

Abstract
Background
Air pollution is a pervasive environmental health hazard that occurs over a lifetime of exposure in individuals from many industrialized societies. However, studies have focused primarily on exposure durations that correspond to only a portion of the lifespan. We therefore tested the hypothesis that exposure over a considerable portion of the lifespan would induce maladaptive cardiovascular responses.
Methods and Results
C57BL/6 male mice were exposed to concentrated ambient particles <2.5 μm (particulate matter, PM or PM2.5) or filtered air (FA), 6 h/d, 5 d/wk, for 9 months. Assessment of cardiac contractile function, coronary arterial flow reserve, isolated cardiomyocyte function, expression of hypertrophic markers, calcium handling proteins, and cardiac fibrosis were then performed. Mean daily concentrations of PM2.5 in the exposure chamber versus ambient daily PM2.5 concentration at the study site were 85.3 versus 10.6 μg/m3 (7.8-fold concentration), respectively. PM2.5 exposure resulted in increased hypertrophic markers leading to adverse ventricular remodeling characterized by myosin heavy chain (MHC) isoform switch and fibrosis, decreased fractional shortening (39.8±1.4 FA versus 27.9±1.3 PM, FS%), and mitral inflow patterns consistent with diastolic dysfunction (1.95±0.05 FA versus 1.52±0.07 PM, E/A ratio). Contractile reserve to dobutamine was depressed (62.3±0.9 FA versus 49.2±1.5 PM, FS%) in response to PM2.5 without significant alterations in maximal vasodilator flow reserve. In vitro cardiomyocyte function revealed depressed peak shortening (8.7±0.6 FA versus 7.0±0.4 PM, %PS) and increased time-to-90% shortening (72.5±3.2 FA versus 82.8±3.2 PM, ms) and relengthening (253.1±7.9 FA versus 282.8±9.3 PM, ms), which were associated with upregulation of profibrotic markers and decreased total antioxidant capacity. Whole-heart SERCA2a levels and the ratio of α/β-MHC were both significantly decreased (P<0.05) in PM2.5-exposed animals, suggesting a switch to fetal programming.
Conclusions
Long-term exposure to environmentally relevant concentrations of PM2.5 resulted in a cardiac phenotype consistent with incipient heart failure.
Keywords: air pollution, particulate matter, cardiovascular, cardiac, remodeling
Epidemiological evidence supports an important association between air pollution exposure and cardiovascular risk. Current evidence suggests that long-term exposure increases cardiovascular risk to an even greater extent than short-term exposure.1 An acute (days) 10 μ g/m3 increase in particulate matter (PM or PM2.5) elevates cardiovascular mortality by 1%, whereas long-term exposure with levels chronically elevated by the same degree increases this risk by ≥10%. This supports the concept that aggregate exposure over time may be an even more important determinant of eventual risk than short-term exposure. Cumulative exposure has the potential of not only influencing adverse events but could also play a fundamental role in organ-specific pathology.
Air pollution exposure occurs over a lifetime in many individuals, and an important consideration is the effect of long-term exposure, or at the very least, over a substantial portion of an individual’s lifespan. Numerous investigations have elucidated potential biological mechanisms whereby exposure to PM2.5 may modulate disease susceptibility, including progression of atherosclerosis, inflammation, and hypertension.2–6 Most of these studies, although considered chronic (≈10–20 weeks), have not explored the effects of exposure over longer periods. Long-term exposure in animal models may provide valuable insights into the resultant phenotype and serve as a foundation to better understand interactive effects of air pollution with other pervasive risk factors, including elevated low-density lipoprotein cholesterol or diabetes. In the present study, we focused on the long-term effects of air pollution exposure over more than half of a rodent’s (mouse) life-span. We hypothesized that air pollution itself, in the absence of other risk factors, may exert discernible effects on the cardiovascular phenotype that are indistinguishable from the protracted effects of other conventional risk factors such as hypertension. This cardiovascular phenotype may explain the propensity to develop complications.
Methods
Animals and Exposure
C57BL/6 male adult mice were exposed for 9 months, starting at 8 weeks of age, to concentrated PM2.5 from the Columbus, OH, region. Concentrated PM2.5 was generated using a versatile aerosol concentration enrichment system modified for long-term exposures (OASIS-1) and has been previously described2,7–9 and characterized.10,11 The ambient mean daily PM2.5 concentration at the study site was 10.6 μg/m3. Mean daily concentration of PM2.5 in the exposure chamber was 85.3 μg/m3 (7.8-fold concentration from the ambient levels), with the control mice exposed to an identical treatment except for a high-efficiency particulate air filter (FA, Pall Life Sciences, East Hills, NY) that was positioned in the inlet valve to the exposure system to remove all of the particles from the airstream. Since the mice were exposed for 6 h/d, 5 d/wk, the equivalent PM2.5 concentration to which the mice were exposed in the chamber normalized over the exposure period (9 months) was 15.2±0.91 μg/m3, which is close to the annual average PM2.5 National Ambient Air Quality Standard (NAAQS) of 15 g/m3.
Blood Pressure
Systolic blood pressure, diastolic blood pressure, and mean arterial blood pressure were measured using a computerized noninvasive tail-cuff manometry system (Visitech IITC model 129 System, Visitech Systems, Apex, NC). To avoid anxiety due to the procedure, mice were trained for 5 consecutive days before the experimental procedure. To minimize procedure-associated effects on blood pressure, the first 10 of 20 blood pressure values recorded during each measurement were not used, and the remaining 10 values were averaged for analyses of the data from each mouse. Blood pressure was recorded daily for 3 days, and the average blood pressure values are presented.
In Vivo Cardiac Assessment
In vivo cardiac function was determined using a VisualSonics Vevo 2100 with a 40-MHz transducer (Visualsonics, Toronto, Ontario, Canada). Mice were anesthetized in a sealed chamber with 2% isoflurane. Mice were then moved to a heated procedure board and anesthesia was sustained with 1.5% isoflurane (delivered in 100% O2) supplied through a nose cone. The limbs were gently taped to ECG electrodes coated with electrode cream (Signa Gel, Parker Labs, Fairfield, NJ) and a rectal thermometer was inserted for maintenance of normothermia (37°C internal temperature). Nair hair removal lotion (Church & Dwight Co, Princeton, NJ) was applied to the chest to remove fur from the imaging location. Next, prewarmed ultrasound gel (Aquasonic, Parker Labs, Fairfield, NJ) was placed on the chest and a 15-MHz probe (optimized and dedicated to rodent studies) was placed in the parasternal, short-axis orientation. LV systolic and diastolic internal dimensions (LVESd and LVEDd) and systolic and diastolic posterior wall thickness (PWTs and PWTd) were recorded. Three loops of M-mode data were captured for each animal, and data were averaged from at least 5 beat cycles per loop. Parameters were detected using the American Society for Echocardiography leading-edge technique by an investigator blinded to group assignment.12 These parameters allow the determination of LV percent fractional shortening (%FS) by the equation . The echo probe was then moved to the subcostal orientation and a 4-chamber apical view was obtained and the mitral valve annulus was visualized. The sample volume was placed at the level of the mitral valve leaflets and pulsed wave Doppler was used to capture the E and A peaks representing passive and active mitral flow, respectively. The relative ratio of the E/A wave from the transmitral valve flow waveform was used to evaluate LV diastolic function.
We adapted the method of Hartley et al13 for evaluation of coronary flow and coronary flow reserve. From the apical 4-chamber view, the probe was moved closer to the surface of the chest (maintaining the same angle as used for the mitral valve flow). Using color flow Doppler, we visualized the left coronary artery (LCA). The isoflurane concentration was adjusted to 1.0% and the animal was allowed to equilibrate for 5 minutes, after which time the sample volume (0.5-mm gate size) was placed on the main LCA branch. Baseline coronary flow data were acquired and 3 traces were analyzed and averaged. Next, adenosine (140 μg/kg per minute) was infused into the tail vein to maximally dilate the coronary vasculature. Maximal hyperemia was obtained within 1 minute. Three traces were analyzed and averaged to calculate the hyperemic coronary flow. Coronary flow velocity reserve (CFVR) was calculated using the following equation .14 All echocardiographic measurements were performed by an investigator blinded to group assignment and analyzed by a second investigator who was also unaware of group assignment.
In Vitro Cardiomyocyte Function
Myocytes were isolated using retrograde perfusion through the aorta with Liberase and cultured until the time of experiment as described previously.15–19 Myocyte mechanics (twitches) and intracellular Ca2+ transients were assessed using video-based sarcomere detection coupled to a fluorescent system (IonOptix, Milton, MA).15–19 Myocytes were plated in small, glass-bottom inserts (Cell MicroControls, Norfolk, VA) and placed into a flow chamber attached to the stage of an inverted Olympus IX-71 microscope. The cells were observed using a 40× objective and superfused with contractile buffer (CB) at 1 mL/min at ≈37°C with a Warner in-line heater and an automatic temperature controller (Warner Instruments, Hamden, CT). The constituents of the CB included (in mol/L, pH 7.4): 131 NaCl, 4 KCl, 10 HEPES, 1 CaCl2, 1 MgCl2, and 10 glucose. The cells were field-stimulated at 1 Hz with 3-ms duration, using a Myopacer Field-Stimulator system (IonOptix, Milton, MA), which uses a setup containing 2 platinum wires on either end of the chamber insert. To measure functional properties of the cells, the Sarclen Sarcomere Length Acquisition Module (IonOptix) was applied. Using the IonOptix video imaging system, sarcomere length was recorded using a Myocam-S Digital CCD camera. The following 5 parameters were recorded: sarcomere peak shortening normalized to baseline length (PS, the maximal percent change of the sarcomere length from the resting state), sarcomere time-to-90% peak shortening (TPS90, time to 90% of cell shortening), time-to-90% relengthening (TR90, time to 90% of cell relaxation), and sarcomere departure and return velocities (±dL/dt, the maximal velocities of cell shortening and relengthening).
Histological Analyses
Frozen and paraffin-embedded hearts were cut from base to apex. The sections below the papillary muscles were identified and 4-μm cross sections were stained with hematoxylin and eosin for morphometric and Picrosirius red (SR) for collagen analyses. Quantitative planimetric analyses were performed on four successive sections per slide, and at least 10 sections from 3 consecutive slides per mouse were examined. Each image was digitized using a digital camera and analyzed under a research microscope (Zeiss Axioscope with Spot I digital camera, Jena, Germany), using NIH Image software (version 1.61). Nuclear and cytosolic areas were quantified, and the results are expressed as ratios of nuclear to cytosolic areas (μm/μm). SR-positive stained area was quantified as a percentage of the total myocardial area (% area). All analyses were performed by an investigator blinded to group assignment.
Western Immunoblotting
Frozen hearts were homogenized in ice-cold 25 mmol/L HEPES, pH 7.4, 50 mmol/L NaCl, 1 mmol/L MgCl2, 2 mmol/L EGTA, 0.1% TritonX-100, 0.1% sodium deoxycholate, 10 mmol/L sodium pyrophosphate, 10 mmol/L sodium fluoride, 1 mmol/L sodium orthovanadate, 0.5 mmol/L phenylmethanesulfonyl fluoride, 10 mg/mL aprotinin, 10 mg/mL leupeptin (≈100 mg tissue/mL lysis buffer), and proteins were separated on SDS-PAGE gels and transferred to PVDF membranes. Membranes were probed with antibodies to SERCA2a ATPase (Biomol, SA-209, Plymouth Meeting, PA), α-MHC (Genway, 20–272–191956, San Diego, CA), β-MHC (Santa Cruz, sc71575, Santa Cruz, CA), collagen I (Abcam, ab292, Cambridge, MA), and collagen III (EMD Millipore, 234189, Billerica, MA). Blots were developed using enhanced chemiluminescence (Pierce ECL Western Blotting Substrate, Thermo Scientific, Rockford, IL) and expression levels were quantified using Image Quant software, version 5.0 (Molecular Dynamics, Sunnydale, CA). The band density of the protein of interest was normalized to β-actin (ab6276, Abcam, Cambridge, MA) or GAPDH (Abcam, ab9485).
Quantitative Real-Time PCR
Total RNA was prepared from heart tissue using an RNeasy Mini kit (Qiagen, Valencia, CA). cDNA was synthesized using a Transcriptor First-Strand cDNA Synthesis Kit (Roche, Indianapolis, IN). Quantitative real-time PCR was performed using the iQ5 Real-Time PCR Detection System (Bio-Rad Laboratories, Hercules, CA). Sequences of primers used for real-time PCR in this study have previously been described.10
Total Antioxidant Capacity
The Cayman Chemical Antioxidant Assay Kit (no. 709001, Cayman Chemical, Ann Arbor, MI) was used to measure total antioxidant capacity of mouse plasma, per the manufacturer’s instructions. The methodology of this assay depends on the ability of antioxidants in the sample to inhibit ABTS oxidation (2,2′-azino-di-[3-ethylbenz-thiazoline sulfonate]) to ABTS • and by metmyoglobin. The ability of antioxidants in the sample to prevent the oxidation of ABTS is compared with Trolox, a water-soluble analog of tocopherol, and is quantified as molar Trolox equivalents.
Statistical Analyses
Data are expressed as mean±SEM. Comparison of continuous variables was performed using a Student t test. P<0.05 was considered statistically significant. Statistical analyses were performed using GraphPad PRISM 5 (La Jolla, CA).
Results
Long-Term PM2.5 Exposure Alters Blood Pressure and Cardiac Morphology
Body weights were not different in PM2.5 exposed mice after 9 months compared with FA controls (exposure averages for each week are shown in Table 1); postmortem analyses confirmed that absolute heart weight and the heart weight/body weight ratio of PM2.5-exposed mice was significantly increased by 8.8% compared with FA mice. Long-term exposure to PM2.5 caused a significant increase in heart rate, systolic blood pressure, diastolic blood pressure, and mean arterial blood pressure of C57BL/6 mice. However, pulse pressure was decreased in PM2.5-exposed compared with FA-exposed mice (Table 2).
Table 1.
Concentrations Weekly Variation in Ambient and PM2.5
| Time, Week | Ambient, μg/m3 | PM2.5, μg/m3 |
|---|---|---|
| 1 | 13.71 | 95.22 |
| 2 | 7.30 | 56.00 |
| 3 | 6.18 | 41.96 |
| 4 | 4.27 | 32.25 |
| 5 | 11.20 | 113.65 |
| 6 | 11.09 | 123.27 |
| 7 | 7.71 | 16.90 |
| 8 | 10.54 | 56.55 |
| 9 | 9.73 | 54.73 |
| 10 | 8.95 | 83.20 |
| 11 | 14.03 | 129.20 |
| 12 | 14.06 | 72.50 |
| 13 | 10.97 | 72.13 |
| 14 | 6.32 | 42.69 |
| 15 | 14.12 | 126.85 |
| 16 | 6.84 | 48.44 |
| 17 | 18.74 | 150.45 |
| 18 | 15.94 | 134.13 |
| 19 | 12.01 | 140.22 |
| 20 | 10.39 | 89.52 |
| 21 | 11.14 | 112.17 |
| 22 | 10.77 | 82.49 |
| 23 | 12.62 | 107.00 |
| 24 | 5.54 | 67.94 |
| 25 | 10.77 | 82.49 |
| 26 | 12.62 | 128.79 |
| 27 | 5.54 | 27.00 |
| 28 | 6.06 | 39.41 |
| 29 | 9.18 | 39.41 |
| 30 | 8.46 | 42.93 |
| 31 | 10.31 | 62.97 |
| 32 | 10.28 | 60.20 |
| 33 | 8.46 | 72.19 |
| 34 | 9.67 | 66.49 |
| 35 | 16.34 | 138.60 |
| 36 | 2.63 | 20.37 |
| 37 | 20.38 | 266.37 |
| 38 | 9.79 | 86.86 |
| 39 | 18.91 | 142.12 |
The particulate matter (PM2.5) data are weekly means for each group (ambient or PM2.5), whereas the concentration ratios were affected by multiple factors, particularly ambient humidity.
Table 2.
Body Weight and Blood Pressure Parameters of Long-Term PM2.5-Exposed Mice
| FA | PM | P Value | n | |
|---|---|---|---|---|
| Body weight, g | 30.6 (±0.3) | 29.7 (±0.4) | 0.103 | FA=10; PM=9 |
| Heart weight, mg | 124.0 (±2.4) | 138. (±3.7) | 0.014 | FA=5; PM=5 |
| Heart/body weight, mg/g | 4.54 (±0.11) | 4.16 (±0.16) | 0.037 | FA=5; PM=4 |
| Heart rate, bpm | 352.5 (±4.4) | 379.4 (±6.3) | 0.008 | FA=5; PM=5 |
| Systolic blood pressure, mm Hg | 103.6 (±1.4) | 112.2 (±1.9) | 0.0015 | FA=10; PM=9 |
| Diastolic blood pressure, mm Hg | 76.17 (±0.5) | 86.9 (±1.8) | <0.0001 | FA=10; PM=9 |
| Mean arterial pressure, mm Hg | 86.6 (±0.6) | 96.3 (±1.4) | <0.0001 | FA=10; PM=9 |
| Pulse pressure, mm Hg | 29.9 (±1.5) | 24.1 (±1.3) | 0.0095 | FA=10; PM=9 |
FA indicates filtered air; PM, particulate matter.
Male adult C57BL/6 mice were exposed to PM2.5 or FA for 9 months.
Data are expressed as mean±SEM and analyzed using a Student t test.
Long-Term PM2.5 Exposure Is Associated With LV Remodeling
Male mice exposed to PM2.5 had considerable cardiac remodeling that was characterized by an increase in both LVESd and LVEDd compared with FA exposed controls. Posterior wall thickness was not different in diastole (PWTd), but systolic posterior wall thickness (PWTs) was decreased in PM2.5-exposed mice. These changes were associated with reduced systolic function as evidenced by decreased percent fractional shortening in the PM2.5-exposed compared with FA-exposed mice. Mitral valve E/A ratio was lower in the PM2.5-exposed compared with FA-exposed mice (Table 3).
Table 3.
Echocardiographic Analyses of Long-Term PM2.5-Exposed Mice
| FA | PM | P Value | n | |
|---|---|---|---|---|
| LVESd, mm | 2.56 (±0.10) | 2.85 (±0.1) | 0.038 | FA=10; PM=9 |
| LVEDd, mm | 4.25 (±0.10) | 3.95 (±0.06) | 0.0319 | FA=10; PM=9 |
| PWTd, mm | 0.73 (±0.01) | 0.69 (±0.03) | 0.2494 | FA=10; PM=9 |
| PWTs, mm | 1.28 (±0.02) | 1.00 (±0.04) | <0.0001 | FA=10; PM=9 |
| FS, % | 39.83 (±1.37) | 27.92 (±1.30) | <0.0001 | FA=10; PM=9 |
| Mitral E/A, ratio | 1.95 (±0.05) | 1.52 (±0.07) | 0.0001 | FA=9; PM=8 |
| Dobutamine HR, bpm | 583.8 (±8.1) | 596.6 (±5.6) | 0.231 | FA=5; PM=5 |
| Dobutamine FS, % | 62.32 (±0.85) | 49.23 (±1.45) | <0.0001 | FA=9; PM=8 |
| Coronary flow velocity, mm/s | 342.9 (±32.6) | 255.6 (±12.5) | 0.0705 | FA=6; PM=4 |
| Coronary flow velocity reserve, mm/s | 800.4 (±41.5) | 745.5 (±21.8) | 0.3771 | FA=5; PM=3 |
FA indicates filtered air; PM, particulate matter; LVESd, left ventricular end-systolic diameter; LVEDd, left ventricular end-diastolic diameter; PWTd, diastolic posterior wall thickness; PWTs, systolic posterior wall thickness; FS, fractional shortening; mitral E/A, mitral flow E-to-A ratio; dobutamine HR, heart rate after dobutamine challenge; and dobutamine FS; fractional shortening after dobutamine challenge.
Data are expressed as mean±SEM and analyzed using a Student t test.
Contractile Reserve in Mice Exposed to Long-Term PM2.5
Chronic systolic dysfunction is often associated with diminished sensitivity of the β-receptor due to receptor desensitization. Thus, we investigated the cardiac response to a β1-adrenergic agonist to assess chronotropic and contractile reserve. Both groups responded to dobutamine (3.65 μg/kg) with a significantly increased HR. However, examination of fractional shortening revealed a reduced contractile response to dobutamine in mice exposed to PM compared with FA controls (Table 3).
Coronary Flow Reserve Is Preserved in Mice Exposed Long-Term to PM2.5
Cardiac hypertrophy and remodeling is frequently linked to changes in coronary flow reserve. On stimulation with the vasodilator adenosine (150 μg/kg per minute), both the PM2.5-exposed and FA-exposed groups had increased coronary flow to a similar degree, indicating preserved coronary flow reserve in PM2.5 exposed mice (Table 3).
Long-Term PM2.5 Exposure Causes Cardiomyocyte Dysfunction In Vitro
We performed functional assessment of isolated single cardiomyocyte function as an additional validation of cardiac-specific PM2.5 effects (representative twitch from FA-exposed and PA-exposed shown in Figure 1A). Isolated myocytes from PM2.5-exposed mice showed a significant decrease in % peak shortening (%PS; Figure 1B), along with an increase in time-to-90% peak shortening (Figure 1C; myocyte systolic dysfunction) and time-to-90% relengthening (Figure 1D; myocyte diastolic dysfunction). These results suggest in vivo cardiac dysfunction is also evident at the myocyte level (cardiomyocyte dysfunction), further confirming the results obtained by echocardiography.
Figure 1.

Isolated myocyte function from mice exposed to filtered air (FA) or particulate matter (PM) for 9 months in the OASIS-1 trailer. A, Representative shortening of FA and PM myocyte contraction. B, Percent peak shortening (%PS) normalized to sarcomere length (SL). C, Time-to-90% peak shortening (TPS90), and D, time-to-90% relengthening (TR90). n=20 to 30 myocytes from 3 to 4 mice per group. Data are expressed as mean±SEM and analyzed using a Student t test. P<0.05 was considered statistically significant.
Cardiomyocyte nucleus/cytoplasm ratio was markedly decreased in PM2.5-exposed mice by 20.9% compared with FA mice (mean [SD], 0.0378 [0.00601] to 0.0299 [0.00281]; P<0.05; Figure 2A and 2B). Atrial natriuretic peptide mRNA expression was increased and α-tubulin mRNA was not significantly different after long-term PM2.5 exposure (Figure 3A and 3B).
Figure 2.
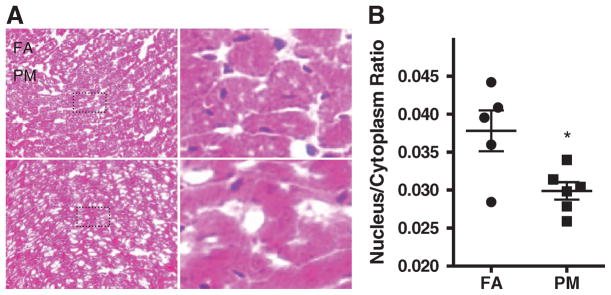
Histological assessments of filtered air (FA) and particulate matter (PM2.5)-exposed hearts. A, Representative heart tissue sections. B, Quantitative analyses of nucleus/cytoplasm ratio. Data are expressed as mean±SEM (n=5) and analyzed using a Student t test. P<0.05 was considered statistically significant.
Figure 3.

Quantitative cardiac mRNA expression of A, Atrial natriuretic peptide (ANP); B, α-tubulin; C, transforming growth factor (TGF)β; D, osteopontin; E, collagen I, and F, collagen III in filtered air (FA)-exposed and particulate matter (PM2.5)-exposed hearts. Data are expressed as mean±SEM (n=5) and analyzed using a Student t test. P<0.05 was considered statistically significant.
Long-Term PM2.5 Exposure Is Associated With Increased Cardiac Collagen Deposition
Picrosirius red staining (Figure 4A and 4B) indicated an increase in cardiac collagen deposition in mice exposed to PM2.5 (166%). Real-time PCR revealed increased transforming growth factor (TGF)-β and collagen I, the major structural collagen in the myocardium, suggesting that PM2.5 exposure altered gene expression that is consistent with a profibrotic phenotype (Figure 3C and 3E). Western blot analyses confirmed increased collagen I in PM2.5-exposed mice (Figure 5A), whereas neither collagen III nor osteopontin were altered (Figure 3D, 3F, and Figure 5B).
Figure 4.
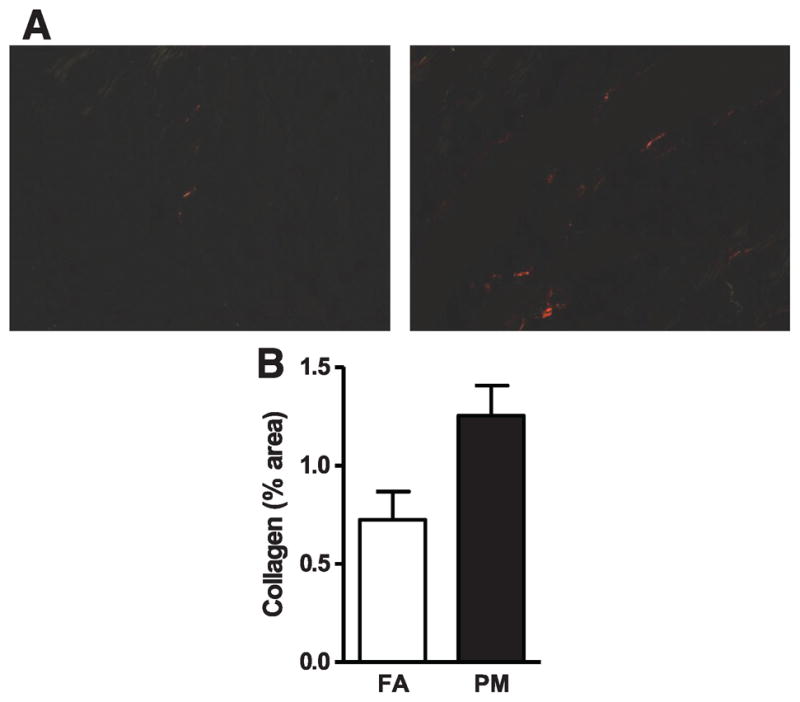
Collagen assessment of filtered air (FA)-exposed and particulate matter (PM2.5)-exposed hearts. A, Representative heart tissue sections stained with Picrosirius red. B, Quantitative assessment of area positive stained for collagen. Data are expressed as mean±SEM (n=5) and analyzed using a Student t test. P<0.05 was considered statistically significant.
Figure 5.

Protein analyses and representative western immune blots from whole heart homogenates of filtered air (FA)-exposed and particulate matter (PM2.5)-exposed mice. A, Collagen I; B, collagen III; C, α-myosin heavy chain (MHC); D, β-MHC; E, ratio α-/β-MHC; and F, SER-CA2a. Data are expressed as mean±SEM (n=5) and analyzed using a Student t test. P<0.05 was considered statistically significant.
Long-Term PM2.5 Exposure Decreases Ca2+ Reuptake Into the Sarcoplasmic Reticulum and Total Antioxidant Capaciy
α-MHC protein levels were not different (Figure 5C); however, β-MHC protein levels were increased and the α/-MHC ratio was decreased in PM2.5-exposed mice (Figure 5D and 5E), a pattern consistent with human and animal models of heart failure. Protein isolated from whole heart homogenates of PM2.5 mice had significantly decreased levels of SERCA-2a (Figure 5F), suggesting reduced mechanisms to promote Ca2+ reuptake into the sarcoplasmic reticulum in response to PM2.5 exposure. Total antioxidant capacity in the plasma was significantly decreased in the plasma of PM2.5mice, suggesting that an imbalance in ROS/antioxidant production occurs after PM2.5 exposure (Figure 6).
Figure 6.
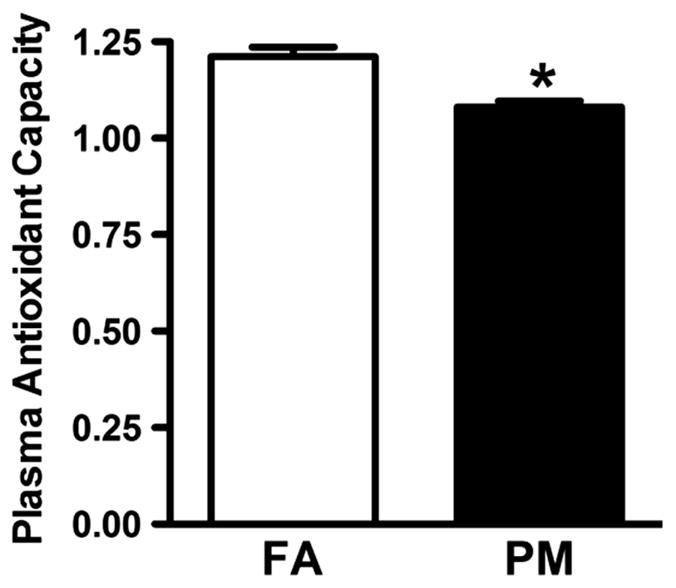
Total plasma antioxidant capacity in filtered air (FA)-exposed and particulate matter (PM2.5)-exposed mice. Data are expressed as mean±SEM (n=6) and analyzed using a Student t test. P<0.05 was considered statistically significant.
Discussion
Cardiac dysfunction and pathophysiologic adaptations consistent with dysfunctional myocardium were observed in mice exposed to environmentally relevant concentrations of PM2.5 over the course of a substantive portion of the lifespan. At the molecular level, these changes were typified by cardiac hypertrophy, cardiac muscle isoform switch, upregulation of profibrotic gene expression, and reduction in Ca2+ reuptake mechanisms, all consistent with an incipient heart failure phenotype.
Epidemiological studies have now confirmed an association of PM2.5 with cardiovascular morbidity and mortality. Short-term elevations over a period of a few hours to days can trigger sudden cardiac death, myocardial infarctions, and heart failure exacerbations.20–25 However, exposure to a pervasive pollutant such as air pollution likely occurs over a lifetime, often at high levels for many individuals living in industrialized countries, including India and China.26–30 Consistent with this concept of integrated exposure over several years, the best available epidemiological data appear to support the idea that the longer the chronicity of exposure, the higher the risk.1 In the Women’s Health Study, which involved exposure over a few years, there was an even more marked increase in this risk (odd ratio for events, 1.76).22 Conversely, regulation of air pollution through mandated controls has been shown to improve life expectancy.31
In the present study, hearts from PM2.5-exposed mice displayed increased systolic, diastolic, and mean arterial blood pressure and decreased pulse pressure, all changes that were consistent with a phenotype of incipient heart dysfunction. Such changes typically follow chronic left ventricular hypertrophy and diastolic dysfunction, which if left unchecked may eventually lead to systolic dysfunction and loss of contractile reserve. We did note evidence of systolic and diastolic dysfunction accompanied by fibrosis of the left ventricle and an increase in gene expression of the major structural collagen types in the myocardium, a finding that has previously been described in conjunction with prohypertensive stimuli.10 Resting LV systolic and diastolic dysfunction were also confirmed at the myocyte level, where abnormal contractile function and relaxation responses were present.
At a molecular level, heart failure is characterized by numerous changes in contractile pathways and is often characterized by a switch to a fetal programming. The myocardium was characterized by abnormally increased β-myosin heavy chain (β-MHC) after long-term PM2.5 exposure. Expression of α- and β-MHC is tightly regulated by both developmental and hormonal factors.32 Expression of these isoforms is often altered in disease states, including cardiac failure or hypertrophy.33 In the failing adult mouse heart, a shift is observed from α-MHC to β-MHC.34 Upregulation of β-MHC transcription is also seen as an early marker of cardiac hypertrophy. Although the adult human myocardium is normally composed of β-MHC, it is believed that minor changes in the expression of MHC isoforms significantly alter power output at the cellular power level.35 β-MHC contains lower adenosine triphosphatase activity and lower filament sliding velocity; however, cross-bridge force is generated with a greater efficiency of energy consumption than α-MHC,36 suggesting that a shift from α- to β-MHC could be an adaptive response to preserve energy. Alternatively, contractile dysfunction can promote the progression of various heart diseases. Therefore, contractile dysfunction due to increased β-MHC levels could outweigh the benefits of improved clinical outcomes. At a molecular level, the downregulation of SERCA2a is indicative of abnormal calcium cycling.37 Decreased SERCA2a expression and activity is found in heart failure regardless of the etiology of the disease.38,39
Mechanistic studies in animals have previously demonstrated direct acute effects of PM2.5 on the myocardium that include generation of reactive oxygen species, spontaneous arrhythmias, alterations in myocardial blood flow, and coronary vascular resistance (in the presence of coronary ischemia).40–43 PM2.5 is also well known to increase blood pressure with several studies demonstrating that acute/subacute air pollution exposure can raise blood pressure.44 Longer-term PM2.5 exposure (over weeks) has been shown to sensitize the vasculature to vasoconstrictive mediators (angiotensin II), probably via reductions in nitric oxide and enhancement of calcium sensitization pathways including Rho/ROCK.5,10 Similarly, blockade of the Rho/ROCK pathway with Fasudil can obviate the exaggerated increase in blood pressure induced by longer durations of PM2.5 exposure in the presence of angiotensin II and decreased cardiac hypertrophy and collagen deposition.10 Although mediators of increased blood pressure probably differ between acute and longer-term exposure, a sustained increase in diastolic and mean arterial blood pressure, as we have demonstrated in this study, may have considerable significance. Interestingly, these effects are similar to those that have been demonstrated to occur in humans, where increases in diastolic blood pressure have been noted.45–47 The effects on blood pressure may make it difficult for conclusions on the relative contribution of direct effects on the myocardium versus blood pressure–mediated effects. Nevertheless, the effects on both systolic and diastolic function at the whole organ and myocyte level were striking and certainly make a case for blood pressure–independent effects that may have stimulated the progression to an incipient heart-failure state.
Although increased life expectancy has occurred after a reduction in air pollution levels, particulate air pollution remains a significant problem in the United States, and the mechanisms by which air pollution directly affects the cardiovascular system are not completely defined.31 An association with heart failure and episodes of air pollution has been previously recognized, although the precise mechanisms remain unclear.23,24 A number of pathways have been hypothesized and include sympathetic activation, increases in blood pressure, and arrhythmogenesis, all of which are well known to occur after acute PM2.5 exposure.44,48 Our results suggest that long-term exposure has direct effects on the myocardium that may have immediate relevance to episodes of heart failure hospitalizations that have been reported after air pollution exposure.23,49 Recurrent heart failure hospitalizations in vulnerable patients is a major cause of morbidity in the United States and worldwide and understanding factors that may play a role at the population level would allow refinement of our current understanding and better inform policy and guidelines.
The concentrations used in this experiment are lower than levels that humans are exposed to in many parts of the world. Daily and annual PM2.5 levels among cities in Latin America, China, and India can average 100 to 150 μ g/m3, approximately 10- to 15-fold higher than concentrations common to major US cities.50–52 This order of magnitude higher exposure encountered routinely in urban regions of developing countries can only be attained in the United States in laboratory environments. Therefore, our results provide additional understanding of the effects of this high level of PM2.5 exposure on functional variables of etiologic/pathogenic/prognostic importance. The pervasive nature of air pollution defines this as a risk factor with considerable population attributable risk. Thus, even small effects are very significant from a societal perspective.53
Our study has several limitations that must be acknowledged. The exposures were not continuous, whereby the animals were not exposed during the weekends. An additional limitation is that we did not record the temporal response of hemodynamic changes over the duration of exposure. Therefore, we cannot comment on the timing of blood pressure increases in response to PM2.5 exposure. Subsets of animals were used for different studies (in vivo versus in vitro), therefore a potential bias could exist regarding the animals chosen for each study. Also, multiple tests were performed on the same animals; however, we consistently observed the same phenotype at the whole organ and cellular levels and therefore it is likely that the constellation of outcomes is responding in concert.
In summary, chronic exposure to PM2.5 over a consideration portion of the mouse lifespan results in adverse cardiac remodeling consistent with an incipient heart failure phenotype. These findings have implications for air pollution as an independent risk factor for the development of cardiovascular disease.
CLINICAL PERSPECTIVE.
Air pollution is a widespread environmental health hazard occurring in many industrialized societies. Current evidence suggests that exposure to air pollution can cause significant cardiovascular risk even within a short time period, and that these effects increase with time. The present study used a mouse model of air pollution exposure to better understand increased cardiovascular risk after exposure to air pollution over a lifespan. Mice were exposed to air pollution or filtered air, 6 h/d, 5 d/wk for 9 months, which is a large portion of the lifespan of a mouse. The mice exposed to air pollution developed both systolic and diastolic dysfunction, as assessed using echocardiography. Interestingly, this dysfunction was evident at the cellular level, as cardiomyocytes isolated from mice exposed to air pollution had decreased function that is translatable to the dysfunction found at the whole heart level. Examination of heart tissue from mice exposed to air pollution also revealed molecular markers of hypertrophy leading to adverse ventricular remodeling. This study showed that long-term exposure to environmentally relevant concentrations of air pollution resulted in cardiovascular dysfunction evident at both the whole heart and cellular level.
Acknowledgments
Sources of Funding
This work was supported by an American Heart Association Scientific Development Grant (0835298N) and NIH R01ES019923 to Dr Wold; NIH ES015146, ES017290, ES017290, and ES019616 to Dr Rajagopalan; NIH ES016588, ES017412, and ES018900 to Dr Sun; NIH R01HL5604-12 to P.A.L., and funds from The Heart Center, Nationwide Children’s Hospital (P.A.L., L.E.W.).
Footnotes
Disclosures
None.
References
- 1.Brook RD, Rajagopalan S, Pope CA, III, Brook JR, Bhatnagar A, Diez-Roux AV, Holguin F, Hong Y, Luepker RV, Mittleman MA, Peters A, Siscovick D, Smith SC, Jr, Whitsel L, Kaufman JD. Particulate matter air pollution and cardiovascular disease: an update to the scientific statement from the American Heart Association. Circulation. 2010;121:2331–2378. doi: 10.1161/CIR.0b013e3181dbece1. [DOI] [PubMed] [Google Scholar]
- 2.Sun Q, Wang A, Jin X, Natanzon A, Duquaine D, Brook RD, Aguinaldo JG, Fayad ZA, Fuster V, Lippmann M, Chen LC, Rajagopalan S. Long-term air pollution exposure and acceleration of atherosclerosis and vascular inflammation in an animal model. JAMA. 2005;294:3003–3010. doi: 10.1001/jama.294.23.3003. [DOI] [PubMed] [Google Scholar]
- 3.Araujo JA, Barajas B, Kleinman M, Wang X, Bennett BJ, Gong KW, Navab M, Harkema J, Sioutas C, Lusis AJ, Nel AE. Ambient particulate pollutants in the ultrafine range promote early atherosclerosis and systemic oxidative stress. Circ Res. 2008;102:589–596. doi: 10.1161/CIRCRESAHA.107.164970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kunzli N, Jerrett M, Garcia-Esteban R, Basagana X, Beckermann B, Gilliland F, Medina M, Peters J, Hodis HN, Mack WJ. Ambient air pollution and the progression of atherosclerosis in adults. PLoS ONE. 2010;5:e9096. doi: 10.1371/journal.pone.0009096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sun Q, Yue P, Ying Z, Cardounel AJ, Brook RD, Devlin R, Hwang JS, Zweier JL, Chen LC, Rajagopalan S. Air pollution exposure potentiates hypertension through reactive oxygen species-mediated activation of rho/rock. Arterioscler Thromb Vasc Biol. 2008;28:1760–1766. doi: 10.1161/ATVBAHA.108.166967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sun Q, Yue P, Deiuliis JA, Lumeng CN, Kampfrath T, Mikolaj MB, Cai Y, Ostrowski MC, Lu B, Parthasarathy S, Brook RD, Moffatt-Bruce SD, Chen LC, Rajagopalan S. Ambient air pollution exaggerates adipose inflammation and insulin resistance in a mouse model of diet-induced obesity. Circulation. 2009;119:538–546. doi: 10.1161/CIRCULATIONAHA.108.799015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen LC, Zhong M, Narciso SP, Kleinman MC, Nadziejko C, Lippmann M. Methods for exposing rodents and cells to concentrated ambient PM using a vaces. American Association for Aerosol Research–Particulate Matter Conference; 2003. [Google Scholar]
- 8.Maciejczyk P, Zhong M, Li Q, Xiong J, Nadziejko C, Chen LC. Effects of subchronic exposures to concentrated ambient particles (caps) in mice, II: the design of a caps exposure system for biometric telemetry monitoring. Inhal Toxicol. 2005;17:189–197. doi: 10.1080/08958370590912743. [DOI] [PubMed] [Google Scholar]
- 9.Sun Q, Yue P, Kirk RI, Wang A, Moatti D, Jin X, Lu B, Schecter AD, Lippmann M, Gordon T, Chen LC, Rajagopalan S. Ambient air particulate matter exposure and tissue factor expression in atherosclerosis. Inhal Toxicol. 2008;20:127–137. doi: 10.1080/08958370701821482. [DOI] [PubMed] [Google Scholar]
- 10.Ying Z, Yue P, Xu X, Zhong M, Sun Q, Mikolaj M, Wang A, Brook RD, Chen LC, Rajagopalan S. Air pollution and cardiac remodeling: a role for rhoa/rhokinase. Am J Physiol Heart Circ Physiol. 2009;296:H1540–H1550. doi: 10.1152/ajpheart.01270.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xu X, Yavar Z, Verdin M, Ying Z, Mihai G, Kampfrath T, Wang A, Zhong M, Lippmann M, Chen LC, Rajagopalan S, Sun Q. Effect of early particulate air pollution exposure on obesity in mice: role of p47phox. Arterioscler Thromb Vasc Biol. 2010;30:2518–2527. doi: 10.1161/ATVBAHA.110.215350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Quinones MA, Otto CM, Stoddard M, Waggoner A, Zoghbi WA. Recommendations for quantification of Doppler echocardiography: a report from the Doppler quantification task force of the nomenclature and standards committee of the American Society of Echocardiography. J Am Soc Echocardiogr. 2002;15:167–184. doi: 10.1067/mje.2002.120202. [DOI] [PubMed] [Google Scholar]
- 13.Hartley CJ, Reddy AK, Madala S, Michael LH, Entman ML, Taffet GE. Effects of isoflurane on coronary blood flow velocity in young, old and ApoE(−/−) mice measured by Doppler ultrasound. Ultrasound Med Biol. 2007;33:512–521. doi: 10.1016/j.ultrasmedbio.2006.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wikstrom J, Gronros J, Gan LM. Adenosine induces dilation of epicardial coronary arteries in mice: relationship between coronary flow velocity reserve and coronary flow reserve in vivo using transthoracic echocardiography. Ultrasound Med Biol. 2008;34:1053–1062. doi: 10.1016/j.ultrasmedbio.2007.12.004. [DOI] [PubMed] [Google Scholar]
- 15.Monreal G, Nicholson LM, Han B, Joshi MS, Phillips AB, Wold LE, Bauer JA, Gerhardt MA. Cytoskeletal remodeling of desmin is a more accurate measure of cardiac dysfunction than fibrosis or myocyte hypertrophy. Life Sci. 2008;83:786–794. doi: 10.1016/j.lfs.2008.09.026. [DOI] [PubMed] [Google Scholar]
- 16.Norby FL, Wold LE, Duan J, Hintz KK, Ren J. IGF-I attenuates diabetes-induced cardiac contractile dysfunction in ventricular myocytes. Am J Physiol Endocrinol Metab. 2002;283:E658–E666. doi: 10.1152/ajpendo.00003.2002. [DOI] [PubMed] [Google Scholar]
- 17.Ren J, Porter JE, Wold LE, Aberle NS, Muralikrishnan D, Haselton JR. Depressed contractile function and adrenergic responsiveness of cardiac myocytes in an experimental model of Parkinson disease, the mptp-treated mouse. Neurobiol Aging. 2004;25:131–138. doi: 10.1016/s0197-4580(03)00035-6. [DOI] [PubMed] [Google Scholar]
- 18.Wold LE, Muralikrishnan D, Albano CB, Norby FL, Ebadi M, Ren J. Insulin-like growth factor I (IGF-1) supplementation prevents diabetes-induced alterations in coenzymes q9 and q10. Acta Diabetol. 2003;40:85–90. doi: 10.1007/s005920300010. [DOI] [PubMed] [Google Scholar]
- 19.Wold LE, Relling DP, Duan J, Norby FL, Ren J. Abrogated leptin-induced cardiac contractile response in ventricular myocytes under spontaneous hypertension: role of jak/stat pathway. Hypertension. 2002;39:69–74. doi: 10.1161/hy0102.100777. [DOI] [PubMed] [Google Scholar]
- 20.Peters A, Dockery DW, Muller JE, Mittleman MA. Increased particulate air pollution and the triggering of myocardial infarction. Circulation. 2001;103:2810–2815. doi: 10.1161/01.cir.103.23.2810. [DOI] [PubMed] [Google Scholar]
- 21.Pope CA, III, Muhlestein JB, May HT, Renlund DG, Anderson JL, Horne BD. Ischemic heart disease events triggered by short-term exposure to fine particulate air pollution. Circulation. 2006;114:2443–2448. doi: 10.1161/CIRCULATIONAHA.106.636977. [DOI] [PubMed] [Google Scholar]
- 22.Miller KA, Siscovick DS, Sheppard L, Shepherd K, Sullivan JH, Anderson GL, Kaufman JD. Long-term exposure to air pollution and incidence of cardiovascular events in women. N Engl J Med. 2007;356:447–458. doi: 10.1056/NEJMoa054409. [DOI] [PubMed] [Google Scholar]
- 23.Morris R, Naumova E, Munasinghe R. Ambient air pollution and hospitalization for congestive heart failure among elderly people in seven large us cities. Am J Public Health. 1995;85:1361–1365. doi: 10.2105/ajph.85.10.1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hoek G, Brunekreef B, Fischer P, van Wijnen J. The association between air pollution and heart failure, arrhythmia, embolism, thrombosis, and other cardiovascular causes of death in a time series study. Epidemiology. 2001;12:355–357. doi: 10.1097/00001648-200105000-00017. [DOI] [PubMed] [Google Scholar]
- 25.Kwon HJ, Cho SH, Nyberg F, Pershagen G. Effects of ambient air pollution on daily mortality in a cohort of patients with congestive heart failure. Epidemiology. 2001;12:413–419. doi: 10.1097/00001648-200107000-00011. [DOI] [PubMed] [Google Scholar]
- 26.Kan H, Chen B, Zhao N, London SJ, Song G, Chen G, Zhang Y, Jiang L. Part 1: a time-series study of ambient air pollution and daily mortality in Shanghai, China. Res Rep Health Eff Inst. 2010:17–78. [PubMed] [Google Scholar]
- 27.Smith KR, Jerrett M, Anderson HR, Burnett RT, Stone V, Derwent R, Atkinson RW, Cohen A, Shonkoff SB, Krewski D, Pope CA, III, Thun MJ, Thurston G. Public health benefits of strategies to reduce greenhouse-gas emissions: health implications of short-lived greenhouse pollutants. Lancet. 2009;374:2091–2103. doi: 10.1016/S0140-6736(09)61716-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Balakrishnan K, Dhaliwal RS, Shah B. Integrated urban-rural frameworks for air pollution and health-related research in India: the way forward. Environ Health Perspect. 2011;119:A12–A13. doi: 10.1289/ehp.1003273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Salvi SS, Barnes PJ. Chronic obstructive pulmonary disease in non-smokers. Lancet. 2009;374:733–743. doi: 10.1016/S0140-6736(09)61303-9. [DOI] [PubMed] [Google Scholar]
- 30.Tielsch JM, Katz J, Thulasiraj RD, Coles CL, Sheeladevi S, Yanik EL, Rahmathullah L. Exposure to indoor biomass fuel and tobacco smoke and risk of adverse reproductive outcomes, mortality, respiratory morbidity and growth among newborn infants in south India. Int J Epidemiol. 2009;38:1351–1363. doi: 10.1093/ije/dyp286. [DOI] [PubMed] [Google Scholar]
- 31.Pope CA, III, Ezzati M, Dockery DW. Fine-particulate air pollution and life expectancy in the united states. N Engl J Med. 2009;360:376–386. doi: 10.1056/NEJMsa0805646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Allen DL, Leinwand LA. Postnatal myosin heavy chain isoform expression in normal mice and mice null for IIb or IId myosin heavy chains. Dev Biol. 2001;229:383–395. doi: 10.1006/dbio.2000.9974. [DOI] [PubMed] [Google Scholar]
- 33.Nadal-Ginard B, Mahdavi V. Molecular basis of cardiac performance: plasticity of the myocardium generated through protein isoform switches. J Clin Invest. 1989;84:1693–1700. doi: 10.1172/JCI114351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Harada K, Sugaya T, Murakami K, Yazaki Y, Komuro I. Angiotensin II type 1a receptor knockout mice display less left ventricular remodeling and improved survival after myocardial infarction. Circulation. 1999;100:2093–2099. doi: 10.1161/01.cir.100.20.2093. [DOI] [PubMed] [Google Scholar]
- 35.Stelzer JE, Brickson SL, Locher MR, Moss RL. Role of myosin heavy chain composition in the stretch activation response of rat myocardium. J Physiol. 2007;579:161–173. doi: 10.1113/jphysiol.2006.119719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Krenz M, Robbins J. Impact of beta-myosin heavy chain expression on cardiac function during stress. J Am Coll Cardiol. 2004;44:2390–2397. doi: 10.1016/j.jacc.2004.09.044. [DOI] [PubMed] [Google Scholar]
- 37.Gwathmey JK, Copelas L, MacKinnon R, Schoen FJ, Feldman MD, Grossman W, Morgan JP. Abnormal intracellular calcium handling in myocardium from patients with end-stage heart failure. Circ Res. 1987;61:70–76. doi: 10.1161/01.res.61.1.70. [DOI] [PubMed] [Google Scholar]
- 38.Hasenfuss G, Reinecke H, Studer R, Meyer M, Pieske B, Holtz J, Holubarsch C, Posival H, Just H, Drexler H. Relation between myocardial function and expression of sarcoplasmic reticulum Ca(2+)-ATPase in failing and nonfailing human myocardium. Circ Res. 1994;75:434–442. doi: 10.1161/01.res.75.3.434. [DOI] [PubMed] [Google Scholar]
- 39.Meyer M, Schillinger W, Pieske B, Holubarsch C, Heilmann C, Posival H, Kuwajima G, Mikoshiba K, Just H, Hasenfuss G. Alterations of sarcoplasmic reticulum proteins in failing human dilated cardiomyopathy. Circulation. 1995;92:778–784. doi: 10.1161/01.cir.92.4.778. [DOI] [PubMed] [Google Scholar]
- 40.Cozzi E, Hazarika S, Stallings HW, III, Cascio WE, Devlin RB, Lust RM, Wingard CJ, Van Scott MR. Ultrafine particulate matter exposure augments ischemia-reperfusion injury in mice. Am J Physiol Heart Circ Physiol. 2006;291:H894–H903. doi: 10.1152/ajpheart.01362.2005. [DOI] [PubMed] [Google Scholar]
- 41.Wellenius GA, Batalha JR, Diaz EA, Lawrence J, Coull BA, Katz T, Verrier RL, Godleski JJ. Cardiac effects of carbon monoxide and ambient particles in a rat model of myocardial infarction. Toxicol Sci. 2004;80:367–376. doi: 10.1093/toxsci/kfh161. [DOI] [PubMed] [Google Scholar]
- 42.Gurgueira SA, Lawrence J, Coull B, Murthy GG, Gonzalez-Flecha B. Rapid increases in the steady-state concentration of reactive oxygen species in the lungs and heart after particulate air pollution inhalation. Environ Health Perspect. 2002;110:749–755. doi: 10.1289/ehp.02110749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bartoli CR, Wellenius GA, Coull BA, Akiyama I, Diaz EA, Lawrence J, Okabe K, Verrier RL, Godleski JJ. Concentrated ambient particles alters myocardial blood flow during acute ischemia in conscious canines. Environ Health Perspect. 2009;117:333–337. doi: 10.1289/ehp.11380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Brook RD, Rajagopalan S. Particulate matter, air pollution, and blood pressure. J Am Soc Hypertens. 2009;3:332–350. doi: 10.1016/j.jash.2009.08.005. [DOI] [PubMed] [Google Scholar]
- 45.van den Hooven EH, de Kluizenaar Y, Pierik FH, Hofman A, van Ratingen SW, Zandveld PY, Mackenbach JP, Steegers EA, Miedema HM, Jaddoe VW. Air pollution, blood pressure, and the risk of hypertensive complications during pregnancy: the generation r study. Hypertension. 2011;57:406–412. doi: 10.1161/HYPERTENSIONAHA.110.164087. [DOI] [PubMed] [Google Scholar]
- 46.Delfino RJ, Gillen DL, Tjoa T, Staimer N, Polidori A, Arhami M, Sioutas C, Longhurst J. Electrocardiographic ST-segment depression and exposure to traffic-related aerosols in elderly subjects with coronary artery disease. Environ Health Perspect. 2011;119:196–202. doi: 10.1289/ehp.1002372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Park SK, Auchincloss AH, O’Neill MS, Prineas R, Correa JC, Keeler J, Barr RG, Kaufman JD, Diez Roux AV. Particulate air pollution, metabolic syndrome, and heart rate variability: the multi-ethnic study of atherosclerosis (mesa) Environ Health Perspect. 2010;118:1406–1411. doi: 10.1289/ehp.0901778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Link MS, Dockery DW. Air pollution and the triggering of cardiac arrhythmias. Curr Opin Cardiol. 2010;25:16–22. doi: 10.1097/HCO.0b013e32833358cd. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Burnett RT, Dales RE, Brook JR, Raizenne ME, Krewski D. Association between ambient carbon monoxide levels and hospitalizations for congestive heart failure in the elderly in 10 Canadian cities. Epidemiology. 1997;8:162–167. doi: 10.1097/00001648-199703000-00007. [DOI] [PubMed] [Google Scholar]
- 50.Li TT, Bai YH, Liu ZR, Liu JF, Zhang GS, Li JL. Air quality in passenger cars of the ground railway transit system in Beijing, China. Sci Total Environ. 2006;367:89–95. doi: 10.1016/j.scitotenv.2006.01.007. [DOI] [PubMed] [Google Scholar]
- 51.Hao J, Wang L. Improving urban air quality in china: Beijing case study. J Air Waste Manag Assoc. 2005;55:1298–1305. doi: 10.1080/10473289.2005.10464726. [DOI] [PubMed] [Google Scholar]
- 52.Liu Y, Chen R, Shen X, Mao X. Wintertime indoor air levels of pm10, pm2.5 and pm1 at public places and their contributions to tsp. Environ Int. 2004;30:189–197. doi: 10.1016/S0160-4120(03)00173-9. [DOI] [PubMed] [Google Scholar]
- 53.Nawrot TS, Perez L, Kunzli N, Munters E, Nemery B. Public health importance of triggers of myocardial infarction: a comparative risk assessment. Lancet. 2011;377:732–740. doi: 10.1016/S0140-6736(10)62296-9. [DOI] [PubMed] [Google Scholar]