

Abstract
The hepatic bile acid uptake transporter Sodium Taurocholate Cotransporting Polypeptide (NTCP) is less well characterized than its ileal paralog, the Apical Sodium Dependent Bile Acid Transporter (ASBT), in terms of drug inhibition requirements. The objectives of this study were a) to identify FDA approved drugs that inhibit human NTCP, b) to develop pharmacophore and Bayesian computational models for NTCP inhibition, and c) to compare NTCP and ASBT transport inhibition requirements. A series of NTCP inhibition studies were performed using FDA approved drugs, in concert with iterative computational model development. Screening studies identified 27 drugs as novel NTCP inhibitors, including irbesartan (Ki =11.9 μM) and ezetimibe (Ki = 25.0 μM). The common feature pharmacophore indicated that two hydrophobes and one hydrogen bond acceptor were important for inhibition of NTCP. From 72 drugs screened in vitro, a total of 31 drugs inhibited NTCP, while 51 drugs (i.e. more than half) inhibited ASBT. Hence, while there was inhibitor overlap, ASBT unexpectedly was more permissive to drug inhibition than was NTCP, and this may be related to NTCP’s possessing fewer pharmacophore features. Findings reflected that a combination of computational and in vitro approaches enriched the understanding of these poorly characterized transporters and yielded additional chemical probes for possible drug-transporter interaction determinations.
Keywords: Sodium Taurocholate Cotransporting Polypeptide (NTCP), Apical Sodium Dependent Bile Acid Transporter (ASBT), pharmacophore, Bayesian, transporter
INTRODUCTION
Human Sodium Taurocholate Cotransporting Polypeptide (NTCP, SLC10A1) is a key bile acid transporter and is predominantly expressed at the basolateral membrane of hepatocytes. Its primary role is to transport bile salts from the portal blood into the liver in a sodium-dependent fashion. This transporter accounts for more than 80% of conjugated bile salts taken up into the liver.1
NTCP also transports some drugs and can impact drug disposition.2–5 For example, the HMG-CoA reductase inhibitor rosuvastatin was the first drug identified as a substrate of human NTCP, contributing 35% of total drug uptake into isolated human hepatocytes.2 Interestingly, this drug is not transported by rat Ntcp, indicating species specificity in substrate affinity. Other statins such as pitavastatin, atorvastatin and fluvastatin are also NTCP substrates in vitro, although NTCP’s contribution to their in vivo hepatic uptake is unknown.3,4 Recently, the antifungal micafungin was shown to be significantly taken up by NTCP (i.e. 45%–50% of total in vitro uptake), while a lesser amount was transported by Organic Anion Transporting Polypeptides (OATPs), which are responsible for sodium-independent bile acid uptake.5 There is growing evidence of NTCP’s role in hepatic drug uptake, including drug-drug interactions due to drug inhibition of this transporter, as exemplified by coadministration of micafungin with cyclosporine A, which mildly increases micafungin AUC exposure in healthy volunteers.6
Because of NTCP-mediated drug-drug interaction potential, it would be advantageous to identify potential inhibitors early in drug development. However, since human NTCP was cloned 18 years ago, very few human NTCP inhibitors have been identified, which include cyclosporine A, ketoconazole, and ritonavir.7,8 Therefore, the first two objectives of the present study were a) to identify FDA approved drugs that inhibit human NTCP and b) to develop pharmacophore and Bayesian computational models for NTCP inhibition.
The two computational modeling approaches, namely pharmacophore and Bayesian models, have been previously successfully developed and applied to identify novel inhibitors for several transporters, including PepT19, P-gp10, MRP111, OCTN212 and MATE113. When there is limited data available, a common feature pharmacophore can be generated as a three dimensional qualitative model that describes the arrangement of the key features essential for biological activity. When more data is available (tens to thousands of compounds), a Bayesian machine learning model can be produced, often as a classification model with a two dimensional fingerprint.13 Both approaches can be used to virtually screen libraries of compounds and predict active and inactive compounds, prior to in vitro verification. Both approaches were applied in this study to identify novel NTCP inhibitors.
The Apical Sodium Dependent Bile Acid Transporter (ASBT, SLC10A2) is the ileal paralog of NTCP with 35% amino acid sequence identity and is responsible for absorbing bile acid in the terminal ileum. It appears widely accepted that NTCP has a broader inhibitor profile than ASBT, based on studies in rabbit with a limited number of inhibitors.14,15 Such studies may however yield a biased conclusion because of small sample size and species specificity. A third objective of this study was to compare human NTCP and ASBT transport inhibition requirements.
Briefly, 31 drugs from various therapeutic classes were found to inhibit human NTCP. Among them, 27 were novel inhibitors that had not previously been reported as NTCP inhibitors. Both the common feature pharmacophore and a Bayesian model were used to screen an FDA approved drug database and were validated by additional in vitro testing. Angiotensin II receptor antagonists were found to be human NTCP inhibitors to varying degrees, with irbesartan being the most potent inhibitor. Interestingly, the inhibitor selectivity for ASBT was more permissive than for NTCP.
EXPERIMENTAL SECTION
Figure 1 illustrates the overall approach to identify human NTCP and ASBT inhibitors. Iterative experimental and computational screening was undertaken. For initial screening, 23 drugs were selected based on commercial availability and whether they were known ASBT inhibitor, as ASBT and NTCP are paralog transporters. A common feature pharmacophore for NTCP inhibition was developed using these observed 11 inhibitors and 12 non-inhibitors, while a Bayesian model was developed from 50 drugs evaluated from initial and secondary screening. All drugs screened for NTCP inhibition were also screened for ASBT inhibition and cytotoxicity in their respective cells.
Figure 1.

Flow diagram of approach to identify drugs that inhibit human NTCP, develop computational models for NTCP inhibition, and compare the drug inhibitor selectivity of NTCP and ASBT. NTCP inhibition studies involved an initial, a secondary, and a tertiary screen for inhibitors.
Materials
[3H] Taurocholate (1 mCi/mL) was purchased from PerkinElmer, Inc (Waltham, MA). Taurocholate was obtained from Sigma-Aldrich (St. Louis, MO). Fetal bovine serum (FBS), penicillin-streptomycin, geneticin, non-essential amino acid, trypsin, and Dulbecco's modified Eagle's medium (DMEM) were purchased from Invitrogen Corporation (Carlsbad, CA). WST-1 reagent was bought from Roche Applied Science (Indianapolis, IN). All drugs and other chemicals were obtained from Sigma-Aldrich (St. Louis, MO), Enzo Life Sciences (Farmingdale, NY), AK Scientific (Mountain View, CA), and LKT Labs (St. Paul, MN).
Cell culture
Stably transfected human NTCP-HEK293 and human ASBT-MDCK cells were cultured, as previous described with minor modifications.16–18 Briefly, NTCP-HEK293 and ASBT-MDCK cells were grown in 37°C, 90% relative humidity and fed every 2 days. The medium consisted of media DMEM supplemented with 10% fetal bovine serum, 100 units/mL penicillin and 100 μg/mL streptomycin. Geneticin (1 mg/mL) was used to maintain selection pressure. For NTCP-HEK293 cells, medium also included 100 μM non-essential amino acid. Cells were passaged after reaching 80% confluence.
Inhibition studies
Inhibition studies were conducted as previously described with minor modifications.16–18 Briefly, NTCP-HEK293 cells were seeded at the density of 300,000 cells/well in 24 well Biocoat plates (2cm2; BD, Franklin Lakes, NJ) for 2 days. ASBT-MDCK cells were seeded at the density of 1.5 million cells/well in 12 well plates (3.8 cm2; Corning, Corning, NY) and grown until 80% confluent. ASBT-MDCK cells were induced by 10 mM sodium butyrate 12–17 hr before each inhibition study to enhance ASBT expression.
All inhibition studies were conducted by exposing cells to donor solution. Donor solution consisted of Hank's Balance Salts Solution (HBSS) and cold taurocholate (10 μM for NTCP studies, or 2.5 μM for ASBT studies), along with 0.5 μCi/mL [3H]-taurocholate. Inhibition studies were either drug screening studies or Ki determination studies. Screening studies employed only one drug inhibitor concentration, while Ki determination studies used seven drug inhibitor concentrations. In screening studies, donor solution also contained drug (typically 100 μM), which was compared to no-drug controls to evaluate whether drug was an inhibitor or not. Taurocholate was incubated for 10 min for ASBT studies, over which taurocholate uptake is linear16. Taurocholate was incubated for 5 min for NTCP studies, as prior studies conducted here showed taurocholate uptake is linear between at least 5 and 20 min. After incubation, buffer was removed, and cells were washed with cold sodium-free buffer. Sodium-free buffer replaced sodium chloride with tetraethylammonium chloride. Cells were lysed using acetonitrile as previously described.19 Lysate was dissolved in phosphate buffered saline (PBS), and aliquots were counted for associated radioactivity using a liquid scintillation counter. A few screening studies employed a drug concentration lower than 100 μM due to drug solubility limitation (i.e. ezetimibe and tioconazole each used 50 μM).
For selected drugs whose screening results showed the drugs to be inhibitors, Ki determination studies were identically performed, except used a range of drug inhibitor concentrations (0–200 μM).
Estimated Ki from screening studies was calculated from three types of taurocholate uptake studies: studies in the presence and absence of sodium, as well as studies in the presence of inhibitor with sodium. Since NTCP and ASBT are sodium-dependent (Figure S1 in Supporting Information), studies without sodium measure passive uptake of taurocholate. Studies with sodium but without inhibitor measure total uptake without inhibitor. Active uptake without inhibitor was calculated by subtracting passive uptake from total uptake without inhibitor, which is represented by . Active uptake with inhibitor was calculated by subtracting passive uptake from total uptake with inhibitor, which is represented by . The ratio of active uptake with inhibitor versus active uptake without inhibitor is:
| eqn 1 |
where I is the inhibitor concentration, S is the taurocholate concentration, Ki is the inhibition constant, and Kt is taurocholate Michaelis-Menten constant. Of note from Figure S1 (Supporting Information), taurocholate uptake kinetic parameters into NTCP-HEK293 cells are Kt = 22.7(±3.4) μM, Vmax = 1.80(±0.03) pmol/sec/cm2, and passive permeability Pp = 1.27(±0.03)x10−6 cm/sec.
Observed Ki differed from estimated Ki in that observed Ki used an inhibition profile over a range of inhibitor concentrations, while estimated Ki used one inhibitor concentration (i.e. from inhibition screening study). In order to determine observed Ki, inhibition profile data was fitted to competitive inhibition model:
| eqn 2 |
where J is the taurocholate flux, and Jmax is maximal flux of taurocholate without inhibitor. Jmax was estimated from taurocholate uptake studies at high taurocholate concentrations where transporter was saturated (i.e. 200 μM). Observed Ki was calculated through nonlinear regression using WinNonlin (Pharsight; Sunnyvale, CA).
Cytotoxicity studies
Cytotoxicity studies were conducted to assess whether drug was cytotoxic to NTCP-HEK293 and ASBT-MDCK cells.20 For each NTCP-HEK293 and ASBT-MDCK cells, cells were seeded at the density of 50,000 cells/well in 96-well Biocoat plates for two days. Cells were washed three times with HBSS buffer and exposed to donor solution of drug (100μM) for 10 min. Donor solution was removed, followed by addition of 10 μL of cell proliferation reagent WST-1 in 100 μL of HBSS and incubated for 4 hr. Absorbance at 440nm was measured using a SpectraMax 384 Plus plate reader (Molecular Devices; Sunnyvale, CA).
Common feature pharmacophore development
A common feature pharmacophore was developed using Catalyst™ in Discovery Studio 2.5.5 (Accelrys; San Diego, CA). Template molecule structures were downloaded from ChemSpider (www.chemspider.com), and conformer generation was carried out using the CAESAR algorithm21 applied to the selected template molecules (maximum of 255 conformations per molecule and maximum energy of 20 kcal/mol). Hydrophobic, hydrogen bond acceptor, hydrogen bond donor, and the positive and negative ionizable features were selected, as well as excluded volumes. The principal value of the inhibitors with estimated Ki less than 100μM were assigned as 2, while principal value of the inhibitors with estimated Ki between 100–300μM were assigned as 1. For the compounds whose estimated Ki was larger than 300μM, the principal value was set as 0. The common features of NTCP inhibitors were extracted from inhibitors with estimated Ki < 300μM, while excluded volumes were added using inhibitors with estimated Ki > 300μM. The van der Waals surface of ezetimibe was applied as a shape restriction to limit the number of hits returned in database searching. The model was subsequently used to screen the Clinician’s Pocket Drug Reference (SCUT) database of 814 frequently used FDA approved drugs, as well as the larger Collaborative Drug Discovery (CDD; Collaborative Drug Discovery, Inc.; Burlingame, CA., www.collaborativedrug.com) database of 2690 FDA approved drugs using the FAST method. We then selected from these compounds molecules which could be purchased and tested. We also selected molecules that were not retrieved by the pharmacophore.
Bayesian model with 2D descriptor development
A Laplacian-corrected Bayesian classifier model was developed using Discovery Studio 2.5.5.22 Molecular function class fingerprints of maximum diameter 6 (FCFP_6), along with eight other descriptors (i.e. AlogP, molecular weight, number of rotatable bonds, number of rings, number of aromatic rings, number of hydrogen bond acceptors, number of hydrogen bond donors, and molecular fractional polar surface area) were calculated from an input sd file using the “calculate molecular properties” protocol. The model was generated by the “create Bayesian model” protocol. Both the leave-one out cross-validation approach and external validation were conducted to evaluate the model, which included using the custom protocol, where 10%, 30%, and 50% of the training set were left out 100 times. The best split was calculated by selecting the split that minimized the sum of the percent misclassified for NTCP inhibitors and for non-inhibitors, using the cross-validated score for each sample.
Principal Component Analysis
Principal Component Analysis (PCA) from Discovery Studio version 3.5 was used to compare the molecular descriptor space for the NTCP and CDD FDA approved drugs data sets (using the descriptors of ALogP, molecular weight, number of hydrogen bond donors, number of hydrogen bond acceptors, number of rotatable bonds, number of rings, number of aromatic rings, and molecular fractional polar surface area). In each case, the respective sets of compounds were combined and used to generate the PCA analysis.
RESULTS
Initial screening for human NTCP inhibition
Twenty-three drugs from various therapeutic classes were screened for human NTCP inhibition. If the drug was an NTCP inhibitor, taurocholate uptake was reduced compared to control and used to calculate estimated Ki (Table 1). Molecules with estimated Ki less than 300μM were denoted NTCP inhibitors. Eleven FDA approved drugs were found to be NTCP inhibitors: bendroflumethiazide, ezetimibe, simvastatin, nitrendipine, rosuvastatin, nefazodone, indomethacin, nifedipine, tioconazole, methylprednisolone and prochlorperazine (Table 1). These drugs reduce taurocholate uptake by at least 19.0% and their estimated Ki ranged from 26.2 μM to 280 μM. These 11 drugs were subsequently subjected to inhibition studies employing a range of seven drug concentrations (0–200μM), yielding an observed Ki (Table 1). Figure 2 shows inhibition of taurocholate uptake into NTCP-HEK293 cells by bendroflumethiazide. As expected, in Table 1, the observed Ki of these 11 drugs were less than 300μM, consistent with their estimated Ki (from a single drug inhibition concentration). Of the 23 drugs, the remaining 12 drugs were denoted as non-inhibitors of NTCP.
Table 1.
Initial NTCP inhibition results. From a single inhibitor concentration, the percent taurocholate uptake compared to no-drug control was measured, from which an estimated Ki was calculated. Additionally, for 14 compounds, an observed Ki was measured from a range of inhibitor concentrations. Compounds are listed in order of estimated Ki.
| Percent taurocholate | Estimated | Observed | |
|---|---|---|---|
| Compound | uptake a | Ki (μM) b | Ki (μM)c |
| Bendroflumethiazide* | 27.6±0.7 | 26.2±1.0 | 53.0±6.8 |
| Ezetimibe* | 62.5±1.4 | 53.8±3.3 | 25.0±3.2 |
| Simvastatin* | 47.6±4.2 | 57.3±11 | 47.9±3.7 |
| Nitrendipine* | 67.7±1.6 | 72.3±5.2 | 111±10 |
| Rosuvastatin* | 59.1±3.0 | 100±12 | 128±13 |
| Nefazodone* | 60.8±1.9 | 100±8 | 126±20 |
| Indomethacin* | 68.4±2.5 | 141±14 | 173±24 |
| Nifedipine* | 67.7±5.8 | 144±43 | 62.6±10 |
| Tioconazole* | 84.3±5.1 | 177±66 | 148±34 |
| Methylprednisolone* | 79.5±3.5 | 255±53 | 238±33 |
| Prochlorperazine* | 81.0±2.5 | 280±37 | 209±27 |
| Chloroquine* | 83.0±4.5 | 336±101 | - |
| Ketoprofen* | 84.6±6.1 | 361±178 | 321±57 |
| Propafenone HCl* | 85.4±3.8 | 402±116 | - |
| Probenecid* | 86.8±3.3 | 435±114 | 544±217 |
| Diltiazem* | 87.5±5.9 | 460±232 | 599±212 |
| Ethosuximide* | 88.2±3.8 | 513±169 | - |
| Abacavir | 94.9±4.4 | 1291±4144 | - |
| Quinine | 95.6±1.8 | 1491±581 | - |
| Thiothixene | 99.2±1.0 | 3000 | - |
| Acarbose | 99.7±4.9 | 3000 | - |
| Aztreonam | 104±4 | 3000 | - |
| Omeprazole | 105±6 | 3000 | - |
Denotes percent of taurocholate uptake in presence of 100 μM of compound (except ezetimibe, nitredipine and tioconazole, which used 50 μM due to solubility limitation), compared to taurocholate uptake in absence of compound. Values denote mean (±SEM).
Estimated Ki was determined from a single inhibitor concentration. A value of 3000μM was assigned if drug did not inhibit. Throughout the manuscript, estimated Ki is an experimentally- determined value from a single inhibitor concentration and is not a result from computational chemistry prediction.
Observed Ki was determined using seven concentrations in the range of 0 to 200μM. Values denote mean (±SEM).
Percent of taurocholate uptake was significantly different from 100% (p < 0.05).
Figure 2.
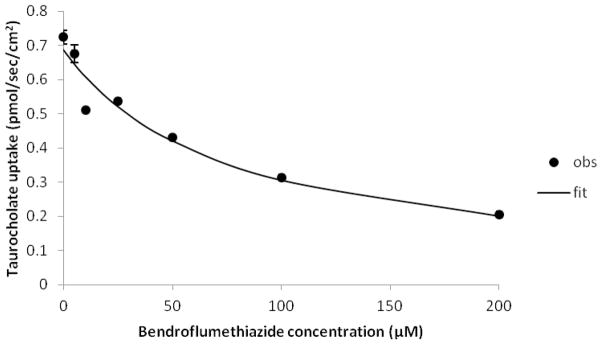
Inhibition of NTCP by bendroflumethiazide. Taurocholate uptake into NTCP-HEK293 cells was reduced in the presence of bendroflumethiazide in a concentration-dependent fashion. Inhibition studies were conducted using seven concentrations of bendroflumethiazide (0–200 μM). Closed circles indicate observed data points, while the solid line indicates model fit. Observed Ki of bendroflumethiazide was 53.0(±6.8) μM.
Common feature pharmacophore for NTCP inhibitors
The 11 inhibitors and 12 non-inhibitors from initial screening were used as a training set to develop a common feature pharmacophore. The resulting model featured two hydrophobes and one hydrogen bond acceptor (Figure 3). Ezetimibe was applied to restrict the pharmacophore to the van der Waals shape. The 12 non-inhibitors were also used to yield ten excluded volumes. The developed pharmacophore was used to screen the SCUT and CDD databases of FDA approved drugs and retrieved 45 and 85 drugs, respectively (Table S1 and S2 in Supporting Information). Fit Value ranged from 0.001 to 2.48 for the SCUT database and from 0.00031 to 2.75 for the CDD database. Eighteen drugs with Fit Values from 0.0012 to 2.58 in the CDD database were screened for inhibition, along with 9 other drugs not retrieved by the pharmacophore. The 18 retrieved drugs were selected since they exhibited a range of Fit Values, which reflected the level of compound mapping to the pharmacophore.
Figure 3.

Common feature pharmacophore of NTCP inhibitors. The pharmacophore employed the 11 most potent inhibitors from initial screening and the 12 inactive compounds were used for developing the excluded volumes. Ezetimibe (shown as stick format) is used as the shape restriction (grey) due to its high potency (i.e. smallest observed Ki of 25.0 μM). Pharmacophore features are two hydrophobes (cyan), one hydrogen bond acceptor (green), and 10 excluded volumes (grey).
Table 2 shows the results of secondary screening of these 27 drugs. Of the 18 drugs retrieved by the pharmacophore, six inhibited NTCP: nateglinide, irbesartan, losartan, olmesartan, fenofibrate, and candesartan. Of the 9 non-retrieved drugs, two inhibited NTCP: doxazosin and budesonide. Observed Ki values were measured from a range of inhibitor concentrations. The most potent inhibitor was irbesartan (Ki = 11.9 μM), which also has a high Fit Value (2.48) (Figure 4). Irbesartan was a more potent inhibitor than ezetimibe (Ki = 25.0 μM) from the initial screen. Interestingly, four of the six angiotensin II receptor antagonists tested were NTCP inhibitors: irbesartan, losartan (Ki = 72.1 μM), olmesartan (Ki = 233 μM), and candesartan (Ki = 233 μM). Valsartan and eprosartan did not inhibit NTCP (Table 2). The second most potent NTCP inhibitor from secondary screening was doxazosin (Ki = 35.3μM), which did not map to this pharmacophore. Interestingly, doxazosin did map to the pharmacophore (Fit Value = 2.12) when the ezetimibe shape restriction was removed.
Table 2.
Secondary NTCP inhibitor results. Of the 85 drugs retrieved from the database of FDA approved drugs, 18 drugs were tested, along with 9 drugs not retrieved. The 18 retrieved drugs that were tested were selected to represent a wide range of Fit Values from Common feature pharmacophore predictions. From a single inhibitor concentration, the percent taurocholate uptake compared to no-drug control was measured, from which an estimated Ki was calculated. Additionally, for 9 compounds, an observed Ki was measured from a range of inhibitor concentrations. Compounds are listed in order of Fit Value. Fit Value for compounds not retrieved by Common feature pharmacophore is denoted “No fit”. Higher Fit Value anticipates a greater congruence to the pharmacophore (i.e. anticipates NTCP inhibition).
| Compounds | Percent taurocholate uptake a | Est Ki (μM) b | Observed Ki (μM) c | Fit Value |
|---|---|---|---|---|
| Yohimbine | 108±4 | 3000 | - | 2.58 |
| Nateglinide* | 62.0±1.4 | 111±7 | 200±32 | 2.49 |
| Irbesartan* | 15.9±1.6 | 12.0±1.6 | 11.9±0.7 | 2.48 |
| Losartan* | 60.7±2.0 | 105±9 | 72.1±5.1 | 2.25 |
| Ropinirole | 133±4 | 3000 | - | 2.24 |
| Sulfinpyrazone | 96.1±3.2 | 1664±2871 | - | 2.10 |
| Bortezomib | 107±4 | 3000 | - | 2.01 |
| Olmesartan* | 86.0±5.5 | 422±204 | 233±43 | 1.84 |
| Fenofibrate* | 76.3±6.0 | 211±60 | 129±20 | 1.80 |
| Valsartan | 101±3 | 3000 | - | 1.72 |
| Cefaclor | 99.1±6.2 | 3000 | - | 1.59 |
| Eletriptan | 104±8 | 3000 | - | 1.07 |
| Nafcillin | 96.7±5.6 | 2006±5836 | - | 0.82 |
| Oseltamivir | 102±9 | 3000 | - | 0.60 |
| Raloxifene HCl* | 90.5±4.6 | 321±139 | 301±131 | 0.58 |
| Eprosartan | 99.9±0.7 | 3000 | - | 0.17 |
| Enalapril | 111±4 | 3000 | - | 0.0048 |
| Candesartan* | 68.0±7.2 | 145±44 | 233±37 | 0.0012 |
| Doxazosin* | 70.8±3.7 | 41.2±7.1 | 35.3±5.4 | No fit |
| Budesonide* | 79.3±6.4 | 264±94 | 220±45 | No fit |
| Econazole* | 92.3±4.5 | 416±624 | - | No fit |
| Miconazole* | 94.6±2.6 | 601±300 | - | No fit |
| Daunorubicin | 107±3 | 3000 | - | No fit |
| Dibucaine | 100±1 | 3000 | - | No fit |
| Oxiconazole | 99.5±2.6 | 3000 | - | No fit |
| Warfarin | 102±2 | 3000 | - | No fit |
| Formoterol | 111±2 | 3000 | - | No fit |
Denotes percent of taurocholate uptake in presence of 100 μM of compound [except doxazosin (25μM); and econazole, miconazole, oxiconazole, and raloxifene (50 μM) due to solubility limitation], compared to taurocholate uptake in absence of compound. Values denote mean (±SEM).
Estimated Ki was determined from a single inhibitor concentration. A value of 3000μM was assigned if drug did not inhibit.
Observed Ki was determined using seven concentrations in the range of 0 to 200μM. Values denote mean (±SEM).
Percent of taurocholate uptake was significantly different from 100% (p < 0.05).
Figure 4.

Inhibition of NTCP by irbesartan. Taurocholate uptake into NTCP-HEK293 cells was reduced in the presence of irbesartan in a concentration-dependent fashion. Inhibition studies were conducted using seven concentrations of irbesartan (0–200 μM). Closed circles indicate observed data points, while the solid line indicates model fit. Observed Ki of irbesartan was 11.9(±0.7) μM.
Bayesian model for NTCP inhibitors
A Bayesian model was developed using results of the 50 drugs from initial and secondary screening (Tables S3-S6 in Supporting Information). The area under the receiver operator curve (ROC) for the leave-one-out cross validation was 0.769. The best split was -0.956, which is the Bayesian score to discriminate NTCP inhibitors from non-inhibitors. Of the 50 compounds, 19 drugs were inhibitors and 31 were non-inhibitors. Of the 19 inhibitors, the Bayesian model classified all these 19 correctly as inhibitors. Of the 31 non-inhibitors, the Bayesian model classified 10 incorrectly as inhibitors (i.e. valsartan, raloxifene, econazole, miconazole, chloroquine, probenecid, ketoprofen, propafenone, diltiazem and ethosuximide). Hence, the model showed good fit to the training set with 80% correct prediction. The model was tested by leaving out 10, 30 and 50% randomly and repeated 100 times (Table S7 in Supporting Information). The ROC values were lower than for leave-one out cross validation, while concordance specificity and selectivity declined as the size of the leave out group increased, suggesting that the model was not stable to large leave out groups (e.g. using 25 molecules for testing and 25 for training).
In order to further assess the model’s ability to discriminate NTCP inhibitors and non-inhibitors, an additional 10 drugs with high Bayesian score (>5) and 12 drugs with low Bayesian score (< -0.956) were tested for NTCP inhibition potency. Seven of the 10 high Bayesian score drugs (70%) were found to be NTCP inhibitors: sulconazole, lovastatin, ketoconazole, cerivastatin, itraconazole, nimodipine and isradipine (Table 3). Meanwhile, seven of 12 low Bayesian score drugs (58%) were identified as NTCP non-inhibitors. However, naproxen, sulfanilamide, reserpine, cyclosporine A and ritonavir each inhibited NTCP. The model showed good ability to predict NTCP inhibitors from non-inhibitors (64% correct overall). In Table 3, it should be noted that sulconazole, itraconazole, reserpine, and cyclosporine A inhibition studies employed lower drug concentrations due to limiting drug solubility, and hence impacted the extent of taurocholate inhibition. Figure 5 shows inhibition of taurocholate uptake into NTCP-HEK293 cells by cyclosporine A
Table 3.
Tertiary NTCP inhibitor results. Of the 1360 FDA-approved drugs (CDD dataset) predicted by the Bayesian model to be inhibitors, 10 drugs were tested, along with 12 additional, non-retrieved drugs. The 10 predicted inhibitors that were tested were selected since their Bayesian scores were five or higher. From a single inhibitor concentration, the percent taurocholate uptake compared to no-drug control was measured, from which an estimated Ki was calculated. Compounds are listed in order of Bayesian score.
| Compounds | Percent taurocholate uptake a | Est Ki (μM) b | Bayesian Score c |
|---|---|---|---|
| Sulconazole* | 86.7±7.2 | 112±115 | 11.62 |
| Prednisolone* | 83.0±8.4 | 335±177 | 9.77 |
| Chlorpromazine* | 91.8±3.0 | 767±268 | 8.14 |
| Lovastatin* | 74.7±3.2 | 202±32 | 7.92 |
| Ketoconazole d,* | 59.1±3.0 | 98.8±12 | 7.81 |
| Cerivastatin* | 68.6±7.4 | 150±52 | 7.66 |
| Itraconazole* | 81.8±5.6 | 77.1±38 | 7.13 |
| Nimodipine d,* | 55.9±1.6 | 86.5±5.4 | 6.77 |
| Nicardipine* | 81.8±6.5 | 309±144 | 6.65 |
| Isradipine* | 59.3±3.2 | 99.5±14 | 5.00 |
| Naproxen* | 76.0±5.2 | 217±69 | −1.37 |
| Sulfanilamide* | 78.4±3.4 | 250±50 | −1.41 |
| Imatinib* | 90.7±1.6 | 673±130 | −1.86 |
| Metronidazole* | 84.3±2.7 | 369±83 | −2.41 |
| Triamterene* | 85.7±4.3 | 411±139 | −2.50 |
| Furosemide* | 81.6±6.9 | 304±132 | −2.61 |
| Cimetidine* | 82.9±2.9 | 332±73 | −2.73 |
| Famotidine* | 84.9±3.6 | 387±130 | −3.16 |
| Cyclosporine Ad,* | 24.0±1.9 | 10.3±1.1 | −6.05 |
| Procainamide HCl* | 84.8±6.7 | 384±185 | −6.14 |
| Ritonavir d,* | 27.0±1.2 | 25.1±1.6 | −6.28 |
| Reserpine* | 83.8±8.3 | 89.1±65 | −15.6 |
Denotes percent of taurocholate uptake in presence of 100 μM of compound [except 25 μM sulconazole, 25 μM itraconazole, 25 μM reserpine, and 50 μM cyclosporine A due to solubility limitation], compared to taurocholate uptake in absence of compound. Values denote mean (±SEM).
Estimated Ki was determined from a single inhibitor concentration.
A Bayesian score higher than -0.956 indicates that the Bayesian model predicts the compound to be an NTCP inhibitor. Lower Bayesian scores predict compound to not be an NTCP inhibitor.
Observed Ki values for ketoconazole, nimodipine, cyclosporine A and ritonavir were 202±48μM, 190±31μM, 7.6±1.1, and 18.4±1.6μM respectively.
Percent of taurocholate uptake was statistically significant difference from 100% (p < 0.05).
Figure 5.
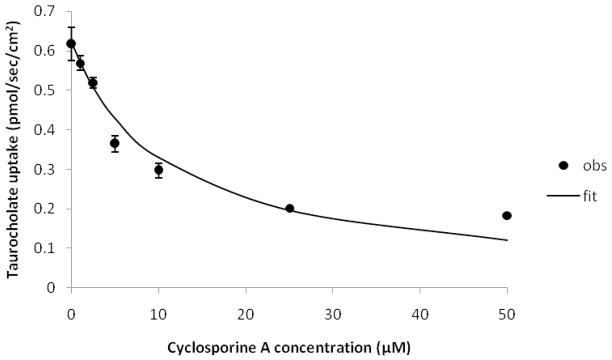
Inhibition of NTCP by cyclosporine A. Taurocholate uptake into NTCP-HEK293 cells was reduced in the presence of cyclosporine A in a concentration-dependent fashion. Inhibition studies were conducted using seven concentrations of irbesartan (0–200 μM). Closed circles indicate observed data points, while the solid line indicates model fit. Observed Ki of cyclosporine A was 7.6(±1.1) μM.
ASBT inhibition
All 72 drugs from initial, secondary, and tertiary NTCP screening were screened for ASBT inhibition, in order to compare the inhibition selectivities of NTCP and ASBT (Table 4 and Table S8 in Supporting Information). ASBT studies used the same drug concentration as NTCP. As with NTCP, a Ki value of 300 μM was applied to differentiate ASBT inhibitors and non-inhibitors. Of the 72 drugs, 51 drugs were ASBT inhibitors, which is much greater than the 31 drugs that inhibited NTCP. ASBT and NTCP share 29 inhibitors and 19 non-inhibitors. However, 22 drugs were ASBT inhibitors but not NTCP inhibitors. Only two drugs, olmesartan and methylprednisolone, inhibited NTCP but not ASBT. Statins, sartans, and calcium channel blockers inhibited both ASBT and NTCP. Meanwhile, antifungal drugs with an imidazole ring were ASBT inhibitors (econazole, itraconazole, ketoconazole, metronidazole, miconazole, oxiconazole, sulconazole, tioconazole), but only a few were NTCP inhibitors (itraconazole, ketoconazole, sulconazole, tioconazole). Figure 6 plots the percent taurocholate uptake across ASBT versus percent taurocholate uptake across NTCP. Each of the 72 data points represents one drug. Fitting a line yielded a slope of 0.439 (±0.135) and r2=0.131. Hence, there was at best a weak correspondence between ASBT inhibition and NTCP inhibition. Relative to the line of unity, 20 drugs were above the line, while 52 drugs were below the line, indicating more drugs were better ASBT inhibitors than NTCP inhibitors. Figure S2 in Supporting Information presents this data in terms of estimated Ki, rather than percent taurocholate uptake.
Table 4.
Comparison of 72 drugs in terms of NTCP and ASBT inhibition. For each NTCP and ASBT, estimated Ki was derived from a single inhibitor concentration. Compounds are listed in order of estimated Ki for NTCP.
| Compounds | Est Ki for NTCP (μM) | Est Ki for ASBT (μM) |
|---|---|---|
| Cyclosporine A | 10.3±1.1 | 41.0±7.4 |
| Irbesartan | 12.0±1.6 | 131±34 |
| Ritonavir | 25.1±1.6 | 47.3±9.7 |
| Bendroflumethiazide | 26.2±1.0 | 168±10 |
| Doxazosin | 41.2±7.1 | 20.9±7.3 |
| Ezetimibe | 53.8±3.3 | 4.61±0.88 |
| Simvastatin | 57.3±11 | 99.1±19 |
| Nitrendipine | 72.3±5.2 | 31.7±6.5 |
| Itraconazole | 77.1±38 | 57.0±25 |
| Nimodipine | 86.5±5.4 | 30.9±1.7 |
| Reserpine | 89.1±65 | 21.6±2.9 |
| Ketoconazole | 98.8±12 | 61.9±27 |
| Isradipine | 99.5±14 | 17.9±2.3 |
| Rosuvastatin | 100±12 | 164±34 |
| Nefazodone | 100±8 | 30.8±3.6 |
| Losartan | 105±9 | 139±58 |
| Nateglinide | 111±7 | 77.0±16 |
| Sulconazole | 112±115 | 29.4±0.6 |
| Indomethacin | 141±14 | 31.3±7.7 |
| Nifedipine | 144±43 | 89.9±34 |
| Candesartan | 145±44 | 67.6±30 |
| Cerivastatin | 150±52 | 220±100 |
| Tioconazole | 177±66 | 11.9±2.7 |
| Lovastatin | 202±32 | 21.7±2.1 |
| Fenofibrate | 211±60 | 121±33 |
| Naproxen | 217±69 | 136±13 |
| Sulfanilamide | 250±50 | 194±24 |
| Methylprednisolone | 255±53 | 3000 |
| Budesonide | 264±94 | 62.6±15 |
| Prochlorperazine | 280±37 | 27.4±4.6 |
| Furosemide | 304±132 | 3000 |
| Nicardipine | 309±144 | 142±3 |
| Raloxifene HCl | 321±139 | 65.8±9.2 |
| Cimetidine | 332±73 | 208±51 |
| Prednisolone | 335±177 | 89.2±32 |
| Chloroquine | 336±101 | 3000 |
| Ketoprofen | 361±178 | 480±609 |
| Metronidazole | 369±83 | 223±90 |
| Procainamide HCl | 384±185 | 3000 |
| Famotidine | 387±130 | 2275±978 |
| Propafenone HCl | 402±116 | 50.3±18 |
| Triamterene | 411±139 | 110±3 |
| Econazole | 416±624 | 18.5±3.9 |
| Olmesartan | 422±204 | 1049±422 |
| Probenecid | 435±114 | 425±155 |
| Diltiazem | 460±232 | 237±21 |
| Ethosuximide | 513±169 | 313±53 |
| Miconazole | 601±300 | 61.3±16 |
| Imatinib | 673±130 | 177±32 |
| Chlorpromazine | 767±268 | 50.7±5.9 |
| Abacavir | 1291±4144 | 1460±5960 |
| Quinine | 1491±581 | 239±117 |
| Sulfinpyrazone | 1664±2871 | 529±600 |
| Nafcillin | 2006±5836 | 3000 |
| Acarbose | 3000 | 318±169 |
| Aztreonam | 3000 | 1419±5313 |
| Bortezomib | 3000 | 811±243 |
| Cefaclor | 3000 | 1886±257 |
| Daunorubicin | 3000 | 3000 |
| Dibucaine | 3000 | 85.8±2.3 |
| Eletriptan | 3000 | 137±48 |
| Enalapril | 3000 | 162±82 |
| Eprosartan | 3000 | 246±119 |
| Formoterol | 3000 | 175±96 |
| Omeprazole | 3000 | 103±23 |
| Oseltamivir | 3000 | 1022±49 |
| Oxiconazole | 3000 | 94.7±38 |
| Ropinirole | 3000 | 30.3±11 |
| Thiothixene | 3000 | 3000 |
| Valsartan | 3000 | 1265±7010 |
| Warfarin | 3000 | 97.4±26 |
| Yohimbine | 3000 | 377±442 |
Figure 6.
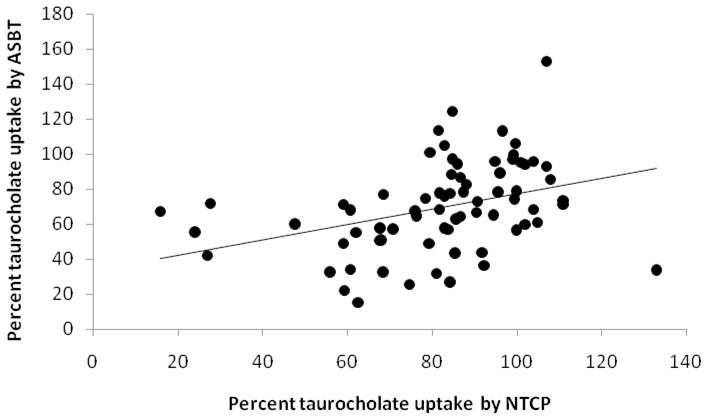
Correlation between NTCP inhibition and ASBT inhibition. The percent taurocholate uptake by ASBT is plotted against percent taurocholate uptake by NTCP for 72 drugs. Linear regression yielded a slope of 0.439 (±0.135) and r2=0.131.
Drug cytotoxicity
Cytotoxicity studies were conducted on each drug using both NTCP-HEK293 and ASBT-MDCK cells in order to exclude false positive results (i.e. to exclude decreased taurocholate uptake due to drug cytotoxicity, rather than NTCP or ASBT uptake inhibition by the drug). Results indicate that no drug was toxic to HEK293 cells under study conditions, with all cell viabilities exceeding 80%.20 (Table S9 in Supporting Information). Meanwhile, fenofibrate and valsartan exerted minor cytotoxicity to MDCK cells, with cell viability being 79.2% and 77.9% respectively. Valsartan did not reduce taurocholate uptake (i.e. was not an inhibitor) into ASBT-MDCK cells, so a false positive was not a concern. Fenofibrate reduced taurocholate uptake into ASBT-MDCK cells to 64.7%, such that cytotoxicity did not appear to solely contribute to fenofibrate’s inhibition.
DISCUSSION
Inhibitor screening
In our study, inhibitors were identified based on the estimated Ki derived from an inhibition screening study (i.e. single concentration of inhibitor study). A benefit of this approach is a requirement for only a minimal amount of drug. However, such an approach may result in less accuracy to assess Ki, potentially yielding false positive or false negative results. Thus, comprehensive inhibition profile studies using seven drug concentrations were conducted on 23 drugs, whose estimated Ki values ranged from 12.0 μM to 460 μM. Results indicate that all drugs whose estimated Ki was less than 300μM (i.e. 18 drugs) were confirmed to be NTCP inhibitors based on observed Ki (Table 5). Only olmesartan was a non-inhibitor based on estimated Ki, but actually was found to be an NTCP inhibitor due to an observed Ki less than 300 μM. Additionally, only five of the 27 compounds had an “observed Ki” that differed from “estimated Ki” by more that 2-fold (i.e. bendroflumethiazide, ezetimibe, nimodipine, ketoconazole and nifedipine).
Table 5.
Comparison of estimated Ki and observed Ki for NTCP. Compounds are listed in order of estimated Ki. Also listed is the percent taurocholate at the highest drug concentration evaluated.
| Compounds | Est Ki for NTCP (μM) | Observed Ki for NTCP (μM) |
|---|---|---|
| Cyclosporine A | 10.3±1.1 | 7.6±1.1 |
| Irbesartan | 12.0±1.6 | 11.9±0.7 |
| Ritonavir | 25.1±1.6 | 18.4±1.6 |
| Bendroflumethiazide | 26.2±1.0 | 53.0±6.8 |
| Doxazosin | 41.2±7.1 | 35.3±5.4 |
| Ezetimibe | 53.8±3.3 | 25.0±3.2 |
| Simvastatin | 57.3±11 | 47.9±3.7 |
| Nitrendipine | 72.3±5.2 | 111±10 |
| Nimodipine | 86.5±5.4 | 190±31 |
| Ketoconazole | 98.8±12 | 202±48 |
| Nefazodone | 100±8 | 126±20 |
| Rosuvastatin | 100±12 | 128±13 |
| Losartan | 105±9 | 72.1±5.1 |
| Nateglinide | 111±7 | 200±32 |
| Indomethacin | 141±14 | 173±24 |
| Nifedipine | 144±43 | 62.6±10 |
| Candesartan | 145±44 | 233±37 |
| Tioconazole | 177±66 | 148±34 |
| Fenofibrate | 211±60 | 129±20 |
| Methylprednisolone | 255±53 | 238±33 |
| Budesonide | 264±94 | 220±45 |
| Prochlorperazine | 280±37 | 209±27 |
| Raloxifene HCl | 321±139 | 301±131 |
| Ketoprofen | 361±178 | 321±57 |
| Olmesartan | 422±204 | 233±43 |
| Probenecid | 435±114 | 544±217 |
| Diltiazem | 460±232 | 599±212 |
Hence, given the favorable consistency between estimated and observed Ki for NTCP, a single concentration inhibition study was found to be reliable. This finding is consistent with several methodological studies on CYP450 and transporter inhibitor screening, which showed that it is reasonable to estimate the Ki from a single drug concentration.23,24 For the 27 drugs where observed Ki was measured for NTCP, there was favorable agreement between Ki estimated from a single drug concentration and observed Ki from seven drug concentrations (r2=0.763, Figure S3 in Supporting Information).
NTCP inhibitors
In the present study, 31 drugs were identified as human NTCP inhibitors, including 27 drugs that are newly identified inhibitors. Most of them (i.e. 20) are antifungal, antihyperlipidemic, antihypertensive, anti-inflammatory or glucocorticoid drugs. Of these, cyclosporine A, ketoconazole, and ritonavir have been previously demonstrated to be a human NTCP inhibitor.8 Reserpine was previously found to inhibit rabbit Ntcp, and also inhibited human NTCP here.15 Rosuvastatin is substrate of human NTCP.2 As expected, it inhibited taurocholate transport here. Interestingly, simvastatin was found here to be a potent inhibitor of NTCP (observed Ki = 47.9 μM), although it is minimally taken up by human NTCP.3 In the present study, nifedipine was a potent NTCP inhibitor (observed Ki = 62.6 μM), although it was previously found not to be an inhibitor when NTCP was transiently expressed in HeLa cells.8
The angiotensin II antagonists showed a structure activity relationship with NTCP, with Ki ranging from 12 to 3000 μM (Table S10 in Supporting Information). Interestingly, candesartan was scored poorly by the ezetimibe shape pharmacophore, perhaps due to candesartan’s carboxylic acid protruding from the molecule shape (Table S10 in Supporting Information), which penalized scoring. The common features across these molecules, namely the two phenyl rings and the tetrazole, are not the sole determinants of activity, as the variable parts of each molecule also impacted activity. For example, valsartan has these features in common with irbesartan, but there is more than a 100-fold difference in activity (Table S10 in Supporting Information). Many of the angiotensin II antagonists were scored highly in the SCUT database (Table S1 in Supporting Information).
It is important to note that we did not test every compound that was retrieved by the pharmacophores, but only sampled based in part on commercial availability and cost. A more exhaustive sampling would require all compounds to be tested. It is possible that additional NTCP inhibitors are missing from the approach outlined in this study. However, the drug space coverage for the 72 compounds tested was excellent, as assessed by principal component analysis with eight simple molecular descriptors (Figure S4 in Supporting Information), indicating a good representation of FDA approved drugs.
Comparison to previous pharmacophores
Ekins et al. previously developed an initial quantitative (HypoGen) pharmacophore based on eight human NTCP inhibitors, which were bile acids and drugs, although the model had not been validated.25 This preliminary model featured two hydrophobes and two hydrogen bond donors. A possible reason for the difference between this previous model and the current model may be a) the previous model employed IC50 values from NTCP-HeLa transiently transfected cells, while our current model was based on estimated Ki values from NTCP-HEK293 stably transfected cells and b) the previous model was quantitative while the current one is qualitative and employed a different algorithm. Interestingly, both models identified two hydrophobic features as important for NTCP inhibition.
Greupink et al. recently developed an NTCP common feature pharmacophore using human NTCP-CHO cells.26 The pharmacophore possessed three hydrophobes and two hydrogen bond acceptors, based on four bile acids and estrone sulfate. In contrast, our pharmacophore reported here possesses two hydrophobes and one hydrogen bond acceptor, based on 23 diverse drugs. Greupink et al. indicate that one or two negative charges are critical for NTCP inhibition, yet their pharmacophore did not possess these features. From a larger and more diverse drug set, we did not observe a negative charge to be required. For example, irbesartan, doxazosin, and simvastatin are potent NTCP inhibitors and do not have functional groups with a negative charge. A further distinction between the present report and findings from Greupink et al. is our focus on FDA approved drugs. The present study involves 72 drugs evaluated for NTCP inhibition. Greupink et al. tested four bile acids, estrone sulfate, and 33 other commercially available chemicals, but no FDA approved drugs. Additionally, we have developed a Bayesian model, which employs 2D molecular fingerprints and allows for more rapid screening, since the pharmacophore approach requires conformation generation for each compound. External testing these models resulted in 64% correct predictions. These findings indicate an overlapping structure activity relationship of inhibitors for ASBT and NTCP, with NTCP being a less permissive transporter than ASBT in terms of susceptibility to inhibition by FDA approved drugs.
It should be noted that experimental and computational approaches here do not evaluate mechanism of inhibition, but rather focus on the identification of novel drug inhibitors. The mechanism of transporter inhibition is poorly elucidated through conventional inhibition data27. Hence, the pharmacophore may reflect more than one binding site, which may not be surprising given the diverse drug molecules tested here.
Pharmacokinetic implications of NTCP inhibition
NTCP is a bile acid transporter which is mainly responsible for transporting bile acid from portal blood into the liver. Recent in vitro studies implied that NTCP may also be involved in rosuvastatin and micafungin hepatic uptake.5 However, even though 35% of rosuvastatin is taken up into liver by NTCP, no drug has been suspected of modifying rosuvastatin disposition via NTCP. Cyclosporine A increases rosuvastatin exposure, which has been attributed to OATP1B1 inhibition.2, 28 Cyclosporine A Ki for OATP1B1 is 0.2 μM 29, while the cyclosporine A Ki for NTCP is 7.6μM (Table 5). Hence, it is possible that cyclosporine A contributes to increased rosuvastatin exposure via NTCP inhibition. Of the 31 in vitro inhibitors observed here, nifedipine, itraconazole, and cyclosporine A showed pharmacokinetic in vivo interaction with micafungin.30 However, a potential mechanism to attenuate a perpetrator drug’s impact via NTCP inhibition is that other influx transporters, such as OATPs, compensate for NTCP inhibition, resulting in no change in rosuvastatin or micafungin pharmacokinetics.
Common feature pharmacophore and Bayesian model
Both common feature pharmacophore and Bayesian models were developed to elucidate NTCP inhibitor requirements. A geometric restriction, based on the van der Waals surface of one of the most active compounds in the training set (i.e. ezetimibe), was added to the common feature pharmacophore when searching the database. This geometric restriction aimed to reduce the number of false positive hits. The model showed that two hydrophobes (e.g. the isobutyl group and the phenyl group in nateglinide) and one hydrogen bond acceptor (e.g. the carbonyl group in nateglinide) were important for NTCP inhibition (Figure 2). Interestingly, cyclosporine A and ritonavir, which were known NTCP inhibitors, were not retrieved by the pharmacophore. This is probably due to no large molecules similar to these being represented in the training set. After removing the shape restriction, cyclosporine A and ritonavir were mapped to the pharmacophore with fit value as 2.08 and 0.45 respectively (Figure S5 in Supporting Information).
The Bayesian model used molecular fingerprint descriptors and identified several molecules that feature benzyl fluorine as important for NTCP inhibition (e.g. ezetimibe, Table S11 in Supporting Information). Both models showed acceptable success in finding new NTCP inhibitors, while the Bayesian model showed better prediction due to less false positive hits. It is still important to note that these models are generated with relatively small datasets, although similar in dataset size to other transporter modeling studies.12, 31
Comparison of the inhibitors between NTCP and ASBT
A previous study by Kramer et al. using rabbit showed that the hepatic bile acid transporter had a much broader specificity for interaction than the ileal transporter.15 A recent study from our group using different bile acid conjugates indicated that human ASBT and NTCP have generally similar inhibition potency.32 For both ASBT and NTCP, inhibition depends on the size of the substituent attached to the bile acid and whether the shape of the steroidal nucleus was distorted. In the present study, 72 drugs were evaluated for human NTCP and ASBT inhibition. Surprisingly, only 31 drugs inhibited NTCP, while 51 drugs inhibited ASBT. Of the 72 drugs, nine drugs reduced taurocholate uptake into NTCP-transfected cells below 50% of control (i.e. more than 50% inhibition): cyclosporine A, ritonavir, bendroflumethiazide, ezetimibe, simvastatin, nefazodone, losartan, nifedipine, irbesartan (Table S12 in Supporting Information). Meanwhile, 15 drugs reduced taurocholate uptake into ASBT-transfected cells below 50% of control: budesonide, chlorpromazine, econazole, ezetimibe, indomethacin, isradipine, ketoconazole, lovastatin, nefazodone, nimodipine, prochlorperazine, propafenone, ritonavir, ropinirole, tioconazole.
Human ASBT showed a broader inhibitor profile than NTCP, in contrast to the conclusion of Kramer et al.15 One possible explanation for this difference is due to species specific differences which has been shown to impact the interaction between bile acid transporter and drug.2,33 For example, rosuvastatin is a human NTCP substrate, but not a substrate for rat Ntcp2; bosentan is a more potent inhibitor for rat Ntcp than human NTCP.33 In the current study, 72 drugs were evaluated, while Kramer evaluated 28 compounds, of which only 19 were drugs.15
Among the 72 drugs tested, based on Ki values in Table 4 and 5, the most selective NTCP inhibitors were irbesartan (NTCP observed Ki = 11.9μM and ASBT estimated Ki =131μM) and methylprednisolone (NTCP observed Ki = 238μM and ASBT estimated Ki =3000μM). Only these two drugs exhibited a 10-fold more potent NTCP inhibition than ASBT inhibition. Meanwhile, 12 drugs showed a 10-fold more potent ASBT inhibition than NTCP inhibition, in rank order: ropinirole, dibucaine, oxiconazole, warfarin, omeprazole, econazole, eletriptan, enalapril, formoterol, chlorpromazine, tioconazole, and eprosartan. For ropinirole, the difference in Ki values was almost 100-fold, where NTCP estimated Ki = 3000μM and ASBT estimated Ki =30.3μM. The compounds selectively inhibiting each transporter could be considered potential chemical probes for mechanistic studies. Several drugs inhibited both NTCP and ASBT. HMG-CoA reductase inhibitors, angiotensin II receptor antagonists, and calcium channel blockers were inhibitors for both ASBT and NTCP. However, antifungal drugs of the imidazole class were ASBT inhibitors, but not all of them were NTCP inhibitors (e.g. econazole, miconazole, oxiconazole).
Eight molecular descriptors (described earlier for Bayesian analysis) were evaluated to differentiate NTCP inhibitors (n = 31) from ASBT inhibitors (n = 51, of which 29 were also NTCP inhibitors). Interestingly, using the t-test, there was no difference in the mean value for each descriptor between NTCP inhibitors and ASBT inhibitors. That is, NTCP inhibitors possessed similar molecular descriptor values to those of ASBT inhibitors, even though about half of the ASBT inhibitors were not NTCP inhibitors.
For NTCP and ASBT, the AlogP of inhibitors were larger than AlogP of non-inhibitors, suggesting that drug lipophilicity favors drug-transporter interaction (and hence inhibition). These results are consistent with prior observations that lipophilicity promotes ASBT inhibition.34,35 Comparing the NTCP pharmacophore from this study to the ASBT pharmacophore from Zheng et al., both pharmacophores possess two hydrophobes in common.35 The ASBT pharmacophore additionally possesses two hydrogen bond acceptors, while the NTCP pharmacophore has one hydrogen bond acceptor instead. Overall, this study showed that ASBT showed wider inhibitor selectivity than NTCP.
In summary, a combination of computational and in vitro approaches was used to identify new NTCP inhibitors. Out of 72 FDA approved drugs screened for both NTCP and ASBT inhibition, 31 inhibited NTCP, while 51 inhibited ASBT. Several novel NTCP inhibitors were identified which fall into a variety of therapeutic classes such as antifungal, antihyperlipidemic, antihypertensive, anti-inflammatory and glucocorticoid drugs.
Supplementary Material
Acknowledgments
This work was supported in part by National Institutes of Health grant DK093406 and FDA grant U01FD004320-01. The authors kindly acknowledge Dr. Keiser (University of Greifswald) for providing the human NTCP-HEK293 cell line used in this study. SE kindly acknowledges Accelrys for providing Discovery Studio.
ABBREVIATIONS USED
- NTCP
sodium taurocholate cotransporting polypeptide
- ASBT
apical sodium dependent bile acid transporter
- OATP
organic anion transporting polypeptides
- HEK
human embryonic kidney
- MDCK
Madin Darby canine kidney
- SAR
structure–activity relationship
- WST
water soluble tetrazolium
- SCUT
Clinician’s Pocket Drug Reference
- CDD
Collaborative Drug Discovery database
Footnotes
Supporting information includes model performance results and principal component analysis results. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Kullak-Ublick GA, Stieger B, Meier PJ. Enterohepatic bile salt transporters in normal physiology and liver disease. Gastroenterology. 2004 Jan;126(1):322–42. doi: 10.1053/j.gastro.2003.06.005. [DOI] [PubMed] [Google Scholar]
- 2.Ho RH, Tirona RG, Leake BF, et al. Drug and bile acid transporters in rosuvastatin hepatic uptake: function, expression, and pharmacogenetics. Gastroenterology. 2006 May;130(6):1793–806. doi: 10.1053/j.gastro.2006.02.034. [DOI] [PubMed] [Google Scholar]
- 3.Choi MK, Shin HJ, Choi YL, Deng JW, Shin JG, Song IS. Differential effect of genetic variants of Na(+)-taurocholate co-transporting polypeptide (NTCP) and organic anion-transporting polypeptide 1B1 (OATP1B1) on the uptake of HMG-CoA reductase inhibitors. Xenobiotica. 2011 Jan;41(1):24–34. doi: 10.3109/00498254.2010.523736. [DOI] [PubMed] [Google Scholar]
- 4.Greupink R, Dillen L, Monshouwer M, Huisman MT, Russel FG. Interaction of fluvastatin with the liver-specific Na+-dependent taurocholate cotransporting polypeptide (NTCP) Eur J Pharm Sci. 2011 Nov 20;44(4):487–96. doi: 10.1016/j.ejps.2011.09.009. [DOI] [PubMed] [Google Scholar]
- 5.Yanni SB, Augustijns PF, Benjamin DK, Jr, et al. In vitro investigation of the hepatobiliary disposition mechanisms of the antifungal agent micafungin in humans and rats. Drug Metab Dispos. 2010 Oct;38(10):1848–56. doi: 10.1124/dmd.110.033811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hebert MF, Townsend RW, Austin S, et al. Concomitant cyclosporine and micafungin pharmacokinetics in healthy volunteers. J Clin Pharmacol. 2005 Aug;45(8):954–60. doi: 10.1177/0091270005278601. [DOI] [PubMed] [Google Scholar]
- 7.McRae MP, Lowe CM, Tian X, et al. Ritonavir, saquinavir, and efavirenz, but not nevirapine, inhibit bile acid transport in human and rat hepatocytes. J Pharmacol Exp Ther. 2006 Sep;318(3):1068–75. doi: 10.1124/jpet.106.102657. [DOI] [PubMed] [Google Scholar]
- 8.Kim RB, Leake B, Cvetkovic M, et al. Modulation by drugs of human hepatic sodium-dependent bile acid transporter (sodium taurocholate cotransporting polypeptide) activity. J Pharmacol Exp Ther. 1999 Dec;291(3):1204–9. [PubMed] [Google Scholar]
- 9.Ekins S, Johnston JS, Bahadduri P, et al. In vitro and pharmacophore-based discovery of novel hPEPT1 inhibitors. Pharm Res. 2005 Apr;22(4):512–7. doi: 10.1007/s11095-005-2505-y. [DOI] [PubMed] [Google Scholar]
- 10.Chang C, Bahadduri PM, Polli JE, Swaan PW, Ekins S. Rapid identification of P-glycoprotein substrates and inhibitors. Drug Metab Dispos. 2006 Dec;34(12):1976–84. doi: 10.1124/dmd.106.012351. [DOI] [PubMed] [Google Scholar]
- 11.Chang C, Ekins S, Bahadduri P, Swaan PW. Pharmacophore-based discovery of ligands for drug transporters. Adv Drug Deliv Rev. 2006 Nov 30;58(12–13):1431–50. doi: 10.1016/j.addr.2006.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Diao L, Ekins S, Polli JE. Novel inhibitors of human organic cation/carnitine transporter (hOCTN2) via computational modeling and in vitro testing. Pharm Res. 2009 Aug;26(8):1890–900. doi: 10.1007/s11095-009-9905-3. Epub 2009 May 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Astorga B, Ekins S, Morales M, Wright SH. Molecular determinants of ligand selectivity for the human multidrug and toxin extruder proteins MATE1 and MATE2-K. J Pharmacol Exp Ther. 2012 Jun;341(3):743–55. doi: 10.1124/jpet.112.191577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dawson PA, Lan T, Rao A. Bile acid transporters. J Lipid Res. 2009 Dec;50(12):2340–57. doi: 10.1194/jlr.R900012-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kramer W, Stengelin S, Baringhaus KH, et al. Substrate specificity of the ileal and the hepatic Na(+)/bile acid cotransporters of the rabbit I. Transport studies with membrane vesicles and cell lines expressing the cloned transporters. J Lipid Res. 1999 Sep;40(9):1604–17. [PubMed] [Google Scholar]
- 16.Balakrishnan A, Sussman DJ, Polli JE. Development of stably transfected monolayer overexpressing the human apical sodium-dependent bile acid transporter (hASBT) Pharm Res. 2005 Aug;22(8):1269–80. doi: 10.1007/s11095-005-5274-8. [DOI] [PubMed] [Google Scholar]
- 17.Balakrishnan A, Wring SA, Polli JE. Interaction of native bile acids with human apical sodium-dependent bile acid transporter (hASBT): influence of steroidal hydroxylation pattern and C-24 conjugation. Pharm Res. 2006 Jul;23(7):1451–9. doi: 10.1007/s11095-006-0219-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Leonhardt M, Keiser M, Oswald S, et al. Hepatic uptake of the magnetic resonance imaging contrast agent Gd-EOB-DTPA: role of human organic anion transporters. Drug Metab Dispos. 2010 Jul;38(7):1024–8. doi: 10.1124/dmd.110.032862. [DOI] [PubMed] [Google Scholar]
- 19.Rais R, Gonzalez PM, Zheng X, et al. Method to screen substrates of apical sodium-dependent bile acid transporter. AAPS J. 2008 Dec;10(4):596–605. doi: 10.1208/s12248-008-9069-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zheng X, Diao L, Ekins S, Polli JE. Why we should be vigilant: drug cytotoxicity observed with in vitro transporter inhibition studies. Biochem Pharmacol. 2010 Oct 1;80(7):1087–92. doi: 10.1016/j.bcp.2010.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li J, Ehlers T, Sutter J, et al. CAESAR: a new conformer generation algorithm based on recursive buildup and local rotational symmetry consideration. J Chem Inf Model. 2007 Sep-Oct;47(5):1923–32. doi: 10.1021/ci700136x. [DOI] [PubMed] [Google Scholar]
- 22.Ekins S, Williams AJ, Xu JJ. A predictive ligand-based Bayesian model for human drug-induced liver injury. Drug Metab Dispos. 2010 Dec;38(12):2302–8. doi: 10.1124/dmd.110.035113. [DOI] [PubMed] [Google Scholar]
- 23.Zheng X, Polli J. Identification of inhibitor concentrations to efficiently screen and measure inhibition Ki values against solute carrier transporters. Eur J Pharm Sci. 2010 Sep 11;41(1):43–52. doi: 10.1016/j.ejps.2010.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gao F, Johnson DL, Ekins S, et al. Optimizing higher throughput methods to assess drug-drug interactions for CYP1A2, CYP2C9, CYP2C19, CYP2D6, rCYP2D6, and CYP3A4 in vitro using a single point IC(50) J Biomol Screen. 2002 Aug;7(4):373–82. doi: 10.1177/108705710200700410. [DOI] [PubMed] [Google Scholar]
- 25.Ekins S, Mirny L, Schuetz EG. A ligand-based approach to understanding selectivity of nuclear hormone receptors PXR, CAR, FXR, LXRalpha, and LXRbeta. Pharm Res. 2002 Dec;19(12):1788–800. doi: 10.1023/a:1021429105173. [DOI] [PubMed] [Google Scholar]
- 26.Greupink R, Nabuurs S, Zarzycka B, et al. In silico identification of potential cholestasis-inducing agents via modeling of Na+-dependent Taurocholate Cotransporting Polypeptide (NTCP) substrate specificity. Toxicol Sci. 2012 May 28; doi: 10.1093/toxsci/kfs188. [DOI] [PubMed] [Google Scholar]
- 27.Kolhatkar V, Polli JE. Reliability of inhibition models to correctly identify type of inhibition. Pharm Res. 2010 Nov;27(11):2433–45. doi: 10.1007/s11095-010-0236-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Simonson SG, Raza A, Martin PD, et al. Rosuvastatin pharmacokinetics in heart transplant recipients administered an antirejection regimen including cyclosporine. Clin Pharmacol Ther. 2004 Aug;76(2):167–77. doi: 10.1016/j.clpt.2004.03.010. [DOI] [PubMed] [Google Scholar]
- 29.Campbell SD, de Morais SM, Xu JJ. Inhibition of human organic anion transporting polypeptide OATP. 1B1 as a mechanism of drug-induced hyperbilirubinemia. Chem Biol Interact. 2004 Nov 20;150(2):179–87. doi: 10.1016/j.cbi.2004.08.008. [DOI] [PubMed] [Google Scholar]
- 30.Carter NJ, Keating GM. Micafungin: a review of its use in the prophylaxis and treatment of invasive Candida infections in pediatric patients. Paediatr Drugs. 2009;11(4):271–91. doi: 10.2165/00148581-200911040-00006. [DOI] [PubMed] [Google Scholar]
- 31.Zheng X, Ekins S, Raufman JP, et al. Computational models for drug inhibition of the human apical sodium-dependent bile acid transporter. Mol Pharm. 2009 Sep-Oct;6(5):1591–603. doi: 10.1021/mp900163d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kolhatkar V, Polli JE. Structural requirements of bile acid transporters: C-3 and C-7 modifications of steroidal hydroxyl groups. Eur J Pharm Sci. 2012 May 12;46(1–2):86–99. doi: 10.1016/j.ejps.2012.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Leslie EM, Watkins PB, Kim RB, Brouwer KL. Differential inhibition of rat and human Na+-dependent taurocholate cotransporting polypeptide (NTCP/SLC10A1) by bosentan: a mechanism for species differences in hepatotoxicity. J Pharmacol Exp Ther. 2007 Jun;321(3):1170–8. doi: 10.1124/jpet.106.119073. [DOI] [PubMed] [Google Scholar]
- 34.Rais R, Acharya C, Tririya G, et al. Molecular Switch Controlling the Binding of Anionic Bile Acid Conjugates to Human Apical Sodium-Dependent Bile Acid Transporter. J Med Chem. 2010 Jun 24;53(12):4749–60. doi: 10.1021/jm1003683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zheng X, Pan Y, Acharya C, et al. Structural requirements of the ASBT by 3D-QSAR analysis using aminopyridine conjugates of chenodeoxycholic acid. Bioconjug Chem. 2010 doi: 10.1021/bc100273w. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.