

Abstract
Introduction
Multiple pharmacological therapies currently in prevalent clinical use for cardiac diseases have antifibrotic properties. Spironolactone, a potent antifibrotic agent, is currently used for advanced heart failure. Therapies such as HMG-CoA reductase inhibitors (statins) and angiotensin-converting enzyme inhibitors (ACEi) also have antifibrotic properties. However, the effect of these medications on the ventricular arrhythmia phenotype is currently unknown. Therefore, we set out to define the ventricular arrhythmia rates in patients actively treated with such therapies.
Methods and Results
We retrospectively evaluated the ventricular tachycardia (VT) rates in patients with structural heart disease actively treated with therapies with antifibrotic properties. VT rates were significantly diminished in patients treated with spironolactone (158 ± 26 beats per minute [bpm], n = 21) compared to patients on no medications (205 ± 22 bpm, n = 13) or those who were on similar heart-failure therapies but not on spironolactone (186 ± 32 bpm, n = 82). In addition, we observed that VT rates showed a significant trend toward lower rates in patients receiving either statins or ACEi, compared to patients on no medical therapy. In multivariate analysis, spironolactone therapy was identified as the single most significant variable for reduced VT rate.
Conclusion
Use of spironolactone in patients with heart failure is associated with reduced VT rate. Similar but less-significant reductions in VT rates were observed with use of other pharmacological agents with antifibrotic properties, such as statins and ACEi. Our findings, at least in part, could account for reduction in sudden cardiac death rates documented with use of these therapies.
Keywords: electrophysiology, ventricular tachycardia, cardiomyopathy, antifibrotic therapies, spironolactone
Introduction
In response to a variety of stressors, such as myocardial infarction and hypertension, the heart undergoes a remodeling process.1 This remodeling process includes alteration in cardiac-gene expression, cardiomyocyte hypertrophy, cell death, and transformation of fibroblasts to myofibroblasts, leading to excessive extracellular matrix (ECM) deposition and interstitial fibrosis. These remodeling events are associated with adverse outcomes, including a markedly increased risk of heart failure, ventricular arrhythmias, and sudden cardiac death.1,2
Excessive cardiac fibrosis contributes to cardiac dysfunction and plays an important role in ventricular arrhythmogenesis.3,4 Myocardial fibrosis promotes ventricular arrhythmogenesis through multiple mechanisms.5–8 Recent work suggests that cardiac fibrosis, long held to be irreversible, may regress under certain conditions.9 Current standard of care for patients with heart failure includes pharmacological therapies known to modulate fibrosis in the structurally remodeled myocardium, including HMG-CoA reductase inhibitors (statins) and angiotensin-converting enzyme inhibitors (ACEi), as well as aldosterone antagonists (spironolactone).10,11 Spironolactone inhibits profibrotic activity,12 reduces cardiac fibrosis,13 and has been shown to reduce the incidence of sudden cardiac death in patients with heart failure.14,15 The mineralocorticoid hormone, aldosterone, contributes to cardiovascular remodeling and morbidity and mortality through multiple mechanisms including cardiac fibrosis,13 which is attenuated by spironolactone.12,13,16 Spironolactone reduces mortality and the risk of sudden cardiac death in patients with dilated cardiomyopathy.14
Several pharmacological agents with antifibrotic properties, including spironolactone, statins, and ACEi are currently in widespread clinical use. The effects of these agents on ventricular arrhythmia burden, rate, and duration have not been investigated. Utilizing an animal model of dilated cardiomyopathy with diminished fibrosis, we recently showed that reduction in fibrosis in the setting of structural heart disease significantly changed the ventricular tachycardia (VT) phenotype. In addition to a diminished burden of VT, animals with reduced fibrosis in the setting of dilated cardiomyopathy had slower rates of inducible VT.17 Although surprising, this is a potentially clinically relevant finding, since slow VT is better tolerated in patients with already diminished cardiac reserve.18 Furthermore, slow VT is more responsive to antitachycardia pacing (ATP) in patients with implantable cardioverter defibrillators (ICD) reducing the number of ICD shocks.19,20 Recent evidence also suggests that patients with VT who are responsive to ATP therapy have less overall mortality compared to patients in whom VT requires shock therapy.21 These data would suggest that therapies that alter the VT phenotype, by reducing the VT rate, might provide significant clinical benefit in patients with heart failure, with a potential reduction in mortality. With this as background, we set out to characterize VT in patients with structural heart disease who were actively treated with the aforementioned therapies that target fibrosis.
Materials and Methods
VT Rate Assessment
Institutional review board (IRB) approval was obtained. We then reviewed data on 103 patients who had a diagnosis of VT in the clinical database at the Veterans Affairs Medical Center in Dallas, Texas, over a 2-year period (April 2006 to November 2008). This included patients who were diagnosed in the emergency room, telemetry units, coronary care unit, ICD clinic, and during electrophysiological studies. In patients with devices or those undergoing electrophysiological study, diagnosis of VT was made using standard electrocardiographic criteria and confirmed either by device interrogations or electrophysiological studies. For others, all patients were seen by cardiac electrophysiology services and diagnosis was based on telemetry strip and electrocardiographic evaluation. More than one physician confirmed this diagnosis on all patients and our review did not indicate that any of these patients were subsequently reported to have a different diagnosis on prospective follow-up. A total of 202 charts were reviewed on the basis of the diagnosis given at the time of initial evaluation. Among this group, 103 patients were selected for analysis of the VT cycle length (CL) after the rest were excluded for the following reasons: VT runs less than 10 beats, inappropriate diagnosis in ICD patients (shocks for supraventricular arrhythmias or ventricular fibrillation), and patients who were treated with antiremodeling medications for less than 6 months. Seventy percent of patients had sustained VT requiring pharmacological or electrical, internal, or external cardioversion. In patients found to have more than one episode of VT during a single encounter, either the last episode of VT was used, or two episodes with similar rates were averaged for this analysis. All patients received pharmacological therapy for at least 6 months.
Statistical Analysis
Summary statistics for VT rate are reported as mean ± standard deviation, with the unpaired Student’s t-test utilized in assessing VT-rate differences among various strata defined by medication use. Box-and-Whisker plots were constructed to visually examine the distribution of VT rate among these groups, with median ± interquartile ranges reported and significance determined via the Wilcoxon-rank-sum test. Univariate linear regression was performed for each of the baseline variables to assess unadjusted associations between VT rate and the particular covariate. A backward selection technique was used to determine significant predictors of VT rate, in which all variables were included in a single model, with the least significant variable at the alpha = 0.15 level removed. This process was repeated until no more variables were removed. Additionally, a prespecified multivariable linear regression model was also fit, controlling for age, ejection fraction, ischemia status, and angiotensin-converting enzyme (ACE) inhibitor, angiotensin receptor blocker (ARB), statin, β-blocker, aldosterone, and amiodarone usage. Significance was defined by two-tailed tests, alpha = 0.05. All statistical analyses were performed using SAS Version 9.1.3 (SAS Institute, Cary, NC, USA).
Results
Patient Characteristics
A total of 103 patients (males n = 102) were included in this analysis. The mean age was 65 ± 12 years and mean left ventricular ejection fraction was 35 ± 13%. Sixty eight (66%) patients had ischemic cardiomyopathy and 35 patients (34%) had nonischemic cardiomyopathy. Baseline patient characteristics are shown in Table I.
Table I.
Baseline Characteristics
| Variable | Overall (n = 103) | No Medications (n = 13) | No Spironolactone (n = 82) | With Spironolactone (n = 21) | P-Value (no Meds vs with Spironolactone) | P-Value (no Spironolactone vs with Spironolactone) |
|---|---|---|---|---|---|---|
| Men | 102 (99%) | 13 (100%) | 81 (99%) | 21 (100%) | n/a | 1.000 |
| African-American | 28 (27%) | 2 (15%) | 21 (26%) | 7 (33%) | 0.327 | 0.824 |
| Hypertension | 95 (92%) | 11 (85%) | 74 (90%) | 21 (100%) | 0.139 | 0.202 |
| Diabetes | 36 (35%) | 3 (23%) | 29 (35%) | 7 (33%) | 0.704 | 1.000 |
| Ischemic | 68 (66%) | 6 (46%) | 55 (67%) | 13 (62%) | 0.484 | 0.797 |
| BMI | 28.2 (7.8) | 28.5 (9.0) | 28.5 (7.9) | 26.9 (7.3) | 0.779 | 0.545 |
| Age | 65.9 (11.4) | 68.0 (17.2) | 66.4 (11.5) | 64.1 (11.2) | 0.230 | 0.199 |
| Ejection fraction | 35.6 (13.4) | 41.4 (12.8) | 37.0 (12.4) | 30.0 (14.1) | 0.054 | 0.043 |
| EF < 30 | 37 (36%) | 4 (31%) | 25 (30%) | 12 (57%) | 0.172 | 0.040 |
| ACEi | 63 (61%) | 0 (0%) | 50 (61%) | 13 (62%) | 0.000 | 1.000 |
| ARB | 14 (14%) | 1 (8%) | 8 (10%) | 6 (29%) | 0.210 | 0.036 |
| β-blocker | 78 (76%) | 12 (92%) | 61 (74%) | 17 (81%) | 0.627 | 0.776 |
| Amiodarone | 15 (15%) | 2 (15%) | 13 (16%) | 2 (10%) | 0.606 | 0.730 |
We defined no medication as no to ACEi, statins, and spironolactone.
BMI = body mass index; EF = ejection fraction; ACEi = angiotensin-converting enzyme inhibitor; ARB = angiotensin-receptor blockers.
VT Characteristics
There was no difference in VT rates between patients with ischemic (180 ± 34 beats per minute [bpm], n = 68) and nonischemic (180 ± 33 bpm, n = 35) cardiomyopathy (Fig. 1, panel A). In comparison, VT rate was significantly lower in patients who were actively treated with spironolactone (158 ± 26 bpm, n = 21) when compared to patients on no medical therapy (205 ± 22 bpm, n = 13) (Fig. 1, panel B), and when compared to patients who were medically treated for heart failure with other antifibrotic drugs with the exception of spironolactone (186 ± 32 bpm, n = 82) (Fig. 1, panel C). In univariate analysis, spironolactone was the only significant predictor of VT rate (Table II). Furthermore, in multivariate analysis, spironolactone was the only pharmacological agent that was an independent predictor of slow VT (Table III). In the backward selection technique, spironolactone had the highest absolute t-value with a P-value of <0.0001 (Table IV). Left ventricular function, β-blocker, or antiarrhythmic therapies were not identified as significant independent variables. Both ACEi (175 ± 33 bpm, n = 63) and statin (176 ± 32 bpm, n = 68) therapy were each associated with a reduction in VT rate (Fig. 2); however, these therapies were not found to be independent predictors of slow VT in multivariate analysis (Table III).
Figure 1.
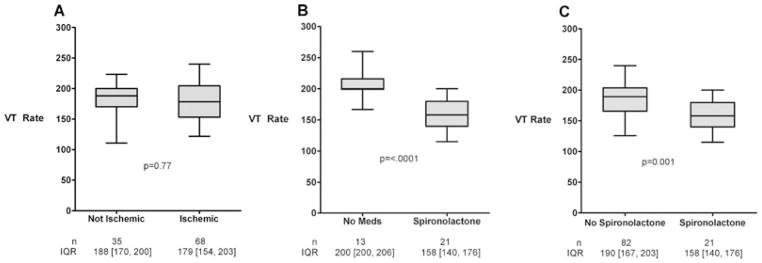
Analysis of ventricular tachycardia rates in ischemic and nonischemic cardiomyopathy showed no significant differences between the two groups in mean rate of ventricular tachycardia (Panel A). However, the average ventricular tachycardia rate was significantly slower in patients who were actively treated with spironolactone as compared to patients on no medical therapy (P < 0.0001) (Panel B) and patients not treated with spironolactone therapy (P < 0.002) (Panel C).
Table II.
Univariate Models; Each Line is a Model with VT Rate as Outcome, Variable as Only Predictor
| Variable | β | t-Value | P-Value |
|---|---|---|---|
| Age | −0.390 | −1.370 | 0.172 |
| EF | 0.291 | 1.200 | 0.235 |
| ACEi | −11.238 | −1.690 | 0.094 |
| ARB | 10.307 | 1.080 | 0.281 |
| Statin | −9.465 | −1.380 | 0.171 |
| β-blocker | −0.223 | −0.030 | 0.977 |
| Spironolactone | −27.513 | −3.590 | 0.001 |
| Amiodarone | 3.958 | 0.430 | 0.671 |
| Ischemic | 0.056 | 0.010 | 0.994 |
| Male sex | 23.275 | 0.700 | 0.487 |
| Black | −0.914 | −0.120 | 0.902 |
| Hypertension | −19.839 | −1.640 | 0.104 |
| Diabetes | −1.164 | −0.170 | 0.866 |
| Ischemic | 0.056 | 0.010 | 0.994 |
| BMI | 0.545 | 1.270 | 0.208 |
EF = ejection fraction; ACEi = angiotensin-converting enzyme inhibitor; ARB = angiotensin-receptor blocker; BMI = body mass index.
Table III.
Multivariable Linear Regression
| Parameter | R2 = 0.22
|
||
|---|---|---|---|
| β | t-Value | P-Value | |
| Intercept | 228.36 | 10.17 | <.0001 |
| EF | −0.62 | −2.24 | 0.028 |
| Age | 0.14 | 0.58 | 0.562 |
| ACEi | −4.56 | −0.6 | 0.548 |
| ARB | 20.95 | 1.94 | 0.056 |
| Statin | −10.57 | −1.5 | 0.138 |
| β-blocker | −0.93 | −0.13 | 0.898 |
| Spironolactone | −31.13 | −3.88 | 0.0002 |
| Amiodarone | −3.47 | −0.38 | 0.703 |
| Ischemic | 2.72 | 0.39 | 0.701 |
Multivariate linear regression model with β coefficients for the variables explaining average VT rate.
All β coefficients represent the change in average VT rate for a 1 unit increase in the particular variable of interest, holding other model parameters fixed.
For categorical variables, the β estimate is used for those on medication or who were ischemic.
The R2 of 0.22 means that 22% of the variance of VT rate is explained by the variables included in the model.
All P-values derived from two-tailed t-distributions of β coefficients, with significance determined using alpha = 0.05.
EF = ejection fraction; ACEi = angiotensin-converting enzyme inhibitor; ARB = angiotensin-receptor blocker.
Table IV.
Backward Selection Technique
| Candidate variables: Age, EF, ACEi, ARB, statin, BB, spironolactone, amiodarone, ischemic, BMI, black race, sex, hypertension, diabetes | |||
|---|---|---|---|
| n = 101 Variable | R2 = 0.209
|
||
| β | t-Value | P-Value | |
| Intercept | 228.45 | 12.36 | <.0001 |
| Age | −0.56 | −2.12 | 0.036 |
| ARB | 23.64 | 2.50 | 0.014 |
| Statin | −11.36 | −1.76 | 0.082 |
| Spironolactone | −32.89 | −4.27 | <.0001 |
EF = ejection fraction, ACEi = angiotensin-converting enzyme inhibitor, BB = β-blockers, ARB = angiotensin receptor blocker; BMI = body mass index.
Figure 2.
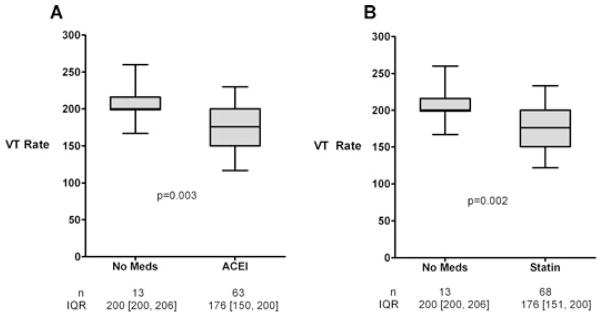
Ventricular tachycardia rates were significantly slower in patients who were actively treated with either ACEi (P < 0.005) (Panel A) or statins (P < 0.005) (Panel B), compared to patients on medical therapy.
Discussion
Cardiac fibrosis is a hallmark of structural remodeling in response to a variety of stressors, and it is central to arrhythmogenesis in the pathologically remodeled heart. The major finding we describe here is that VT rate (bpm) is altered in patients on pharmacological therapies with antifibrotic properties. Specifically, VT rates are significantly slower in patients on spironolactone therapy, and a similar but less-significant reduction was observed in patients on therapies such as statins and ACEi. Together, these results suggest a previously unknown, and clinically beneficial, electrophysiological effect of therapies that modulate cardiac fibrosis.
Fibrosis in the heart contributes to both diastolic and systolic contractile dysfunction as well as arrhythmogenesis.5,10 It is well established that increased amounts of fibrotic tissue in the heart strongly correlate with an increased incidence of atrial and ventricular tachyarrhythmias and sudden cardiac death.11,22 The collagenous septa and ventricular myofibroblasts have both been shown conclusively to facilitate the arrhythmic phenotype of the remodeled myocardium.6–8 Therefore, it is not surprising that recent investigations have targeted the fibrotic scar of the heart to manipulate the structure and arrhythmic phenotype of the remodeled heart.23–25 Current therapies that reduce the amount of fibrosis as a component of their mechanism, such as spironolactone, have already been shown to reduce sudden cardiac death rates.14,26 This reduction in sudden cardiac death correlates with reduction in serum markers of ECM.15 In fact, both specific and nonspecific inhibitors of aldosterone have been shown to reduce markers of ECM and mortality.15,27 The exact mechanism in the reduction of observed arrhythmic events and sudden cardiac deaths are not yet known.
In addition to the findings presented here, we have recently demonstrated that arrhythmias in a fibrosis-deficient model of cardiomyopathy propagate for longer durations and have slower rates as compared to those in a pressure-overload model of cardiomyopathy.17 More importantly, we report in this study that ventricular arrhythmias manifest longer CLs (reduced rate in bpm) in humans who are actively treated with a therapy with significant antifibrotic activity, namely, spironolactone. This is an important finding as new antifibrotic therapies are developed, and further evidence of the significant impact fibrosis has on induction and propagation of conduction emerges.28 Since slower rates of VT are better tolerated clinically,18 this finding may in part explain the significant reduction in mortality and sudden cardiac death observed in patients taking antifibrotic therapies such as spironolactone. In the RALES trial, the maximum benefit in sudden cardiac death was observed in patients with reductions in markers of fibrosis,15 which may be related to decreased or better tolerated arrhythmic events.
Besides the potential role in reduction in sudden cardiac death from antifibrotic therapies, our findings have other important clinical implications. In this era of prevalent ICD use, along with adjunctive use of pharmacological therapy, recurrent ventricular arrhythmias requiring ICD shocks remain a major source of morbidity in patients with cardiovascular disease. Most VTs in failing hearts have fast rates (short CLs) and are nonsustained, self-terminating events; however, when sustained they require defibrillation for termination, which is painful and can lead to induction of sustained life-threatening arrhythmias. Slower VT is not only better clinically tolerated, but is more easily terminated with painless therapies, such as ATP therapy.29 Altering the VT phenotype from fast VT to slow is a novel approach to treating this particular subset of patients and provides important adjunctive therapy in the management of patients with VT due to cardiac remodeling, including fibrosis. Recent evidence also suggests that VT that can be terminated with ATP therapies is associated with less mortality as compared to VT treated with ICD shock.21 On the basis of our previous work17 and our findings in this study, we propose that altering the VT phenotype utilizing antifibrotic agents, in particular spironolactone, provides significant clinical benefit. These therapies should be considered in patients in whom such effects are desired. Further validation of our findings should be confirmed in a prospective randomized fashion.
Conclusion
From this retrospective study, spironolactone therapy is associated with a slower rate, in bpm (longer CL), of spontaneous VT episodes. Use of pharmacological agents with antifibrotic properties, in patients with or at risk of VT, may result in clinically better tolerated VTs and potentially fewer ICD shocks.
Limitations
The major limitation of this study is its retrospective nature. Although our findings are substantiated by powerful statistical methods, a causal relationship cannot be established. Additionally, while statin and ACEi therapies show slower VT rates, these two therapies were not identified as statistically significant factors in our multivariate analysis. In this retrospective analysis, only three patients with VT were actively taking amiodarone; therefore, a lack of relationship with this therapy and the VT phenotype is not adequately addressed, given the patient population. Furthermore, we have not evaluated the VT burden in these patients, or other characteristics of VT.
Acknowledgments
Source of Funding
This work was supported by the Heart American Heart Association 0705170Y, 0830313N (RHN), 0640084N (JAH), and National Institutes of Health Grants HL-075173, HL-090842, and HL-080144 (to J.A.H.).
Footnotes
Conflict of Interest: None.
References
- 1.Hill JA, Olson EN. Cardiac plasticity. N Engl J Med. 2008;358:1370–1380. doi: 10.1056/NEJMra072139. [DOI] [PubMed] [Google Scholar]
- 2.McLenachan JM, Henderson E, Morris KI, Dargie HJ. Ventricular arrhythmias in patients with hypertensive left ventricular hypertrophy. N Engl J Med. 1987;317:787–792. doi: 10.1056/NEJM198709243171302. [DOI] [PubMed] [Google Scholar]
- 3.Manabe I, Shindo T, Nagai R. Gene expression in fibroblasts and fibrosis: Involvement in cardiac hypertrophy. Circ Res. 2002;91:1103–1113. doi: 10.1161/01.res.0000046452.67724.b8. [DOI] [PubMed] [Google Scholar]
- 4.Weber KT. Fibrosis in hypertensive heart disease: Focus on cardiac fibroblasts. J Hypertens. 2004;22:47–50. doi: 10.1097/00004872-200401000-00011. [DOI] [PubMed] [Google Scholar]
- 5.Zeppenfeld K, Stevenson WG. Ablation of ventricular tachycardia in patients with structural heart disease. Pacing Clin Electrophysiol. 2008;31:358–374. doi: 10.1111/j.1540-8159.2008.00999.x. [DOI] [PubMed] [Google Scholar]
- 6.Spach MS, Boineau JP. Microfibrosis produces electrical load variations due to loss of side-to-side cell connections: A major mechanism of structural heart disease arrhythmias. Pacing Clin Electrophysiol. 1997;20:397–413. doi: 10.1111/j.1540-8159.1997.tb06199.x. [DOI] [PubMed] [Google Scholar]
- 7.Miragoli M, Gaudesius G, Rohr S. Electrotonic modulation of cardiac impulse conduction by myofibroblasts. Circ Res. 2006;98:801–810. doi: 10.1161/01.RES.0000214537.44195.a3. [DOI] [PubMed] [Google Scholar]
- 8.Miragoli M, Salvarani N, Rohr S. Myofibroblasts induce ectopic activity in cardiac tissue. Circ Res. 2007;101:755–758. doi: 10.1161/CIRCRESAHA.107.160549. [DOI] [PubMed] [Google Scholar]
- 9.Zeisberg EM, Tarnavski O, Zeisberg M, Dorfman AL, McMullen JR, Gustafsson E, Chandraker A, et al. Endothelial-to-mesenchymal transition contributes to cardiac fibrosis. Nat Med. 2007;13:952–961. doi: 10.1038/nm1613. [DOI] [PubMed] [Google Scholar]
- 10.Spinale FG. Myocardial matrix remodeling and the matrix metalloproteinases: Influence on cardiac form and function. Physiol Rev. 2007;87:1285–1342. doi: 10.1152/physrev.00012.2007. [DOI] [PubMed] [Google Scholar]
- 11.Everett THt, Olgin JE. Atrial fibrosis and the mechanisms of atrial fibrillation. Heart Rhythm. 2007;4:S24–S27. doi: 10.1016/j.hrthm.2006.12.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lea WB, Kwak ES, Luther JM, Fowler SM, Wang Z, Ma J, Fogo AB, et al. Aldosterone antagonism or synthase inhibition reduces end-organ damage induced by treatment with angiotensin and high salt. Kidney Int. 2009;75:936–944. doi: 10.1038/ki.2009.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brilla CG, Matsubara LS, Weber KT. Anti-aldosterone treatment and the prevention of myocardial fibrosis in primary and secondary hyperaldosteronism. J Mol Cell Cardiol. 1993;25:563–575. doi: 10.1006/jmcc.1993.1066. [DOI] [PubMed] [Google Scholar]
- 14.Pitt B, Zannad F, Remme WJ, Cody R, Castaigne A, Perez A, Palensky J, et al. The effect of spironolactone on morbidity and mortality in patients with severe heart failure. Randomized Aldactone Evaluation Study Investigators. N Engl J Med. 1999;341:709–717. doi: 10.1056/NEJM199909023411001. [DOI] [PubMed] [Google Scholar]
- 15.Zannad F, Alla F, Dousset B, Perez A, Pitt B. Limitation of excessive extracellular matrix turnover may contribute to survival benefit of spironolactone therapy in patients with congestive heart failure: insights from the randomized aldactone evaluation study (RALES). Rales Investigators. Circulation. 2000;102:2700–2706. doi: 10.1161/01.cir.102.22.2700. [DOI] [PubMed] [Google Scholar]
- 16.Van Den Borne SW, Isobe S, Zandbergen HR, Li P, Petrov A, Wong ND, Fujimoto S, et al. Molecular imaging for efficacy of pharmacologic intervention in myocardial remodeling. JACC Cardiovasc Imaging. 2009;2:187–198. doi: 10.1016/j.jcmg.2008.11.011. [DOI] [PubMed] [Google Scholar]
- 17.Massare J, Berry JM, Luo X, Rob F, Johnstone JL, Shelton JM, Bassel-Duby R, et al. Diminished cardiac fibrosis in heart failure is associated with altered ventricular arrhythmia phenotype. J Cardiovasc Electrophysiol. 2010 doi: 10.1111/j.1540-8167.2010.01736.x. (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sadoul N, Mletzko R, Anselme F, Bowes R, Schöls W, Kouakam C, Casteigneau G, et al. Incidence and clinical relevance of slow ventricular tachycardia in implantable cardioverter-defibrillator recipients: An international multicenter prospective study. Circulation. 2005;112:946–953. doi: 10.1161/CIRCULATIONAHA.105.533513. [DOI] [PubMed] [Google Scholar]
- 19.Wathen MS, DeGroot PJ, Sweeney MO, Stark AJ, Otterness MF, Adkisson WO, Canby RC, et al. Prospective randomized multicenter trial of empirical antitachycardia pacing versus shocks for spontaneous rapid ventricular tachycardia in patients with implantable cardioverter-defibrillators: Pacing Fast Ventricular Tachycardia Reduces Shock Therapies (PainFREE Rx II) trial results. Circulation. 2004;110:2591–2596. doi: 10.1161/01.CIR.0000145610.64014.E4. [DOI] [PubMed] [Google Scholar]
- 20.Wathen MS, Sweeney MO, DeGroot PJ, Stark AJ, Koehler JL, Chisner MB, Machado C, et al. Shock reduction using antitachycardia pacing for spontaneous rapid ventricular tachycardia in patients with coronary artery disease. Circulation. 2001;104:796–801. doi: 10.1161/hc3101.093906. [DOI] [PubMed] [Google Scholar]
- 21.Sweeney MO, Sherfesee L, DeGroot PJ, Wathen MS, Wilkoff BL. Differences in effects of electrical therapy type for ventricular arrhythmias on mortality in implantable cardioverter-defibrillator patients. Heart Rhythm. 2010;7:353–360. doi: 10.1016/j.hrthm.2009.11.027. [DOI] [PubMed] [Google Scholar]
- 22.Varnava AM, Elliott PM, Mahon N, Davies MJ, McKenna WJ. Relation between myocyte disarray and outcome in hypertrophic cardiomyopathy. Am J Cardiol. 2001;88:275–279. doi: 10.1016/s0002-9149(01)01640-x. [DOI] [PubMed] [Google Scholar]
- 23.Laflamme MA, Murry CE. Regenerating the heart. Nat Biotechnol. 2005;23:845–856. doi: 10.1038/nbt1117. [DOI] [PubMed] [Google Scholar]
- 24.Duffy HS. Cardiac connections–The antiarrhythmic solution? N Engl J Med. 2008;358:1397–1398. doi: 10.1056/NEJMcibr0708922. [DOI] [PubMed] [Google Scholar]
- 25.Roell W, Lewalter T, Sasse P, Tallini YN, Choi BR, Breitbach M, Doran R, et al. Engraftment of connexin 43-expressing cells prevents post-infarct arrhythmia. Nature. 2007;450:819–824. doi: 10.1038/nature06321. [DOI] [PubMed] [Google Scholar]
- 26.Brown RD, Ambler SK, Mitchell MD, Long CS. The cardiac fibroblast: Therapeutic target in myocardial remodeling and failure. Annu Rev Pharmacol Toxicol. 2005;45:657–687. doi: 10.1146/annurev.pharmtox.45.120403.095802. [DOI] [PubMed] [Google Scholar]
- 27.Iraqi W, Rossignol P, Angioi M, Fay R, Nuée J, Ketelslegers JM, Vincent J, et al. Extracellular cardiac matrix biomarkers in patients with acute myocardial infarction complicated by left ventricular dysfunction and heart failure: Insights from the Eplerenone Post-Acute Myocardial Infarction Heart Failure Efficacy and Survival Study (EPHESUS) study. Circulation. 2009;119:2471–2479. doi: 10.1161/CIRCULATIONAHA.108.809194. [DOI] [PubMed] [Google Scholar]
- 28.Ten Tusscher KH, Panfilov AV. Influence of diffuse fibrosis on wave propagation in human ventricular tissue. Europace. 2007;9(Suppl 6):vi38–vi45. doi: 10.1093/europace/eum206. [DOI] [PubMed] [Google Scholar]
- 29.Schaumann A, von zur Muhlen F, Herse B, Gonska BD, Kreuzer H. Empirical versus tested antitachycardia pacing in implantable cardioverter defibrillators: a prospective study including 200 patients. Circulation. 1998;97:66–74. doi: 10.1161/01.cir.97.1.66. [DOI] [PubMed] [Google Scholar]