

Abstract
The Krebs cycle is of fundamental importance for the generation of the energetic and molecular needs of both prokaryotic and eukaryotic cells. Both enantiomers of metabolite 2-hydroxyglutarate are directly linked to this pivotal biochemical pathway and are found elevated not only in several cancers, but also in different variants of the neurometabolic disease 2-hydroxyglutaric aciduria. Recently we showed that cancer-associated IDH2 germline mutations cause one variant of 2-hydroxyglutaric aciduria. Complementary to these findings, we now report recessive mutations in SLC25A1, the mitochondrial citrate carrier, in 12 out of 12 individuals with combined D-2- and L-2-hydroxyglutaric aciduria. Impaired mitochondrial citrate efflux, demonstrated by stable isotope labeling experiments and the absence of SLC25A1 in fibroblasts harboring certain mutations, suggest that SLC25A1 deficiency is pathogenic. Our results identify defects in SLC25A1 as a cause of combined D-2- and L-2-hydroxyglutaric aciduria.
Main Text
D-2-hydroxyglutarate (D-2-HG) and L-2-hydroxyglutarate (L-2-HG) are associated with the Krebs (tricarboxylic acid/citrate) cycle,1 with rare disorders,2 and with certain types of cancer.3–5 D-2-hydroxyglutaric aciduria (D-2-HGA), characterized by elevated levels of D-2-HG, is either autosomal recessive, resulting from mutations in D2HGDH6 (type I [MIM 600721]), or is caused by recurrent usually de novo dominant gain-of-function mutations in IDH27 (type II [MIM 613657]). Manifestations of both types of D-2-HGA include developmental delay, hypotonia, and seizures.2 Recessive mutations in L2HGDH result in L-2-hydroxyglutaric aciduria8,9 (L-2-HGA [MIM 236792]), and predominant signs include macrocephaly, developmental delay, epilepsy, and cerebellar ataxia. Combined D-2- and L-2-hydroxyglutaric aciduria (D,L-2-HGA), characterized by elevated levels of both D-2-HG and L-2-HG in body fluids, is not caused by mutations in any of the above-mentioned genes and mainly manifests in a severe neonatal epileptic encephalopathy, absence of developmental progress, and often early death10 (Table S1 available online).
Elevated D-2-HG levels are also found in gliomas and myeloid leukemias3,11 and are associated with recurrent mutations in the isocitrate dehydrogenase (IDH) genes IDH1 or IDH2. These mutations disable the normal ability of IDH to convert isocitrate to α-ketoglutarate and result in a new function: converting α-ketoglutarate to D-2-HG.3 However, elevated D-2-HG and L-2-HG levels are also found in thyroid carcinomas without IDH mutations,4 caused by still unknown mechanisms. The exact role of “oncometabolite” 2-hydroxyglutarate (2-HG) in cancer is not yet fully understood, but recent studies indicate an important role in epigenetics.12–14 Identifying the genetic cause of combined D,L-2-HGA could therefore not only improve our knowledge of the pathophysiology of this neurometabolic disorder, but also provide further insight into its role in cancer.
We performed whole-exome sequencing to identify the genetic basis of combined D,L-2-HGA in DNA of three affected individuals and the unaffected parents of one of them (subject 1). A total of 13 affected individuals diagnosed with combined D,L-2-HGA from 12 unrelated families (Arabic, European, or Latin-American descent) were included in this study. They all showed elevated levels of D-2-HG and L-2-HG in urine (Table S2). Written consent was obtained from participants and/or their guardians for the exome sequencing analysis; for all other participants informed consent was waived because of the retrospective nature of the study. The study received Institutional Review Board approval from the VU University Medical Center Amsterdam (registered with the US Office of Human Research Protections under number IORG0002436). Genomic DNA was extracted from blood, and 3 μg was fragmented via Covaris technology. Fragments were ligated with adaptors (Illumina), amplified by ligation-mediated PCR, and enriched with SureSelect Human All Exon v.2 Kit (Agilent). Sequencing was performed on a HiSeq2000 (Illumina) and generated paired-end 100 bp reads. Mapping of the reads to the human reference genome (UCSC Genome Browser hg19) was done with Burrows-Wheeler Aligner.15 VarScan16 software was used for calling SNPs and indels. Annotation of the obtained variants was performed with ANNOVAR.17 We obtained a mean coverage depth of the exomes ranging from 83× to 109× and searched for nonsynonymous single-nucleotide variants (SNVs) in the family of subject 1, not present in dbSNP (132) or 1000 Genomes, and with a SIFT18 prediction score < 0.05. Assuming a model in which mutations have a recessive effect,10 we identified nonsynonymous SNVs in three genes (OR52K1, OR51I2, and SLC25A1 [RefSeq accession number NM_005984.2, MIM 190315]) that were homozygous (100% of the reads) in the affected individual and heterozygous (50% of the reads) in either parent with >40× coverage depth. Of these three genes, only SNVs in SLC25A1, with the above-mentioned criteria, were identified in the other two affected individuals (subjects 2 and 3). A similar search for small insertions or deletions did not identify any additional genes. Sanger sequencing of the ORF of SLC25A1 confirmed the mutations in these three individuals and their families and revealed mutations in nine additional unrelated affected subjects (Table 1). In total, 12 different mutations were detected (Figure 1A): eight missense, one nonsense, two frameshift (of which one was recurrent in four unrelated individuals), and one mutation resulting in a splicing error. The latter mutation, c.821C>T, found in three unrelated individuals, is located at the most 3′ end of exon 8 and introduces a splice donor site, resulting in a deletion in mRNA (r.820_821del), and is predicted to cause a frameshift at the protein level (p.Ala274Ilefs∗24). Sequencing of mRNA from fibroblasts derived from subject 4 confirmed a 2 bp deletion (Figure S2). All missense mutations were predicted with SIFT to be damaging (Table 1).
Table 1.
Homozygous or Compound Heterozygous SLC25A1 Mutations Found in Combined D,L-2-HGA Cases
| Subject | Nucleotide Change | Deduced Effect | Exon | SIFT Scorea |
|---|---|---|---|---|
| 1b | c.578C>G | p.Ser193Trp | 6 | 0.00 |
| 2b | c.844C>G | p.Arg282Gly | 9 | 0.00 |
| 3c | c.844C>T | p.Arg282Cys | 9 | 0.00 |
| 4d | c.821C>T | r.820_821del (p.Ala274Ilefs∗24) | 8 | – |
| 5 | c.18_24dup | p.Ala9Profs∗82 | 1 | – |
| c.499G>A | p.Gly167Arg | 5 | 0.00 | |
| 6 | c.18_24dup | p.Ala9Profs∗82 | 1 | – |
| c.134C>T | p.Pro45Leu | 2 | 0.00 | |
| 7b | c.18_24dup | p.Ala9Profs∗82 | 1 | – |
| c.768C>G | p.Tyr256∗ | 8 | – | |
| 8b | c.430G>C | p.Glu144Gln | 4 | 0.00 |
| 9 | c.18_24dup | p.Ala9Profs∗82 | 1 | – |
| 10b | c.605T>C | p.Met202Thr | 6 | 0.01 |
| c.890A>G | p.Tyr297Cys | 9 | 0.00 | |
| 11 | c.821C>T | r.820_821del (p.Ala274Ilefs∗24) | 8 | – |
| 12b | c.517_526del | p.Arg173Glyfs∗2 | 5 | – |
| c.821C>T | r.820_821del (p.Ala274Ilefs∗24) | 8 | – |
The primers used for PCR amplification and sequencing are summarized in Table S3. SLC25A1 RefSeq NM_005984.2.
All missense mutations were predicted with SIFT to be damaging (p < 0.05).
Homozygosity or compound heterozygosity was confirmed by DNA sequencing of the parents.
Homozygous mutation was also found with DNA sequencing in affected sibling.
Mutation was confirmed in mRNA isolated from fibroblasts.
Figure 1.
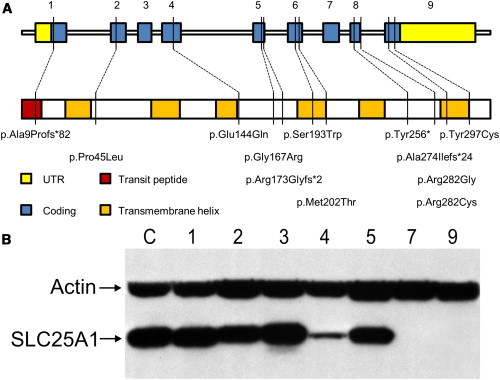
Schematic Overview of Genomic Organization and Protein Domain Structure of SLC25A1
(A) SLC25A1 contains nine exons that encode a 311 amino acid long product. The first 13 amino acids are a transit peptide (red) for mitochondrial targeting, and the mature protein contains six membrane-spanning helices (orange). Locations of the 12 different identified mutations are shown. An alignment of the involved amino acids with different species is shown in Figure S1.
(B) Immunoblot analysis of SLC25A1 accumulation in fibroblasts derived from control “C” and affected subjects 1–5, 7, and 9. SLC25A1 is immunodetected in control, subjects 1–3 (homozygous missense mutations), and subject 5 with mutation c.18_24dup (p.Ala9Profs∗82) and c.499G>A (p.Gly167Arg). Low detection in subject 4 (homozygous mutation c.821C>T [p.Ala274Ilefs∗24]) and no detection in subject 7 with mutation c.18_24dup (p.Ala9Profs∗82) and c.768C>G (p.Tyr256∗) and subject 9 (homozygous mutation c.18_24dup [p.Ala9Profs∗82]) is consistent with the presence of truncating mutations. The whole-blot image and a blot with a lower exposure time are shown in Figure S3.
With immunoblot analysis, we examined SLC25A1 in fibroblasts available from seven affected individuals (Figures 1B and S3). SLC25A1 was detected in cells from control subjects and all subjects harboring homozygous or compound heterozygous missense mutations. However, in cells from subjects containing two alleles with truncating mutations (subjects 7 and 9), no protein was detected, probably as a consequence of nonsense-mediated decay. The homozygous splice site mutation c.821C>T (subject 4; p.Ala274Ilefs∗24) had reduced levels of SLC25A1. This could be explained by some authentic spliced protein product (p.Ala274Val), or alternatively, because the frameshift occurs at the C-terminal end, this protein could still be immunodetected but is degraded at higher rate.
The location of SLC25A1 within the human genome was previously described to be involved in other disorders.19 SLC25A1 is located on chromosome 22q11 within the critical region deleted in DiGeorge syndrome20 (MIM 188400). DiGeorge syndrome is a rather common congenital disorder (prevalence of 1:4,000) caused by a hemizygous deletion of chromosome 22q11.2 (containing ∼45 genes), with a wide variety of clinical abnormalities including heart defects, velopharyngeal inadequacies, recurrent infections, and learning disabilities.21 Consequently, individuals with DiGeorge syndrome are heterozygous carriers of SLC25A1 deficiency.
Expression of SLC25A1 mRNA is high in liver, kidney, and pancreas.22 It encodes a protein of 311 amino acids containing six transmembrane helices that is located in the inner mitochondrial membrane. SLC25A1 mediates the efflux of mitochondrial citrate or isocitrate in exchange for cytosolic malate.23 Citrate and isocitrate are intermediates of the Krebs cycle, which occurs in the matrix of mitochondria and is responsible for energy production. Citrate exported by SLC25A1 out of the mitochondria into the cytosol is converted into acetyl coenzyme A (acetyl-CoA), which is essential for fatty acid and sterol synthesis.24 Furthermore, cytosolic citrate is an important regulator of glycolysis, because it is an allosteric inhibitor of 6-phosphofructokinase (PFK1), the most important regulator enzyme of glycolysis.24
To examine the effect of SLC25A1 deficiency, we retrospectively measured the urinary citrate and isocitrate levels of individuals with combined D,L-2-HGA by using liquid chromatography-tandem mass spectrometry. Compared to age-matched controls (n = 40), significantly lower levels of both metabolites were detected (Figures 2A and 2B; Table S2). Interestingly, the eldest affected individual (subject 10) in our cohort showed the highest levels of citrate and isocitrate (Table S2). To further study SLC25A1 activity, we incubated combined D,L-2-HGA-affected fibroblasts (n = 7) and controls (n = 2) with [U-13C6]glucose-enriched medium. [U-13C6]glucose is metabolized via glycolysis and enters the Krebs cycle predominantly as [13C2]acetyl-CoA. The condensation reaction of [13C2]acetyl-CoA and oxaloacetate results in intramitochondrial [13C2]citrate. In combined D,L-2-HGA-affected fibroblasts, we detected lower [13C2]citrate levels in the culture medium than in controls (Figure 2C; Table S2). This demonstrates that SLC25A1 mutations result in impaired citrate efflux. Interestingly, fibroblasts from subject 7 (c.18_24dup and c.768C>G) and subject 9 (c.18_24dup) showed ∼7% efflux as compared to controls, indicating that citrate can leave to a lesser extent the mitochondrial matrix via other means than SLC25A1 or is formed in the cytosol.
Figure 2.
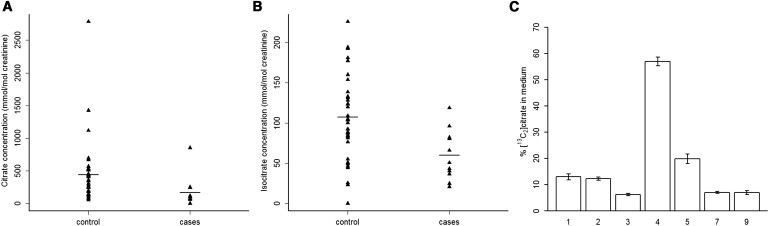
Decreased Citrate and Isocitrate Levels in Combined D,L-2-HGA
(A and B) Urinary levels of (A) citrate and (B) isocitrate were measured with LC-MS/MS in samples of controls (n = 40) and of individuals with combined D,L-2-HGA (n = 11) with SLC25A1 mutations. Horizontal lines indicate the group sample means. The differences observed between controls and combined D,L-2-HGA-affected individuals were statistically significant by Mann-Whitney’s test for both citrate (p = 0.0011) and isocitrate (p = 0.0015) levels (p < 0.05 was considered significant). Urinary levels of an unrelated metabolite (α-aminoadipic acid) was not significantly different between individuals with combined D,L-2-HGA and controls (Figure S4).
(C) Fibroblast cultures derived from affected subjects (n = 7) were given [U-13C6]glucose, which is metabolized by glycolysis predominantly to [13C2]acetyl-CoA. [13C2]acetyl-CoA is converted to [13C2]citrate within mitochondria via the Krebs cycle. Levels of [13C2]citrate were measured after 72 hr incubation in culture medium and are expressed as percentage compared to control fibroblasts (n = 2). Error bars indicate standard error of mean of replicate measurements (n = 3).
Individuals with combined D,L-2-HGA excrete increased levels of several Krebs cycle intermediates including α-ketoglutarate, malate, fumarate, and succinate, accompanied with decreased levels of citrate and isocitrate (Table S2). We speculate that the increased urinary excretion of D-2-HG and L-2-HG is caused by the inability of mitochondria to export citrate and isocitrate into the cytosol. This would probably result in increased mitochondrial concentrations, and subsequently higher excretion, of Krebs cycle metabolites downstream of isocitrate. We therefore measured the above-mentioned metabolites in homogenates of fibroblasts from controls and affected individuals (Table S4). Compared to controls, the levels of D-2-HG and L-2-HG in affected individuals are increased, which is in agreement with previously reported data.25 Additionally, citrate levels were lower in affected fibroblasts, but isocitrate and the other metabolites were normal compared to controls.
In conclusion, SLC25A1 mutations cause combined D,L-2-HGA as well as altered levels of several Krebs cycle intermediates. This finding provides the basis for an understanding of the pathophysiology and future therapeutic research concerning this disease that may include citrate supplementation. SLC25A1 deficiency may be underdiagnosed and inclusion in the differential diagnosis of mitochondrial disorders should be considered. Further research is needed to unravel how SLC25A1 defects cause elevation of 2-HG, a cancer-associated metabolite. Although no cancer was diagnosed in our combined D,L-2HGA cohort (Table S1), an important role for citrate or SLC25A1 has previously been implied in epigenetics or cancer biology.26–30 The absence of cancer cases in our combined D,L-2-HGA cohort might be explained by the early age of death. Another explanation might be that the L-2-HG levels in our cohort are not as high as in L-2-HGA-affected individuals. Only L-2-HGA has previously been associated with higher incidences of cancer.5 In both types of D-2-HGA, no higher incidence of cancer has been found.2 Moreover, certain cancer types show elevated levels of 2-HG but contain no mutations in IDH1 or IDH2.4 It would therefore be interesting to further investigate the role of citrate and SLC25A1 in these cancer types.
Acknowledgments
We thank the families that participated in this study; Chris Mühlhausen (Universitätsklinikum Hamburg-Eppendorf, Germany) and Nathalie Leporrier (CHU de Caen, France) for their help and suggestions on this topic; Stefan Dentro, Daoud Sie, Najim Ameziane, Ingrid Bakker, and Nanne Aben (VUmc Amsterdam, the Netherlands) for assistance with the exome sequencing; Ovidiu Pop (VUmc VU University, Amsterdam) for help with the immunoblot; and Mirjam Wamelink, Jiddeke van de Kamp, and Joe Ndika (VUmc) for valuable discussions. Special thanks to Eric Wever (VUmc) for correct preparation of the figures.
Contributor Information
Benjamin Nota, Email: benjamin.nota@gmail.com.
Gajja S. Salomons, Email: g.salomons@vumc.nl.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
1000 Genomes, http://browser.1000genomes.org
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org/
UCSC Genome Browser, http://genome.ucsc.edu
References
- 1.Raimundo N., Baysal B.E., Shadel G.S. Revisiting the TCA cycle: signaling to tumor formation. Trends Mol. Med. 2011;17:641–649. doi: 10.1016/j.molmed.2011.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kranendijk M., Struys E.A., Salomons G.S., Van der Knaap M.S., Jakobs C. Progress in understanding 2-hydroxyglutaric acidurias. J. Inherit. Metab. Dis. 2012;35:571–587. doi: 10.1007/s10545-012-9462-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dang L., White D.W., Gross S., Bennett B.D., Bittinger M.A., Driggers E.M., Fantin V.R., Jang H.G., Jin S., Keenan M.C. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature. 2009;462:739–744. doi: 10.1038/nature08617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rakheja D., Boriack R.L., Mitui M., Khokhar S., Holt S.A., Kapur P. Papillary thyroid carcinoma shows elevated levels of 2-hydroxyglutarate. Tumour Biol. 2011;32:325–333. doi: 10.1007/s13277-010-0125-6. [DOI] [PubMed] [Google Scholar]
- 5.Aghili M., Zahedi F., Rafiee E. Hydroxyglutaric aciduria and malignant brain tumor: a case report and literature review. J. Neurooncol. 2009;91:233–236. doi: 10.1007/s11060-008-9706-2. [DOI] [PubMed] [Google Scholar]
- 6.Struys E.A., Salomons G.S., Achouri Y., Van Schaftingen E., Grosso S., Craigen W.J., Verhoeven N.M., Jakobs C. Mutations in the D-2-hydroxyglutarate dehydrogenase gene cause D-2-hydroxyglutaric aciduria. Am. J. Hum. Genet. 2005;76:358–360. doi: 10.1086/427890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kranendijk M., Struys E.A., van Schaftingen E., Gibson K.M., Kanhai W.A., van der Knaap M.S., Amiel J., Buist N.R., Das A.M., de Klerk J.B. IDH2 mutations in patients with D-2-hydroxyglutaric aciduria. Science. 2010;330 doi: 10.1126/science.1192632. 336–336. [DOI] [PubMed] [Google Scholar]
- 8.Rzem R., Veiga-da-Cunha M., Noël G., Goffette S., Nassogne M.-C., Tabarki B., Schöller C., Marquardt T., Vikkula M., Van Schaftingen E. A gene encoding a putative FAD-dependent L-2-hydroxyglutarate dehydrogenase is mutated in L-2-hydroxyglutaric aciduria. Proc. Natl. Acad. Sci. USA. 2004;101:16849–16854. doi: 10.1073/pnas.0404840101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Topçu M., Jobard F., Halliez S., Coskun T., Yalçinkayal C., Gerceker F.O., Wanders R.J.A., Prud’homme J.-F., Lathrop M., Özguc M., Fischer J. L-2-hydroxyglutaric aciduria: identification of a mutant gene C14orf160, localized on chromosome 14q22.1. Hum. Mol. Genet. 2004;13:2803–2811. doi: 10.1093/hmg/ddh300. [DOI] [PubMed] [Google Scholar]
- 10.Muntau A.C., Röschinger W., Merkenschlager A., van der Knaap M.S., Jakobs C., Duran M., Hoffmann G.F., Roscher A.A. Combined D-2- and L-2-hydroxyglutaric aciduria with neonatal onset encephalopathy: a third biochemical variant of 2-hydroxyglutaric aciduria? Neuropediatrics. 2000;31:137–140. doi: 10.1055/s-2000-7497. [DOI] [PubMed] [Google Scholar]
- 11.Dang L., Jin S., Su S.M. IDH mutations in glioma and acute myeloid leukemia. Trends Mol. Med. 2010;16:387–397. doi: 10.1016/j.molmed.2010.07.002. [DOI] [PubMed] [Google Scholar]
- 12.Lu C., Ward P.S., Kapoor G.S., Rohle D., Turcan S., Abdel-Wahab O., Edwards C.R., Khanin R., Figueroa M.E., Melnick A. IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature. 2012;483:474–478. doi: 10.1038/nature10860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Turcan S., Rohle D., Goenka A., Walsh L.A., Fang F., Yilmaz E., Campos C., Fabius A.W.M., Lu C., Ward P.S. IDH1 mutation is sufficient to establish the glioma hypermethylator phenotype. Nature. 2012;483:479–483. doi: 10.1038/nature10866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Koivunen P., Lee S., Duncan C.G., Lopez G., Lu G., Ramkissoon S., Losman J.A., Joensuu P., Bergmann U., Gross S. Transformation by the (R)-enantiomer of 2-hydroxyglutarate linked to EGLN activation. Nature. 2012;483:484–488. doi: 10.1038/nature10898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li H., Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Koboldt D.C., Chen K., Wylie T., Larson D.E., McLellan M.D., Mardis E.R., Weinstock G.M., Wilson R.K., Ding L. VarScan: variant detection in massively parallel sequencing of individual and pooled samples. Bioinformatics. 2009;25:2283–2285. doi: 10.1093/bioinformatics/btp373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang K., Li M., Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38:e164. doi: 10.1093/nar/gkq603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ng P.C., Henikoff S. Predicting deleterious amino acid substitutions. Genome Res. 2001;11:863–874. doi: 10.1101/gr.176601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Heisterkamp N., Mulder M.P., Langeveld A., ten Hoeve J., Wang Z., Roe B.A., Groffen J. Localization of the human mitochondrial citrate transporter protein gene to chromosome 22Q11 in the DiGeorge syndrome critical region. Genomics. 1995;29:451–456. doi: 10.1006/geno.1995.9982. [DOI] [PubMed] [Google Scholar]
- 20.Stoffel M., Karayiorgou M., Espinosa R., 3rd, Beau M.M.L. The human mitochondrial citrate transporter gene (SLC20A3) maps to chromosome band 22q11 within a region implicated in DiGeorge syndrome, velo-cardio-facial syndrome and schizophrenia. Hum. Genet. 1996;98:113–115. doi: 10.1007/s004390050169. [DOI] [PubMed] [Google Scholar]
- 21.Oskarsdóttir S., Vujic M., Fasth A. Incidence and prevalence of the 22q11 deletion syndrome: a population-based study in Western Sweden. Arch. Dis. Child. 2004;89:148–151. doi: 10.1136/adc.2003.026880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Huizing M., Ruitenbeek W., van den Heuvel L.P., Dolce V., Iacobazzi V., Smeitink J.A.M., Palmieri F., Trijbels J.M. Human mitochondrial transmembrane metabolite carriers: tissue distribution and its implication for mitochondrial disorders. J. Bioenerg. Biomembr. 1998;30:277–284. doi: 10.1023/a:1020501021222. [DOI] [PubMed] [Google Scholar]
- 23.Palmieri F. The mitochondrial transporter family (SLC25): physiological and pathological implications. Pflugers Arch. 2004;447:689–709. doi: 10.1007/s00424-003-1099-7. [DOI] [PubMed] [Google Scholar]
- 24.Mycielska M.E., Patel A., Rizaner N., Mazurek M.P., Keun H., Patel A., Ganapathy V., Djamgoz M.B.A. Citrate transport and metabolism in mammalian cells: prostate epithelial cells and prostate cancer. Bioessays. 2009;31:10–20. doi: 10.1002/bies.080137. [DOI] [PubMed] [Google Scholar]
- 25.Struys E.A., Verhoeven N.M., Roos B., Jakobs C. Disease-related metabolites in culture medium of fibroblasts from patients with D-2-hydroxyglutaric aciduria, L-2-hydroxyglutaric aciduria, and combined D/L-2-hydroxyglutaric aciduria. Clin. Chem. 2003;49:1133–1138. doi: 10.1373/49.7.1133. [DOI] [PubMed] [Google Scholar]
- 26.Morciano P., Carrisi C., Capobianco L., Mannini L., Burgio G., Cestra G., De Benedetto G.E., Corona D.F.V., Musio A., Cenci G. A conserved role for the mitochondrial citrate transporter Sea/SLC25A1 in the maintenance of chromosome integrity. Hum. Mol. Genet. 2009;18:4180–4188. doi: 10.1093/hmg/ddp370. [DOI] [PubMed] [Google Scholar]
- 27.Catalina-Rodriguez O., Kolukula V.K., Tomita Y., Preet A., Palmieri F., Wellstein A., Byers S., Giaccia A.J., Glasgow E., Albanese C., Avantaggiati M.L. The mitochondrial citrate transporter, CIC, is essential for mitochondrial homeostasis. Oncotarget. 2012;3:1220–1235. doi: 10.18632/oncotarget.714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Brière J.-J., Favier J., Gimenez-Roqueplo A.-P., Rustin P. Tricarboxylic acid cycle dysfunction as a cause of human diseases and tumor formation. Am. J. Physiol. Cell Physiol. 2006;291:C1114–C1120. doi: 10.1152/ajpcell.00216.2006. [DOI] [PubMed] [Google Scholar]
- 29.Icard P., Poulain L., Lincet H. Understanding the central role of citrate in the metabolism of cancer cells. Biochim. Biophys. Acta. 2012;1825:111–116. doi: 10.1016/j.bbcan.2011.10.007. [DOI] [PubMed] [Google Scholar]
- 30.Wellen K.E., Hatzivassiliou G., Sachdeva U.M., Bui T.V., Cross J.R., Thompson C.B. ATP-citrate lyase links cellular metabolism to histone acetylation. Science. 2009;324:1076–1080. doi: 10.1126/science.1164097. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.