

Abstract
Proteases are one of the most important and historically utilized classes of drug targets. To effectively interrogate this class of proteins, which encodes nearly 2% of the human proteome, it is necessary to develop effective and cost-efficient methods that report on their activity both in vitro and in vivo. We have developed a robust reporter of caspase proteolytic activity, called caspase-activatable green fluorescent protein (CA-GFP). The caspases play central roles in homeostatic regulation, as they execute programmed cell death, and in drug design, as caspases are involved in diseases ranging from cancer to neurodegeneration. CA-GFP is a genetically encoded dark-to-bright fluorescent reporter of caspase activity in in vitro, cell-based, and animal systems. Based on the CA-GFP platform, we developed reporters that can discriminate the activities of caspase-6 and -7, two highly related proteases. A second series of reporters, activated by human rhinovirus 3C protease, demonstrated that we could alter the specificity of the reporter by reengineering the protease recognition sequence. Finally, we took advantage of the spectrum of known fluorescent proteins to generate green, yellow, cyan, and red reporters, paving the way for multiplex protease monitoring.
Introduction
Proteases are at the center of several of the most critical cellular cycles and pathways and are among the most common targets for drug design (for review see (1)]. Although they have been widely studied and targeted, a full understanding of the unique biological functions of related proteases has been very difficult to achieve. Tools to monitor and catalog the unique biological roles of all cellular proteases in a spatiotemporal manner are essential to a complete understanding of proteases and the degradome they control.
The use of genetically encoded fluorescent reporters has become increasingly important in the study of proteolytic activity both in vitro and in vivo because activity can be monitored as a function of both time and location in living organisms (for review, see Turk (2)). Current technologies for studying protease activity in a native cellular context also include fluorescence resonance energy transfer, luciferase, and positron emission reporters; however, each approach comes with certain limitations to its applications. To address some of these shortcomings, we recently developed a genetically encoded, dark-to-bright reporter of caspase proteolytic activity (3). This reporter is multifunctional in that it can be used both in vitro and in vivo and in bacterial and mammalian systems to indicate the activity of apoptotic caspases. Although the fluorescent signal was shown to be significantly higher in the bacterial system, the 3-fold increase in signal over background in mammalian cells is higher than that observed for comparable genetically encoded reporters. We designed a caspase-activatable (CA) reporter by fusing green fluorescent protein (GFP; S65T) through a flexible linker containing the caspase-3 and -7 recognition sequence DEVD to a hydrophobic 27-amino-acid peptide on the C-terminus (Fig. 1). The presence of this peptide inhibits maturation of the chromophore in GFP until it is removed in the presence of an active caspase. The presence of active caspases even in healthy cell cultures does lead to higher levels of background; nevertheless, the significant difference in signal from cells undergoing apoptosis even over this background makes CA-GFP a powerful tool for observing cell death.
Figure 1.
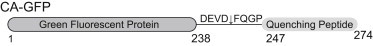
Construct of CA-GFP showing the GFP fused through the flexible, cleavable linker to the quenching peptide.
Given the functional success of CA-GFP for reporting caspase activity in global apoptosis, we sought to take advantage of this platform and develop a robust series of reporters that would enable a number of new protease reporting applications. We designed a new series of reporters to distinguish the activity of closely related caspases and found that these reporters could also be adapted to functionally monitor the activity of unrelated proteases. Initially we targeted two closely related caspases, caspase-6 and caspase-7 (both clan CD proteases), as well as the human rhinovirus-14 3C (hRV3C) protease (clan CB).
The caspases are central to the critical process of apoptosis or programmed cell death, but some caspases are also involved in inflammation and other biological processes. Thus, there has been significant interest in harnessing the cell-killing power of apoptotic proteases for applications such as treatment of cancer. If any one caspase or subset of caspases is to be effectively targeted therapeutically without unwanted effects, it is essential to ensure that the unique roles of that particular caspase have been unambiguously uncovered. Up to now, it has been impossible to fully distinguish the location and activity of each individual caspase; however, this information potentially can be collected using the genetically encoded reporters we describe here. When the apoptotic cascade is activated, initiator caspases (caspase-8 and -9) cleave the zymogen executioner caspases (caspase-3, -6, and -7) to form active proteases. Activation of the executioners is the last irreversible step in the apoptotic cascade, ultimately leading to cell death. Caspase-6 was recently suggested to play casual roles in both Alzheimer’s and other neurodegerative diseases. Thus it appears that although caspases-3, -6, and -7 play overlapping roles in apoptosis, it is their unique roles (e.g., caspase-6’s role in neurodegeneration) that may be of the greatest therapeutic interest. Tools that can distinguish the roles of individual related proteases are therefore of great importance.
Viral proteases are important drug targets as well, but their function must be mapped independently of any cellular proteases before they can be used therapeutically. Human rhinovirus is the major viral agent in the common cold. Currently there are no effective drugs to kill this virus, and patients only receive symptomatic treatments as the body fights the infection. The replication and maturation of the human rhinovirus requires the function of the 3C protease (hRV3C), which therefore represents a major drug target (6).
Although many biologically important proteolytic processes occur as part of a propagating proteolytic chain, to date, few robust technologies exist that allow simultaneous monitoring of the activity of multiple proteolytic events. An ideal system for monitoring proteolytic behavior would be one that could be adapted to any protease of interest and could be made in any color to allow for simultaneous monitoring of various members of a proteolytic cascade. In this work, we demonstrate the steps involved in engineering CA-GFP to be recognized by both homologous proteases and distally related proteases. We also describe our ability to change the color of the fluorescent reporter. These capabilities position this technology uniquely to fill a substantial void in protease profiling, particularly in whole, intact organisms.
Materials and Methods
Molecular cloning
Caspase-6 and hRV3C protease reporters
CA-GFP in pBB75 contains a DEVDFQGP linker as previously described (3). All of the caspase-6 and hRV3C protease reporter variations, unless otherwise specified, are based on this construct. We mutated the DEVDF sequence in CA-GFP to corresponding sequences by site-directed mutagenesis using overlapping inverted primers and amplification of the entire plasmid, in a manner similar to the QuikChange (Agilent Technologies Inc., Santa Clara, CA) approach. In all constructs denoted as GFPe (Table 1), we introduced an additional D27E mutation by site-directed mutagenesis to eliminate the internal caspase-6 recognition site (VELDG, residues 24–28; Fig. 2 A) in GFP.
Table 1.
GFP-Based Protease Reporters
| Version of GFP reporter | Linker sequence | Intended protease | Additional mutations |
|---|---|---|---|
| CA-GFP | DEVD↓FQGP | caspase-7 | |
| C7A-GFPe or DEVDF-GFPe | DEVD↓FQGP | caspase-7 | D27E |
| VEIDF-GFP | VEID↓FQGP | caspase-6 | |
| VEIDF-GFPe | VEID↓FQGP | caspase-6 | D27E |
| C6A-GFPe or VEIDG-GFPe | VEID↓GQGP | caspase-6 | D27E |
| VEIDE-GFPe | VEID↓EQGP | caspase-6 | D27E |
| VEIDK-GFPe | VEID↓KQGP | caspase-6 | D27E |
| LEVLF-GFP | LEVLFQ↓GP | hRV3C | |
| LEVLG-GFP | LEVLGQ↓GP | hRV3C | |
| DEVLF-GFP | DEVLFQ↓GP | hRV3C | |
| DEVLG-GFP | DEVLGQ↓GP | hRV3C | |
| EEVLF-GFP | EEVLFQ↓GP | hRV3C | |
| DEVEF-GFP | DEVEFQ↓GP | hRV3C | |
| DEVEG-GFP | DEVEGQ↓GP | hRV3C | |
| DEVCF-GFP | DEVCFQ↓GP | hRV3C | |
| DEVSF-GFP | DEVSFQ↓GP | hRV3C | |
| VEIDF-GFP | VEIDFQ↓GP | hRV3C | |
| PEHDF-GFP | PEHDFQ↓GP | hRV3C | |
| LEHDF-GFP | LEHDFQ↓GP | hRV3C | |
| LEHDG-GFP | LEHDFQ↓GP | hRV3C | |
| HRA-ΔYKGFP | LEVLFQ↓GP | hRV3C | Y273, K274 deleted |
| DEVD∗ | DEVD∗ | ||
| DEVDFQ∗ | DEVDFQ∗ | ||
| LEVL∗ | LEVL∗ | ||
| LEVLFQ∗ | LEVLFQ∗ | ||
| DEVL∗ | DEVL∗ | ||
| DEVLFQ∗ | DEVLFQ∗ | ||
| EEVLFQ∗ | EEVLFQ∗ | ||
| EEVEFQ∗ | EEVEFQ∗ | ||
| VEID∗ | VEID∗ | ||
| CA-CerFP | DEVD↓FQGP | caspase-7 | F64L, Y66W, S72A, Y145A, H148D |
| CA-CitFP | DEVD↓FQGP | caspase-7 | T65G, V68L, Q69M, S72A, T203Y |
| CA-mNeptune | DEVD↓FQGP | caspase-7 | Quenching peptide on the N-terminus of mNeptune fluorescent protein |
The caspase reporters were designed to have an overlapping hRV3C cleavage site as a secondary control for reporter function. The nomenclature GFPe indicates mutation of D27E to remove the caspase-6 cleavage at the N-terminus of GFP; ↓ indicates a cleavage site, ∗ indicates a stop codon.
Figure 2.
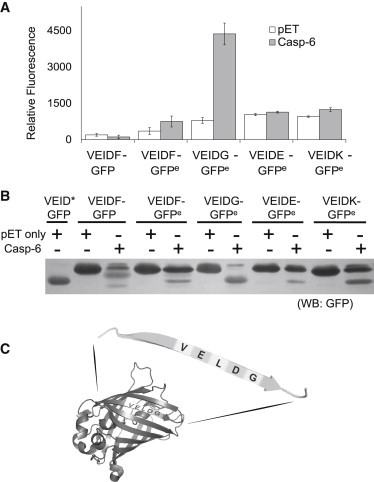
Engineering CA-GFP into C6A-GFP. (A) Comparison of the whole-cell GFP fluorescence of various C6A-GFP constructs. C6A-GFP constructs cotransformed into E. coli with either active caspase-6 (gray) or empty pET vector as control (white). The nomenclature GFPe indicates the mutation of D27E to remove the caspase-6 cleavage at the N-terminus of GFP. Whole-cell GFP fluorescence was measured. (B) Western blot of various C6A-GFP constructs against GFP antibody. (C) GFP contains a caspase-6 recognizable site (VELD↓G) at residue 27 near the N-terminus.
The CA-CerFP construct was developed by replacing the codons for the following residues in CA-GFP: F64L, S72A, Y66W, Y145A, and H148D. These substitutions were made through multiple rounds of single-point mutagenesis using a modified QuikChange (Agilent) protocol with CA-GFP in pET21b as the template plasmid. The CA-CitFP construct was also developed using CA-GFP in pET21b as the template plasmid for replacing resides T65G, V68L, Q69M, S72A, and T203Y through multiple rounds of modified QuikChange mutagenesis.
The CA-mNeptune expression construct was generated by amplifying the peptide (M2) region from the CA-GFP gene separately from the mNeptune gene using two primers to amplify M2 and two primers to amplify mNeptune. The mNeptune parent was generated by replacing the codons for M41G, A45V, S61C, A158C, and Y197F in the mKate2 gene in the pmKate2C vector (Evrogen Joing Stock Company, Moscow, Russia). The first primer (P1) annealed to the N-terminal region of the mNeptune gene with the linker sequence included N-terminal to the mNeptune gene. The second primer (P2) annealed to the C-terminus of mNeptune and included a stop codon (UAA) and the restriction site for XhoI. The third primer (P3) included an NheI restriction enzyme site as well as a 6His sequence and annealed to the N-terminal region of the M2 portion of the CA-GFP gene. The last primer (P4) annealed to the C-terminus of M2 and included the same sequence for the linker as P1, giving primers P1 and P4 a 24 bp overlapping region. The mNeptune fragment was then amplified using primers P1 and P2, and the M2 portion was amplified using primers P3 and P4. After gel purification of the amplified fragments, they were combined and allowed to anneal through the overlapping region for five PCR cycles before the addition of primers P2 and P3, which then amplified the full-length gene. The gene was then ligated into pACYCDuet vector in the NheI and XhoI sites. The final sequence of the N-terminal peptide and linker, MHHHHHHMCNDSSDPLVVAASIIGILHLILWILDRLDEVDFQGP, was appended to the N-terminus of mNeptune. The full-length construct was then further amplified using PCR and ligated into the EcoRI and NotI sites of pET21b.
Caspase constructs
A constitutive two-chain expression system for caspase-6 as described in Vaidya et al. (7) was used for all coexpressions with active caspase-6. The expression construct consists of an Escherichia coli optimized codon sequence of the full-length caspase-6 in the pET11a vector. The gene is separated by a stop codon, the ribosome-binding site TATACATATG, and a start codon inserted after position 179. This allows for homogeneous expression of active caspase-6. A similar construct was developed for caspase-7, as reported in Witkowski and Hardy (8), in which the caspase-7 gene contained in the plasmid pET23b is separated by a stop codon, ribosome-binding site, and start codon inserted after residue 198. This caspase-7 constitutive two-chain sequence was then amplified by PCR and ligated into the NdeI and SacI restriction enzyme sites of pBB75. The inactive caspase-7 (C186A) expression construct consists of the full-length human caspase-7 gene with the single-point mutation at the active site in the pET23b vector. This gene was also amplified via PCR and ligated into the NdeI and SacI restriction enzyme sites of pBB75.
Fluorescence assays in whole cells
Expression constructs for caspase-6 or hRV3C protease reporters contained in the pBB75 vector (kanamycin (Kan)) were cotransformed into the BL21(DE3) strain of E. coli with either an active constitutively two-chain version of caspase-6 (7) in pET11b (ampicillin (Amp)), an hRV3C protease in GEX-vector (Amp; a gift from M. Romanowski), or an empty pET21b (Amp) vector as a control. Autoinduction media cultures (2 mL) were inoculated from a dense 100 μL overnight lysogeny broth (LB; Amp/Kan) culture with 100 μg/mL Amp and 40 μg/mL Kan, and incubated until they reached an OD600 of 0.6–0.9. The cultures were then incubated for 18 h at 16°C. Cells from each sample were rinsed with PBS, resuspended, and diluted in PBS to a final OD600 of 2.0. Then 200 μL of this diluted suspension was measured for GFP fluorescence (excitation (Ex.) 475/emission (Em.) 512) in a costar 96-well black plate on a Spectramax M5 spectrophotometer (Molecular Devices).
On-plate fluorescence assays
Protease reporters in pBB75 vector (Kan resistant) were cotransformed with the corresponding protease constructs in pET vectors (Amp resistant) and incubated in a 2 mL LB culture overnight. Then 0.5 μL of this culture was inoculated onto a nitrocellulose membrane. The membrane was first incubated on an LB agar plate containing both Amp and Kan overnight at 37°C. The nitrocellulose membrane was then transferred onto a LB agar induction plate with 100 μg/ml Amp, 40 μg/ml Kan, and 500 μM isopropylthio-β-galactoside (IPTG), and incubated for 6 h at 25°C and 24 h at 4°C (9). The result was recorded using an InGenius (SynGene, Cambridge, United Kingdom) gel dock under long-wavelength UV light, and a white illumination picture was also taken as a control.
Western blotting
After the whole-cell fluorescence analysis described above was completed, samples of the E. coli cell suspension were run on an SDS-PAGE gel. The gel was then transferred to a PVDF membrane (Millipore, Billerica, MA) and blotted with an anti-GFP (Millipore) monoclonal mouse primary antibody. The Western blots were then treated with anti-mouse IgG alkaline phosphatase produced in goat (Sigma-Aldrich, St. Louis, MO) and visualized using 1-Step NBT/BCIP (Thermo Fisher Scientific Inc., Waltham, MA).
Fluorescence assays in cell lysates
Expression constructs for the fluorescent protein reporters CA-GFP, CA-CerFP, CA-CitFP, and CA-mNeptune (each in the pET21b vector, which also contains an Amp resistance gene) were cotransformed via electroporation in the BL21(DE3) strain E. coli with expression constructs for an active constitutively two-chain version of caspase-7 or a full-length inactive version of caspase-7, in which the active-site cysteine had been mutated to alanine (C186A) in the vector pBB75, which also contains a Kan resistance gene. Then 50 mL LB cultures supplemented with 100 μg/mL Amp and 40 μg/mL Kan were inoculated with 50 μL from a dense 5 mL overnight culture and incubated until reaching an OD600 of 0.6. The cultures were then induced with 1 mM IPTG for 18 h at 25°C. From each sample, 800 μL aliquots were taken in duplicate, pelleted by centrifugation, and resuspended in a solution of 0.5 mg/mL lysozyme and 2 U DNAase. After lysis with four cycles of freeze-thaw, the supernatant of each sample was measured for fluorescence. CA-GFP (Ex. 475/Em. 512 nm), CA-CerFP (Ex. 433/Em. 475 nm), and CA-mNeptune (Ex. 580/Em. 650 nm) fluorescence measurements were taken in a costar 96-well black plate on a Molecular Devices Spectramax M5 spectrophotometer. CA-CitFP (Ex. 515/Em. 529 nm) fluorescence was measured on a JASCO FP-6500 spectrofluorometer using a quartz cell. Although the excitation and emission wavelengths used are not identical to published values, they were determined to be the optimum wavelengths for measurements using the instruments indicated. Fluorescence values were normalized to the total number of cells in culture, which was estimated from the relative A600 of each culture.
Results and Discussion
We have previously shown that CA-GFP is a robust, genetically encoded reporter of caspase activity in apoptosis (3). Here we sought to leverage the desirable attributes of CA-GFP as a tunable platform to build a family of protease activatable reporters that could distinguish the activities of various proteases simultaneously.
Development of a caspase-6-activatable GFP
Until very recently, caspase-6 was viewed as being very structurally and functionally similar to other executioner caspases, including caspase-3 and -7. However, more recent studies have shown that caspase-6 not only populates structurally unique conformations (7,10,11) but also may have unique nonapoptotic roles in neurodegenerative diseases (see 4, and see 5). To harness the unique capabilities of these caspases effectively, it is essential to assess and distinguish their individual activities in a spatiotemporal manner. CA-GFP has been shown to be a robust in vivo reporter for apoptotic caspases both in E. coli and in mammalian cells (3). Therefore, we thought that if we could adapt CA-GFP to distinguish caspase-6 activity from caspase-7 activity, we would be able to further understand and better harness the unique therapeutic potential of these caspases.
To build a caspase-6-activatable-GFP reporter (C6A-GFP), we replaced the DEVD linker sequence in CA-GFP with the caspase-6 recognition sequence VEID (VEIDF-GFP; Table 1). The resulting construct was dark (nonfluorescent) before cleavage by caspase-6, indicating that the quenching peptide functioned as expected. However, when VEIDF-GFP was cotransformed with active caspase-6 in E. coli, there was no observable fluorescence (Fig. 2 A). Although we had previously shown that mature GFP is resistant to cleavage by caspase-6 in vitro (12), Western blot analysis using an anti-GFP antibody showed a predominant cleavage event that was not at the expected linker cleavage site (Fig. 2 B). We compared the cleavage pattern observed with that of a construct in which a stop codon was inserted after the VEID in the linker (VEID∗; Table 1). We found that one band resulted from cleavage of the VELD↓G sequence at residue 27 near the N-terminus of GFP (Fig. 2 C), whereas the other band was the result of a double cleavage at both the N-terminal VELD↓G site and linker VEID↓F site. Interestingly, no protein band with a migration pattern similar to that of VEID∗, corresponding to a single cleavage solely at the VEID↓F linker, was observed. This indicates that caspase-6 cleaves the N-terminal VELD↓G site before cleaving the VEID↓F linker. GFP is very sensitive to truncations at either terminus (13), so it was not surprising that no fluorescence was observed when the VELD↓G was cleaved. Interestingly, we did not observe cleavage of the N-terminal site at residue 27 when apoptosis was induced in mammalian cells (3). This is likely due to the robust activity of caspase-3 and -7 relative to the much lower level of caspase-6 during staurosporine-induced apoptosis.
To remove this inactivating cleavage site, Asp-27 was substituted by Glu (D27E). The resulting VEIDF-GFPe was also dark before cleavage occurred. When cotransformed with active caspase-6, VEIDF-GFPe showed a small fluorescence increase (Fig. 2 A). Western blotting with an anti-GFP antibody showed that VEIDF-GFPe was not efficiently cleaved in the experimental conditions (Fig. 2 B). In prior VEIDF-GFP/caspase-6 coexpression experiments, the caspase-6 cleavage of the VEID↓F linker was much less efficient than cleavage of the N-terminal VELD↓G. The fact that we did not observe any bands representing a single cleavage at the VEID↓F linker strongly suggested that caspase-6 cleaves the N-terminal VELD↓G site first and that caspase-6 prefers a glycine at the P1′ subsite in our reporter construct. This observation reflects prior observations made against peptide substrates in which caspase-6 showed a preference for Gly at P1′ (14). In light of this, we modified the VEID↓F linker in VEIDF-GFPe into VEID↓G. The resulting VEIDG-GFPe reporter yielded a much stronger fluorescent signal increase upon coexpression with active caspase-6 (Fig. 2 B). When we coexpressed VEIDG-GFPe with an empty pET vector, we found that the replacement of Phe with Gly led to the increase of the basal fluorescent level in the reporter. Therefore, we also explored other options for P1′ substitution. Residues including Glu and Lys are frequently found in the P1′ subsite of caspase-6 protein substrates, but not in caspase-3 and -7 substrates (15). We hypothesized that substitution of the Phe in the P1′ site with either of these residues would improve the reporter specificity toward caspase-6 over caspase-7. Introducing Glu or Lys at the P1′ neither improved the signal strength nor decreased the basal fluorescence of the reporter (Fig. 2 B). Therefore, although P1′ Gly increases the basal fluorescent level in our reporter coexpressed with an empty pET vector, VEIDG-GFPe is still the best caspase-6 reporter overall due to the strong increase in signal in the presence of caspase-6 activity, and we will refer to it from this point on as C6A-GFPe.
C6A-GFPe and CA-GFP distinguish caspase-6 and -7 activities
Although all caspases have very similar catalytic dyad geometries during substrate cleavage, they each show unique cleavage site preferences due to the composition of residues that define the peptide recognition subsites (S1–S4) (15). The classic substrate recognition sequence for caspase-6 is VEID, and that for caspase-7 is DEVD. Here we applied caspase-6 and -7 to CA-GFP (DEVDF-GFP), C7A-GFPe (DEVDF-GFPe), and C6A-GFPe (VEIDG-GFPe) to assess the responsiveness of the reporters (Fig. 3 A). Western blot analysis using an anti-GFP antibody showed that caspase-7 does not efficiently cleave the VEID recognition sequence in C6A-GFPe (Fig. 3 B). Thus, it appears that C6A-GFPe is a reasonably good reporter for caspase-6 activity because caspase-6 triggers a 3- to 4-fold greater response than caspase-7. As negative controls for this system, we developed reporters such as DEVLF-GFP, which is activated by an orthogonal protease (hRV3C) but is not activated by caspase-6 or -7 (Fig. S1 in the Supporting Material).
Figure 3.
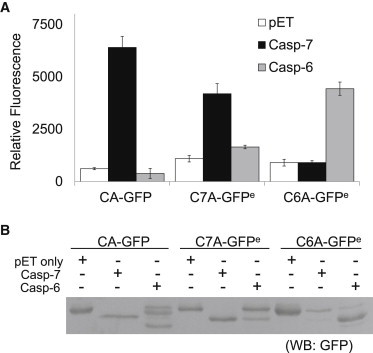
CA-GFP, C7A-GFPe, and C6A-GFPe distinguish caspase-6 and caspase-7 activity. The CA-GFP, C7A-GFPe, and C6A-GFPe reporters were cotransformed into E. coli with empty pET vector (white), active caspase-7 (black), and active caspase-6 (gray). (A) Whole-cell fluorescence measurement of reporters. (B) Western blot analysis is shown using anti-GFP antibody to determine the cleavage efficiency of the reporters.
In the presence of caspase-7, CA-GFP clearly showed a more pronounced fluorescent increase than C7A-GFPe (Fig. 3 A). We should note that under our previously reported conditions, CA-GFP was coexpressed with caspase-7 in LB media at 25°C, which led to a 45-fold fluorescent signal increase (3). To consistently coexpress different proteases and GFP-reporters in E. coli at the same time, here we found it necessary to optimize the expression conditions for all components. In this work, we performed expression at 16°C in autoinduction media. We observed similar intensity from the positive signals, though at this lower temperature the background fluorescence level increased, which led to a slightly lower increase in fluorescence (∼10-fold here). The D27E mutation in C7A-GFPe apparently affects the signal strength of the reporter. Although CA-GFP and C6A-GFPe are most responsive to their cognate enzymes, caspase-6 cleaves a fraction of CA-GFP and C7A-GFPe at the linker, and caspase-7 also cleaves a small fraction of the C6A-GFPe at the linker (Fig. 3 B). This is as expected, because caspases are known to be less selective toward the P2 and P4 substrate subsite residues. It is worth noting that in the case of coexpression of caspase-6 and CA-GFP, the fluorescence is much lower compared with that observed for coexpression with an empty pET vector. This is because removal of the N-terminal 27 residues in CA-GFP by caspase-6 prevents the fluorescence of GFP. We note that this property can be put to use to increase the stringency and selectivity of caspase reporters. Caspase-7 cleaves CA-GFP at the linker, increasing fluorescence, whereas caspase-6 cleaves at residue 27, which prevents fluorescence. This dual recognition could prove useful for identifying the ratio of caspase-6 to caspase-7 activity. This characteristic could be extraordinarily useful in protease engineering, where the ratiometric dual recognition could serve as a built-in counterscreen, dramatically aiding the recovery of proteins with the desired phenotype. We could also apply both the CA-GFP and C6A-GFPe in this case, where a lowering of signal from the C7A-GFPe and an elevated C6A-GFPe signal would increase the detectability of caspase-6 activity. Thus, in addition to their ability to robustly distinguish caspase-6 from caspase-7 activity, these reporters also have other, more nuanced applications.
The CA-GFP family of reporters has exciting applications in mammalian cells and whole organisms, as we have shown previously (3). Apoptotic executioner caspases are capable of functionally substituting for one another in knockout and knockdown conditions. This property complicates our ability to characterize the activity of caspase-6 and -7 in mammalian cells, which is why we chose to work in the clean background afforded by bacteria heterologously expressing capases. Other investigators and we are actively pursuing the development of small-molecule inhibitors that are specific for caspase-6 or caspase-7. We look forward to a time, hopefully in the near future, when small-molecule inhibitors with sufficient specificity for caspase-6 and -7 are available to allow accurate benchmarking of C6A-GFP and C7A-GFP in mammalian systems.
Engineering new proteolytic recognition: hRV3C
By successfully engineering a caspase-6 reporter, we were able to gain insight into how the CA-GFP concept could be adapted to report on other biomedically important proteases. We next introduced the hRV3C protease recognition sequence into the linker to generate a reporter for hRV3C (Fig. 4 A). hRV3C has a preferred substrate sequence of LEVLFQGP (16). Our initial design changed the DEVDFQGP sequence in CA-GFP to LEVLFQGP, and thus the sequences of the CA-GFP and LEVLF-GFP reporters are identical except for the substitution of two aspartates (D) for the two leucines (L) in the linker. This LEVLF-GFP reporter showed a moderate (8-fold) increase in fluorescence upon hRV3C protease coexpression compared with expression with an empty vector or with caspase-7, which, as expected, was unable to cleave this linker (Fig. 4 B). Although the fluorescence in the cleaved hRV3C reporter is significantly higher than background, the fluorescence increase is much lower than that observed for CA-GFP coexpressed with caspase-7. The overall fluorescence after cleavage is also very low in comparison with CA-GFP or C6A-GFPe. We found that this was not due to a difference in reporter expression levels or to the completeness of the protease digestion (data not shown).
Figure 4.
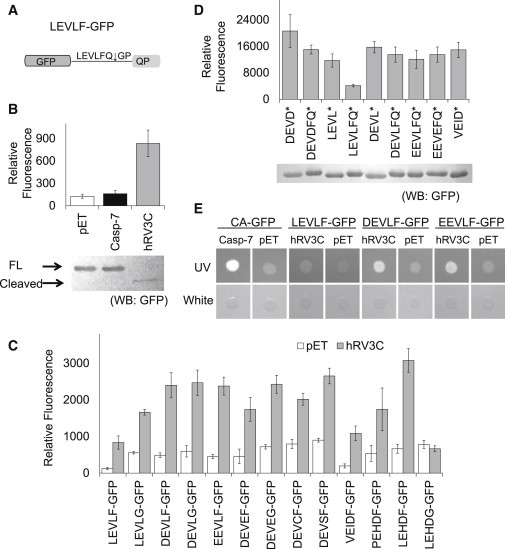
Engineering CA-GFP into an hRV3C protease-activatable GFP reporter. (A) Construct LEVLF-GFP is similar to CA-GFP but has a LEVLFQ↓GP linker that can be recognized by hRV3C protease. (B) Whole-cell fluorescence of LEVLF-GFP cotransformed with empty pET vector (white), active caspase-7 (black), and hRV3C protease (gray). Western blot analysis is shown using an anti-GFP antibody to determine cleavage efficiency. Thick arrow: full-length reporter; thin arrow: cleaved reporter. (C) Comparison of the whole-cell fluorescence of a spectrum of hRV3C protease-activatable GFP constructs. Each construct was cotransformed into E. coli with empty vector (white) or hRV3C protease (gray). Whole-cell fluorescence was measured. (D) Comparison of the whole-cell fluorescence of C-terminally truncated GFP reporter constructs. A stop codon (∗) was introduced into selected GFP reporters to generate GFPs with different linker remnants. These truncated reporters were cotransformed with an empty pET vector and whole-cell fluorescence was measured. Western blot analysis with anti-GFP antibody is shown for evaluation of relative expression levels. (E) Comparison of the signal strengths of CA-GFP, LEVLF-GFP, DEVLF-GFP, and EEVLF-GFP in an on-plate fluorescence assay. CA-GFP was cotransformed with active caspase-7. Other reporters were cotransformed with hRV3C protease. The fluorescence of E. coli colonies coexpressing the reporters and their corresponding proteases (or empty pET vectors as controls) was monitored under a long-wavelength UV light. White-light illumination pictures were taken as controls.
We reasoned that the difference in fluorescence after cleavage might be due to the remaining linker residues that differ from those in CA-GFP. Due to the position of the cleavage point within the recognition sequence, caspase-7 generates a DEVD C-terminus in CA-GFP, whereas hRV3C protease yields a LEVLFQ C-terminus, resulting in a C-terminal remnant that is two residues longer. We first investigated the impact of the length of the C-terminal remnant. We removed Tyr and Lys residues just before the LEVLFQ sequence (HRA-ΔYKGFP; Table 1) to yield a C-terminus of equal length to the DEVD-containing remnant in CA-GFP, and we did not observe a significant improvement in signal (data not shown). We then focused on the chemical composition of the remaining linker residues. To increase the reporter detectability while keeping its specificity against hRV3C protease, we made a spectrum of variations in the linker residues and assessed their ability to function as hRV3C reporters (Fig. 4 C). We were especially interested in the LEVLFQ linker. The two leucines represent the only sequence difference between CA-GFP and LEVLF-GFP, indicating that they play a prominent role in the diminished fluorescence of the engineered reporter. We replaced the leucines in LEVLF-GFP with negatively charged or hydrophilic residues. Replacing the two leucines indeed led to a significant increase in fluorescent signal in all of the resulting reporters (Fig. 4 C). Two of our best reporters, DEVLF-GFP and EEVLF-GFP, showed a strong increase in fluorescence upon coexpression of hRV3C protease while still maintaining a recognition site that was sufficient for robust cleavage, making them the best candidates for an hRV3C reporter. Other linker variants had either a lower signal strength or higher background in the dark state. LEHDF-GFP shows a signal/noise ratio similar to that of DEVLF-GFP or EEVLF-GFP and an even stronger signal strength; however, LEHD is a caspase-9 recognized sequence and therefore is likely to have lower global specificity than the DEVLF-GFP or EEVLF-GFP reporters.
We next sought to understand the bases of the difference in fluorescent signal of the various candidate hRV3C reporters. We hypothesized that the amount of fluorescence recovery observed could be due to the intrinsic fluorescent properties of GFP with various C-terminal remnants, as we previously showed that the ability to fold properly after removal of the quenching peptide is central to the mechanism of the CA-GFP reporter (12). To probe the influence of various C-terminal remnants, we introduced stop codons either after the glutamine in the linker (to mimic the C-terminus generated by hRV3C cleavage) or before the phenylalanine (to mimic the length of the remnant in CA-GFP cleavage by caspase-7; Fig. 4 D). The C-terminal LEVLFQ remnant strongly suppressed the fluorescent signal of GFP, likely due to the clustering of four hydrophobic residues, which may lead to aggregation and prevent GFP chromophore maturation. Thus it is clear that one important aspect in the design of CA-GFP-derived reporters is the consideration of the C-terminal remnant after cleavage. To decrease the hydrophobicity of the C-terminal remnant, we replaced phenylalanine with glycine. As we anticipated, this improved the fluorescence of the cleaved reporter, indicating that the presence of the bulky, hydrophobic phenylalanine interferes with the fluorescence recovery of GFP (Fig. 4 C). Unfortunately, replacing the phenylalanine with glycine also led to increased background fluorescence in the uncleaved state. Therefore, this substitution is not necessarily beneficial for generating a better reporter overall. Replacing phenylalanine with glycine in a different context could become productive, though, because it leads to diminished recognition by the hRV3C protease in the LEHDG-GFP reporter while retaining the recognition sequence for caspase-9 (Fig. 4 C). Thus, it is clear that one must balance specificity for the protease and optimal chemical characteristics of the resulting peptide tail when designing an optimal reporter.
From this panel of variants we sought to identify the hRV3C reporter with detection properties similar to those of CA-GFP. In an on-plate assay, E. coli colonies coexpressing DEVLF-GFP or EEVLF-GFP with hRV3C protease demonstrated clearly improved fluorescence detectability over that of the original LEVLF-GFP constructs. This level of fluorescence allows for obvious detection in on-plate screening applications of the reporter (Fig. 4 E).
Engineering various colors of reporters for multiprotease profiling
To enable simultaneous monitoring of multiple protease reporters, we took advantage of the spectrum of fluorescent proteins available. Several color-shifted versions of GFP result from only a few point mutations, so we hypothesized that the color of the fluorescent protein in CA-GFP could be shifted to cyan and yellow versions without disrupting the mechanism of the dark state. We designed CA-Cerulean (17) (CA-CerFP) and CA-Citrine (18) (CA-CitFP) versions of our reporter, both of which still contain the caspase-7 recognition sequence DEVD in the linker (Fig. 5 A). These fluorescent proteins have minimally overlapping excitation and emission wavelengths, and both have the highest quantum yield for their color class (19), giving them the most potential for the best signal/noise ratio. Both versions are in a dark state when coexpressed in E. coli with the inactive caspase-7 C186A and have a significant increase in fluorescence when coexpressed with active caspase-7 (Fig. 5 B).
Figure 5.
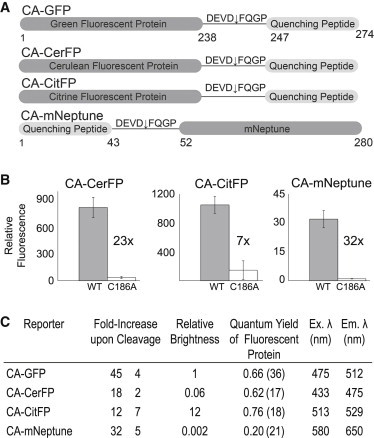
(A) Design of CA-CerFP, CA-CitFP, and CA-mNeptune. The constructs for CA-CerFP and CA-CitFP are identical to CA-GFP with the exception of the color-shifting point mutations in the GFP portion of the reporter. CA-mNeptune has the quenching peptide fused to the N-terminus of the mNeptune fluorescent protein derived from Entacmaea. (B) Increase in relative fluorescence of each reporter in response to active caspase-7 (gray) or an inactivated version of caspase-7 (C186A, white) in which the active-site cysteine nucleophile has been replaced by alanine. (C) The properties of each reporter, including the increase in fluorescence, the relative brightness (which was calculated as the fluorescence of each reporter relative to CA-GFP, with the total fluorescence yield of CA-GFP being normalized to one), the reported quantum yield (17,18,21,36), and the optimal excitation and emission wavelengths for each fluorescent protein are shown.
Red-shifted fluorescent proteins are becoming more widely used because their emission properties fall within the critical window for whole-organism imaging due to the deep-tissue penetrance for red wavelengths of light and the absence of background autofluorescence in tissues in this spectral region. Red fluorescent proteins (RFPs) are derived from Entacmaea quadricolor (20). Although they have a different protein sequence compared with GFPs from Aequorea, the two families show strikingly similar protein folding. We selected mNeptune (21) for its improved brightness and spectral properties in the near-infrared region, which are increasing its popularity for in vivo imaging. In our first design of CA-mNeptune, we fused the M2 peptide through a linker identical to that used in CA-GFP to the C-terminus of mNeptune; however, this protein showed a considerable level of fluorescence before cleavage. Because we had already observed that an N-terminal fusion of M2 to GFP resulted in a dark state before cleavage (nCA-GFP (12)), we reasoned that mNeptune might be similarly quenched from the N-terminus. The resulting N-terminal fusion, CA-mNeptune, has a very low fluorescent background. Therefore, although the overall fluorescence intensity is low, cleavage of CA-mNeptune by active caspase-7 results in a >30-fold increase in signal in E. coli (Fig. 5 B), demonstrating that the quenching peptide works on other nonGFP-derived fluorescent proteins and at various locations.
All three reporters can clearly be activated and show an improved signal/noise ratio relative to that of comparable reporters (22–30). Nevertheless, the CA-mNeptune, CA-CerFP, and CA-CitFP reporters showed a lower overall fluorescence intensity and a lower relative increase in fluorescence over the background than CA-GFP (Fig. 5 C). We hypothesize that this is due not only to differences in the published quantum yields of the mNeptune (21), Cerulean (17), and Citrine (18) variants but also to differences in the folding kinetics of the different fluorescent proteins (12,31,32).
The four fluorescent proteins described here—GFP, Cerulean, Citrine, and mNeptune—constitute a panel of spectrally distinct fluorescent proteins that have been individually engineered for improved brightness in their respective families of color (cyan, yellow, and red). Each can be used individually to monitor specific proteolytic events or together to watch several events using multichannel imaging. CA-mNeptune in particular has the potential for effective use in whole-animal models, because mNeptune’s fluorescence falls in the near-infrared window that minimizes background from water and hemoglobin in tissue. The fact that the quenching peptide used in CA-GFP and its derivatives can effectively quench fluorescence in GFP, Cerulean, Citrine, and mNeptune suggests that this type of reporter may be feasible with other even more distantly related fluorescent proteins, such as IRFP (33).
Conclusions
CA-GFP has proved to be a useful and effective reporter of caspase activity and apoptosis in a variety of systems. We have now shown that CA-GFP can be engineered to change the protease specificity and the color of fluorescence. With this expansion of the color palette, these new reporters can ultimately be used in multiplex microscopy, fluorescence-activated cell sorting, and other applications that require the detection of protease activity changes. Although challenges remain in the effort to engineer optimal reporters for additional enzymes, in this work we have demonstrated a series of engineering steps by which new reporters can be developed. One challenge for any protease reporter is to achieve specificity for one protease over other related proteases. Of course, the specificity of any synthetic reporter is limited by the inherent properties of the parent protease it is designed to monitor. The most important factor in the design is the unique characteristics of the protease. One must consider all potential processing sites, as well as the propensity of the protease to cleave in specific secondary structure conformations, such as loops. The ability of multiple proteases to cleave a similar substrate is also a major limitation; for example, LEHDF-GFP, which can be cleaved by hRV3C protease, is also the substrate for caspase-9. Potentially, one could ameliorate this overlapping specificity by using small-molecule inhibitors for one class of proteases while fluorescently monitoring the activity of the desired protein.
To identify the activity of a specific caspase independently of other caspases, it is imperative to ensure that the reporter only responds effectively to the selected protease. We may be able to use the ability of caspases to cleave secondary sites (as we found in caspase-6) to our advantage if we can design a reporter that is activated in the presence of a particular caspase but is inactivated by secondary cleavage if multiple caspases are active. This dual recognition, combined with multicolor reporting, could be applied to increase the detectability of a specific caspase, such as caspase-6, in both apoptotic and nonapoptotic events. When caspase-6 is active, it will increase the fluorescence of the caspase-6 reporter and at the same time repress the signal from a caspase-7 reporter. This reporter would also necessitate a multicolored reporter system in which multiple signals could be monitored at the same time.
We observed that, especially in the case of hRV3C protease-activatable GFP reporters, the reported optimized substrate sequence was often not the one that gave the best signal. The size and active-site conformations of some proteases, as well as the accessibility of the cleavable linker in the reporter, could all contribute to the interaction between them and affect the efficiency of the reporter. Emerging data suggest that the small peptide substrates that have been widely used to characterize protease activity may not accurately reflect the actual selectivity properties of proteases. When a protease is presented with a protein substrate, the steric interactions and kinetics are significantly different from those observed with a peptide substrate. This may prove to be advantageous for the CA-GFP family of reporters, which are protein rather than peptide substrates. Although caspase-3 and -7 share the same P1–P4 specificity, the P6, P5, P2′, and P3′ residues also contribute to caspase-7 specificity in substrates (34), for example. The small tetrapeptide substrates frequently used in caspase activity measurement and profiling might lead one to overlook the contribution of exosites. Boucher and co-workers (35) identified a caspase-7 exosite that is critical for substrate recognition and differentiation of caspase-7 substrates from caspase-3 substrates. The fact that CA-GFP is a protein-based reporter provides an advantage in designing specificity, particularly as more details about the extended recognition preferences of caspases come to light. In summary, we have demonstrated that CA-GFP is a modular platform that allows substitution of both the fluorescent and recognition modules. This modular construction allows one to tune the reporter to meet a wide range of desired specifications.
Acknowledgments
We thank Jessica Bauer, Genevieve Abbruzzese, Jun Chu, Charnell Chasten, and Richard Boehnke for preliminary work on this project. We also thank Jun Chu for construction of the CA-mNeptune gene in the pACYCDuet vector.
This work was supported by the National Institutes of Health (GM80532).
Supporting Material
References
- 1.Turk B. Targeting proteases: successes, failures and future prospects. Nat. Rev. Drug Discov. 2006;5:785–799. doi: 10.1038/nrd2092. [DOI] [PubMed] [Google Scholar]
- 2.Deu E., Verdoes M., Bogyo M. New approaches for dissecting protease functions to improve probe development and drug discovery. Nat. Struct. Mol. Biol. 2012;19:9–16. doi: 10.1038/nsmb.2203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nicholls S.B., Chu J., Hardy J.A. Mechanism of a genetically encoded dark-to-bright reporter for caspase activity. J. Biol. Chem. 2011;286:24977–24986. doi: 10.1074/jbc.M111.221648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Galvan V., Gorostiza O.F., Bredesen D.E. Reversal of Alzheimer’s-like pathology and behavior in human APP transgenic mice by mutation of Asp664. Proc. Natl. Acad. Sci. USA. 2006;103:7130–7135. doi: 10.1073/pnas.0509695103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Graham R.K., Deng Y., Hayden M.R. Cleavage at the caspase-6 site is required for neuronal dysfunction and degeneration due to mutant huntingtin. Cell. 2006;125:1179–1191. doi: 10.1016/j.cell.2006.04.026. [DOI] [PubMed] [Google Scholar]
- 6.Wanga Q.M., Chen S.-H. Human rhinovirus 3C protease as a potential target for the development of antiviral agents. Curr. Protein Pept. Sci. 2007;8:19–27. doi: 10.2174/138920307779941523. [DOI] [PubMed] [Google Scholar]
- 7.Vaidya S., Velázquez-Delgado E.M., Hardy J.A. Substrate-induced conformational changes occur in all cleaved forms of caspase-6. J. Mol. Biol. 2011;406:75–91. doi: 10.1016/j.jmb.2010.11.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Witkowski W.A., Hardy J.A. A designed redox-controlled caspase. Protein Sci. 2011;20:1421–1431. doi: 10.1002/pro.673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kim W., Hecht M.H. Mutations enhance the aggregation propensity of the Alzheimer’s A β peptide. J. Mol. Biol. 2008;377:565–574. doi: 10.1016/j.jmb.2007.12.079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Baumgartner R., Meder G., Renatus M. The crystal structure of caspase-6, a selective effector of axonal degeneration. Biochem. J. 2009;423:429–439. doi: 10.1042/BJ20090540. [DOI] [PubMed] [Google Scholar]
- 11.Wang X.J., Cao Q., Su X.D. Crystal structures of human caspase 6 reveal a new mechanism for intramolecular cleavage self-activation. EMBO Rep. 2010;11:841–847. doi: 10.1038/embor.2010.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nicholls S.B., Hardy J.A. Structural basis of fluorescence quenching in caspase activatable-GFP. Protein Sci. 2012 doi: 10.1002/pro.2188. Nov 8 [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li X., Zhang G., Huang C.C. Deletions of the Aequorea victoria green fluorescent protein define the minimal domain required for fluorescence. J. Biol. Chem. 1997;272:28545–28549. doi: 10.1074/jbc.272.45.28545. [DOI] [PubMed] [Google Scholar]
- 14.Rawlings N.D., Barrett A.J., Bateman A. MEROPS: the database of proteolytic enzymes, their substrates and inhibitors. Nucleic Acids Res. 2012;40(Database issue):D343–D350. doi: 10.1093/nar/gkr987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Stennicke H.R., Renatus M., Salvesen G.S. Internally quenched fluorescent peptide substrates disclose the subsite preferences of human caspases 1, 3, 6, 7 and 8. Biochem. J. 2000;350:563–568. [PMC free article] [PubMed] [Google Scholar]
- 16.Cordingley M.G., Callahan P.L., Colonno R.J. Substrate requirements of human rhinovirus 3C protease for peptide cleavage in vitro. J. Biol. Chem. 1990;265:9062–9065. [PubMed] [Google Scholar]
- 17.Rizzo M.A., Springer G.H., Piston D.W. An improved cyan fluorescent protein variant useful for FRET. Nat. Biotechnol. 2004;22:445–449. doi: 10.1038/nbt945. [DOI] [PubMed] [Google Scholar]
- 18.Griesbeck O., Baird G.S., Tsien R.Y. Reducing the environmental sensitivity of yellow fluorescent protein. Mechanism and applications. J. Biol. Chem. 2001;276:29188–29194. doi: 10.1074/jbc.M102815200. [DOI] [PubMed] [Google Scholar]
- 19.Shaner N.C., Steinbach P.A., Tsien R.Y. A guide to choosing fluorescent proteins. Nat. Methods. 2005;2:905–909. doi: 10.1038/nmeth819. [DOI] [PubMed] [Google Scholar]
- 20.Shcherbo D., Merzlyak E.M., Chudakov D.M. Bright far-red fluorescent protein for whole-body imaging. Nat. Methods. 2007;4:741–746. doi: 10.1038/nmeth1083. [DOI] [PubMed] [Google Scholar]
- 21.Lin M.Z., McKeown M.R., Tsien R.Y. Autofluorescent proteins with excitation in the optical window for intravital imaging in mammals. Chem. Biol. 2009;16:1169–1179. doi: 10.1016/j.chembiol.2009.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Promega. 2011. Caspase-Glo 3/7 Assay. Technical bulletin. http://www.promega.com/∼/media/Files/Resources/Protocols/Technical Bulletins/101/Caspase-Glo 3 7 Assay Protocol.pdf. Accessed February 21, 2013.
- 23.Park K., Kang H.J., Kim M. A potent reporter applicable to the monitoring of caspase-3-dependent proteolytic cleavage. J. Biotechnol. 2008;138:17–23. doi: 10.1016/j.jbiotec.2008.07.1999. [DOI] [PubMed] [Google Scholar]
- 24.Yamaguchi Y., Shinotsuka N., Miura M. Live imaging of apoptosis in a novel transgenic mouse highlights its role in neural tube closure. J. Cell Biol. 2011;195:1047–1060. doi: 10.1083/jcb.201104057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Takemoto K., Nagai T., Miura M. Spatio-temporal activation of caspase revealed by indicator that is insensitive to environmental effects. J. Cell Biol. 2003;160:235–243. doi: 10.1083/jcb.200207111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chu J., Wang L., Luo Q., Zhang Z. Simultaneous imaging of two initiator caspases during cisplatin-induced HeLa apoptosis. Proc. SPIE. 2008;6857:68570R. [Google Scholar]
- 27.Shcherbo D., Souslova E.A., Hudakov D.M.C. Practical and reliable FRET/FLIM pair of fluorescent proteins. BMC Biotechnol. 2009;9:9. doi: 10.1186/1472-6750-9-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bardet P.-L., Kolahgar G., Vincent J.P. A fluorescent reporter of caspase activity for live imaging. Proc. Natl. Acad. Sci. USA. 2008;105:13901–13905. doi: 10.1073/pnas.0806983105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lee P., Beem E., Segal M.S. Marker for real-time analysis of caspase activity in intact cells. Biotechniques. 2002;33:1284–1287. doi: 10.2144/02336rr02. 1289–1291. [DOI] [PubMed] [Google Scholar]
- 30.Ray P., De A., Gambhir S.S. Monitoring caspase-3 activation with a multimodality imaging sensor in living subjects. Clin. Cancer Res. 2008;14:5801–5809. doi: 10.1158/1078-0432.CCR-07-5244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nagai T., Ibata K., Miyawaki A. A variant of yellow fluorescent protein with fast and efficient maturation for cell-biological applications. Nat. Biotechnol. 2002;20:87–90. doi: 10.1038/nbt0102-87. [DOI] [PubMed] [Google Scholar]
- 32.Hsu S.T., Blaser G., Jackson S.E. The folding, stability and conformational dynamics of B-barrel fluorescent proteins. Chem. Soc. Rev. 2009;38:2951–2965. doi: 10.1039/b908170b. [DOI] [PubMed] [Google Scholar]
- 33.Shu X., Royant A., Tsien R.Y. Mammalian expression of infrared fluorescent proteins engineered from a bacterial phytochrome. Science. 2009;324:804–807. doi: 10.1126/science.1168683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Demon D., Van Damme P., Vandenabeele P. Proteome-wide substrate analysis indicates substrate exclusion as a mechanism to generate caspase-7 versus caspase-3 specificity. Mol. Cell. Proteomics. 2009;8:2700–2714. doi: 10.1074/mcp.M900310-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Boucher D., Blais V., Denault J.-B. Caspase-7 uses an exosite to promote poly(ADP ribose) polymerase 1 proteolysis. Proc. Natl. Acad. Sci. USA. 2012;109:5669–5674. doi: 10.1073/pnas.1200934109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Heim R., Tsien R.Y. Engineering green fluorescent protein for improved brightness, longer wavelengths and fluorescence resonance energy transfer. Curr. Biol. 1996;6:178–182. doi: 10.1016/s0960-9822(02)00450-5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.