

Abstract
Objective
Acyl-CoA:cholesterol acyltransferase (ACAT) converts cholesterol to cholesteryl esters in plaque foam cells. Complete deficiency of macrophage ACAT has been shown to increase atherosclerosis in hypercholesterolemic mice due to cytotoxicity from free cholesterol accumulation, while we previously showed that partial ACAT inhibition by Fujirebio compound F1394 decreased early atherosclerosis development. In this report, we tested F1394 effects on pre-established, advanced lesions of apoE-/- mice.
Methods & Results
ApoE-/- mice on Western diet for 14 weeks developed advanced plaques, and were either sacrificed (“Baseline”), or continued on Western diet without or with F1394 and sacrificed after 14 more weeks. F1394 was not associated with systemic toxicity. Compared to the baseline group, lesion size progressed in both groups; however, F1394 significantly retarded plaque progression, and reduced plaque macrophage, free and esterified cholesterol, and tissue factor contents compared to the untreated group. Apoptosis of plaque cells was not increased, consistent with the decrease in lesional free cholesterol, plaque necrosis was not increased, and efferocytosis (phagocytic clearance of apoptotic cells) was not impaired. The effects of F1394 were independent of changes in plasma cholesterol levels.
Conclusions
Partial ACAT inhibition by F1394 lowered plaque cholesterol content and had other antiatherogenic effects in advanced lesions in apoE-/- mice without overt systemic or plaque toxicity, suggesting the continued potential of ACAT inhibition for the clinical treatment of atherosclerosis in spite of recent trial data.
Introduction
Acyl-CoA:cholesterol acyltransferase (ACAT) converts cholesterol to cholesteryl esters and plays important roles in lipoprotein assembly, dietary cholesterol absorption, and intracellular cholesterol metabolism 1. ACAT exists in 2 forms, ACAT1 and ACAT2. ACAT2 is expressed in the liver, and indirectly contributes to coronary artery disease by influencing the content of cholesteryl ester (CE) on the atherogenic lipoprotein particles VLDL and LDL 2-4. Another important role for ACAT in cardiovascular disease is that in macrophages and smooth muscle cells in the arterial wall, cholesteryl esters produced by ACAT1 accumulate leading to formation of foam cells, whose presence is a hallmark of atherosclerotic lesions and whose deranged metabolism exacerbate the inflammatory milieu within a plaque 1, 5.
Complete deficiency of macrophage ACAT results in increased atherosclerotic lesions in hypercholesterolemic mouse models (LDL receptor or apolipoprotein E deficient (apoE-/-) mice) due to the cytotoxicity from free cholesterol (FC) accumulation in cells and tissues 6. Early studies attributed this toxicity to either damage to the plasma membrane (because excess membrane FC, which ordinarily can be safely stored in the more inert cytosolic CE lipid droplets, was now retained), or from the formation of FC intracellular crystals 7, 8. More recent studies, first by Tabas and colleagues and then others 9-14 have shown that a component of the cellular cytotoxicity is ER-stress from the accumulation of FC in the ER membrane, ultimately resulting in apoptosis in vitro and in vivo. In recent reviews, it has been proposed that in advanced plaques, there is diminution of ACAT activity and apoptotic cell clearance, so that in the face of persistent hyperlipidemia, foam cells become progressively overloaded with FC, undergo ER-stress and apoptosis and then add to the necrotic core 15-17. Implicit in this scenario is that the rate of efflux of FC is insufficient to maintain sub-toxic cellular levels.
In previous studies, we showed that for predominately foam cell-rich lesions partial ACAT inhibition by Fujirebio compound F1394 was non-toxic and decreased atherosclerosis progression in apoE-/- mice when they were treated prior to lesion initiation 5. To simulate the scenario summarized above for more advanced plaques, in the present study, we have allowed plaques to develop to a more advanced stage (so that they contain cholesterol clefts and lipid cores). Then the mice were fed either western diet (WD) or WD+F1394 to partially inhibit ACAT for the next 14 weeks. As will be described, F1394 treatment led to a delay in further progression of atherosclerotic lesions, as well as to other benefits including a decrease in lesional free cholesterol, without evidence of increased foam cell apoptosis, impaired efferocytosis, greater plaque necrosis, or signs of systemic toxicity.
Methods
Experimental Design and Animals
All experimental procedures in animals were performed with protocols approved by either the Mount Sinai School of Medicine or the NYU School of Medicine Animal Care and Use Committee. Compound F1394 was supplied by Fujirebio Inc (Tokyo, Japan) 5. Male apoE-/- mice (n=47) were weaned at 4 weeks of age onto a 21% (wt/wt) fat, 0.15% cholesterol “Western-type diet” (WD; catalogue No. 100244, Dyets Inc) 5 and fed this diet for 14 weeks to develop in the aortic roots advanced lesions containing necrotic lipid cores and cholesterol clefts (AHA class IV 18). These mice were then divided into 3 groups: one group was sacrificed to obtain lesion status before F1394 diet began (N= 16, “Baseline”), and the other two groups were continued on WD with (N = 15, “Treatment”, 900 mg/kg diet; dose based on our previous study 5) or without (N = 16, “No Treatment”; i.e., control) supplementation of F1394 for additional 14 weeks and sacrificed.
Measurement of plasma lipids and lipoproteins
Plasma samples were collected at sacrifice for all groups. The HDL fraction was isolated by ultracentrifugation as previously described 19. Total cholesterol and HDL-cholesterol were then analyzed using commercial kits (Sigma cholesterol-reagent kit).
Two plasma samples (pooled from 2 mice) from the F1394 and No Treatment group were separated via fast protein liquid chromatography (FPLC). The lipoproteins eluted approximately at fractions: 29-38 for VLDL, 39-55 for LDL, 58-67 for HDL and 68-77 for plasma proteins. The respective fractions of VLDL, LDL, and HDL were pooled to determine CE fatty acid composition by fatty acid methyl ester analysis as previously described 20, 21. Briefly, the lipids were extracted in chloroform and methanol as described by Bligh and Dyer 22. Lipid classes were separated by thin layer chromatography (TLC) on silica gel G plates. The band corresponding to CE was removed, saponified, and the resulting fatty acids were methylated. Fatty acid composition was determined after separation of methyl esters by gas chromatography.
Aortic Lipid Analysis
The TC, FC, and CE content of aortas (N=6 or 7) were measured according to methods described by Willner et al 23. Briefly, all adherent adipose and connective tissue was removed from the formalin preserved aortas. The lipids were extracted in 2:1 chloroform:methanol with 5α-chloestane added as an internal standard. Gas chromatography yielded the concentration of FC. After saponification, TC was determined by GC. Aortic CE was subsequently determined by subtracting FC from TC and multiplying the difference by 1.6724. Protein was determined by the method of Lowry on the delipidated aortas following digestion in 1 N sodium hydroxide.
ACAT Assays
Flash frozen liver samples from the “Treatment” group (N=3) and the “No Treatment” group (N=3) were used to determine ACAT activity. Microsomes were isolated from the liver samples as previously described by Carr et al 25. Microsomal protein concentration was measured by the method of Lowry et al 26. A 50 ug aliquot of microsomal protein was used to determine ACAT activity as previously described 2, 25. Briefly, microsomal protein, 1 mg of BSA, and 50 nmol of free cholesterol in 45%(w/v) β-cyclodextrin were mixed together and subsequently incubated for 30 min at 37°C. Then, 30 nmol of [14C]oleoyl-CoA was added to the mix and incubated for 10 min. The reaction was stopped by the addition of 2:1 chloroform-methanol. After the phases separated, the organic phase was removed and applied to a TLC plate. The CE band was scraped, suspended in scintillation fluid, and counted for 14C radioactivity.
ACAT2 activity was specifically determined by comparing the results of F-1394 on total ACAT activity to those obtained with the potent ACAT 2 inhibitor Pyripyropene A (PPPA), as done before 27. As shown in Supplemental Figure 1, F-1394 added to the diet reduced ACAT2 activity by ∼50% in the hepatic microsomes isolated from the treated mice.
Tissue processing, histology, immunohistochemistry, and morphometry
For harvesting tissue specimens, mice were exsanguinated and perfusion-fixed in 4% paraformaldehyde (at 100 mmHg, 5 min). The heart was transected at the lower poles of the atria and processed for paraffin embedding. Serial 5μm sections (4 per mouse for all section-based assays) were taken from paraffin-embedded aortic roots and stained with Combined Masson's trichome-elastic (CME) stain 19, 28.
For immunohistochemical analyses, 5μm thick formalin fixed, paraffin embedded sections were deparaffinized and hydrated with PBS, then incubated with 3% hydrogen peroxide, washed with PBS and subsequently exposed to blocking serum (normal horse serum, normal rabbit serum, normal goat serum; Vector Laboratories, Burlingame, CA). The primary antibody (rat anti-mouse CD68 ABD Serotec; rabbit monoclonal CCR7 (Y59); rat anti-mouse VCAM-1/CD106 Southern Biotechnology) was applied overnight at 4°C as previously described 19. Immunodetection was performed using biotinylated horse anti-mouse secondary (Vector Laboratories, Burlingame, CA), peroxidase-labeled streptavidin (Jackson Immuno Research, West Grove, PA), and visualized using the Nova Red substrate (Vector Laboratories, Burlingame, CA). The slides were then counterstained in hematoxylin. The appropriate positive control was used and the antibody was omitted for the negative control.
For TUNEL staining to detect apoptotic cells, the hydrated sections were incubated with 3% hydrogen peroxide and subsequently treated following the protocol of an apoptosis detection kit (ApopTag® Peroxidase in situ apoptosis detection kit; Millipore, Billerica, MA). Staining was visualized using DAB (DAKO) and slides were counterstained with methyl green.
Apoptotic cells in atherosclerotic lesions were detected by TUNEL (Tdt-mediated dUTP nick-end labeling) after proteinase K treatment. Stringency methods of Kockx were followed to avoid non-specific staining 29. Nuclei were counterstained and the sections were imaged and quantified by microscopy and Image J software in a blinded manner. For percent TUNEL positive cells, TUNEL + nuclei were divided by total intimal nuclei that were counted from serial sections along the aortic root. Percent necrosis was quantified as hematoxylin and eosin negative intimal area as previously described 30.
Microscopic images from stained sections were captured by a Sony 3CCD Video camera attached to a Zeiss (Axioscop) light microscope, digitized, and analyzed by Image-Pro Plus software (Media Cybernetics, Silver Spring, MD) as previously described 19. Morphometric measurement of the total lesion area or the area stained by a specific antibody was determined and averaged from 3 to 4 serial sections (separated by 50–60μm) per animal.
During tissue processing (i.e., de-paraffinization and CME staining) in ethanol and xylene, 95-99% of plaque lipid was dissolved, leaving clear areas visible by light microscopy 31. As described previously 32,33, such transparent areas in the plaque was quantified and used to represent areas in which lipid was present prior to processing 31, 34.
In Situ Efferocytosis Assay
The efferocytosis assay was performed on frozen aortic root sections from five mice in the Treatment group and five mice in the No Treatment group. The assay was conducted using procedures previously described35-39. Briefly, in situ efferocytosis was assessed by counting free and macrophage-associated apoptotic (TUNEL positive) nuclei (Hoechst). Macrophage-associated apoptotic cells followed the criteria of TUNEL positive nuclei surrounded by or in contact with neighboring F4/80+ macrophages. “Free” apoptotic cells exhibited nuclear condensation, loss of antibody F4/80 reactivity, and were not in contact with neighboring Mφs. The quantitative analysis was conducted by an observer that was blinded to the identity of the samples.
Statistical analysis
Numerical data were expressed as means ± standard error of the mean (SEM). The means between different experimental groups were compared by One-way ANOVA with Bonferroni post hoc analysis. P values <0.05, <0.01, and <0.001 were considered significant. For the calculation of Spearman r-values, the software package Prism (GraphPad Software, San Diego) was used, with statistical significance determined by 2-tailed t-testing. Additional testing by a professional statistician is described at the end of Results.
Results
General effects of ACAT inhibition
ApoE-/- mice had comparable body weights after 14 weeks on WD among the 3 groups prior to F1394 treatment (not shown). After 14 more weeks, body weights between mice continued on WD (“No Treatment”) and WD plus F1394 (“Treatment”) remained comparable (35.0±1.8 and 35.6±1.6 g, respectively, NS). No mice died during the treatment. The dermatologic changes in ACAT1 knockout mice (alopecia and dermal hypertrophy40) were not observed in the F1394 group.
Effects of ACAT inhibition on plasma lipids
ApoE-/- mice develop hypercholesterolemia spontaneously, and this is further increased by WD feeding 41,42. After 14 weeks on WD, total cholesterol (TC) level in the Baseline group reached ∼1300 mg/dL, and HDL cholesterol (HDL-C) was 26.8 mg/dL (Table 1). Continuation of WD for 14 more weeks did not significantly change TC or HDL-C levels. Compared to the Baseline and No Treatment, F1394 treatment tended to decrease TC modestly, though this was not statistically significant (Table 1). F1394 treatment had no effect on the plasma levels of HDL-C.
Table 1. Plasma Cholesterol Values in the Different Experimental Groups.
| Pretreatment | No F1394 | F1394 | |
|---|---|---|---|
| Total Cholesterol (mg/dL) | 1313±138 | 1400±132 | 1061±104 |
| HDL Cholesterol (mg/dL) | 26.8±3.3 | 30.3±6.0 | 30.2±2.8 |
Shown are mean (+/-SE) values. N=9 for each group. By ANOVA, the differences among the groups did not achieve statistical significance (P=0.1618).
More detailed analyses of plasma lipoprotein cholesteryl ester (CE) content are summarized on Supplemental Figures 2&3. There were no obvious differences in the pattern of fatty acid-type esterified to cholesterol related to treatment in the different lipoprotein classes (Supplemental Figure 2). Specifically, there was no difference in the % of CE that was cholesteryl oleate (thought to be particularly atherogenic 43) in any lipoprotein class (Supplemental Figure 3).
Effects of ACAT inhibition on the progression of pre-existing atherosclerotic lesion size and lipid content
After 14 weeks on WD, apoE-/- mice developed advanced atherosclerotic lesions in the aortic root containing necrotic lipid cores and cholesterol clefts, as shown in Figure 1A. WD-feeding for 14 more weeks led to further progression of the lesion as reflected by the ∼3-fold increase in size (Figure 1B) and the ∼8 fold increase in lipid content (Figure 1C). As shown (Figure 1A&B), F1394 treatment retarded the progression in lesion size by about 30% (P<0.05) and in lipid content, as determined histologically (Figure 1C), by ∼50% (P<0.05).
Figure 1. Histology of atherosclerotic lesions in experimental groups and measurements of lesion size and lipid content.
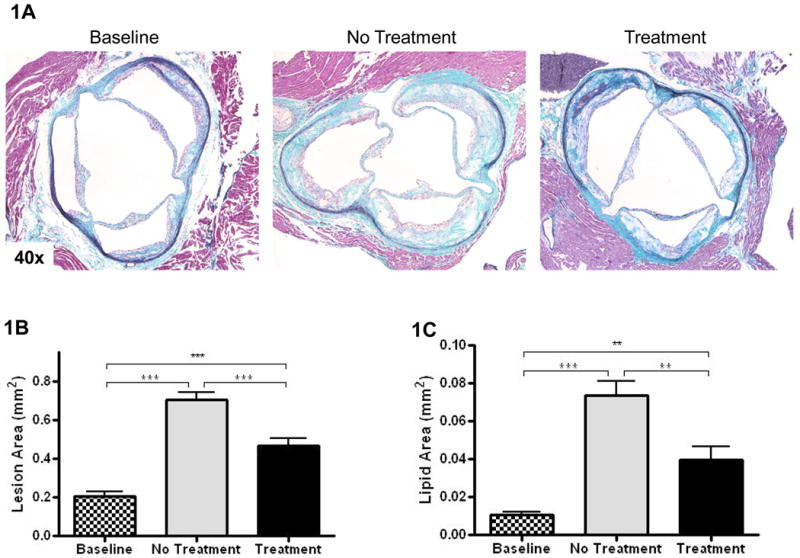
A) Representative CME-stained slides from the indicated groups are shown at 40×. B) and C) Bar graphs represent lesion sizes and lipid contents, respectively. Baseline N=9, No Treatment N=12, and Treatment N=12. Data analyzed by Bonferroni's Multiple Comparison test. Data are expressed as Mean ± SEM. Statistically significant differences (P < 0.05, P < 0.01, and P < 0.001) are represented by *, **, and ***, respectively.
Changes in aortic lipid content were also measured chemically and the results are summarized in Figure 2. As shown, at the end of the 28 week study, aortic total cholesterol, free cholesterol, and cholesteryl ester contents were all statistically lower in the treatment vs. no treatment group.
Figure 2. Effects of ACAT inhibition on aortic free cholesterol (FC), cholesteryl ester (CE), and total cholesterol (TC) content.

Aortic FC (A), CE (B), and TC (C) contents in baseline, untreated (“No Treatment”), and treated with F1394 ACAT inhibitor (“Treatment”) mouse arches were measured by gas-liquid chromatography. Results expressed as μg FC, CE, or TC per g of aortic protein. Baseline N=7, No Treatment N=7, and Treatment N=6. Data analyzed by Bonferroni's Multiple Comparison test. Data are expressed as Mean ± SEM. Statistically significant differences (P < 0.05, P < 0.01, and P < 0.001) are represented by *, **, and ***, respectively.
Effects of ACAT inhibition on plaque contents of macrophages and tissue factor
As shown in Figure 3A, macrophage marker Mac2-positive areas were localized mainly to the subendothelial area in all experimental groups. Compared to the baseline group, over the next 14 weeks of WD feeding, without ACAT inhibition there was an increase in the Mac-2-positive area, which was prevented by F1394 treatment (Figure 3A and B).
Figure 3. Effects of ACAT inhibition on lesion macrophage content.
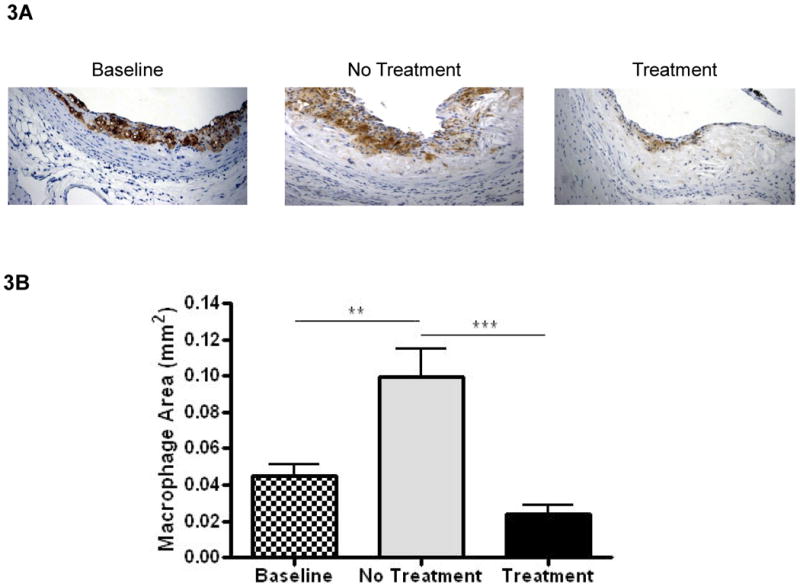
A) Representative sections stained for macrophages using the Mac2 and CD68 antibody were visualized at 200×. B) Bar graph represents measurements of stained areas for Mac2 and CD68. Baseline N=9, No Treatment N=12, and Treatment N=12. Data analyzed by Bonferroni's Multiple Comparison test. Data are expressed as Mean ± SEM. Statistically significant differences (P < 0.05, P < 0.01, and P < 0.001) are represented by *, **, and ***, respectively.
Tissue factor (TF) content of plaques is considered to be the major culprit in the formation of the occlusive thrombus that follows plaque rupture. Though all 3 of the major cell types (endothelial cells, macrophages, and smooth muscle cells) can produce TF, we have found that the majority of it is associated with macrophages 19, 44. Consistent with this association, as shown in Figures 4A and 4B, the distribution of TF staining and its quantitative changes were parallel to the corresponding data for Mac-2. In other words, without ACAT inhibition, TF content rose in plaques in the apoE-/- mice continued on the WD for an additional 14 weeks, but were maintained at the pretreatment levels in the mice fed the WD containing F1394.
Figure 4. Effects on ACAT inhibition on lesion tissue factor content.
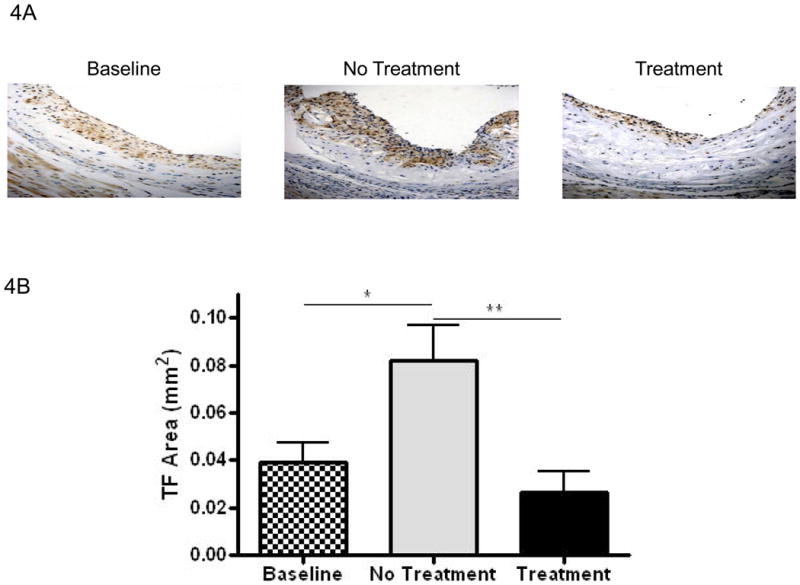
A) Representative sections stained for tissue factor visualized at 200×. B) Bar graph represents measurements of stained areas for tissue factor. Baseline N=8, No Treatment N=7, and Treatment N=8. Data analyzed by Bonferroni's Multiple Comparison test. Data are expressed as Mean ± SEM. Statistically significant differences (P < 0.05, P < 0.01, and P < 0.001) are represented by *, **, and ***, respectively.
Effects of ACAT inhibition on apoptosis, plaque necrosis, and efferocytosis of cells in atherosclerotic lesions
Complete deficiency of ACAT1, the major macrophage ACAT isoform, has been shown to cause increased lesional free cholesterol, macrophage death, and tissue inflammation associated with increased atherosclerotic lesions 6. In studies using macrophage cell lines in which ACAT was inhibited (by Sandoz compound 58035) at the same time cells were loaded with cholesterol, it was shown that one basis for the macrophage cell death in ACAT1-deficient mice was the accumulation of FC in the ER membrane, triggering an ER-stress response that culminated in apoptosis 13.
Given that under the present experimental conditions the ACAT inhibitor actually decreased lesional free cholesterol, not increased it, we predicted that lesional macrophage apoptosis would not be increased. As shown in Figure 5A, TUNEL-positive areas were present in most animals from all experimental groups. As expected 15-17, the positive areas were close to the necrotic cores. Compared to the baseline group, with continued feeding of the WD, TUNEL staining increased as lesions further advanced (Figure 5B). As predicted by the decrease in lesional free cholesterol, partial ACAT inhibition was associated with decreased TUNEL staining (Figure 5B). The pattern of TUNEL data was independent of whether the area of staining or the % of nuclei positive for the staining (Figure 5C) was assessed. While the decrease in apoptosis is perfectly consistent with the decrease in plaque free cholesterol, this finding indicates that the decrease in lesional macrophages cannot be explained by an increase in macrophage death. In addition, while there was an increase in the necrotic areas of the plaques associated with continued WD feeding, necrosis was not greater in the F1394-treated group relative to the untreated group; in fact the trend was for a smaller increase with treatment (Figure 5D). There was also no difference in efferocytosis (the clearance of dead cells) in the plaques of mice treated or not treated with F1394 (Figure 6).
Figure 5. Effects of ACAT inhibition on measurements of apoptosis and necrosis in atherosclerotic lesions.
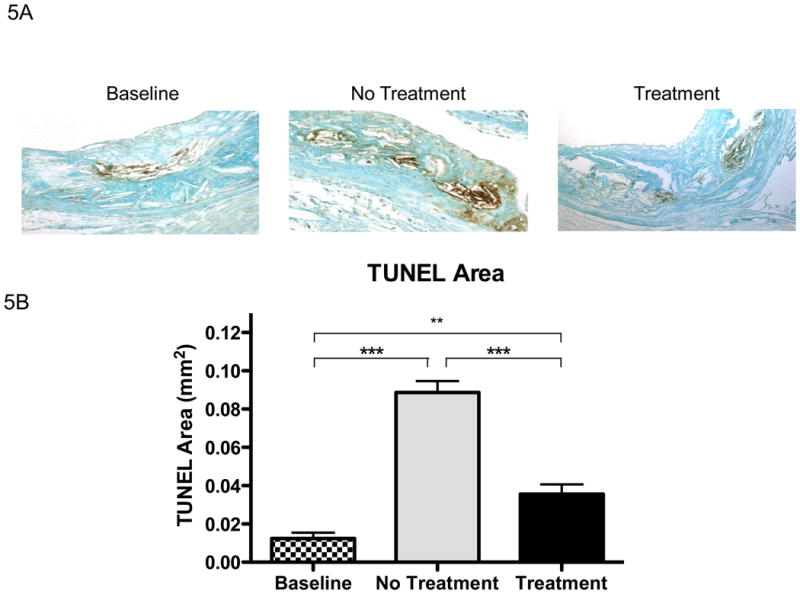

A) Representative sections stained for apoptosis using TUNEL staining and visualized at 200×. B) Bar graph represents measurements of stained areas positive for apoptosis; Baseline N=7, No Treatment N=8, and Treatment N=8. C) % of nuclei that were TUNEL positive in the plaques in each group; Baseline N=7, No Treatment N=8, and Treatment N=8. D) Area of plaque that was necrotic in each group; same N′s as in panel C. Data analyzed by ANOVA, followed by Bonferroni's Multiple Comparison test. Data are expressed as Mean ± SEM. Statistically significant differences (P < 0.05, P < 0.01, and P < 0.001) are represented by *, **, and ***, respectively.
Figure 6. Effects of ACAT inhibition on phagocytosis efficiency.

Free-to-macrophage-associated ratios of apoptotic (TUNEL-positive) cells in Apoe-/- aortic root lesions from mice untreated (“No Treatment”) and treated (“Treatment”) with F1394 ACAT inhibitor. There was no significant change in phagocytosis efficiency of apoptotic cells between the two groups, as indicated by the similar ratios. N=5 for each group. Data analyzed by two-tailed t-test. Data are expressed as Mean ± SEM.
Effects of ACAT inhibition were independent of plasma lipid levels
As shown in Table 1, ACAT inhibition resulted in a modest decrease in the plasma levels of non-HDL cholesterol, as expected from its effects on intestinal cholesterol absorption and VLDL cholesteryl ester content 3. Nonetheless, the levels in the F1394 group remained high and in the range known to be highly atherogenic. To exclude that the effects of ACAT inhibition on lesion area or macrophage content were not indirect through changes in plasma cholesterol levels, as before 5, correlation analyses were performed. The Spearman r-values for the correlations between plasma total cholesterol (TC) and the macrophage content and plaque area were 0.23 (P=0.24) and 0.20 (P=0.31), respectively, indicating that the effects of ACAT inhibition were independent of plasma lipid levels.
To further support the finding that plasma TC was not statistically related to any of the outcomes and does not account for the differences in them among the groups, analyses of covariance (ANCOVA) were performed to compare the outcomes among the three study groups (baseline, treatment, no treatment) adjusting for plasma TC. There were still significant differences in the outcome measures among the three groups when adjusting for TC. In other words, adjustment for plasma TC was not significantly associated with the outcome in any analysis. In a separate analysis in which we normalized for plaque size, the patterns we reported for changes in macrophage and tissue factor content in the treatment group were also not changed.
Discussion
In a previous report 5, we showed that partial inhibition of ACAT reduced the initiation and progression of early atherosclerosis in apoE-/- mice without evidence of obvious toxicity, in agreement with a recent report in which a specific ACAT 2 inhibitor was used in these mice 45. In the present study, we have focused on a more clinically relevant scenario- the effects of partial ACAT inhibition on established atherosclerotic plaques that progressed beyond the foam cell stage. Compared to the baseline group, over the next 14 weeks, the plaque size increased, but the progression of increase was significantly retarded in the F1394 group. ACAT inhibition was also associated with beneficial changes in plaque composition. These included fewer macrophages and reductions in the content of tissue factor or neutral lipid in general, and total cholesterol, free cholesterol, and cholesteryl ester in particular, important factors in the pathophysiology of atherothrombosis. Furthermore, these effects were not associated with toxicity that was systemic (e.g., normal weight gain and no dermal pathology) or in the plaques themselves, as assessed by the level of apoptosis, efferocytosis, or signs of necrosis. The lack of an increase in apoptosis is perfectly consistent with the drug-induced decrease in lesional free cholesterol.
As expected, ACAT inhibition tended to have a modest impact on plasma cholesterol levels (Table 1), though it was not statistically significant. As in our previous study 5, there were no statistically significant correlations between the major findings and plasma cholesterol levels. As before, even in the F1394 group, plasma cholesterol levels (>1000 mg/dL) were far in excess of those known to accelerate plaque progression in apoE-/- mice (e.g., 46). Thus, the benefits of partial ACAT inhibition on plaque progression and composition were in the face of persistent, severe, hyperlipidemia.
Given that plaque macrophage content decreased without an increase in apoptosis, it is interesting to consider the mechanisms for this decrease. The two major kinetic arms of interest would seem to be the recruitment of monocytes to and the egress of macrophages from plaques. Regression of atherosclerosis attributable to both arms has been reported (e.g., 47, 48). In preliminary studies, we have evidence to support both kinetic possibilities. For example, in the F1394 treated mice, there appeared to be less endothelial, as well as sub-endothelial, expression of a major monocyte recruitment factor, VCAM-1 (Supplemental Figure 4). VCAM-1 expression on endothelial cells mediates the adhesion of monocytes and promotes their subsequent entry into the intimal space, and its decreased expression results in less atherosclerosis in apoE-/- mice 49. In addition, we have recently reported 50 that the promoter of the chemokine receptor CCR7, a macrophage egress factor during atherosclerosis regression 48, is regulated in vitro and in vivo by a sterol responsive element (SRE), which is likely to be stimulated in the F1394-treated plaque macrophages, given the reduction in sterol content we observed (Figure 2).
The potential of ACAT inhibitors as anti-atherosclerotic agents has been a hotly contested issue. On one hand, ACAT has been considered to be atherogenic by promoting plaque growth by allowing excess macrophage cholesterol to be stored in the form of ester, as well as by increasing the CE content of hepatic lipoproteins, the conveyors of lipids to the arterial wall. Thus, if ACAT were inhibited in macrophages and foam cells, assuming sufficient local acceptors of free cholesterol, excess cholesterol would not accumulate, thereby protecting against foam cell formation, expansion, activation, and apoptosis. Indeed, consistent with the theoretical benefits of ACAT inhibition on atherosclerosis are a number of animal studies in which a variety of inhibitors have delayed progression of disease and favorably altered plaque composition 5, 51, 52.
Counterbalancing the potential benefits of ACAT inhibition is the literature that partial or complete deficiency of ACAT activity is pro-atherogenic. For example, mice with complete deficiency of macrophage ACAT (ACAT1-/- mice) have evidence of large, inflamed plaques and skin pathology attributed to the deposition of crystals of free cholesterol 6. While it is tempting to speculate that the difference between this study and the beneficial outcomes in many pre-clinical inhibitor studies was the degree of toxicity, two clinical studies 52,53 also showed that ACAT inhibition did not reduce plaque volume as measured by intravascular ultrasound (IVUS), although IVUS would not have detected the changes in plaque composition as the histological assays we used can. As recently reviewed 54, a plausible explanation for these apparent clinical failures includes the accumulation of free cholesterol in macrophages and foam cells, which, in addition to the formation of toxic cholesterol crystals, can also lead to ER stress, activation of the inflammasome, apoptosis, and impaired efferocytosis of dead cells 9-17, 39, 55-57.
The mechanisms(s) for the apparent discrepancies between the present results and the clinical trial data will require further study. A key feature of the study here was a decrease in lesional free cholesterol, which is thought to be a key determinant of advanced lesional macrophage death and plaque necrosis. Thus, in settings where plaque free cholesterol increases, such as in the course of normal advanced plaque progression or in the setting of complete ACAT1 inhibition, advanced lesional macrophage apoptosis and plaque necrosis ensue. Accordingly, we propose that under conditions of partial ACAT inhibition by the drug used in this study, either enough HDL particles were available to prevent surpassing a critical threshold level of free cholesterol in the macrophages and foam cells or the residual level of ACAT activity (estimated to be ∼50%;5) was sufficiently protective.
In summary, we have shown that when mice with established plaques are treated with ACAT inhibitor F1394, there are major beneficial results characterized by slower plaque progression and decreased contents of macrophage foam cells, lipids, and tissue factor. Taking into account the previous animal and clinical data, these results argue that the inhibition of ACAT is still a viable clinical target to retard plaque progression, but that the dosing may be the critical issue. Too much or too little inhibition is likely to be undesirable because of cellular toxicity or lack of efficacy, respectively. While this is an important practical issue to resolve, until current imaging techniques are sufficiently sensitive to changes in plaque composition, important potential benefits of ACAT inhibition that are effected through changes in plaque composition, may go undetected, making the determination of efficacy difficult to assess.
Supplementary Material
Acknowledgments
These studies were supported by NIH grants HL61814, HL70524, HL084312 (E.A.F), HL073364 (M.B.T), HL49373 (L.L.R), and HL54591 & HL75662 (I.T.). J.E.F was supported by NIH grant fellowship AG029748, and E.T. was supported by an NIH K99/R00 grant (HL097021) and by an Irving Institute for Clinical and Translational Research Pilot Grant.
Footnotes
None of the authors have disclosures to declare.
References
- 1.Buhman KF, Accad M, Farese RV. Mammalian acyl-coa:Cholesterol acyltransferases. Biochim Biophys Acta. 2000;1529:142–154. doi: 10.1016/s1388-1981(00)00144-x. [DOI] [PubMed] [Google Scholar]
- 2.Lee RG, Willingham MC, Davis MA, Skinner KA, Rudel LL. Differential expression of acat1 and acat2 among cells within liver, intestine, kidney, and adrenal of nonhuman primates. J Lipid Res. 2000;41:1991–2001. [PubMed] [Google Scholar]
- 3.Rudel LL, Lee RG, Cockman TL. Acyl coenzyme a: Cholesterol acyltransferase types 1 and 2: Structure and function in atherosclerosis. Curr Opin Lipidol. 2001;12:121–127. doi: 10.1097/00041433-200104000-00005. [DOI] [PubMed] [Google Scholar]
- 4.Rudel LL, Lee RG, Parini P. Acat2 is a target for treatment of coronary heart disease associated with hypercholesterolemia. Arterioscler Thromb Vasc Biol. 2005;25:1112–1118. doi: 10.1161/01.ATV.0000166548.65753.1e. [DOI] [PubMed] [Google Scholar]
- 5.Kusunoki J, Hansoty DK, Aragane K, Fallon JT, Badimon JJ, Fisher EA. Acyl-coa:Cholesterol acyltransferase inhibition reduces atherosclerosis in apolipoprotein e-deficient mice. Circulation. 2001;103:2604–2609. doi: 10.1161/01.cir.103.21.2604. [DOI] [PubMed] [Google Scholar]
- 6.Fazio S, Major AS, Swift LL, Gleaves LA, Accad M, Linton MF, Farese RV., Jr Increased atherosclerosis in ldl receptor-null mice lacking acat1 in macrophages. J Clin Invest. 2001;107:163–171. doi: 10.1172/JCI10310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kellner-Weibel G, Geng YJ, Rothblat GH. Cytotoxic cholesterol is generated by the hydrolysis of cytoplasmic cholesteryl ester and transported to the plasma membrane. Atherosclerosis. 1999;146:309–319. doi: 10.1016/s0021-9150(99)00155-0. [DOI] [PubMed] [Google Scholar]
- 8.Kellner-Weibel G, Yancey PG, Jerome WG, Walser T, Mason RP, Phillips MC, Rothblat GH. Crystallization of free cholesterol in model macrophage foam cells. Arterioscler Thromb Vasc Biol. 1999;19:1891–1898. doi: 10.1161/01.atv.19.8.1891. [DOI] [PubMed] [Google Scholar]
- 9.Colgan SM, Tang D, Werstuck GH, Austin RC. Endoplasmic reticulum stress causes the activation of sterol regulatory element binding protein-2. Int J Biochem Cell Biol. 2007;39:1843–1851. doi: 10.1016/j.biocel.2007.05.002. [DOI] [PubMed] [Google Scholar]
- 10.Devries-Seimon T, Li Y, Yao PM, Stone E, Wang Y, Davis RJ, Flavell R, Tabas I. Cholesterol-induced macrophage apoptosis requires er stress pathways and engagement of the type a scavenger receptor. J Cell Biol. 2005;171:61–73. doi: 10.1083/jcb.200502078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dickhout JG, Colgan SM, Lhotak S, Austin RC. Increased endoplasmic reticulum stress in atherosclerotic plaques associated with acute coronary syndrome: A balancing act between plaque stability and rupture. Circulation. 2007;116:1214–1216. doi: 10.1161/CIRCULATIONAHA.107.728378. [DOI] [PubMed] [Google Scholar]
- 12.Zhou J, Lhotak S, Hilditch BA, Austin RC. Activation of the unfolded protein response occurs at all stages of atherosclerotic lesion development in apolipoprotein e-deficient mice. Circulation. 2005;111:1814–1821. doi: 10.1161/01.CIR.0000160864.31351.C1. [DOI] [PubMed] [Google Scholar]
- 13.Feng B, Yao PM, Li Y, Devlin CM, Zhang D, Harding HP, Sweeney M, Rong JX, Kuriakose G, Fisher EA, Marks AR, Ron D, Tabas I. The endoplasmic reticulum is the site of cholesterol-induced cytotoxicity in macrophages. Nat Cell Biol. 2003;5:781–792. doi: 10.1038/ncb1035. [DOI] [PubMed] [Google Scholar]
- 14.Li Y, Gerbod-Giannone MC, Seitz H, Cui D, Thorp E, Tall AR, Matsushima GK, Tabas I. Cholesterol-induced apoptotic macrophages elicit an inflammatory response in phagocytes, which is partially attenuated by the mer receptor. J Biol Chem. 2006;281:6707–6717. doi: 10.1074/jbc.M510579200. [DOI] [PubMed] [Google Scholar]
- 15.Tabas I. Apoptosis and plaque destabilization in atherosclerosis: The role of macrophage apoptosis induced by cholesterol. Cell Death Differ. 2004;11(1):S12–16. doi: 10.1038/sj.cdd.4401444. [DOI] [PubMed] [Google Scholar]
- 16.Tabas I. Consequences and therapeutic implications of macrophage apoptosis in atherosclerosis: The importance of lesion stage and phagocytic efficiency. Arterioscler Thromb Vasc Biol. 2005;25:2255–2264. doi: 10.1161/01.ATV.0000184783.04864.9f. [DOI] [PubMed] [Google Scholar]
- 17.Tabas I. Apoptosis and efferocytosis in mouse models of atherosclerosis. Curr Drug Targets. 2007;8:1288–1296. doi: 10.2174/138945007783220623. [DOI] [PubMed] [Google Scholar]
- 18.Stary HC, Chandler AB, Dinsmore RE, Fuster V, Glagov S, Insull W, Jr, Rosenfeld ME, Schwartz CJ, Wagner WD, Wissler RW. A definition of advanced types of atherosclerotic lesions and a histological classification of atherosclerosis. A report from the committee on vascular lesions of the council on arteriosclerosis, american heart association. Circulation. 1995;92:1355–1374. doi: 10.1161/01.cir.92.5.1355. [DOI] [PubMed] [Google Scholar]
- 19.Rong JX, Li J, Reis ED, Choudhury RP, Dansky HM, Elmalem VI, Fallon JT, Breslow JL, Fisher EA. Elevating high-density lipoprotein cholesterol in apolipoprotein e-deficient mice remodels advanced atherosclerotic lesions by decreasing macrophage and increasing smooth muscle cell content. Circulation. 2001;104:2447–2452. doi: 10.1161/hc4501.098952. [DOI] [PubMed] [Google Scholar]
- 20.Lee RG, Kelley KL, Sawyer JK, Farese RV, Jr, Parks JS, Rudel LL. Plasma cholesteryl esters provided by lecithin:Cholesterol acyltransferase and acyl-coenzyme a:Cholesterol acyltransferase 2 have opposite atherosclerotic potential. Circ Res. 2004;95:998–1004. doi: 10.1161/01.RES.0000147558.15554.67. [DOI] [PubMed] [Google Scholar]
- 21.Liu KS. Preparation of fatty acid methyl esters for gas-chromatographic analysis of lipids in biological materials. J Am Oil Chem Soc. 1994:1179–1188. [Google Scholar]
- 22.Bligh EG, Dyer WJ. A rapid method of total lipid extraction and purification. Can J Biochem Physiol. 1959;37:911–917. doi: 10.1139/o59-099. [DOI] [PubMed] [Google Scholar]
- 23.Willner EL, Tow B, Buhman KK, Wilson M, Sanan DA, Rudel LL, Farese RV., Jr Deficiency of acyl coa:Cholesterol acyltransferase 2 prevents atherosclerosis in apolipoprotein e-deficient mice. Proc Natl Acad Sci U S A. 2003;100:1262–1267. doi: 10.1073/pnas.0336398100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Temel RE, Lee RG, Kelley KL, Davis MA, Shah R, Sawyer JK, Wilson MD, Rudel LL. Intestinal cholesterol absorption is substantially reduced in mice deficient in both abca1 and acat2. J Lipid Res. 2005;46:2423–2431. doi: 10.1194/jlr.M500232-JLR200. [DOI] [PubMed] [Google Scholar]
- 25.Carr TP, Parks JS, Rudel LL. Hepatic acat activity in african green monkeys is highly correlated to plasma ldl cholesteryl ester enrichment and coronary artery atherosclerosis. Arterioscler Thromb. 1992;12:1274–1283. doi: 10.1161/01.atv.12.11.1274. [DOI] [PubMed] [Google Scholar]
- 26.Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. Protein measurement with the folin phenol reagent. J Biol Chem. 1951;193:265–275. [PubMed] [Google Scholar]
- 27.Lada AT, Davis M, Kent C, Chapman J, Tomoda H, Omura S, Rudel LL. Identification of acat1- and acat2-specific inhibitors using a novel, cell-based fluorescence assay: Individual acat uniqueness. J Lipid Res. 2004;45:378–386. doi: 10.1194/jlr.D300037-JLR200. [DOI] [PubMed] [Google Scholar]
- 28.Reis ED, Li J, Fayad ZA, Rong JX, Hansoty D, Aguinaldo JG, Fallon JT, Fisher EA. Dramatic remodeling of advanced atherosclerotic plaques of the apolipoprotein e-deficient mouse in a novel transplantation model. J Vasc Surg. 2001;34:541–547. doi: 10.1067/mva.2001.115963. [DOI] [PubMed] [Google Scholar]
- 29.Kockx MM. Apoptosis in the atherosclerotic plaque: Quantitative and qualitative aspects. Arterioscler Thromb Vasc Biol. 1998;18:1519–1522. doi: 10.1161/01.atv.18.10.1519. [DOI] [PubMed] [Google Scholar]
- 30.Thorp E, Li G, Seimon TA, Kuriakose G, Ron D, Tabas I. Reduced apoptosis and plaque necrosis in advanced atherosclerotic lesions of apoe-/- and ldlr-/- mice lacking chop. Cell metabolism. 2009;9:474–481. doi: 10.1016/j.cmet.2009.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mitchinson MJ. Insoluble lipids in human atherosclerotic plaques. Atherosclerosis. 1982;45:11–15. doi: 10.1016/0021-9150(82)90167-8. [DOI] [PubMed] [Google Scholar]
- 32.Moreno PR, Lodder RA, Purushothaman KR, Charash WE, O'Connor WN, Muller JE. Detection of lipid pool, thin fibrous cap, and inflammatory cells in human aortic atherosclerotic plaques by near-infrared spectroscopy. Circulation. 2002;105:923–927. doi: 10.1161/hc0802.104291. [DOI] [PubMed] [Google Scholar]
- 33.Choudhury RP, Rong JX, Trogan E, Elmalem VI, Dansky HM, Breslow JL, Witztum JL, Fallon JT, Fisher EA. High-density lipoproteins retard the progression of atherosclerosis and favorably remodel lesions without suppressing indices of inflammation or oxidation. Arterioscler Thromb Vasc Biol. 2004;24:1904–1909. doi: 10.1161/01.ATV.0000142808.34602.25. [DOI] [PubMed] [Google Scholar]
- 34.Guyton JR, Klemp KF. Transitional features in human atherosclerosis. Intimal thickening, cholesterol clefts, and cell loss in human aortic fatty streaks. Am J Pathol. 1993;143:1444–1457. [PMC free article] [PubMed] [Google Scholar]
- 35.Aprahamian T, Rifkin I, Bonegio R, Hugel B, Freyssinet JM, Sato K, Castellot JJ, Jr, Walsh K. Impaired clearance of apoptotic cells promotes synergy between atherogenesis and autoimmune disease. J Exp Med. 2004;199:1121–1131. doi: 10.1084/jem.20031557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Potter PK, Cortes-Hernandez J, Quartier P, Botto M, Walport MJ. Lupus-prone mice have an abnormal response to thioglycolate and an impaired clearance of apoptotic cells. J Immunol. 2003;170:3223–3232. doi: 10.4049/jimmunol.170.6.3223. [DOI] [PubMed] [Google Scholar]
- 37.Schrijvers DM, De Meyer GR, Kockx MM, Herman AG, Martinet W. Phagocytosis of apoptotic cells by macrophages is impaired in atherosclerosis. Arterioscler Thromb Vasc Biol. 2005;25:1256–1261. doi: 10.1161/01.ATV.0000166517.18801.a7. [DOI] [PubMed] [Google Scholar]
- 38.Taylor PR, Carugati A, Fadok VA, Cook HT, Andrews M, Carroll MC, Savill JS, Henson PM, Botto M, Walport MJ. A hierarchical role for classical pathway complement proteins in the clearance of apoptotic cells in vivo. J Exp Med. 2000;192:359–366. doi: 10.1084/jem.192.3.359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Thorp E, Cui D, Schrijvers DM, Kuriakose G, Tabas I. Mertk receptor mutation reduces efferocytosis efficiency and promotes apoptotic cell accumulation and plaque necrosis in atherosclerotic lesions of apoe-/- mice. Arterioscler Thromb Vasc Biol. 2008 doi: 10.1161/ATVBAHA.108.167197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Accad M, Smith SJ, Newland DL, Sanan DA, King LE, Jr, Linton MF, Fazio S, Farese RV., Jr Massive xanthomatosis and altered composition of atherosclerotic lesions in hyperlipidemic mice lacking acyl coa:Cholesterol acyltransferase 1. J Clin Invest. 2000;105:711–719. doi: 10.1172/JCI9021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Breslow JL. Mouse models of atherosclerosis. Science. 1996;272:685–688. doi: 10.1126/science.272.5262.685. [DOI] [PubMed] [Google Scholar]
- 42.Nakashima Y, Plump AS, Raines EW, Breslow JL, Ross R. Apoe-deficient mice develop lesions of all phases of atherosclerosis throughout the arterial tree. Arterioscler Thromb. 1994;14:133–140. doi: 10.1161/01.atv.14.1.133. [DOI] [PubMed] [Google Scholar]
- 43.Degirolamo C, Shelness GS, Rudel LL. Ldl cholesteryl oleate as a predictor for atherosclerosis: Evidence from human and animal studies on dietary fat. J Lipid Res. 2009;(50):S434–439. doi: 10.1194/jlr.R800076-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rong JX, Kusunoki J, Oelkers P, Sturley SL, Fisher EA. Acyl-coenzymea (coa):Cholesterol acyltransferase inhibition in rat and human aortic smooth muscle cells is nontoxic and retards foam cell formation. Arterioscler Thromb Vasc Biol. 2005;25:122–127. doi: 10.1161/01.ATV.0000148202.49842.3b. [DOI] [PubMed] [Google Scholar]
- 45.Ohshiro T, Matsuda D, Sakai K, Degirolamo C, Yagyu H, Rudel LL, Omura S, Ishibashi S, Tomoda H. Pyripyropene a, an acyl-coenzyme a:Cholesterol acyltransferase 2-selective inhibitor, attenuates hypercholesterolemia and atherosclerosis in murine models of hyperlipidemia. Arterioscler Thromb Vasc Biol. 2011;31:1108–1115. doi: 10.1161/ATVBAHA.111.223552. [DOI] [PubMed] [Google Scholar]
- 46.Plump AS, Smith JD, Hayek T, Aalto-Setala K, Walsh A, Verstuyft JG, Rubin EM, Breslow JL. Severe hypercholesterolemia and atherosclerosis in apolipoprotein e-deficient mice created by homologous recombination in es cells. Cell. 1992;71:343–353. doi: 10.1016/0092-8674(92)90362-g. [DOI] [PubMed] [Google Scholar]
- 47.Potteaux S, Gautier EL, Hutchison SB, van Rooijen N, Rader DJ, Thomas MJ, Sorci-Thomas MG, Randolph GJ. Suppressed monocyte recruitment drives macrophage removal from atherosclerotic plaques of apoe-/- mice during disease regression. J Clin Invest. 2011;121:2025–2036. doi: 10.1172/JCI43802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Trogan E, Feig JE, Dogan S, Rothblat GH, Angeli V, Tacke F, Randolph GJ, Fisher EA. Gene expression changes in foam cells and the role of chemokine receptor ccr7 during atherosclerosis regression in apoe-deficient mice. Proc Natl Acad Sci U S A. 2006;103:3781–3786. doi: 10.1073/pnas.0511043103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dansky HM, Barlow CB, Lominska C, Sikes JL, Kao C, Weinsaft J, Cybulsky MI, Smith JD. Adhesion of monocytes to arterial endothelium and initiation of atherosclerosis are critically dependent on vascular cell adhesion molecule-1 gene dosage. Arterioscler Thromb Vasc Biol. 2001;21:1662–1667. doi: 10.1161/hq1001.096625. [DOI] [PubMed] [Google Scholar]
- 50.Feig JE, Shang Y, Rotllan N, Vengrenyuk Y, Wu C, Shamir R, Torra IP, Fernandez-Hernando C, Fisher EA, Garabedian MJ. Statins promote the regression of atherosclerosis via activation of the ccr7-dependent emigration pathway in macrophages. PloS one. 2011;6:e28534. doi: 10.1371/journal.pone.0028534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Terasaka N, Miyazaki A, Kasanuki N, Ito K, Ubukata N, Koieyama T, Kitayama K, Tanimoto T, Maeda N, Inaba T. Acat inhibitor pactimibe sulfate (cs-505) reduces and stabilizes atherosclerotic lesions by cholesterol-lowering and direct effects in apolipoprotein e-deficient mice. Atherosclerosis. 2007;190:239–247. doi: 10.1016/j.atherosclerosis.2006.03.007. [DOI] [PubMed] [Google Scholar]
- 52.Tardif JC, Gregoire J, L'Allier PL, Anderson TJ, Bertrand O, Reeves F, Title LM, Alfonso F, Schampaert E, Hassan A, McLain R, Pressler ML, Ibrahim R, Lesperance J, Blue J, Heinonen T, Rodes-Cabau J. Effects of the acyl coenzyme a:Cholesterol acyltransferase inhibitor avasimibe on human atherosclerotic lesions. Circulation. 2004;110:3372–3377. doi: 10.1161/01.CIR.0000147777.12010.EF. [DOI] [PubMed] [Google Scholar]
- 53.Nissen SE, Tuzcu EM, Brewer HB, Sipahi I, Nicholls SJ, Ganz P, Schoenhagen P, Waters DD, Pepine CJ, Crowe TD, Davidson MH, Deanfield JE, Wisniewski LM, Hanyok JJ, Kassalow LM. Effect of acat inhibition on the progression of coronary atherosclerosis. N Engl J Med. 2006;354:1253–1263. doi: 10.1056/NEJMoa054699. [DOI] [PubMed] [Google Scholar]
- 54.Fazio S, Linton M. Failure of acat inhibition to retard atherosclerosis. N Engl J Med. 2006;354:1307–1309. doi: 10.1056/NEJMe068012. [DOI] [PubMed] [Google Scholar]
- 55.Mak BC, Wang Q, Laschinger C, Lee W, Ron D, Harding HP, Kaufman RJ, Scheuner D, Austin RC, McCulloch CA. Novel function of perk as a mediator of force-induced apoptosis. J Biol Chem. 2008 doi: 10.1074/jbc.M803194200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lim WS, Timmins JM, Seimon TA, Sadler A, Kolodgie FD, Virmani R, Tabas I. Signal transducer and activator of transcription-1 is critical for apoptosis in macrophages subjected to endoplasmic reticulum stress in vitro and in advanced atherosclerotic lesions in vivo. Circulation. 2008;117:940–951. doi: 10.1161/CIRCULATIONAHA.107.711275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Duewell P, Kono H, Rayner KJ, Sirois CM, Vladimer G, Bauernfeind FG, Abela GS, Franchi L, Nunez G, Schnurr M, Espevik T, Lien E, Fitzgerald KA, Rock KL, Moore KJ, Wright SD, Hornung V, Latz E. Nlrp3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature. 2010;464:1357–1361. doi: 10.1038/nature08938. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.