

Abstract
The Rad51 ATPase plays central roles in DNA homologous recombination. Yeast Rad51 dimer structure in the active form of the filament was constructed using homology modeling techniques, and all-atom molecular dynamics (MD) simulations were performed using the modeled structure. We found two crucial interaction networks involving ATP: one is among the γ-phosphate of ATP, K+ ions, H352, and D374; the other is among the adenine ring of ATP, R228, and P379. Multiple MD simulations were performed in which the number of bound K+ ions was changed. The simulated structures suggested that K+ ions are indispensable for the stabilization of the active dimer and resemble the arginine and lysine fingers of other P-loop containing ATPases and GTPases. MD simulations also showed that the adenine ring of ATP mediates interactions between adjacent protomers. Furthermore, in MD simulations starting from a structure just after ATP hydrolysis, the opening motion corresponding to dissociation from DNA was observed. These results support the hypothesis that ATP and K+ ions function as glue between protomers.
Introduction
Homologous recombination, in which DNA strands are exchanged between a pair of homologous sequences, is crucial for both the repair of damaged DNA and the maintenance of genomic diversity. The DNA strand exchange reaction in homologous recombination is mainly catalyzed by ATPases that are known as recombinases. The processes in the DNA strand exchange that involve recombinases are evolutionally conserved (1). After double-strand breaks in DNA, a free 3′ end of single-stranded DNA (ssDNA) is generated by a nuclease that degrades the 5′ end of the complementary strand. Recombinases bind to ssDNA and form a nucleo-protein filament called a presynaptic filament that searches for the homologous intact double-stranded DNA (dsDNA). Next, strand invasion of the ssDNA into dsDNA occurs, and DNA strands are subsequently exchanged between the ssDNA and dsDNA. Therefore, a presynaptic filament comprising recombinases and ssDNA is key in the DNA strand exchange reaction.
The recombinase protein family consists of Rad51 for eukaryotes, RadA for archaea, and RecA for bacteria. The primary sequence of Rad51 is more homologous to RadA than to RecA. The sequence identities between Rad51 and RadA and between Rad51 and RecA are ∼40% and ∼20%, respectively (2). All three recombinases share a core domain exhibiting ATPase activity. RecA has an additional C-terminal DNA-binding domain that is absent in Rad51 and RadA. In contrast, Rad51 and RadA have an extra N-terminal DNA-binding domain that is completely different from the C-terminal domain of RecA.
After the first report of an x-ray crystallographic structure of RecA (3), a number of recombinase structures have been reported for two decades (4–7). Electron microscopy (EM) studies revealed that the recombinase filament adopts two different forms; one is extended with a helical pitch of ∼90–100 Å, and the other is compressed with a helical pitch of ∼70–80 Å (4,8). The extended form is observed in the presence of DNA and ATP and corresponds to the active presynaptic form. In contrast, in the presence of ADP or in the absence of nucleotides, recombinases form the compressed inactive filament. Most of the crystal structures of RecA exhibit a helical pitch of ∼70–80 Å, corresponding to the compressed inactive form. In those inactive structures, the nucleotide-binding site is located at the side of the filament, not at the interface between protomers. An exception is a recently determined structure of the DNA-RecA complex, which adopts the extended active form (9). In the structure, ATP (actually, ADP and AlF4 are bound in the crystal structure) is located at the interface of protomers and functions as glue connecting the RecA protomers in the recombinase filament. The phosphates of ATP are bound to the Walker A or P-loop motif of one protomer and to two lysine side chains of the other adjacent protomer (Fig. S1 in the Supporting Material). The recognition of the ATP γ-phosphate by two lysine residues of the adjacent protomer is similar to that of the arginine fingers of ATP synthase (10) and Ras-GAP complex (11). However, because the two lysine residues are specific for RecA (Fig. S1), Rad51 and RadA adopt another method for recognizing the ATP γ-phosphate.
The crystal structure of yeast Rad51 exhibits a significantly more extended form with a helical pitch of ∼130 Å (12,13), which is unlikely to be the active form. In addition, no nucleotide is bound to the structure. Recently, using site-specific linear dichroism spectroscopy, a filament model of human Rad51 was constructed (14). Because this study focused on overall arrangements of Rad51 protomers in the filament, the ATP-binding site was not investigated in detail. Thus, at present, the recognition mechanism of ATP in Rad51 is not understood in a detailed fashion. In this study, we investigate ATP recognition in Rad51 using homology modeling and molecular dynamics (MD) simulations with explicit solvent. In homology modeling, we used a crystal structure of RadA (5) as a reference structure. The RadA crystal structures adopt a properly extended form with a helical pitch of ∼90–100 Å, and either an ATP analog or ADP is bound to the interface of protomers (6). Depending on the solvent conditions, one or two potassium ions or one calcium ion is located in the vicinity of the ATP γ-phosphate (5–7). Therefore, in RadA, these cations are likely to act as the lysine finger in RecA. In this study, by referencing a monomeric structure in the yeast Rad51 crystal structure and the relative arrangement of protomers in the RadA crystal structure, we constructed a dimer model of yeast Rad51 as a minimal unit of an active form of the Rad51 filament. Next, multiple MD simulations were performed of the modeled structures under different conditions, including the presence or absence of cations in the vicinity of the γ-phosphate. We address the questions of what condition stabilizes the Rad51 filament, whether cations also play roles in recognition of the ATP γ-phosphate in Rad51, and what are other factors in the role of ATP as a glue connecting protomers. Finally, we discuss effects of ATP hydrolysis on the active form of the Rad51 filament.
Methods
The yeast Rad51 dimer structure in the active form of the filament was modeled using the homology modeling techniques. For reference structures, the structures of a protomer and dimer were extracted from the crystal structure of the yeast Rad51 filament (PDBID: 3LDA) and the archaeal RadA filament (PDBID: 1XU4), respectively. Sequence alignments between Rad51 and RadA were performed using the program BLAST (Fig. S2) (15). The identity and similarity of the alignment were 40% and 59%, respectively. Because gap residues in the alignment (4%) were located at the surface far from the interface of the dimer, the insertions and deletions have little impact on the modeling. We compared the BLAST results with the alignment generated using ClustalX (16), T-COFFEE (17), and MUSCLE (18). All of the alignments were essentially the same, and the slight difference in the alignments (e.g., terminal residues) did not affect homology modeling. Thirty dimer structures were built with the program MODELER (19) using multiple templates, i.e., two monomeric Rad51 crystal structures, and a dimeric RadA crystal structure. Among the 30 built structures, we selected three candidate structures in which the side-chain configurations of the five important interface residues, D280, S192, R228, H352, and D374 resembled those of the RadA crystal structure and the side chain of R188 did not overlap with the ATP. From those candidates, we then chose the best structure in terms of the MODELER score function. The geometrical quality of the model evaluated using PROCHECK (20) was similar to that of the crystal structures (Table S1). ATP, magnesium, and potassium ions were inserted into the modeled Rad51 structure by referencing the RadA crystal structure.
The modeled structure of the yeast Rad51 dimer was subjected to all-atom MD simulations with explicit water. The conditions of the MD simulations are summarized in Table 1. All of the MD simulations were performed with the program MARBLE (21) using CHARMM22/CMAP for proteins (22), CHARMM27 for nucleotides and ions (23), and TIP3P for water (24) as the force-field parameters. Electrostatic interactions were calculated using the particle-mesh Ewald method. The Lennard-Jones potential was smoothly switched to zero over the range 8–10 Å. The symplectic integrator for rigid bodies was used for constraining the bond lengths and angles involving hydrogen atoms. The time step was 2.0 fs. The procedures of the MD simulations were as follows: The initial structures were immersed in a water box. K+ and Cl− ions were added to the systems such that the KCl concentration of the resulting system was 150 mM. The resulting systems contained ∼66,000 atoms. For equilibration, the systems were gradually heated to 293 K for 100 ps under the NVT ensemble with constraints on the positions of solute atoms. Subsequent MD simulations were performed for 100 ps under the NPT ensemble using the same constraints. Next, the constraints on the L1 and L2 loops in Rad51 were gradually decreased over a period of 100 ps, and finally, the constraints on all other solute atoms were gradually removed over a period of 100 ps. After the equilibration, 100 ns product runs were performed at 1 atm and 293 K. Each simulation was repeated as shown in Table 1.
Table 1.
Summary of MD simulations
| Simulation | Number of K+ | Nucleotide | Initial structure | Simulation times | Repeat |
|---|---|---|---|---|---|
| K2NATPIM | 2 | ATP | Model | 100 ns | 2 |
| K1NATPIM | 1 | ATP | Model | 100 ns | 2 |
| K0NATPIM | 0 | ATP | Model | 100 ns | 3 |
| K2NADP+PiIM | 2 | ADP, Pi | Model | 100 ns | 2 |
| K2NADPIM | 2 | ADP | Model | 100 ns | 2 |
| K2N-IM | 2 | – | Model | 100 ns | 2 |
| K2NATPIC | 2 | ATP | Crystal | 100 ns | 2 |
| K0NATPIR357 | 0 | ATP | Modela | 100 ns | 1 |
The side chain of R357 was oriented toward the γ-phosphate of ATP.
To analyze the opening motions in ADP-bound simulations, principal component analysis (PCA) was performed. First, structures in the trajectory of simulation K2NATPIM were averaged, and snapshots of simulations K2NATPIM, K2NADP+PiIM, and K2NADPIM were aligned to the average structure using least square fits of the core domain of protomer A (see Fig. 1). A covariance matrix of the core domain of protomer B from the average structure was then calculated and diagonalized to obtain principal modes. Therefore, principal modes represent the major motions of protomer B relative to protomer A. To estimate how much principal modes resemble random diffusion, their cosine contents were calculated. Hess has shown that for random diffusion on a flat potential surface, the first few principal components are represented by cosines with the number of periods equal to half the principal component index (25). The cosine content ci of principal component i is given by the following equation:
where T is the length of the simulation, and pi(t) is the amplitude of the motion along principal mode i at time t. The cosine content is in the range from 0 (no similarity to a cosine) to 1 (a perfect cosine).
Figure 1.
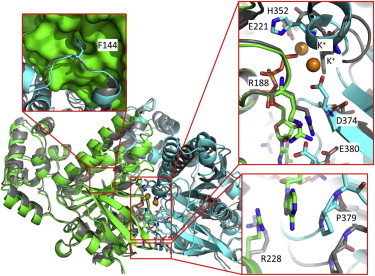
Homology modeling of yeast Rad51 dimer. The modeled structure (green (protomer A); cyan (protomer B)) was superimposed onto the crystal structure (gray). The binding mode of F144 in the polymerization motif is unchanged between the modeled and crystal structures (upper left). Protomer B pivots relative to protomer A, resulting in the movements of residues (H352, D374, P379, and E380) of protomer B at the ATP-binding site (right). In the modeled structure, the side chain of H352 in protomer B interacts with the ATP γ-phosphate, and the two K+ mediate interactions between the ATP and D374 of protomer B and between ATP and the main chain of H352 (upper right). P379 of protomer B and R228 of protomer A sandwiched the adenine ring of ATP (lower left).
To complement the MD simulations, electrostatic energies of the modeled structures with different numbers of bound K+ ions were calculated using the Poisson-Boltzmann equation method implemented in CHARMM (26). The ion concentration in the Poisson-Boltzmann calculations was set at 100 mM. The protein and water dielectric constants were 1 and 80, respectively. For comparison, electrostatic energies of the P-loop containing ATPases and GTPases (RecA, RadA, Ras_RasGAP, and F1ATPase) were also calculated using the same procedure. The PDB structures of RecA, RadA, Ras_RasGAP, and F1ATPase used were 3CMW, 1XU4, 1WQ1, and 2JDI, respectively. To evaluate the effects of positive charges supplied by the arginine and lysine fingers of adjacent protomers in RecA, Ras_RasGAP, and F1ATPase, electrostatic energies of arginine or lysine mutants to alanine were compared with those of their wild-types. The mutant structures were generated by simply removing side chains beyond Cβ. In RadA, electrostatic energies of structures in which one or two bound K+ ions removed were compared with those of the original crystal structure.
Results
Homology modeling of yeast Rad51 dimer
An active dimer model of yeast Rad51 was constructed with homology modeling techniques combining a monomeric structure extracted from the crystal structure of yeast Rad51 (PDB ID: 3LDA) and the relative arrangement of a dimeric structure from the archaeal RadA crystal filament in the active form (PDB ID: 1XU4) (Fig. 1). Because each protomer of the target yeast Rad51 dimer has the same sequence as the reference yeast Rad51 crystal structure, except for the missing L1 and L2 loops and H352 mutations in the crystal structure, the modeled protomer structures closely resemble the Rad51 crystal structure. Cα-root mean-square deviations (RMSDs) between the modeled protomer and crystal structure were 0.67 ± 0.16 Å. In contrast, the relative arrangements of two protomers in the modeled dimer structures were more similar to that of the RadA crystal structure than the Rad51 crystal structure, because the dimer of the RadA crystal structure was used as one of the template structures, and in the monomeric Rad51 crystal structure used as another template, there was no information on the relative arrangement of two protomers in the dimer. Cα-RMSDs of the core domain in the modeled dimer to the corresponding part of the RadA crystal structure were 0.97 ± 0.01 Å, whereas those to the Rad51 crystal structure were 1.52 ± 0.03 Å.
Because no nucleotide is bound to the crystal structure of yeast Rad51, the ATP-binding site of Rad51 was modeled with reference to that of RadA. The protomer in which ATP is bound to the P-loop motif is referred to as protomer A, and the other protomer is referred to as protomer B. Although the original crystal structure of yeast Rad51 is more extended than the active form observed in EM studies, the helical pitch of the modeled structure is ∼109 Å corresponding to the helical pitch of the active form. The binding mode of the polymerization motif (e.g., F144), which is a hot spot of interactions between adjacent protomers (27) is unchanged between the original crystal and modeled structures (Fig. 1). In the modeled structure, protomer B pivots about the polymerization motif relative to protomer A in contrast to the original crystal structure. Accordingly, the binding sites of the γ-phosphate (e.g., H352 and D374) and the adenine ring (e.g., P379) are spatially shifted from the crystal structure (Fig. 1).
In the crystal structures of RadA, one or two potassium ions are found at the binding site of the γ-phosphate of ATP (5,6). In the modeled structure of Rad51, we placed one or two potassium ions at the same positions as those of RadA (Fig. 1). Next, we performed multiple MD simulations with and without the potassium ions to examine how the potassium ions stabilize the dimer structure of Rad51 (Table 1).
Potassium ions stabilize Rad51 dimer
Throughout the MD simulations, individual domains, i.e., the N-terminal and core domains, of the Rad51 dimer were stable (Fig. S3). In contrast, the relative arrangements of the two protomers in the dimer exhibited significant variation depending on simulation conditions. The RMSDs of the two core domains in the dimer structure from the initial model are plotted in Fig. 2. In the simulations containing two K+ ions (simulation K2NATPIM in Table 1) and one K+ ion (simulation K1NATPIM), the relative arrangements of the two protomers were stable during the simulations. In simulation K2NATPIM, an interaction network involving two K+ ions was retained throughout the simulation (Fig. 3 A). Specifically, two K+ ions mediated the interactions of the γ-phosphate with the backbone of H352 and with the side chain of D374, and the side chain of H352 formed a direct or water-mediated hydrogen bond to the γ-phosphate (Figs. 3 A and 4 A). Thus, in Rad51, two K+ ions and the phosphate of ATP functions as glue between adjacent protomers through interactions with the P-loop of protomer A and two residues, H352 and D374, of protomer B. In simulation K1NATPIM, because one of the K+ ions was missing, the K+-mediated interaction involving the γ-phosphate became weak. Therefore, the protomer interface in the vicinity of the γ-phosphate became open, as indicated by the distance between the Cα atoms of D374 in protomer B and K191 (the central residue of the P-loop motif) in protomer A (Figs. 3 B and 4 B). In contrast, the K+-unbound simulation, K0NATPIM, showed that the binding site of the γ-phosphate became substantially open (Fig. 3 C). The relative arrangement of the two protomers also changed significantly (Fig. 2), whereas the binding at the polymerization motif was rigidly retained (Fig. 4 C). This large displacement was probably a result of the absence of mediation by K+ ions for the interaction between the phosphate and D374. In simulation K0NATPIM, D374 formed a new salt bridge with R188 (the fourth residue of the P-loop motif, GXXXXGKT/S) of protomer A (Fig. S4). Because R188 initially formed a salt bridge with E380, R188 switched an interaction partner from E380 to D374. The salt bridge between R188 and D374 is also found in the crystal structure of yeast Rad51, suggesting that the salt bridge is a stable configuration in the K+-unbound state. Consequently, the simulations with and without K+ ions indicate that K+ ions lead to the stable interactions of the ATP phosphate with the adjacent protomer of Rad51.
Figure 2.
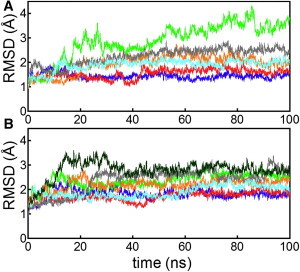
RMSD of the core domains of the Rad51 dimer from the initial structures in the first run (panel A) and second run (panel B) of simulations K2NATPIM (blue), K1NATPIM (red), K0NATPIM (green), K2NADP+PiIM (cyan), K2NADPIM (orange), and K2N-IM (gray). The dark green line in panel B indicates the third run of K0NATPIM. In simulations containing K+ ions, core domains were stable. In contrast, the K+-free simulations exhibited large deviations from the modeled structure.
Figure 3.
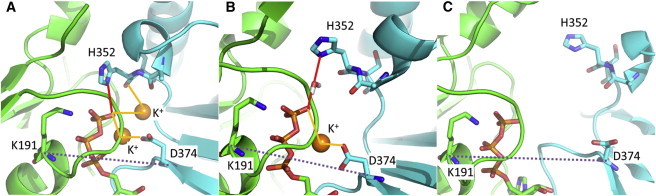
Effects of K+ on the stability of the ATP-γ-phosphate binding site. The final structures of the ATP-γ-phosphate binding site in the first runs of simulations K2NATPIM (A), K1NATPIM (B), and K0NATPIM (C) are shown. The dashed line indicates the distance between the Cα atoms of D374 and K191 that is used as an indicator of the opening of the interface. In panel A, a hydrogen bond between H352 and the γ-phosphate is shown with the red line, and interactions involving K+ ions are shown in yellow lines. In simulations containing two K+ ions, the ATP-binding site was tight. In the case of one K+ ion, the side chain of H352 formed a water-mediated hydrogen bond with the γ-phosphate. In contrast, the K+-free simulation exhibited a large opening motion at the ATP-binding site.
Figure 4.
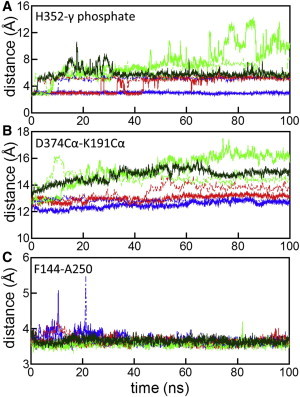
Effects of K+ on the interactions between the two protomers of the dimer. In panel A, distances between the side chain of H352 and the γ-phosphate of ATP are shown. In panel B, distances between the Cα atoms of D374 and K191 are shown as an indicator of the opening of the dimer interface. In panel C, distances between the side chains of F144 and A250 in the polymerization-motif binding site are shown. The colors of the lines in all panels indicate simulations K2NATPIM (blue), K1NATPIM (red), and K0NATPIM (green). Solid and dashed lines represent the results of the first and second runs, respectively. The third run of simulation K0NATPIM is indicated by solid dark green lines.
As another factor that impacts on stabilization of ATP-Rad51 interactions, the side chain of R357 was examined. By simple alternation of the side chain rotamer, the charged moiety of R357 can occupy the K+ location. To examine this possibility, we modified the rotamer of R357 so that the side chain was oriented toward the ATP γ-phosphate in the modeled structure (Fig. S5). We then carried out MD simulations of the modified model without bound K+ ions. In the MD simulations, the opening motion at the ATP-phosphate binding site was observed in a similar manner to the K+-free simulation K0NATPIM, and the side chain of R357 tended to return to the original configuration, possibly due to interactions with E182 (Fig. S5). These results also indicate crucial roles of K+ ions in the stabilization of the active form of Rad51.
To complement the MD simulations, electrostatic energies were calculated for the modeled structures with and without the bound K+ ions. Clearly, the bound K+ ions stabilize the electrostatic energies. Compared with the K+-unbound structure, the K+ ions decrease ∼250 kcal/mol of electrostatic energies. In contrast, the modification of the R357 rotamer only slightly stabilizes the electrostatic energies. These results were consistent with the MD simulations. For comparison, we also calculated the electrostatic energies of other P-loop containing ATPases and GTPases. The details are described in the Discussion section.
Interestingly, E221, which is conserved among P-loop-containing ATPases and is supposed to play a role in activating a water molecule in ATP hydrolysis (28), occasionally formed a salt bridge with R308 in MD simulations (Fig. S6). Due to the salt bridge, the side chain of E221 was oriented away from the γ-phosphate of ATP. This interaction may cause the reduction of ATPase activity. In the RecA crystal structure, the arginine residue corresponding to R308 participates in interactions with the phosphate of the DNA backbone. Upon DNA binding, the salt bridge between E221 and R308 should be broken, and free E221 may increase ATPase activity. Thus, this salt bridge may be an explanation of low ATPase activity in the absence of DNA. However, to confirm the hypothesis, quantum mechanics/molecular mechanics studies for ATP hydrolysis and/or mutagenesis experiments should be performed.
Adenine ring of ATP also mediate dimer interactions
The adenine ring of ATP also mediates the interactions between adjacent protomers of Rad51. Both sides of the adenine ring are packed by R228 of protomer A and P379 of protomer B (Fig. 1). In all of the nucleotide-K+-bound simulations starting from the modeled structure, the interactions of the adenine ring were stable throughout the simulations (Fig. 5, A and B). In contrast, in the nucleotide-free simulation K2N-IM, the position of P379 of protomer B changed significantly (Fig. 5 C and Fig. S7), suggesting that interactions of the adenine ring are crucial for the relative arrangement of the two protomers in the active form of the Rad51. To confirm this result, an additional simulation (K2NATPIC) was conducted starting from the crystal structure of Rad51, in which P379 is shifted and apart from the adenine ring (Fig. 1). After ∼40 ns in simulation K2NATPIC (∼10 ns in the second run), P379 was spontaneously in contact with the adenine ring of ATP (Fig. 5 D and Fig. S8). Consequently, our simulations suggest the interaction between P379 and the adenine ring contributes to the formation of the active filament of Rad51.
Figure 5.
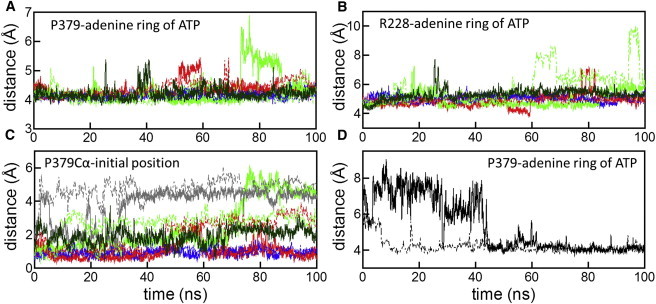
The adenine ring of ATP mediates interactions between adjacent protomers. In panel A, distances between the centers of mass of the P379 side chain (protomer B) and of the adenine ring of ATP are shown. In panel B, distances between the centers of mass of the R228 side chain (protomer A) and of the adenine ring of ATP are shown. In panel C, displacements of the Cα of P379 (protomer B) from the initial position during simulations are shown. In panels A–C, the colors of the lines indicate simulations K2NATPIM (blue), K1NATPIM (red), K0NATPIM (green), and K2N-IM (gray). In panel D, in simulation K2NATPIC starting from the crystal structure, distances between the centers of mass of the P379 side chain (protomer B) and of the adenine ring of ATP are shown. The spontaneous binding of P379 to the adenine ring was observed. Solid and dashed lines represent the results of the first and second runs, respectively. The third run of simulation K0NATPIM is indicated by solid dark green lines.
ATP hydrolysis destabilizes Rad51 dimer
Next, to investigate the effects of ATP hydrolysis on dimer stability, we conducted two MD simulations with ADP + Pi (K2NADP+PiIM) and ADP (K2NADPIM). In the initial model of simulation K2NADP+PiIM, which approximated the nucleotide state immediately after ATP hydrolysis, we replaced ATP with ADP and Pi; for this replacement, the quantum mechanics/molecular mechanics study of ATP hydrolysis in the F1-ATPase that belongs to the same superfamily as Rad51 was consulted (28). For comparison, we performed an MD simulation including only ADP without Pi. The RMSD of the two core domains in simulations K2NADP+PiIM and K2NADPIM were slightly larger than those of the ATP and K+-bound simulations (K2NATPIM and K1NATPIM) (Fig. 2), and the distances between D374 and K191 were larger than those of simulation K2NATPIM (Figs. 4 B and 6 A). These findings indicate that the interprotomer interface at the γ-phosphate-binding site became open (Fig. 6 B). To characterize the opening motions in the ADP-bound simulations, we performed PCA for the trajectories of simulations K2NATPIM, K2NADP+PiIM, and K2NADPIM. Because the core domain of protomer A was used for the structural alignment and the core domain of protomer B was used for calculation of the covariance matrix as described in the Methods, the principal modes in this analysis represent major motions of protomer B relative to protomer A. In this PCA, we do not intend to extract breathing motions during the simulations but to characterize the opening motions in K2NADP+PiIM and K2NADPIM. Fig. 7 A shows that the first principal component (PC1) representing the largest motions discriminates the post-hydrolysis trajectories (K2NADP+PiIM and K2NADPIM) from the prehydrolysis trajectory (K2NATPIM). Structural transitions in posthydrolysis trajectories took place during the first 10–20 ns. The cosine contents of PC1 of trajectories K2NADP+PiIM (9%) and K2NADPIM (41%) in the first run are not very high, indicating that the motions of posthydrolysis trajectories are different from the random diffusion on a flat potential surface. However, the directions of PC1 and PC2 in the second run were swapped in comparison to the directions in the first run (Fig. S9), suggesting that the opening motions are stochastic rather than deterministic. The directions of PC1 and PC2 are depicted in Fig. 7 C. To visualize the PC1 and PC2 motions of the Rad51 protomer in the Rad51-dsDNA filament, the filament model was constructed by superimposing the average dimer structure onto the crystal structure of the RecA-dsDNA complex and replacing RecA protomers with Rad51 protomers. The directions of the PC modes appear to correspond to dissociation directions of a terminal Rad51 protomer from DNA. Because the phosphate of the bound ATP is oriented toward the DNA (see Fig. 7 C), the opening motion at the ATP-phosphate binding site may initiate dissociation of a terminal protomer from the nucleoprotein filament. This observation is consistent with single-molecule experiments in which Rad51 protomers dissociate from the terminus of the filament after ATP hydrolysis (29). However, because our simulations did not include DNA, effects of ATP hydrolysis on Rad51-DNA binding should be investigated using simulations of the Rad51-DNA complex in the future.
Figure 6.
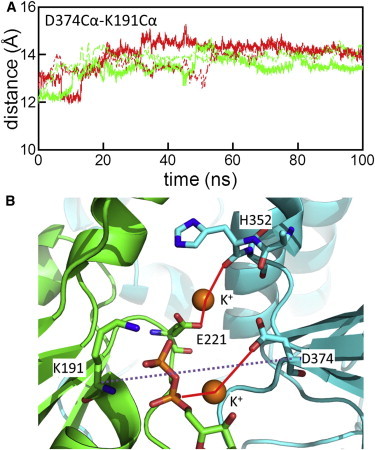
Opening of the dimer interface in ADP-bound simulations. In panel A, distances between the Cα atoms of D374 and K191 in simulations K2NADP+PiIM (green) and K2NADPIM (red) are shown. Solid and dashed lines represent the results of the first and second runs, respectively. In panel B, the ATP-binding site of the final structure in simulation K2NADPIM of the first run is shown. Red lines indicate interactions involving K+ ions. The dashed line is drawn to indicate the distance between D374 and K191.
Figure 7.
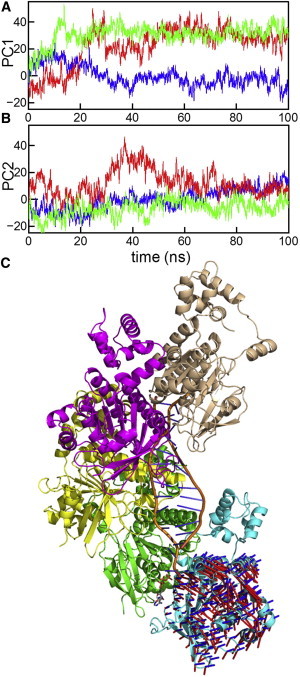
PCA for trajectories of the ADP-bound simulations. Projections of snapshots onto PC1 and PC2 in the first run of simulations K2NATPIM (blue), K2NADP+PiIM (green), and K2NADPIM (red) are shown in panels A and B, respectively. PC1 discriminates the posthydrolysis trajectories (K2NADP+PiIM and K2NADPIM) from the prehydrolysis trajectory (K2NATPIM). The cosine contents of PC1 in K2NATPIM, K2NADP+PiIM, and K2NADPIM are 34%, 9%, and 41%, respectively. The cosine contents of PC2 in K2NATPIM, K2NADP+PiIM, and K2NADPIM are 0.2%, 6%, and 28%, respectively. In panel C, the directions of PC1 (red lines) and PC2 (blue lines) are shown in the Rad51-dsDNA filament (3 × square root of eigen values). The filament model was constructed by superimposing the average dimer structure onto the crystal structure of the RecA-dsDNA complex and replacing RecA protomers with Rad51 protomers. The bound ATP in the dimer was shown in stick representation.
Discussion
Our simulation results indicated that one or two K+ ions stabilize the Rad51 active form and are consistent with experimental data. Although Rad51 exhibits weak strand exchange activity compared with RecA in vitro, salts can stimulate the strand exchange activity of human Rad51 (30). This strand exchange stimulation in human Rad51 is dependent on the cation component of salts. The NH4+ or K+ cations can stimulate the strand exchange, whereas the Na+ cation is incapable of stimulating the strand exchange. In a recent EM study, the human Rad51 filament exhibits the relatively short helical pitch of ∼86 Å without salts. In contrast, by adding salts containing the NH4+ or K+ cation, the helical pitch is extended to ∼110 Å, which corresponds to the active form, suggesting that the cations stabilize the active form of human Rad51 (31). Yeast Rad51 also exhibits a requirement of KCl in its strand exchange reaction (32). In addition to monovalent cations, the divalent Ca2+ cation also stimulates the strand exchange reaction of human Rad51 (33) by stabilizing the active form of the human Rad51 filament (34). However, Ca2+ does not stimulate the strand exchange activity of yeast Rad51, although most of the residues in the ATP-binding site are conserved between the two species.
The ATPase activity is also affected by the cation component. In archaeal recombinase RadA, DNA-stimulated ATPase activity requires the K+ cation (5). No ATPase activity was observed in the presence of salts containing the Na+ cation. However, human Rad51 exhibits the ATPase activity even in the presence of NaCl (30). It should be noted that the increase in ATPase activity does not always enhance the strand exchange activity. For instance, Ca2+ reduces the ATPase activity of human Rad51 but enhances the strand exchange activity (33). Ca2+ appears to maintain the active form of the Rad51 filament by reducing the ATP hydrolysis rate.
DNA binding also affects the ATPase activity of Rad51 and RadA (5,30). Although our simulation did not include DNA, our simulation results suggest possible linkages between the DNA- and ATP-binding sites. In our simulations, K+ ions stabilized interactions between the γ-phosphate of ATP and the H352-containing α-helix in the downstream region of the L2 loop that is crucial for DNA binding. In the RadA crystal structures with low K+ concentrations or in the presence of ADP instead of AMP-PNP, the corresponding α-helix is disordered (6). By stabilizing the L2 loop and the downstream α-helix, DNA binding may affect the ATPase activity of Rad51. In addition, our simulation suggests that R308 could potentially regulate ATP activity by the salt bridge with E221 that activates a water molecule in ATP hydrolysis as described in the Results section.
In vivo, in addition to cations, other factors such as mediators have effects on Rad51 strand exchange activity. For instance, the protein complex, Swi5-Sfr1, which is conserved from fission yeast to human, stabilizes the active form of fission yeast Rad51 and enhances the ATPase activity of Rad51 (35–37).
In our simulations, one of the K+ ions mediated interactions between the γ-phosphate of ATP and D374, belong to the adjacent protomer. The other K+ ion interacted with both the ATP γ-phosphate and the H352 backbone of the adjacent protomer. Recognition of the ATP phosphate by the adjacent protomer is a common feature among P-loop-containing ATPases and GTPases (Fig. S1). In the bacterial recombinase RecA, two lysine side chains of the adjacent protomer recognize the phosphate of ATP (Fig. 8) (9). Similarly, in ATP synthase, an arginine side chain of an adjacent subunit directly interacts with the phosphate of ATP (10). In Rasp21, which functions in signal transduction, an arginine side chain of a binding partner, GTPase-activating protein (GAP), binds to the phosphate of ATP (11). Thus, recognition of the nucleotide phosphate by a moiety of an adjacent protomer is shared among these P-loop containing ATPases and GTPases. In the case of Rad51 as well as RadA, two K+ ions, H352 and D374 play roles in recognition of the nucleotide phosphate. To confirm stabilization of ATP-protein interactions by supplying positive charges, we counted the number of charged residues of these P-loop containing ATPases in the vicinity of the ATP phosphate. Even including the supplied positive charges of adjacent protomers and cations, the net charge in the vicinity of the ATP phosphate is slightly negative, suggesting that the supplied positive charges contribute to stabilizing ATP-protein interactions (Table S2). Electrostatic energies calculated using the Poisson-Boltzmann equation also clearly indicate that the supplied positive charges stabilized the complex structure (Fig. 9), consistent with our MD simulations. Interestingly, in paralogs of human Rad51, D316, which corresponds to D374 of yeast Rad51, is mutated to lysine (31). EM of the human Rad51 D316K mutant showed that even without salts, the helical pitch is ∼110 Å, corresponding to the active form. In the crystal structure of the RadA D302K mutant, in which D302 corresponds to D374 of yeast Rad51 and D316 of human Rad510, K302 directly interacts with the ATP γ-phosphate. These experimental results are clearly consistent with our simulation results.
Figure 8.
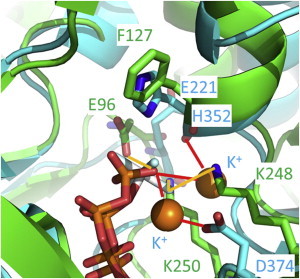
Comparison between the ATP-binding sites of the final structure of the K2NATPIM simulation (cyan) and RecA (PDB code 3CMW, green). The K+ ions (orange spheres) in the simulation structure and the lysine side chains of RecA are located in similar positions.
Figure 9.
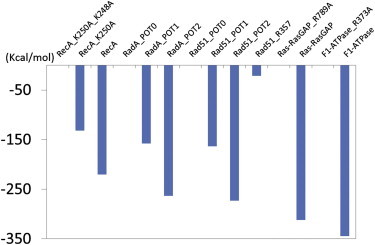
Electrostatic energies of P-loop containing ATPases and GTPases decreased due to positive charges supplied by the arginine and lysine fingers of adjacent protomers and K+ ions. Relative energies to those without the supplied positive charges are shown. Electrostatic energies were calculated using the Poisson-Boltzmann equation. In RecA, Ras_RasGAP, and F1ATPase, mutants of lysine and arginine fingers were compared with their wild-types. In RadA and Rad51, structures with one or two K+ ions in the ATP-binding site were compared with those in the absence of bound K+ ions. Rad51 with the modified configuration of R357 in the absence of the K+ ion was also compared.
Very recently, molecular modeling and MD simulations of human Rad51 were reported (34). This work focused on Ca2+ and Mg2+ ions rather than the K+ ions we studied for this work. The researchers found that when a Ca2+ or Mg2+ ion was missing from the vicinity of the γ-phosphate of ATP, local structures close to the γ-phosphate became unstable. Considering that the charge of a Ca2+ ion is the same as that of two K+ ions, their results are in good agreement with our results. However, Ca2+ does not stimulate the strand exchange activity of yeast Rad51 in contrast to human Rad51 (33), as described previously. Interestingly, in RadA, a crystal structure in which one Ca2+ ion is bound to the same position as two K+ ions has been reported (7).
In summary, we constructed a dimer model of Rad51 in the active form of the filament using homology modeling techniques, and performed MD simulations from the modeled structures with explicit solvent. We found two crucial interaction networks involving ATP: one is an interaction network among the γ-phosphate of ATP, K+ ions, H352, and D374; the other is among the adenine ring of ATP, R228, and P379. We also investigated how ATP hydrolysis affects the connection of the two protomers in the active form of Rad51. In the MD simulation starting from a structure mimicking the state immediately after ATP hydrolysis, the interaction network involving the phosphate of ATP was significantly perturbed, resulting in an opening motion at the dimer interface. This opening motion occurred in the direction in which the terminal protomer dissociated from DNA. These results support that one of the functional roles of ATP γ-phosphate and K+ is as glue between the protomers in the active form of Rad51.
Acknowledgments
We thank Hiroshi Iwasaki for helpful discussions.
This study was supported by Grants-in-Aids for Scientific Research on Innovative Areas from the Ministry of Education, Culture, Sports, Science, and Technology of Japan (MEXT) and for Scientific Research (B); by the Grand Challenges in Next-Generation Integrated Simulation of Living Matter, which is part of the Development and Use of the Next-Generation Supercomputer Project of MEXT; by Platform for Drug Design, Informatics and Structural Life Sciences (MEXT); by X-ray Free Electron Laser Priority Strategy Program (MEXT).
Footnotes
This is an Open Access article distributed under the terms of the Creative Commons-Attribution Noncommercial License (http://creativecommons.org/licenses/by-nc/2.0/), which permits unrestricted noncommercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Supporting Material
References
- 1.Bianco P.R., Tracy R.B., Kowalczykowski S.C. DNA strand exchange proteins: a biochemical and physical comparison. Front. Biosci. 1998;3:D570–D603. doi: 10.2741/a304. [DOI] [PubMed] [Google Scholar]
- 2.Brendel V., Brocchieri L., Karlin S. Evolutionary comparisons of RecA-like proteins across all major kingdoms of living organisms. J. Mol. Evol. 1997;44:528–541. doi: 10.1007/pl00006177. [DOI] [PubMed] [Google Scholar]
- 3.Story R.M., Weber I.T., Steitz T.A. The structure of the E. coli recA protein monomer and polymer. Nature. 1992;355:318–325. doi: 10.1038/355318a0. [DOI] [PubMed] [Google Scholar]
- 4.Bell C.E. Structure and mechanism of Escherichia coli RecA ATPase. Mol. Microbiol. 2005;58:358–366. doi: 10.1111/j.1365-2958.2005.04876.x. [DOI] [PubMed] [Google Scholar]
- 5.Wu Y., Qian X., Luo Y. Crystal structure of an ATPase-active form of Rad51 homolog from Methanococcus voltae. Insights into potassium dependence. J. Biol. Chem. 2005;280:722–728. doi: 10.1074/jbc.M411093200. [DOI] [PubMed] [Google Scholar]
- 6.Qian X., Wu Y., Luo Y. Crystal structure of Methanococcus voltae RadA in complex with ADP: hydrolysis-induced conformational change. Biochemistry. 2005;44:13753–13761. doi: 10.1021/bi051222i. [DOI] [PubMed] [Google Scholar]
- 7.Qian X., He Y., Luo Y. Calcium stiffens archaeal Rad51 recombinase from Methanococcus voltae for homologous recombination. J. Biol. Chem. 2006;281:39380–39387. doi: 10.1074/jbc.M607785200. [DOI] [PubMed] [Google Scholar]
- 8.VanLoock M.S., Yu X., Egelman E.H. ATP-mediated conformational changes in the RecA filament. Structure. 2003;11:187–196. doi: 10.1016/s0969-2126(03)00003-0. [DOI] [PubMed] [Google Scholar]
- 9.Chen Z., Yang H., Pavletich N.P. Mechanism of homologous recombination from the RecA-ssDNA/dsDNA structures. Nature. 2008;453:489–494. doi: 10.1038/nature06971. [DOI] [PubMed] [Google Scholar]
- 10.Bowler M.W., Montgomery M.G., Walker J.E. Ground state structure of F1-ATPase from bovine heart mitochondria at 1.9 Å resolution. J. Biol. Chem. 2007;282:14238–14242. doi: 10.1074/jbc.M700203200. [DOI] [PubMed] [Google Scholar]
- 11.Scheffzek K., Ahmadian M.R., Wittinghofer A. The Ras-RasGAP complex: structural basis for GTPase activation and its loss in oncogenic Ras mutants. Science. 1997;277:333–338. doi: 10.1126/science.277.5324.333. [DOI] [PubMed] [Google Scholar]
- 12.Conway A.B., Lynch T.W., Rice P.A. Crystal structure of a Rad51 filament. Nat. Struct. Mol. Biol. 2004;11:791–796. doi: 10.1038/nsmb795. [DOI] [PubMed] [Google Scholar]
- 13.Chen J., Villanueva N., Morrical S.W. Insights into the mechanism of Rad51 recombinase from the structure and properties of a filament interface mutant. Nucleic Acids Res. 2010;38:4889–4906. doi: 10.1093/nar/gkq209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Reymer A., Frykholm K., Nordén B. Structure of human Rad51 protein filament from molecular modeling and site-specific linear dichroism spectroscopy. Proc. Natl. Acad. Sci. USA. 2009;106:13248–13253. doi: 10.1073/pnas.0902723106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Altschul S.F., Madden T.L., Lipman D.J. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Thompson J.D., Gibson T.J., Higgins D.G. The CLUSTAL_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 1997;25:4876–4882. doi: 10.1093/nar/25.24.4876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Notredame C., Higgins D.G., Heringa J. T-Coffee: a novel method for fast and accurate multiple sequence alignment. J. Mol. Biol. 2000;302:205–217. doi: 10.1006/jmbi.2000.4042. [DOI] [PubMed] [Google Scholar]
- 18.Edgar R.C. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sali A., Blundell T.L. Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 1993;234:779–815. doi: 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- 20.Laskowski R.A., MacArthur M.W., Thornton J.M. PROCHECK: a program to check the stereochemical quality of protein structures. J. Appl. Cryst. 1993;26:283–291. [Google Scholar]
- 21.Ikeguchi M. Partial rigid-body dynamics in NPT, NPAT and NPgammaT ensembles for proteins and membranes. J. Comput. Chem. 2004;25:529–541. doi: 10.1002/jcc.10402. [DOI] [PubMed] [Google Scholar]
- 22.Mackerell A.D., Jr., Feig M., Brooks C.L., 3rd Extending the treatment of backbone energetics in protein force fields: limitations of gas-phase quantum mechanics in reproducing protein conformational distributions in molecular dynamics simulations. J. Comput. Chem. 2004;25:1400–1415. doi: 10.1002/jcc.20065. [DOI] [PubMed] [Google Scholar]
- 23.MacKerell A.D., Jr., Banavali N., Foloppe N. Development and current status of the CHARMM force field for nucleic acids. Biopolymers. 2000-2001;56:257–265. doi: 10.1002/1097-0282(2000)56:4<257::AID-BIP10029>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- 24.Jorgensen W.L., Chandrasekhar J., Klein M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983;79:926–935. [Google Scholar]
- 25.Hess B. Similarities between principal components of protein dynamics and random diffusion. Phys. Rev. E Stat. Phys. Plasmas Fluids Relat. Interdiscip. Topics. 2000;62(6 Pt B):8438–8448. doi: 10.1103/physreve.62.8438. [DOI] [PubMed] [Google Scholar]
- 26.Brooks B.R., Bruccoleri R.E., Karplus M. CHARMM: a program for macromolecular energy, minimization, and dynamics calculations. J. Comput. Chem. 1983;4:187–217. [Google Scholar]
- 27.Pellegrini L., Yu D.S., Venkitaraman A.R. Insights into DNA recombination from the structure of a RAD51-BRCA2 complex. Nature. 2002;420:287–293. doi: 10.1038/nature01230. [DOI] [PubMed] [Google Scholar]
- 28.Hayashi S., Ueno H., Noji H. Molecular mechanism of ATP hydrolysis in F1-ATPase revealed by molecular simulations and single-molecule observations. J. Am. Chem. Soc. 2012;134:8447–8454. doi: 10.1021/ja211027m. [DOI] [PubMed] [Google Scholar]
- 29.van Mameren J., Modesti M., Wuite G.J. Counting RAD51 proteins disassembling from nucleoprotein filaments under tension. Nature. 2009;457:745–748. doi: 10.1038/nature07581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shim K.S., Schmutte C., Fishel R. Defining the salt effect on human RAD51 activities. DNA Repair (Amst.) 2006;5:718–730. doi: 10.1016/j.dnarep.2006.03.006. [DOI] [PubMed] [Google Scholar]
- 31.Amunugama R., He Y., Fishel R. RAD51 protein ATP cap regulates nucleoprotein filament stability. J. Biol. Chem. 2012;287:8724–8736. doi: 10.1074/jbc.M111.239426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rice K.P., Eggler A.L., Cox M.M. DNA pairing and strand exchange by the Escherichia coli RecA and yeast Rad51 proteins without ATP hydrolysis: on the importance of not getting stuck. J. Biol. Chem. 2001;276:38570–38581. doi: 10.1074/jbc.M105678200. [DOI] [PubMed] [Google Scholar]
- 33.Bugreev D.V., Mazin A.V. Ca2+ activates human homologous recombination protein Rad51 by modulating its ATPase activity. Proc. Natl. Acad. Sci. USA. 2004;101:9988–9993. doi: 10.1073/pnas.0402105101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fornander L.H., Frykholm K., Nordén B. Ca2+ improves organization of single-stranded DNA bases in human Rad51 filament, explaining stimulatory effect on gene recombination. Nucleic Acids Res. 2012;40:4904–4913. doi: 10.1093/nar/gks140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Haruta N., Akamatsu Y., Iwasaki H. Fission yeast Swi5 protein, a novel DNA recombination mediator. DNA Repair (Amst.) 2008;7:1–9. doi: 10.1016/j.dnarep.2007.07.004. [DOI] [PubMed] [Google Scholar]
- 36.Kokabu Y., Murayama Y., Ikeguchi M. Fission yeast Swi5-Sfr1 protein complex, an activator of Rad51 recombinase, forms an extremely elongated dogleg-shaped structure. J. Biol. Chem. 2011;286:43569–43576. doi: 10.1074/jbc.M111.303339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kuwabara N., Murayama Y., Shimizu T. Mechanistic insights into the activation of Rad51-mediated strand exchange from the structure of a recombination activator, the Swi5-Sfr1 complex. Structure. 2012;20:440–449. doi: 10.1016/j.str.2012.01.005. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.