion sequence not present in GRCh37. We identified ten previously uncharacterized IGHV alleles from functional and ORF genes identified in the fosmid sequences (Table S7). In all cases for which alleles were described at IGHV genes present in GRCh37, variants were represented by SNPs in dbSNP135 and/or 1KG data sets. In addition, we characterized 5′ regulatory regions and 3′ RS sequences for IGHV genes identified in the fosmid data that previously lacked complete descriptions of these elements;39 in all cases, sequences matched those of other IGHV genes (Table S6).
Figure 2.
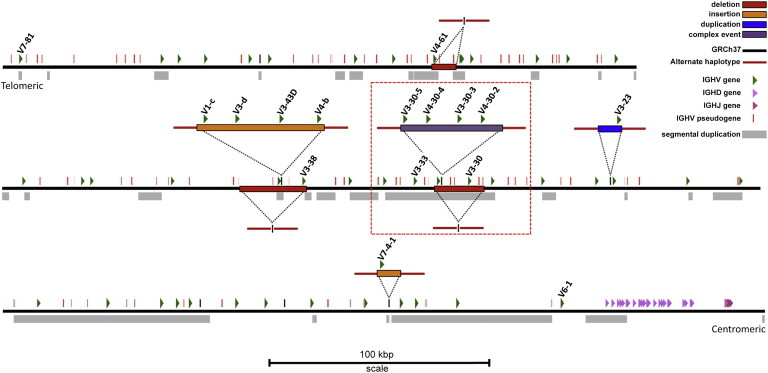
Map of IGHV CNVs Identified by Complete Sequencing of Fosmid Clones
The positions of three deletions, two insertions, one duplication, and one complex event characterized from fosmid alternative haplotypes are shown mapped to the GRCh37 IGH reference (black line; chr14:105,928,955–107,289,540) with the same parameters as in Figure 1B. The locus is presented in the same orientation as that depicted by IMGT. Functional and ORF IGHV, IGHD, and IGHJ genes (not to scale) as well as IGHV pseudogenes are shown (to scale); the names of IGHV genes involved in the characterized structural variants are indicated. Segmental duplications82,83 downloaded from the UCSC genome browser are shown below GRCh37. The large red box indicates a hotspot region of recurrent mutation (see Figure 4 for additional haplotypes associated with this hotspot). The deletion of IGHV4-61 was identified by Mills et al.5 (chr14:107,084,861–107,096,738) and was also included in our analyses.
Analysis of CNV Breakpoints and Inference of Mutational Mechanisms
By using previously described methods,43 we assessed the breakpoints of the CNVs described here, as well as previously identified breakpoints of the IGHV4-61 deletion5 (Tables 1 and 2; Figures 1 and 2; Figures S6–S14). Despite being able to determine event breakpoints, we were not able to infer the mutational mechanism underlying the complex CNV involving the genes IGHV1-8, IGHV3-9, IGHV5-a, and IGHV3-64D because this variant was not consistent with mechanisms typically associated with simple insertion, deletion, or duplication events (Figure S6). For all other events, we searched for and identified stretches of extended sequence homology flanking or spanning the breakpoints—a characteristic feature of nonallelic homologous recombination (NAHR).43 An example is shown in Figure 3 for the duplication event involving IGHV3-23 [MIM 611939], one of the most highly expressed genes in naive antibody repertoires.29,54 We identified two 5.3 kbp repeat segments (86% similarity) in GRCh37, suggesting that this duplication likely arose as a result of NAHR.
Table 2.
Analysis of CNV Breakpoints
| CNV Type | IGHV Genes in Event | Shared Sequence at Breakpointa(bp) | Extended Homology (∼kbp) | Sequence Identityb | IGHV Genes in Regions of Extended Homology |
|---|---|---|---|---|---|
| Insertion | V1-69D, V1-f, V3-h, V2-70D | 13 | 38 | 0.94 | V2-70, V1-69D, V2-70D, V1-69 |
| Deletionc | V4-61 | 58 | 5.6 | 0.95 | V3-62P, V3-60P |
| Insertion | V1-c, V3-d, V3-43D, V4b | nd | 11.7 | 0.93 | V4-39, V4-b |
| Deletion | V4-39, V4-38 | 24 | 0.73 | 0.79 | V3-41P, V3-38 |
| Complex event | V4-31, V3-30-5, V4-30-4, V3-30-3d | 47 | 25 | 0.92 | V3-30-3, V4-30-2, V3-30, V4-28 |
| Complex event (NA18555 haplotype A) | V3-30-5, V4-30-4, V3-30-3, V4-30-2 | nde | 25 | 0.94 | V3-33, V4-31, V3-30-5, V4-30-4 |
| Deletion (NA18555 haplotype B) | V4-31, V3-30 | 249 | 25 | 0.94 | V3-33, V4-31, V3-30, V4-28 |
| Complex event | V3-30, V4-30-2c | 61 | 25 | 0.94 | V3-33, V4-31, V3-30, V4-28 |
| Complex event | V3-33, V4-31c | nd | 25 | 0.92 | V3-33, V4-31, V3-30-3, V4-30-2 |
| Duplication | V3-23D | 373 | 5.3 | 0.86 | V3-23, V3-22P |
| Insertion | V7-4-1 | nd | 0.86 | 0.97f | No IGHV genes |
nd, not determined.
Length of shared sequence at predicted breakpoint with greater than 98% sequence identity.
Percent sequence identity between regions of extended homology spanning event breakpoints.
Deletion identified by Mills et al.5 (chr14: 107,084,861–107,096,738).
Genes that are deleted in the context of haplotype A (see Figure 4)—genes listed for other events (insertion/deletions, duplications) represent changes in the context of GRCh37.
Although this haplotype could be defined as an insertion based on a direct comparison to GRCh37 and CH17, due to the complicated duplication structure in the region, the exact mechanisms underlying this event could not be determined based on available haplotypes (see Figure S1A).
Calculated from CH17 haplotype.
Figure 3.
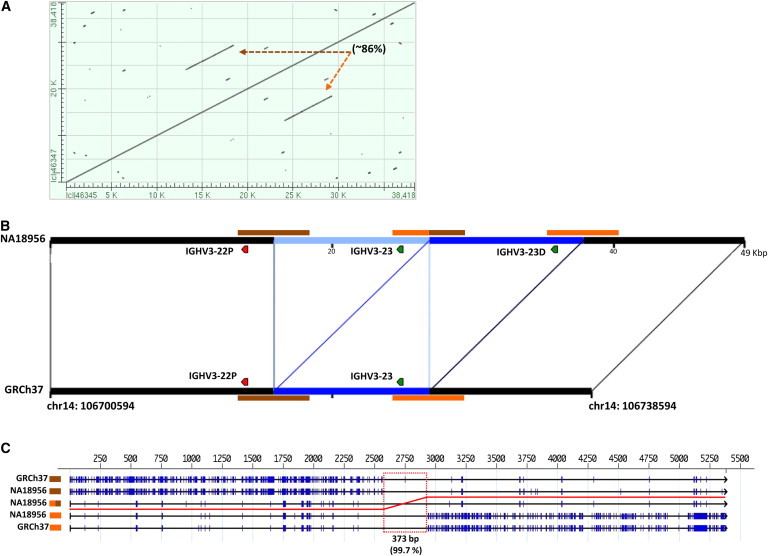
Breakpoint Analysis of the IGHV3-23 Duplication
(A) Pairwise BLAST alignment of 38 kbp region surrounding IGHV3-23 in GRCh37 (chr14:106700594–106738594). Brown and orange dotted arrows point to ∼5.3 kbp repeat sequences (extended homology) suspected to have mediated the IGHV3-23 duplication. Repeat sequences show 86% sequence identity.
(B) Sequence harboring the IGHV3-23 duplication identified in individual NA18956 (clones AC244473 and AC206018) is compared to GRCh37 (chr14:106700594–106738594). Regions of similarity between the two haplotypes are connected by black lines. Segments colored in blue in both haplotypes indicate the locations of the 10.8 kbp duplicates. Brown and orange bars above the NA18956 haplotype and below the GRCh37 haplotype indicate ∼5.3 kbp repeat sequences identified by BLAST in (A). Labeled IGHV genes and pseudogenes are depicted by green and red chevrons, respectively.
(C) A five-way alignment of ∼5.3 kbp repeat sequences from both haplotypes. Alignment of base positions is shown along the top of the diagram. Each repeat sequence (three from NA18956 and two from GRCh37) is represented by a single horizontal black line. Blue tick marks on each line indicate nucleotide (nt) differences and gaps observed between the aligned sequences. The red line tracks the most similar alignment of the middle NA18956 repeat sequence to the other four sequences. Based on nt similarity, the event breakpoint is presumed to have occurred within the 373 bp region (red box) in which all aligned sequences share 99.7% sequence identity (372/373 nt).
The lengths of repeat segments (extended homology) involved in each of the analyzed CNVs varied, ranging from 600 bp to 38 kbp in size (Table 2). Sequence identities between repeat segments also varied, but in most cases where crossover events were suspected to have occurred, we observed shorter stretches of sequence with almost perfect sequence identity (>98%) within the predicted breakpoint regions (e.g., Figure 3C). However, in several instances, likely due to the accumulation of mutations (some of which might be population-specific) following the formation of the CNV, we were not able to predict with certainty where the breakpoints occurred based on the sequences available (e.g., Figures S10 and S14; Table 2). With the exception of the IGHV7-4-1 insertion, all predicted event-mediating (referred to as extended homology) repeats contained IGHV genes or pseudogenes (Table 2). Notably, within the region spanning IGHV4-28 to IGHV4-34, which has previously been shown to exhibit extreme haplotype variation24,25,55,56 and has been implicated in several autoimmune disorders,24,31,33,57,58 we identified evidence of an NAHR hotspot. Five distinct CNVs were characterized, all of which were predicted to be mediated by crossover events involving large ∼25 kbp segmental duplications (Figure 4). Each of these duplicated blocks includes two functional IGHV genes. Compared to GRCh37, which contains two ∼25 kbp segmental duplicates, one of the haplotypes identified in NA18555 in this region contained two additional repeated ∼25 kbp segmental duplications (Figure 4). The exact mechanisms mediating this expanded haplotype could not be determined (Figure S1); however, based on sequence similarity, it was clear that several events were mediated by segmental duplication blocks found in this haplotype, rather than by those represented in GRCh37 (Figure 4; Figures S1 and S12–S14). The haplotypes identified here in this region are corroborated by previous data,27 but this represents a comprehensive description of these variants at nucleotide resolution.
Figure 4.
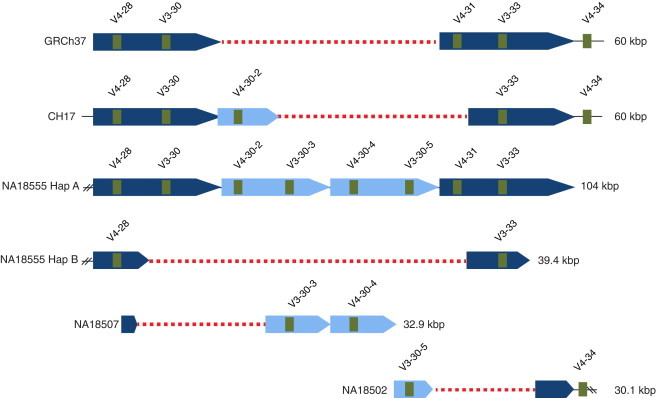
A Hotspot of IGH Structural Polymorphism
Each of the five identified haplotypes harboring diverse CNVs is shown relative to GRCh37. Each haplotype is labeled with a sample identifier, and the length (kbp) is indicated at the right of each haplotype in parentheses. Two haplotypes in this region were identified from the individual NA18555 (haplotypes A and B). One to four ∼25 kbp segmental duplication sequence blocks (or partial blocks), depending on the haplotype, are depicted by shaded blue bars. Deleted regions identified in five of the haplotypes, including GRCh37, are indicated by red dotted lines. The positions and names of functional IGHV genes (green boxes) are shown in each haplotype. The partial haplotype identified in this region from individual NA19240 (AC234301), which overlapped that of NA18555 haplotype A and included the genes IGHV4-30-2, IGHV3-30-3, IGHV4-30-4, and IGHV3-30-5, is not depicted but was included in the analysis and allowed for the placement of the NA18502 haplotype (also see Figures S1 and S12–S14).
It is also important to note that although the sequence identity between characterized allelic 25 kbp segments in this region was higher than that observed between paralogous segments, point mutations were also observed between homologous segments from different haplotypes. Thus, further sequencing of this region in more individuals will be required to fully catalog the range of nucleotide variation between homologous and paralogous segmental duplicates, which will be essential for the development of effective genotyping tools and more precise delineation of event breakpoints. Additional resequencing is likely to yield the discovery of novel structurally variant haplotypes, which might shed more light on the history of these events. For example, the gene IGHV4-30-1 is also known to be present in this region24,25,55,56 but was not identified as part of the CNVs characterized here.
Population Stratification of IGHV CNVs
To assess the global frequency of a subset of the discovered IGHV CNVs, we genotyped 4 of these in a total of 425 individuals from 9 diverse populations. For three of the CNVs (the IGHV1-c, IGHV3-d, IGHV3-43D, and IGHV4-b insertion; the IGHV3-64D, IGHV5-a, IGHV3-9, and IGHV1-8 complex event; and the IGHV7-4-1 insertion), we designed haplotype-specific primers for standard PCR. For the fourth CNV corresponding to the IGHV1-69D, IGHV1-f, IGHV3-h, and IGHV2-70D insertion, we used a TaqMan Copy Number Assay, in addition to haplotype-specific standard PCR assays designed near IGHV1-f. The allele frequencies for each event are listed in Table 3. We examined population stratification by using Fst. In contrast to genome-wide average pairwise Fst values for common autosomal SNPs, which are typically less than 0.2,59 all four CNVs showed significant population stratification, with population pairwise Fst values reaching 0.5 for some variants (Figure 5). For example, the ∼61 kbp insertion containing IGHV1-c, IGHV3-d, IGHV3-43D, and IGHV4-b was highly stratified between African and Asian/European populations (Figure 5A). Additionally, copy number of the IGHV1-69 and IGHV2-70 haplotype was also highly stratified between the three major continental groups, with Asians having fewer copies than Africans (Figure 5B; Table 3). We observed that all individuals with more than three copies of IGHV2-70 and IGHV1-69 in their genome also carry IGHV1-f; however, this gene was observed in several individuals for which only two IGHV1-69/IGHV2-70 copies were predicted, suggesting that some variability in our custom TaqMan assay exists, which has likely resulted in an underestimation of copy number in some individuals.
Table 3.
Allele Frequencies of Genotyped CNVs
| Population | N | Abbreviation | Region | V7-4-1 Insertion | V5-a and V3-64D (CH17) | V4-b, V3-43D, V3-d, and V1-c Insertion | V1-69, V2-70, V1-f Insertiona,1 | V1-69, V2-70, V1-f Insertion2 |
|---|---|---|---|---|---|---|---|---|
| Luyha | 48 | LWK | Africa | 0.51 | 0.15 | 0.65 | 0.57 | 0.41 |
| Maasai | 46 | MKK | Africa | 0.42 | 0.16 | 0.45 | 0.42 | 0.34 |
| Yoruba | 48 | YRI | Africa | 0.34 | 0.03 | 0.56 | 0.52 | 0.45 |
| Han Chinese | 45 | CHB | Asia | 0.79 | 0.20 | 0.23 | 0.08 | 0.04 |
| Japanese | 46 | JPT | Asia | 0.78 | 0.21 | 0.08 | 0.02 | 0.00 |
| Finnish | 48 | FIN | Europe | 0.75 | 0.47 | 0.13 | 0.19 | 0.18 |
| British | 48 | GBR | Europe | 0.65 | 0.48 | 0.12 | 0.22 | 0.16 |
| Iberian | 48 | IBS | Europe | 0.54 | 0.34 | 0.12 | 0.16 | 0.15 |
| Toscani | 48 | TSI | Europe | 0.62 | 0.46 | 0.14 | 0.24 | 0.22 |
| Chimpanzee | 5 | PTR | NA | 0.80 | 0.00 | 0.80 | 0.00 | NT |
| Gorilla | 5 | GGO | NA | 1.00 | 0.00 | 1.00 | 1.00 | NT |
| Orangutan | 5 | PPY | NA | 0.00 | 1.00 | 0.40 | 0.00 | NT |
Figure 5.
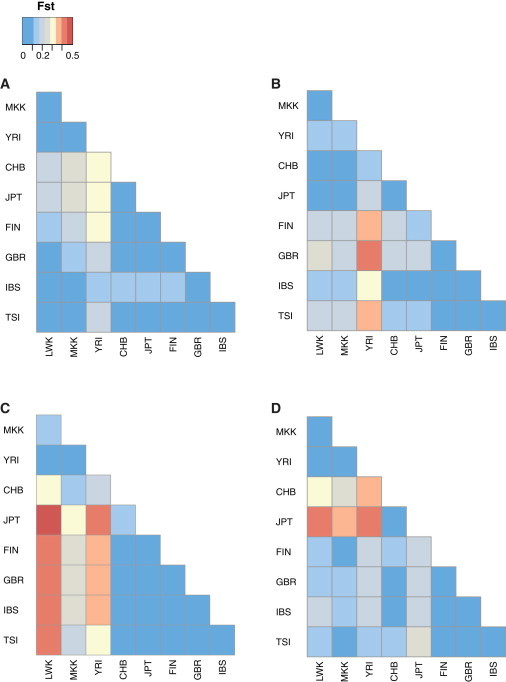
Pairwise Fst of Copy-Number Variant Loci in Nine Human Populations
Heatmaps representing pairwise Fst values between populations calculated based on CNV allele frequency data generated at four loci are as follows: (A) IGHV7-4-1 insertion; (B) IGHV3-9, IGHV1-8, IGHV5-a, and IGHV3-64D complex event; (C) IGHV1-c, IGHV3-d, IGHV3-43D, and IGHV4-b insertion; and (D) IGHV1-69D, IGHV1-f, IGHV3-h, and IGHV2-70D insertion. Population abbreviations are shown on both map axes. Colors in each square correspond to a given Fst value range as indicated by the key.
We also genotyped these four CNVs in five chimpanzees (Pan troglodytes), five gorillas (Gorilla gorilla), and five orangutans (two Bornean, Pongo pygmaeus, and three Sumatran, Pongo abelii) (Table 3). Strikingly, the presence of each CNV was noted in at least one nonhuman primate, suggesting that none of these variants are unique to humans. Interestingly, the IGHV5-a and IGHV3-64D haplotype was observed in orangutan in the absence of IGHV3-9 and IGHV1-8, whereas the converse was noted in chimpanzee and gorilla. Likewise, the haplotype containing the insertion of IGHV1-69D, IGHV1f, IGHV3-h, and IGHV2-70D was amplified in gorilla, indicating that a duplication of IGHV1-69 and IGHV2-70 could have preceded the split of gorillas from the chimpanzee/human lineage. The fact that this variant was not detected in chimpanzees or orangutans could indicate an actual difference between species or simply reflect effects of potential species-specific variants on assay performance or the limited number of individuals screened. Further to this point, in a number of instances we found that CNV allele frequency was less than 1.0 (Table 3); however, these are the result of a lack of PCR product generated from some individuals for assays of both alleles at a given CNV locus (e.g., IGHV7-4-1 insertion in chimpanzee). Thus, the extent to which the loci screened here are polymorphic and stratified within nonhuman primate species will require additional genotyping in a larger panel of samples.
To investigate whether IGHV CNVs were in LD with SNPs found on commercial arrays, we downloaded genotypes for Affymetrix 6.0 and the Illumina Omni 1 Quad array SNPs in individuals from 1KG data sets for seven of the nine populations screened with the four IGHV CNV assays. We assessed LD between these SNPs and alleles at CNV loci using r2 (Table 4). This analysis revealed that, in general, the genotyped CNVs were poorly represented (r2 < 0.8) by commercial arrays in all of the populations screened. LD was stronger between IGHV CNVs and SNPs found on the Illumina Omni 1Quad, which was likely a result of the increased density of SNPs in IGH on this array (noted previously).16 Furthermore, analogous to trends observed genome-wide,60 we observed that LD estimates between IGHV CNVs and array SNPs were lowest in African populations.
Table 4.
Commercial Array SNPs in Linkage Disequilibrium with IGHV CNVs
| Population | Abbreviation | Region | V7-4-1 Insertion | V5-a, V3-64D, V3-9, V1-8 Complex Event | V4-b, V3-43D, V3-d, V1-c Insertion | V1-69, V2-70, V1-f Insertiona |
|---|---|---|---|---|---|---|
| Luyha | LWK | Africa | 0.28 (rs8005760) | 0.30 (rs12586543) | 0.29 (rs11847718) | 0.36 (rs10129255) |
| Yoruba | YRI | Africa | 0.09 (rs17646414) | 0.34 (rs4774001) | 0.10 (rs2337470) | 0.13 (rs17112644) |
| Han Chinese | CHB | Asia | 0.10 (rs885883) | 0.17 (rs8010605) | 0.37 (rs41471651) | 0.63 (rs17113366) |
| Japanese | JPT | Asia | 0.05 (rs10141701) | 0.23 (rs17113281) | 0.76 (rs41471651) | NA |
| Finnish | FIN | Europe | 0.21 (rs8005760) | 0.21 (rs4774028) | 0.19 (rs41471651) | 0.49 (rs17672538) |
| British | GBR | Europe | 0.14 (rs6576127) | 0.29 (rs10150642) | 0.30 (rs17646414) | 0.81 (rs17672538) |
| Toscani | TSI | Europe | 0.09 (rs4774001) | 0.18 (rs17737576) | 0.43 (rs17646414) | 0.69 (rs17672538) |
| Affymetrix 6.0 SNP Chip | ||||||
| Population | Abbreviation | Region | V7-4-1 Insertion | V5-a, V3-64D, V3-9, V1-8 Complex Event | V4-b, V3-43D, V3-d, V1-c Insertion | V1-69, V2-70, V1-f Insertiona |
| Luyha | LWK | Africa | 0.26 (rs17112739) | 0.68 (rs2077170) | 0.27 (rs12886451) | 0.38 (rs6576220) |
| Yoruba | YRI | Africa | 0.22 (rs12365) | 0.34 (rs12588113) | 0.40 (rs2027916) | 0.27 (rs11848687) |
| Han Chinese | CHB | Asia | 0.57 (rs7144717) | 0.49 (rs2077170) | 0.84 (rs7400983) | 0.16 (rs4774166) |
| Japanese | JPT | Asia | 0.55 (rs12884400) | 0.72 (rs2077170) | 0.76 (rs4774143) | NA |
| Finnish | FIN | Europe | 0.65 (rs1985733) | 0.92 (rs2077170) | 0.42 (rs2106002) | 0.47 (rs10141910) |
| British | GBR | Europe | 0.67 (rs7144717) | 0.81 (rs2077170) | 0.37 (rs7146845) | 0.71 (rs10141910) |
| Toscani | TSI | Europe | 0.53 (rs7144717) | 0.73 (rs2077170) | 0.61 (rs7400983) | 0.69 (rs10141910) |
| Illumina Omni 1 Quad | ||||||
SNPs with the highest LD values for a given CNV and population are shown; r2 values are indicated for each SNP. NA, not applicable.
Genotype data used from TaqMan assay.
Discussion
The primary motivation of this study was to produce a set of alternative haplotypes that better represent standing genetic diversity in the IGH locus, such that these sequences could aid future investigations of the relationships between IGH germline polymorphisms, antibody expression and function, and disease susceptibility. To this aim, we have generated an alternative reference genome assembly for the IGHV, IGHD, and IGHJ gene clusters from a hydatidiform mole BAC library and analyzed sequence from 36 fosmid clones from a diverse set of ethnic backgrounds. The CH17 assembly represents the most complete haplotype upon which IGHV CNVs can be mapped. From these data, we have analyzed 12 large CNVs ranging in size from 6.5 kbp to 61.1 kbp (Table 1), including descriptions of polymorphisms implicated in human disease, at nucleotide resolution. As a result, we have characterized 222 kbp of insertion sequence not presently found in the human reference genome, effectively increasing the length of available reference sequence in the IGHV locus by over 20% and providing a resource for future genotyping and association studies.
Annotations of these sequences allowed for the identification of 15 IGHV gene alleles. The fact that we identified 15 previously uncharacterized alleles from a targeted survey in a relatively small sample of individuals suggests that additional sampling of IGHV gene allelic variation will be required, especially in less studied populations.
With the exception of the duplication of IGHV3-43 and deletion of IGHV3-38, each of the CNVs identified here included genes previously known or suspected to vary in copy number; however, these events have not previously been fully sequenced and characterized at the genomic level. For example, the presence of the 9.5 kbp insertion including IGHV7-4-1 had been previously identified by using restriction fragment length polymorphism (RFLP) analysis in Japanese and European cohorts,61 but the complete sequence of this insertion was not known. Prior to this study, up to 28 IGHV functional or ORF genes were suspected to vary in copy number, either from 0–1 copies (n = 19) or 1–2 copies (n = 9) per haploid genome.16 Importantly, 12 of the 19 IGHV genes that vary from 0–1 haploid copies are not represented in the human reference genome.38 We identified four of these 12 genes in the CH17 haplotype and an additional six in the fosmid haplotypes described here. In addition, the pseudogene, IGHV3-h, which is also not represented in GRCh37 but has previously been observed in 58% of Danish individuals,62 was identified in CH17. Of the remaining seven “0–1” haploid copy genes, all were included in CNVs analyzed in this study. We also described CNVs that included duplications of IGHV genes represented in GRCh37, including four of the nine IGHV genes that are suspected to vary from 1–2 haploid copies, as well as a duplication of IGHV3-43 (IGHV3-43D). In summary, we have characterized haplotypes involving 22 of the known 28 above mentioned copy-number-variable IGHV genes. Thus, although our sequencing survey facilitated the characterization of an overwhelming majority of suspected CNVs in IGHV, our results suggest that, similar to IGHV coding variation, more work will be necessary to complete descriptions of CNVs in the region. Importantly, the IGH haplotypes generated here will provide a starting point from which additional CNVs can be identified.
In addition to confirming previously reported CNVs in IGH, our data also allowed for IGHV genes involved in the same CNV polymorphism to be definitively ordered into genomic haplotypes, either validating or improving the characterization of previously suspected variants inferred by PCR, RFLP, or expressed IGHV gene analysis. For example, a deletion of IGHV3-9 and IGHV1-8 had been reported in some haplotypes;24,26,27,29 our data confirmed this but also revealed that rather than these genes occurring as a simple deletion variant, they were replaced by two unrelated genes, IGHV5-a and IGHV3-64D, not previously known to co-occur on the same haplotype. It is interesting to note that recent descriptions of putative IGHV haplotypes based on the analysis of expressed antibody repertoires showed that IGHV5-a is present only on individual haplotypes lacking IGHV1-8 and IGHV3-9.24,26,27,29 Our genotyping data also support this finding because all individuals lacking the IGHV1-8 and IGHV3-9 haplotype were positive for the IGHV5-a and IGHV3-64D haplotype. Despite the fact that these data, taken together, support the notion that the IGHV5-a and IGHV3-64D variant in CH17 is genuine, further sequence validation of this complex event in additional haplotypes should be conducted. Similarly, IGHV1-69 and IGHV2-70 have also been suggested to occur as part of the same duplication, which we confirmed from the CH17 haplotype. However, our data revealed that this 38 kbp duplication also included the previously unmapped pseudogene IGHV3-h 62 and was associated with the presence of IGHV1-f.
The repetitive nature of the IGHV locus, which is primarily the result of IGHV gene duplication,20 is presumed to have facilitated the high frequency of structural variation found throughout the locus.25 Because complete descriptions of CNVs in IGHV have remained limited until now, an assessment of the mutational mechanisms directing haplotype diversity across the locus has not been possible. Our data provide strong evidence that NAHR appears to be the dominating mechanism underlying haplotype diversity in the region. For 10 of the 11 CNVs for which NAHR was suspected, event-mediating sequences were closely associated with IGHV genes or pseudogenes, suggesting that the evolutionary expansion of the IGHV genes and the generation of associated segmental duplications have provided substrate for the formation of the majority of CNVs in the locus. Similar to IGHV, complex and repetitive genomic architectures are also known to underlie CNVs in the IGHC locus17–19,21,22,52,53 and in other immune system gene loci, such as killer immunoglobulin receptor (KIR) genes and beta-defensins.63–66
Extensive overlap of CNVs with segmental duplications has been observed in both human and primate genomes.67,68 In fact, the recent discovery that an increase in segmental duplications occurred genome-wide in the ancestor of humans and African great apes implicates such regions in the formation of shared and species-specific CNVs.69 We found that four of the CNV-containing haplotypes characterized in this study (three insertions and one complex event) were also present in at least one of three nonhuman primates. Two of the insertions included large segmental duplications encompassing functional IGHV genes that mediated the deletions observed in the human genome reference assembly. Although we did not observe direct evidence that any of the four loci were polymorphic within chimpanzee, gorilla, or orangutan, variants at two of the loci were observed in only one of the three species, suggesting the potential for fixed differences between species; however, it is important to note that we screened only a very small number of individuals (n = 5 per species), and thus we cannot rule out effects of limited sample size or species-specific sequence variability on assay performance. We found evidence of the insertion haplotype that included IGHV1-69D and IGHV2-70D in gorilla, but not in chimpanzee or orangutan. Specific IGHV1-69 alleles have been shown to be important in neutralizing antibodies isolated from three human populations against particular influenza epitopes,70–73 leading to the hypothesis that IGHV1-69 evolved to respond quickly during the initial stages of infection.73 Furthermore, extensions of this hypothesis suggest a potential role for infection-driven B cell clonal expansion in the development of chronic lymphocytic leukemias [MIM 151400],73 motivated by the observation that these cancers are often associated with B cell usage of particular IGHV genes, especially IGHV1-69.74–76 Interestingly, usage of IGHV1-69 is not commonly found in individuals with B cell lymphoma from Asian populations.76 It is worth noting that investigations of IGHV1-69 provide the only example explicitly connecting IGHV gene copy number to gene usage,77 which might be important considering that we found Asians to have low IGHV1-69 copy number compared to African and European populations.
In fact, we observed evidence of population stratification for all of the CNVs screened here, because each locus had pairwise Fst values above 0.3 for at least two of the populations surveyed, with some values reaching upward of 0.5. Population differences had been noted previously for one of the CNVs (IGHV7-4-1 insertion61), although this report included fewer individuals and populations. However, prior to this study, the remaining three CNVs had only been screened in individuals of European descent; thus, our data represent the most extensive survey of IGHV CNVs to date. Our findings seem somewhat striking given that we surveyed only four CNVs but observed differences in population allele frequencies at each locus. For example, this is in contrast to a previous examination of Fst at 190 insertion-deletion loci in three populations, also identified from discordant fosmid clones, which showed that only 20 of these polymorphisms exhibited Fst values above 0.35.3 The high Fst values observed for the IGHV CNV screened here might reflect the fact that these particular variants associate with segmental duplication and include functional genes—two features that have been suspected to influence selection.14 In addition, population differences have also been noted for CNVs involving genes related to the immune response, and in many instances these genes have been implicated in disease risk and progression.11,12,78
Ultimately, it will be important to understand the role of IGHV germline CNVs and allelic polymorphisms in antibody expression and the potential influences of these variants on human disease. IGHV gene copy number differences and point mutations in IGHV gene regulatory sequences are likely to be important factors underlying individual differences in IGHV gene usage in expressed antibody repertoires.77,79 This point has recently been bolstered by observations of strong correlations between IGHV gene usage in naive antibody repertoires of monozygotic twins, suggesting the involvement of heritable factors.54 With respect to copy-number variation, we also observed haplotypes containing deletions of IGHV genes that have been shown to be absent in expressed naive antibody repertoires in some individuals.24,26,27,29,54 The development of robust genotyping assays provided for by the sequence presented here will now allow for more direct connections between IGHV CNVs and gene expression—an important step toward understanding the potential roles of these polymorphisms in disease.
To date there have been few human diseases linked to IGHV polymorphisms. For example, only one putative association has been reported as part of standard GWAS (Kawasaki disease [MIM 611775]32), and of the associations that have been reported from candidate-gene studies, few have been robustly replicated. We propose that the lack of GWAS associations might be the result of poor SNP coverage and fragmented LD in the region, which can vary depending on ethnicity. Our analysis of four IGHV CNVs and SNPs from two commercial arrays supports this, because most of the CNVs were poorly tagged by surrogate SNPs in the populations surveyed, particularly those of African descent. For example, only 7% of the possible CNVs and population intersections showed significant association with a tag SNP (r2 > 0.8). Similar observations have also been shown for IGHV gene allelic SNPs.16 It is also important to note that our LD analysis excluded CNVs in the IGHV4-28 to IGHV4-34 gene region, in which we identified five previously uncharacterized CNV-containing haplotypes in addition to GRCh37. Several of these variants, although unique, included insertions and deletions of the same genes. For example, IGHV4-31 and IGHV3-30 were deleted in more than one haplotype; likewise, duplicates of IGHV3-30 (IGHV3-30-3 and IGHV3-30-5) were also observed on several haplotypes (Figure 4). Consistent with previous findings,24,27,55,56 these data indicate that diploid copy number of IGHV3-30 and related genes can range from zero to six. Importantly, genotypes lacking these genes have been associated to rheumatoid arthritis [MIM 180300], chronic idiopathic thrombocytopenic purpura [MIM 188030], and systemic lupus erythematosus [MIM 152700].31,33,57,58 The importance of this point is that the occurrence of recurrent CNVs is known to have significant impacts on correlations between such variants and neighboring SNPs.80 Thus, given the presence of recurrent but overlapping events in this region, it would not be surprising if CNVs involving IGHV3-30 and other genes between IGHV4-28 and IGHV4-34 were also poorly represented by neighboring SNPs and might explain why previous disease associations in the region have not been replicated using high-throughput techniques. Such considerations would also be important for CNVs that include insertions and deletions of functional IGHV genes with known allelic polymorphisms, for example, IGHV1-69 and IGHV1-69D for which at least 12 CNV haplotypes and 14 alleles are known.56,81 In such instances, it might be essential to take into account the effects of both types of variation for studies of disease association.
In summary, we have undertaken the most extensive genomic sequencing study in the IGH locus to date, from which we have generated sequence and characterized a significant portion of “missing” IGHV genes, as well as an overwhelming majority of IGHV genes previously reported to be involved in structural variation. These data provide a useful foundation for continued efforts toward the establishment of a more complete genomic map of this complex region of the genome. The sequence generated here will also undoubtedly aid the development of more effective genomic tools in the IGH locus, which will be essential for exploring the evolutionary history of IGH genes and their diversity in human populations, and will ultimately provide a stronger framework for understanding the potential role of IGH genetic diversity in antibody expression and human disease. In a broader context, our findings can also serve as an illustrative example for those studying other structurally complex regions of the genome for which genomic reference sequence data remain limited. The CH17 BAC resource, for example, has already been used to discover and describe missing sequence including previously uncharacterized genes.34 Many other stretches of highly homologous sequence are now being identified as a result of this haploid resource. In addition, for loci that are somatically rearranged, including immunoglobulin and T cell receptor loci, the germline nature of this clone resource allows for the characterization of a single haplotype unaffected by somatic rearrangement.
Acknowledgments
We are grateful to T. Brown for assistance with manuscript preparation. We are grateful to Marie-Paule Lefranc and to the IMGT Nomenclature Committee for their help in defining IGHV genes and alleles and for providing the IMGT standardized rules for descriptions of CNVs. C.T.W. was supported in part by a President’s Research Stipend and graduate fellowship awarded by Simon Fraser University. K.M.S. was supported by a Ruth L. Kirschstein National Research Service Award (NRSA) training grant to the University of Washington (T32HG00035) and an individual NRSA Fellowship (F32GM097807). This work was supported by the US National Institutes of Health (grants HG002385 and HG004120 to E.E.E.) and a National Science and Engineering Research Council of Canada grant to F.B. E.E.E. is an Investigator of the Howard Hughes Medical Institute. E.E.E. is on the scientific advisory boards for Pacific Biosciences, SynapDx, and DNAnexus.
Contributor Information
Evan E. Eichler, Email: eee@gs.washington.edu.
Felix Breden, Email: breden@sfu.ca.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
BACPAC Resources Center: http://bacpac.chori.org
dbVAR: www.ncbi.nlm.nih.gov/dbvar
IMGT, the International ImMunoGeneTics Information System, www.imgt.org
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org/
UCSC Genome Browser, http://genome.ucsc.edu
Accession Numbers
The GenBank accession numbers for the large-insert clone sequences and IGHV genes reported in this paper are AC244483, AC244478, AC244410, AC244412, AC244459, AC244496, AC244411, AC244460, AC244396, AC244491, AC244399, AC244400, AC244487, AC244397, AC244398, AC206018, AC244473, AC244395, AC244480, AC244470, AC244495, AC244482, AC241513, AC244463, AC244430, AC234301, AC233755, AC234135, AC244405, AC244467, AC244500, AC244481, AC244464, AC231260, AC244456, AC244477, AC245090, AC244490, AC244484, AC244494, AC244486, AC244488, AC244492, AC244476, AC244497, AC244468, AC245243, AC244393, AC244450, AC244449, AC241995, AC234225, AC246787, AC244226, AC247036, AC245085, AC245166, AC244452, AC245369, AC245023, AC245094, KC162924, KC162925, KC162926, KC713934, KC713935, KC713936, KC713937, KC713938, KC713939, KC713940, KC713941, KC713942, KC713943, KC713944, KC713945, KC713946, KC713947, KC713948, KC713949, and KC713950. The CNVs characterized in this study have been submitted to dbVar under the accession number nstd76.
References
- 1.Tuzun E., Sharp A.J., Bailey J.A., Kaul R., Morrison V.A., Pertz L.M., Haugen E., Hayden H., Albertson D., Pinkel D. Fine-scale structural variation of the human genome. Nat. Genet. 2005;37:727–732. doi: 10.1038/ng1562. [DOI] [PubMed] [Google Scholar]
- 2.Kidd J.M., Cooper G.M., Donahue W.F., Hayden H.S., Sampas N., Graves T., Hansen N., Teague B., Alkan C., Antonacci F. Mapping and sequencing of structural variation from eight human genomes. Nature. 2008;453:56–64. doi: 10.1038/nature06862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kidd J.M., Sampas N., Antonacci F., Graves T., Fulton R., Hayden H.S., Alkan C., Malig M., Ventura M., Giannuzzi G. Characterization of missing human genome sequences and copy-number polymorphic insertions. Nat. Methods. 2010;7:365–371. doi: 10.1038/nmeth.1451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sudmant P.H., Kitzman J.O., Antonacci F., Alkan C., Malig M., Tsalenko A., Sampas N., Bruhn L., Shendure J., Eichler E.E., 1000 Genomes Project Diversity of human copy number variation and multicopy genes. Science. 2010;330:641–646. doi: 10.1126/science.1197005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mills R.E., Walter K., Stewart C., Handsaker R.E., Chen K., Alkan C., Abyzov A., Yoon S.C., Ye K., Cheetham R.K., 1000 Genomes Project Mapping copy number variation by population-scale genome sequencing. Nature. 2011;470:59–65. doi: 10.1038/nature09708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Perry G.H., Dominy N.J., Claw K.G., Lee A.S., Fiegler H., Redon R., Werner J., Villanea F.A., Mountain J.L., Misra R. Diet and the evolution of human amylase gene copy number variation. Nat. Genet. 2007;39:1256–1260. doi: 10.1038/ng2123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Koolen D.A., Vissers L.E., Pfundt R., de Leeuw N., Knight S.J., Regan R., Kooy R.F., Reyniers E., Romano C., Fichera M. A new chromosome 17q21.31 microdeletion syndrome associated with a common inversion polymorphism. Nat. Genet. 2006;38:999–1001. doi: 10.1038/ng1853. [DOI] [PubMed] [Google Scholar]
- 8.Sharp A.J., Hansen S., Selzer R.R., Cheng Z., Regan R., Hurst J.A., Stewart H., Price S.M., Blair E., Hennekam R.C. Discovery of previously unidentified genomic disorders from the duplication architecture of the human genome. Nat. Genet. 2006;38:1038–1042. doi: 10.1038/ng1862. [DOI] [PubMed] [Google Scholar]
- 9.Mefford H.C., Yendle S.C., Hsu C., Cook J., Geraghty E., McMahon J.M., Eeg-Olofsson O., Sadleir L.G., Gill D., Ben-Zeev B. Rare copy number variants are an important cause of epileptic encephalopathies. Ann. Neurol. 2011;70:974–985. doi: 10.1002/ana.22645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mamtani M., Anaya J.M., He W., Ahuja S.K. Association of copy number variation in the FCGR3B gene with risk of autoimmune diseases. Genes Immun. 2010;11:155–160. doi: 10.1038/gene.2009.71. [DOI] [PubMed] [Google Scholar]
- 11.Mamtani M., Matsubara T., Shimizu C., Furukawa S., Akagi T., Onouchi Y., Hata A., Fujino A., He W., Ahuja S.K., Burns J.C. Association of CCR2-CCR5 haplotypes and CCL3L1 copy number with Kawasaki Disease, coronary artery lesions, and IVIG responses in Japanese children. PLoS ONE. 2010;5:e11458. doi: 10.1371/journal.pone.0011458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pelak K., Need A.C., Fellay J., Shianna K.V., Feng S., Urban T.J., Ge D., De Luca A., Martinez-Picado J., Wolinsky S.M., NIAID Center for HIV/AIDS Vaccine Immunology Copy number variation of KIR genes influences HIV-1 control. PLoS Biol. 2011;9:e1001208. doi: 10.1371/journal.pbio.1001208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Levy S., Sutton G., Ng P.C., Feuk L., Halpern A.L., Walenz B.P., Axelrod N., Huang J., Kirkness E.F., Denisov G. The diploid genome sequence of an individual human. PLoS Biol. 2007;5:e254. doi: 10.1371/journal.pbio.0050254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Campbell C.D., Sampas N., Tsalenko A., Sudmant P.H., Kidd J.M., Malig M., Vu T.H., Vives L., Tsang P., Bruhn L., Eichler E.E. Population-genetic properties of differentiated human copy-number polymorphisms. Am. J. Hum. Genet. 2011;88:317–332. doi: 10.1016/j.ajhg.2011.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lefranc M.P., Lefranc G. Academic Press; London: 2001. The Immunoglobulin FactsBook. [Google Scholar]
- 16.Watson C.T., Breden F. The immunoglobulin heavy chain locus: genetic variation, missing data, and implications for human disease. Genes Immun. 2012;13:363–373. doi: 10.1038/gene.2012.12. [DOI] [PubMed] [Google Scholar]
- 17.Keyeux G., Lefranc G., Lefranc M.P. A multigene deletion in the human IGH constant region locus involves highly homologous hot spots of recombination. Genomics. 1989;5:431–441. doi: 10.1016/0888-7543(89)90006-2. [DOI] [PubMed] [Google Scholar]
- 18.Lefranc M.P., Lefranc G., de Lange G., Out T.A., van den Broek P.J., van Nieuwkoop J., Radl J., Helal A.N., Chaabani H., van Loghem E. Instability of the human immunoglobulin heavy chain constant region locus indicated by different inherited chromosomal deletions. Mol. Biol. Med. 1983;1:207–217. [PubMed] [Google Scholar]
- 19.Lefranc M.P., Lefranc G., Rabbitts T.H. Inherited deletion of immunoglobulin heavy chain constant region genes in normal human individuals. Nature. 1982;300:760–762. doi: 10.1038/300760a0. [DOI] [PubMed] [Google Scholar]
- 20.Matsuda F., Ishii K., Bourvagnet P., Kuma Ki., Hayashida H., Miyata T., Honjo T. The complete nucleotide sequence of the human immunoglobulin heavy chain variable region locus. J. Exp. Med. 1998;188:2151–2162. doi: 10.1084/jem.188.11.2151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wiebe V., Helal A., Lefranc M.P., Lefranc G. Molecular analysis of the T17 immunoglobulin CH multigene deletion (del A1-GP-G2-G4-E) Hum. Genet. 1994;93:520–528. doi: 10.1007/BF00202816. [DOI] [PubMed] [Google Scholar]
- 22.Lefranc M.P., Lefranc G. Human Gm, Km, and Am allotypes and their molecular characterization: a remarkable demonstration of polymorphism. Methods Mol. Biol. 2012;882:635–680. doi: 10.1007/978-1-61779-842-9_34. [DOI] [PubMed] [Google Scholar]
- 23.Cook G.P., Tomlinson I.M., Walter G., Riethman H., Carter N.P., Buluwela L., Winter G., Rabbitts T.H. A map of the human immunoglobulin VH locus completed by analysis of the telomeric region of chromosome 14q. Nat. Genet. 1994;7:162–168. doi: 10.1038/ng0694-162. [DOI] [PubMed] [Google Scholar]
- 24.Chimge N.O., Pramanik S., Hu G., Lin Y., Gao R., Shen L., Li H. Determination of gene organization in the human IGHV region on single chromosomes. Genes Immun. 2005;6:186–193. doi: 10.1038/sj.gene.6364176. [DOI] [PubMed] [Google Scholar]
- 25.Pramanik S., Cui X., Wang H.Y., Chimge N.O., Hu G., Shen L., Gao R., Li H. Segmental duplication as one of the driving forces underlying the diversity of the human immunoglobulin heavy chain variable gene region. BMC Genomics. 2011;12:78. doi: 10.1186/1471-2164-12-78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pramanik S., Li H. Direct detection of insertion/deletion polymorphisms in an autosomal region by analyzing high-density markers in individual spermatozoa. Am. J. Hum. Genet. 2002;71:1342–1352. doi: 10.1086/344713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kidd M.J., Chen Z., Wang Y., Jackson K.J., Zhang L., Boyd S.D., Fire A.Z., Tanaka M.M., Gaëta B.A., Collins A.M. The inference of phased haplotypes for the immunoglobulin H chain V region gene loci by analysis of VDJ gene rearrangements. J. Immunol. 2012;188:1333–1340. doi: 10.4049/jimmunol.1102097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Conrad D.F., Bird C., Blackburne B., Lindsay S., Mamanova L., Lee C., Turner D.J., Hurles M.E. Mutation spectrum revealed by breakpoint sequencing of human germline CNVs. Nat. Genet. 2010;42:385–391. doi: 10.1038/ng.564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Boyd S.D., Gaëta B.A., Jackson K.J., Fire A.Z., Marshall E.L., Merker J.D., Maniar J.M., Zhang L.N., Sahaf B., Jones C.D. Individual variation in the germline Ig gene repertoire inferred from variable region gene rearrangements. J. Immunol. 2010;184:6986–6992. doi: 10.4049/jimmunol.1000445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Field L.L., Larsen Z., Pociot F., Nerup J., Tobias R., Bonnevie-Nielsen V. Evidence for a locus (IDDM16) in the immunoglobulin heavy chain region on chromosome 14q32.3 producing susceptibility to type 1 diabetes. Genes Immun. 2002;3:338–344. doi: 10.1038/sj.gene.6363857. [DOI] [PubMed] [Google Scholar]
- 31.Olee T., Yang P.M., Siminovitch K.A., Olsen N.J., Hillson J., Wu J., Kozin F., Carson D.A., Chen P.P. Molecular basis of an autoantibody-associated restriction fragment length polymorphism that confers susceptibility to autoimmune diseases. J. Clin. Invest. 1991;88:193–203. doi: 10.1172/JCI115277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tsai F.J., Lee Y.C., Chang J.S., Huang L.M., Huang F.Y., Chiu N.C., Chen M.R., Chi H., Lee Y.J., Chang L.C. Identification of novel susceptibility loci for kawasaki disease in a Han chinese population by a genome-wide association study. PLoS ONE. 2011;6:e16853. doi: 10.1371/journal.pone.0016853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cho M.L., Chen P.P., Seo Y.I., Hwang S.Y., Kim W.U., Min D.J., Park S.H., Cho C.S. Association of homozygous deletion of the Humhv3005 and the VH3-30.3 genes with renal involvement in systemic lupus erythematosus. Lupus. 2003;12:400–405. doi: 10.1191/0961203303lu385oa. [DOI] [PubMed] [Google Scholar]
- 34.Dennis M.Y., Nuttle X., Sudmant P.H., Antonacci F., Graves T.A., Nefedov M., Rosenfeld J.A., Sajjadian S., Malig M., Kotkiewicz H. Evolution of human-specific neural SRGAP2 genes by incomplete segmental duplication. Cell. 2012;149:912–922. doi: 10.1016/j.cell.2012.03.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Giudicelli V., Chaume D., Lefranc M.P. IMGT/GENE-DB: a comprehensive database for human and mouse immunoglobulin and T cell receptor genes. Nucleic Acids Res. 2005;33(Database issue):D256–D261. doi: 10.1093/nar/gki010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pallarès N., Lefebvre S., Contet V., Matsuda F., Lefranc M.P. The human immunoglobulin heavy variable genes. Exp. Clin. Immunogenet. 1999;16:36–60. doi: 10.1159/000019095. [DOI] [PubMed] [Google Scholar]
- 37.Ruiz M., Pallarès N., Contet V., Barbi V., Lefranc M.P. The human immunoglobulin heavy diversity (IGHD) and joining (IGHJ) segments. Exp. Clin. Immunogenet. 1999;16:173–184. doi: 10.1159/000019109. [DOI] [PubMed] [Google Scholar]
- 38.Lefranc M.P. IMGT, the international ImMunoGeneTics database. Nucleic Acids Res. 2001;29:207–209. doi: 10.1093/nar/29.1.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lefranc M.P. Nomenclature of the human immunoglobulin genes. Curr. Protoc. Immunol. Appendix. 2001;1(Appendix):1P. doi: 10.1002/0471142735.ima01ps40. [DOI] [PubMed] [Google Scholar]
- 40.Altschul S.F., Gish W., Miller W., Myers E.W., Lipman D.J. Basic local alignment search tool. J. Mol. Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 41.Brochet X., Lefranc M.P., Giudicelli V. IMGT/V-QUEST: the highly customized and integrated system for IG and TR standardized V-J and V-D-J sequence analysis. Nucleic Acids Res. 2008;36(Web Server issue):W503–W508. doi: 10.1093/nar/gkn316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Giudicelli V., Brochet X., Lefranc M.P. IMGT/V-QUEST: IMGT standardized analysis of the immunoglobulin (IG) and T cell receptor (TR) nucleotide sequences. Cold Spring Harb Protoc. 2011;2011:695–715. doi: 10.1101/pdb.prot5633. [DOI] [PubMed] [Google Scholar]
- 43.Kidd J.M., Graves T., Newman T.L., Fulton R., Hayden H.S., Malig M., Kallicki J., Kaul R., Wilson R.K., Eichler E.E. A human genome structural variation sequencing resource reveals insights into mutational mechanisms. Cell. 2010;143:837–847. doi: 10.1016/j.cell.2010.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Parsons J.D. Miropeats: graphical DNA sequence comparisons. Comput. Appl. Biosci. 1995;11:615–619. doi: 10.1093/bioinformatics/11.6.615. [DOI] [PubMed] [Google Scholar]
- 45.Purcell S., Neale B., Todd-Brown K., Thomas L., Ferreira M.A., Bender D., Maller J., Sklar P., de Bakker P.I., Daly M.J., Sham P.C. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rice P., Longden I., Bleasby A. EMBOSS: the European Molecular Biology Open Software Suite. Trends Genet. 2000;16:276–277. doi: 10.1016/s0168-9525(00)02024-2. [DOI] [PubMed] [Google Scholar]
- 47.Thompson J.D., Higgins D.G., Gibson T.J. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Abecasis G.R., Altshuler D., Auton A., Brooks L.D., Durbin R.M., Gibbs R.A., Hurles M.E., McVean G.A., 1000 Genomes Project Consortium A map of human genome variation from population-scale sequencing. Nature. 2010;467:1061–1073. doi: 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lefranc M.P. WHO-IUIS Nomenclature Subcommittee for immunoglobulins and T cell receptors report. Immunogenetics. 2007;59:899–902. doi: 10.1007/s00251-007-0260-4. [DOI] [PubMed] [Google Scholar]
- 50.Lefranc M.P., WHO-IUIS Nomenclature Subcommittee for immunoglobulins and Tcell receptors WHO-IUIS Nomenclature Subcommittee for immunoglobulins and T cell receptors report August 2007, 13th International Congress of Immunology, Rio de Janeiro, Brazil. Dev. Comp. Immunol. 2008;32:461–463. doi: 10.1016/j.dci.2007.09.008. [DOI] [PubMed] [Google Scholar]
- 51.Lefranc M.P. From IMGT-ONTOLOGY IDENTIFICATION axiom to IMGT standardized keywords: for immunoglobulins (IG), T cell receptors (TR), and conventional genes. Cold Spring Harb. Protoc. 2011;2011:604–613. doi: 10.1101/pdb.ip82. [DOI] [PubMed] [Google Scholar]
- 52.Lefranc G., Chaabani H., Van Loghem E., Lefranc M.P., De Lange G., Helal A.N. Simultaneous absence of the human IgG1, IgG2, IgG4 and IgA1 subclasses: immunological and immunogenetical considerations. Eur. J. Immunol. 1983;13:240–244. doi: 10.1002/eji.1830130312. [DOI] [PubMed] [Google Scholar]
- 53.Lefranc M.P., Hammarström L., Smith C.I., Lefranc G. Gene deletions in the human immunoglobulin heavy chain constant region locus: molecular and immunological analysis. Immunodefic. Rev. 1991;2:265–281. [PubMed] [Google Scholar]
- 54.Glanville J., Kuo T.C., von Büdingen H.C., Guey L., Berka J., Sundar P.D., Huerta G., Mehta G.R., Oksenberg J.R., Hauser S.L. Naive antibody gene-segment frequencies are heritable and unaltered by chronic lymphocyte ablation. Proc. Natl. Acad. Sci. USA. 2011;108:20066–20071. doi: 10.1073/pnas.1107498108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Walter G., Tomlinson I.M., Cook G.P., Winter G., Rabbitts T.H., Dear P.H. HAPPY mapping of a YAC reveals alternative haplotypes in the human immunoglobulin VH locus. Nucleic Acids Res. 1993;21:4524–4529. doi: 10.1093/nar/21.19.4524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Milner E.C., Hufnagle W.O., Glas A.M., Suzuki I., Alexander C. Polymorphism and utilization of human VH Genes. Ann. N Y Acad. Sci. 1995;764:50–61. doi: 10.1111/j.1749-6632.1995.tb55806.x. [DOI] [PubMed] [Google Scholar]
- 57.Mo L., Leu S.J., Berry C., Liu F., Olee T., Yang Y.Y., Beardsley D.S., McMillan R., Woods V.L., Jr., Chen P.P. The frequency of homozygous deletion of a developmentally regulated Vh gene (Humhv3005) is increased in patients with chronic idiopathic thrombocytopenic purpura. Autoimmunity. 1996;24:257–263. doi: 10.3109/08916939608994718. [DOI] [PubMed] [Google Scholar]
- 58.Yang P.M., Olsen N.J., Siminovitch K.A., Olee T., Kozin F., Carson D.A., Chen P.P. Possible deletion of a developmentally regulated heavy-chain variable region gene in autoimmune diseases. Proc. Natl. Acad. Sci. USA. 1990;87:7907–7911. doi: 10.1073/pnas.87.20.7907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Altshuler D.M., Gibbs R.A., Peltonen L., Altshuler D.M., Gibbs R.A., Peltonen L., Dermitzakis E., Schaffner S.F., Yu F., Peltonen L., International HapMap 3 Consortium Integrating common and rare genetic variation in diverse human populations. Nature. 2010;467:52–58. doi: 10.1038/nature09298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.McCarroll S.A., Kuruvilla F.G., Korn J.M., Cawley S., Nemesh J., Wysoker A., Shapero M.H., de Bakker P.I., Maller J.B., Kirby A. Integrated detection and population-genetic analysis of SNPs and copy number variation. Nat. Genet. 2008;40:1166–1174. doi: 10.1038/ng.238. [DOI] [PubMed] [Google Scholar]
- 61.Shin E.K., Matsuda F., Ozaki S., Kumagai S., Olerup O., Ström H., Melchers I., Honjo T. Polymorphism of the human immunoglobulin variable region segment V1-4.1. Immunogenetics. 1993;38:304–306. doi: 10.1007/BF00188810. [DOI] [PubMed] [Google Scholar]
- 62.Ohm-Laursen L., Larsen S.R., Barington T. Identification of two new alleles, IGHV3-23∗04 and IGHJ6∗04, and the complete sequence of the IGHV3-h pseudogene in the human immunoglobulin locus and their prevalences in Danish Caucasians. Immunogenetics. 2005;57:621–627. doi: 10.1007/s00251-005-0035-8. [DOI] [PubMed] [Google Scholar]
- 63.Ballana E., González J.R., Bosch N., Estivill X. Inter-population variability of DEFA3 gene absence: correlation with haplotype structure and population variability. BMC Genomics. 2007;8:14. doi: 10.1186/1471-2164-8-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hollox E.J., Armour J.A. Directional and balancing selection in human beta-defensins. BMC Evol. Biol. 2008;8:113. doi: 10.1186/1471-2148-8-113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hollox E.J., Huffmeier U., Zeeuwen P.L., Palla R., Lascorz J., Rodijk-Olthuis D., van de Kerkhof P.C., Traupe H., de Jongh G., den Heijer M. Psoriasis is associated with increased beta-defensin genomic copy number. Nat. Genet. 2008;40:23–25. doi: 10.1038/ng.2007.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Traherne J.A., Martin M., Ward R., Ohashi M., Pellett F., Gladman D., Middleton D., Carrington M., Trowsdale J. Mechanisms of copy number variation and hybrid gene formation in the KIR immune gene complex. Hum. Mol. Genet. 2010;19:737–751. doi: 10.1093/hmg/ddp538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Gazave E., Darré F., Morcillo-Suarez C., Petit-Marty N., Carreño A., Marigorta U.M., Ryder O.A., Blancher A., Rocchi M., Bosch E. Copy number variation analysis in the great apes reveals species-specific patterns of structural variation. Genome Res. 2011;21:1626–1639. doi: 10.1101/gr.117242.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sharp A.J., Locke D.P., McGrath S.D., Cheng Z., Bailey J.A., Vallente R.U., Pertz L.M., Clark R.A., Schwartz S., Segraves R. Segmental duplications and copy-number variation in the human genome. Am. J. Hum. Genet. 2005;77:78–88. doi: 10.1086/431652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Marques-Bonet T., Kidd J.M., Ventura M., Graves T.A., Cheng Z., Hillier L.W., Jiang Z., Baker C., Malfavon-Borja R., Fulton L.A. A burst of segmental duplications in the genome of the African great ape ancestor. Nature. 2009;457:877–881. doi: 10.1038/nature07744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kashyap A.K., Steel J., Oner A.F., Dillon M.A., Swale R.E., Wall K.M., Perry K.J., Faynboym A., Ilhan M., Horowitz M. Combinatorial antibody libraries from survivors of the Turkish H5N1 avian influenza outbreak reveal virus neutralization strategies. Proc. Natl. Acad. Sci. USA. 2008;105:5986–5991. doi: 10.1073/pnas.0801367105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Throsby M., van den Brink E., Jongeneelen M., Poon L.L., Alard P., Cornelissen L., Bakker A., Cox F., van Deventer E., Guan Y. Heterosubtypic neutralizing monoclonal antibodies cross-protective against H5N1 and H1N1 recovered from human IgM+ memory B cells. PLoS ONE. 2008;3:e3942. doi: 10.1371/journal.pone.0003942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sui J., Hwang W.C., Perez S., Wei G., Aird D., Chen L.M., Santelli E., Stec B., Cadwell G., Ali M. Structural and functional bases for broad-spectrum neutralization of avian and human influenza A viruses. Nat. Struct. Mol. Biol. 2009;16:265–273. doi: 10.1038/nsmb.1566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Lerner R.A. Rare antibodies from combinatorial libraries suggests an S.O.S. component of the human immunological repertoire. Mol. Biosyst. 2011;7:1004–1012. doi: 10.1039/c0mb00310g. [DOI] [PubMed] [Google Scholar]
- 74.Johnson T.A., Rassenti L.Z., Kipps T.J. Ig VH1 genes expressed in B cell chronic lymphocytic leukemia exhibit distinctive molecular features. J. Immunol. 1997;158:235–246. [PubMed] [Google Scholar]
- 75.Hamblin T.J., Davis Z., Gardiner A., Oscier D.G., Stevenson F.K. Unmutated Ig V(H) genes are associated with a more aggressive form of chronic lymphocytic leukemia. Blood. 1999;94:1848–1854. [PubMed] [Google Scholar]
- 76.Chen L., Zhang Y., Zheng W., Wu Y., Qiao C., Fan L., Xu W., Li J. Distinctive IgVH gene segments usage and mutation status in Chinese patients with chronic lymphocytic leukemia. Leuk. Res. 2008;32:1491–1498. doi: 10.1016/j.leukres.2008.02.005. [DOI] [PubMed] [Google Scholar]
- 77.Sasso E.H., Johnson T., Kipps T.J. Expression of the immunoglobulin VH gene 51p1 is proportional to its germline gene copy number. J. Clin. Invest. 1996;97:2074–2080. doi: 10.1172/JCI118644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Gonzalez E., Kulkarni H., Bolivar H., Mangano A., Sanchez R., Catano G., Nibbs R.J., Freedman B.I., Quinones M.P., Bamshad M.J. The influence of CCL3L1 gene-containing segmental duplications on HIV-1/AIDS susceptibility. Science. 2005;307:1434–1440. doi: 10.1126/science.1101160. [DOI] [PubMed] [Google Scholar]
- 79.Feeney A.J., Atkinson M.J., Cowan M.J., Escuro G., Lugo G. A defective Vkappa A2 allele in Navajos which may play a role in increased susceptibility to haemophilus influenzae type b disease. J. Clin. Invest. 1996;97:2277–2282. doi: 10.1172/JCI118669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Schrider D.R., Hahn M.W. Lower linkage disequilibrium at CNVs is due to both recurrent mutation and transposing duplications. Mol. Biol. Evol. 2010;27:103–111. doi: 10.1093/molbev/msp210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Lefranc M.P. From IMGT-ONTOLOGY CLASSIFICATION Axiom to IMGT standardized gene and allele nomenclature: for immunoglobulins (IG) and T cell receptors (TR) Cold Spring Harb Protoc. 2011;2011:627–632. doi: 10.1101/pdb.ip84. [DOI] [PubMed] [Google Scholar]
- 82.Bailey J.A., Gu Z., Clark R.A., Reinert K., Samonte R.V., Schwartz S., Adams M.D., Myers E.W., Li P.W., Eichler E.E. Recent segmental duplications in the human genome. Science. 2002;297:1003–1007. doi: 10.1126/science.1072047. [DOI] [PubMed] [Google Scholar]
- 83.Bailey J.A., Yavor A.M., Massa H.F., Trask B.J., Eichler E.E. Segmental duplications: organization and impact within the current human genome project assembly. Genome Res. 2001;11:1005–1017. doi: 10.1101/gr.187101. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.