

Summary
Signaling pathways controlled by reversible protein phosphorylation (catalyzed by kinases and phosphatases) in the malaria parasite Plasmodium are of great interest, for both increased understanding of parasite biology and identification of novel drug targets. Here, we report a functional analysis in Plasmodium of an ancient bacterial Shewanella-like protein phosphatase (SHLP1) found only in bacteria, fungi, protists, and plants. SHLP1 is abundant in asexual blood stages and expressed at all stages of the parasite life cycle. shlp1 deletion results in a reduction in ookinete (zygote) development, microneme formation, and complete ablation of oocyst formation, thereby blocking parasite transmission. This defect is carried by the female gamete and can be rescued by direct injection of mutant ookinetes into the mosquito hemocoel, where oocysts develop. This study emphasizes the varied functions of SHLP1 in Plasmodium ookinete biology and suggests that it could be a novel drug target for blocking parasite transmission.
Graphical Abstract

Highlights
► SHLP1 has phosphatase activity insensitive to phosphoprotein phosphatase inhibitors ► SHLP1 deletion results in reduced ookinete and microneme development ► SHLP1 is contributed by the female gamete ► SHLP1 deletion results in oocyst ablation and is critical for parasite transmission
Reversible protein phosphorylation (catalyzed by protein kinases and phosphatases) in the malaria parasite Plasmodium is of great interest. Here, Tewari and colleagues report the first functional analysis of a Shewanella-like protein phosphatase (SHLP1) in rodent Plasmodium. SHLP1 is an enzyme that was reported previously only in bacteria, fungi, protists, and plants. Deletion of the gene results in reduced development of zygotes (ookinetes) and disruption of organelles (micronemes) associated with invasion into the mosquito gut, thus resulting in complete ablation of parasite transmission.
Introduction
Unicellular parasites of the genus Plasmodium are the causative agent of malaria, with annual fatalities close to 1.25 million (Murray et al., 2012). Development of mammalian Plasmodium species proceeds via asexual exoerythrocytic proliferation and intraerythrocytic multiplication occurring in mammalian liver hepatocytes and erythrocytes, respectively, whereas sexual development and sporogony occur in the mosquito. Plasmodium belongs to the phylum Apicomplexa, which is characterized by the presence of distinct apical organelles consisting of micronemes, dense granules, and rhoptries that are used by the parasite for host invasion and gliding motility (Bannister and Sherman, 2009). Of the three invasive stages (sporozoites, merozoites, and ookinetes), the ookinete uniquely lacks rhoptries and dense granules.
In most organisms, processes involved in cell cycle, differentiation, and development are regulated by reversible phosphorylation of proteins. While kinases are well recognized as important drug targets, the study of protein phosphatases (PPs) has only recently begun to identify them as potential therapeutic targets (McConnell and Wadzinski, 2009; Moorhead et al., 2007).
Bioinformatic analyses have identified ∼80 kinases and∼30 PP catalytic subunits encoded in the P. falciparum genome (Ward et al., 2004; Wilkes and Doerig, 2008), compared to 518 kinases and 147 PP catalytic subunits identified in the human genome (Moorhead et al., 2007). Recent systematic functional studies have uncovered the important roles of kinases during signal transduction and various developmental processes in Plasmodium (Solyakov et al., 2011; Tewari et al., 2010).
Although complementary functional studies of the Plasmodium phosphatome are lacking, its bioinformatic analysis has revealed the presence of nonconventional PPs containing kelch-like motifs as found in plant PPs and two bacterial Shewanella-like PPs (Shelphs or SHLP), both lacking orthologs in humans (Wilkes and Doerig, 2008). SHLPs are related to the phosphoprotein phosphatase (PPP) family of serine/threonine PPs (STPs), span the eukaryote-prokaryote boundary, and are also present in plants, heterokonts, fungi, and some Protozoa (Andreeva and Kutuzov, 2004; Uhrig and Moorhead, 2011).
In Plasmodium, the expression of its two SHLP isoforms in late schizont and merozoite stages has been reported (Hu et al., 2010). Although the γ-proteobacterium Shewanella spp. SHLP has been utilized to analyze enzymatic catalysis at low temperatures (Tsuruta et al., 2008), no function of any SHLP is known to date (Kutuzov and Andreeva, 2012).
In this study, we have used the rodent malaria P. berghei to elucidate the function of Plasmodium SHLP1 (PBANKA_133240) during the parasite life cycle using reverse genetics, biochemical and cell biological approaches.
Results and Discussion
Bioinformatics of Shewanella-like Protein Phosphatases in Plasmodium
We confirmed the presence of two genes in the P. berghei genome encoding Shewanella-like PPs: PBANKA_133240, which has a signal peptide and apicoplast targeting sequence and PBANKA_060470, which were named Shewanella-like protein phosphatases SHLP1 and SHLP2, respectively.
In eukaryotes other than in Apicomplexa, SHLPs have been found in, Archaeplastida, some fungi, and some Chromalveolates and are structurally related to a class of bacterial PPPs first identified in the psychrophilic bacteria Shewanella and Colwellia (Tsuruta et al., 2008). Consequently, a phylogenetic tree was constructed from multiple alignments of 47 SHLP-related amino acid sequences (Figure 1A; Table S1) that confirmed the presence of SHLPs in the examined species in addition to flowering plants (Kutuzov and Andreeva, 2012; Uhrig and Moorhead, 2011). Both PbSHLP1 and PbSHLP2 show higher identity to AtSLP2 than AtSLP1 (Table S1) and form a distinct phylogenetic group in Chromalveolata.
Figure 1.

Phylogenetic Analysis and Expression of SHLP1 during Parasite Development
(A) Multiple alignment of the catalytic domains of SHLP proteins excluding positions with a gap in any sequence was used to construct a phylogenetic tree by the neighbor-joining method and using Kimura’s empirical correction for multiple hits with 100 bootstrapped replicates implemented in ClustaW2. The PP catalytic domains were identified by the conserved PPP specific motifs “GD(L/I/V)HG” at the N-terminal and the “(I/L/V)D(S/T/G)” at the C-terminal region. A. thaliana PPP family PPs were used as an out group in the analysis. Species and gene abbreviations are outlined in Table S1. Trypanosome T. brucei (Tb), T. cruzi (Tc), and L. major (Lmj) PP domains are indicated by systematic gene IDs. Bootstrap values of 50% and above for the nodes are shown. The scale bar indicates 0.1 substitutions per site.
(B) SHLP1-GFP expression in transgenic parasites. A Cy3-conjugated antibody recognizing P28 on the surface of activated females, zygotes, and ookinetes was used in the sexual stages. Scale bar, 5 μm.
(C) Costaining with ER-tracker Red shows association of SHLP1-GFP with the ER. Scale bar, 10 μm.
(D) WT-GFP and SHLP1-GFP subcellular fractionation. Anti-GFP (left two panels) and anti-GAP45 (right panel) western blot of soluble (S), peripheral membrane (PM), and integral membrane (IM) fractions.
See also Figure S1.
Plasmodium spp. contained two SHLPs, while Toxoplasma and Cryptosporidium have only one SHLP mostly resembling SHLP2. Sequence analysis of Plasmodium SHLPs revealed that only the highly conserved STP catalytic residues (GD[I/V/T/L]HG, GD[L/Y/F]V[D/A]RG, GNHE, HGG) and metal ion binding sites (underlined) are largely conserved. The rest of the conserved STP residues, okadaic acid, and microcystin LR inhibitor binding sites, as well as PP1 regulatory subunit binding sites (KIF, EFF) and PP1 substrate binding sites are only partially or not conserved (Figure S1). Amino acids involved in PP2A trimeric holoenzyme formation were also absent (Figure S1).
Figure S1.

Sequence Alignment of the Plasmodium spp SHLPs, Related to Figure 1
(A) The invariant residues conserved in protein phosphatases are highlighted in gray and metal ion binding sites are marked with solid circles. The amino acids with role in inhibitor binding are highlighted: Inhibitor-2 binding amino acids (1.-7. blue squares); microcystin binding amino acids (1.-4. orange squares); okadaic acid and microcystin binding amino acids (1.-5. green squares); inhibitor-2, microcystin and okadaic acid binding amino acid (1.-2. blue boxes). A. thaliana type 1 protein phosphatase (TOPP1) was included in the alignment as marker for the conserved STP residues. The NCBI/GenBank identifiers of the sequences used to construct the alignment can be found in Table S1.
(B) High resolution 2D projection of SHLP1-GFP asexual blood stage and gametocyte co-stained with ER tracker. Bar = 10 μM.
SHLP1-GFP Is Present in All the Parasite Developmental Stages Examined, Is Preferentially Localized to the Endoplasmic Reticulum, and Is Present in Membrane Fractions
To study its expression during the P. berghei life cycle, we generated a C-terminal GFP fusion protein using endogenous shlp1 (PBANKA_133240) and single crossover recombination (Figures S2A–S2C). SHLP1-GFP showed diffuse fluorescence staining in all parasite stages examined by microscopy with prominent staining in the asexual stages, the male gamete, and oocyst (Figure 1B). However, despite the presence of a predicted apicoplast targeting signal, the protein was found distributed nonuniformly throughout the parasite body in all stages analyzed (Figure 1B) and colocalized with ER tracker Red, suggesting that SHLP1-GFP is located in the endoplasmic reticulum (ER) (Figure 1C). This was further confirmed by high-resolution imaging (Figure S1B). Furthermore, subcellular fractionation of asexual blood stages using hypotonic lysis and carbonate solubility recovered most of the SHLP1-GFP protein in the carbonate soluble (peripheral membrane) and carbonate insoluble (integral membrane proteins) fractions, suggesting a membrane localization of the protein in the ER. GFP alone in the control was in the soluble (cytosolic) fraction and GAP45 control was found in the integral membrane fraction (Figure 1D).
Figure S2.

gfp Tagging of the Endogenous shlp1 Locus and Knockout of shlp1, Related to Figure 3
(A) Schematic representation of the gene targeting strategy used for gene tagging the endogenous locus with gfp via single homologous recombination. The C-fusion tag construct contains an insert (white box) homologous to the 3′ end of the shlp1 ORF fused to gfp. A human dehydrofolate reductase selectable marker (hdhfr) allows for selection of transgenic parasites. Arrows 1 and 2 indicate primers used for diagnostic PCR.
(B) Diagnostic integration PCR showing the expected 1.4 kb band and confirming successful integration of the tagging construct.
(C) Western blot analysis using an anti-GFP (Invitrogen) antibody against protein extracted from gametocytes of WT P. berghei ANKA 507 clone 1 constitutively expressing GFP (wt) and transgenic (tag) parasites showing bands of expected sizes of 29 kDa for wild-type-GFP and 71 kDa for SHLP1-GFP.
(D) Schematic representation of the endogenous shlp1 locus, the knockout construct and the recombined shlp1 locus following double cross-over recombination. The knockout construct contains a Toxoplasma gondii dehydrofolate reductase/thymidylate synthase (tg dhfr/ts) cassette with a Pbdhfr 3′ UTR for selection of transgenic parasites with pyrimethamine. Arrows 1, 2, 3 and 4 indicate binding sites for primers used in integration PCR and knockout PCR. EcoRV restriction sites and probe binding sites used for Southern blot analysis are shown.
(E) Genotypic analysis by integration PCR and knockout PCR. Presence of a 735 bp band using integration specific primers 1 and 2 on gDNA of Δshlp1 mutants cl3 and cl7 GFP generated in 507 clone 1 indicates correct integration of the knockout construct. Absence of the 343 bp WT specific band amplified by primers 3 and 4 in both mutants shows loss of the wild-type shlp1 locus. WT gDNA was used as control.
(F) Genotypic analysis by Southern blot. gDNA of WT parasites, wild-type 507 clone 1 (GFPcon) and Δshlp1 mutants cl3 and cl7 GFP was analyzed by Southern blot following EcoRV digestion. A probe homologous to the shlp1 3′UTR recognizes a 7.8 kb fragment for the endogenous locus and a > 10 kb fragment for the recombined locus.
(G) Pulse-field gel electrophoresis blot (PFGE) hybridized with a Pbdhfr 3′UTR probe. The probe hybridizes to the endogenous dhfr locus on chromosome 7 and the disrupted locus on chromosome 14. (H) Bar graph showing relative expression of endogenous shlp1 in Δshlp1 mutants using qRT-PCR compared to wild-type. Error bars represent ±SEM, n = 3 from three separate experiments in both clone 3 and clone 7 GFP.
A cytosolic localization has recently been described for AtSLP2 (At1g18480), whereas AtSLP1 was located in the chloroplast (Uhrig and Moorhead, 2011); P. falciparum SHLP2 (PF3D7_1206000) was recently identified at the apical end of the merozoite (Hu et al., 2010).
SHLP1 Has Phosphatase Enzyme Activity and Is Not Itself Phosphorylated in Schizonts or Activated Gametocytes
We investigated whether SHLP1 is an active phosphatase in P. berghei using the SHLP1-GFP protein expressed in vivo and 3-O-methylfluorescein phosphate (MFP) as a substrate. SHLP1-GFP displayed phosphatase activity proportional to the amount of lysate in the assay; wild-type (WT) parasite extracts not expressing GFP and WT and SHLP1-GFP controls without MFP substrate produced almost no signal. The basal fluorescence level observed in GFP control lysates was likely due to GFP fluorescence, as the signal was still present in the absence of MFP substrate (Figure 2A, upper panel). Unlike classical eukaryotic PPP, which are specific for Ser and Thr residues, bacterial PPPs may have broader substrate specificity, and the cold-active phosphatase originally described in Shewanella is a Tyr PP (Tsuruta et al., 2008). MFP is hydrolyzed by both Ser/Thr- and Tyr-specific PPs, and, since SHLP1 is related to the STP family of PPs, we tested for activity in the presence of the PPP inhibitors okadaic acid and microcystin-LR. SHLP1-GFP phosphatase activity was not affected by either inhibitor (Figure 2A, lower panel left), consistent with previous studies with AtSLPs (Uhrig and Moorhead, 2011) and predictions from our bioinformatic analysis (Figure S1) that Plasmodium SHLP1 is divergent in the residues involved in inhibitor binding to type 1 PPs. A PP2A enzyme was used as a positive control in the same assay conditions, and its activity was almost abolished by both inhibitors (Figure 2A, lower panel right).
Figure 2.

SHLP1-GFP Phosphatase Activity and In Vivo Phosphorylation Levels
(A) Upper panel: phosphatase activity in immunoprecipitates from lysates of SHLP1-GFP, WT-GFP, and WT parasite lines with or without MFP substrate and anti-GFP western blot showing amounts of SHLP1-GFP and WT-GFP retained from the corresponding lysates. Lower panel: phosphatase activity of the same immunoprecipitates (left, from 30 μg total protein) and of recombinant PP2A control (right, 0.1 U) in the presence or absence of okadaic acid (OA, 120 nM) and microcystin-LR (MC-LR, 10 nM). Error bar ±SEM, n = 3.
(B) Upper panel: autoradiograph of 32P-labeled total protein lysates before immunoprecipitation in activated gametocytes (AG) and schizonts (Sch) from WT-GFP, SHLP1-GFP, and PPKL-GFP (+C) parasites. Middle panel: autoradiograph showing phosphorylation of GFP-TRAP immunoprecipitated lysates. Lower panel: corresponding western blot using anti-GFP antibody on the immunoprecipitates. Arrow shows the position of SHLP1-GFP on the gels, and the star marks the position of phosphorylated PPKL-GFP (Guttery et al., 2012b).
As a number of PPs are known to be regulated by phosphorylation (Mochida and Hunt, 2012), we analyzed SHLP1-GFP phosphorylation in vivo. Although activated gametocytes and schizonts from WT-GFP- and SHLP1-GFP-expressing parasite lines incorporated 32P-orthophosphate into the total complement of proteins (Figure 2B, upper panel), SHLP1-GFP was not phosphorylated at either stage (Figure 2B, middle panel, arrow), though detectable by western blot (Figure 2B lower panel), consistent with data from previous global phosphoproteomics studies of P. falciparum schizonts (Lasonder et al., 2012).
SHLP1 Is Critical to Ookinete Formation, Ookinete-to-Oocyst Transition, and Parasite Transmission
To elucidate the function of SHLP1 during the Plasmodium life cycle, we replaced the endogenous shlp1 gene with a T. gondii dhfr/ts selectable marker using double homologous recombination (Figures S2D–S2H). Analysis of two deletion mutant clones from two independent transfections identified no phenotypic differences from WT parasites during asexual stages or gametocytogenesis, as assessed on blood smears (data not shown).
Microgametogenesis as assessed by exflagellation was comparable to that of WT parasites (Figure 3A). Analysis of zygote formation and ookinete development postfertilization in vitro revealed a significant reduction (55%–60%) in ookinete conversion in Δshlp1 parasites (Figure 3B). Time-course analysis revealed that ookinete formation and differentiation was reduced from the onset in Δshlp1 mutants (9 hr postfertilization) and thus was not a result of parasite death or dedifferentiation of ookinetes before they reached full maturity at 24 hr (Figure 3C), in contrast to a previously described phosphodiesterase (pdeδ)-deficient parasite (Moon et al., 2009). This suggests that SHLP1 plays an important (though not essential) role at an early stage in ookinete development and differentiation.
Figure 3.

Phenotypic Analyses of Δshlp1 Mutants: Ookinete Formation and Oocyst Development Are Affected
(A) Microgametogenesis in WT and Δshlp1 mutant lines measured as number of exflagellation centers (ns = not significant, Mann-Whitney U test, p > 0.05).
(B) Ookinete conversion in Δshlp1 mutants compared to WT (Mann-Whitney U test, ∗∗∗p < 0.001). Data shown as mean ±SD.
(C) Developing ookinete stages shown as the percentage of female-derived cells for WT and Δshlp1 with the correct shape at either 9 hr (stages I–III) or 24 hr (fully differentiated ookinete) postfertilization. Data shown as mean ±SD.
(D) Representative frames from time-lapse movies of WT (upper panels) and Δshlp1 (lower panels) ookinetes in Matrigel. Arrow indicates the apical end of the ookinete. Bar = 10 μm. Velocity of individual WT or Δshlp1 ookinetes from 24 hr cultures was measured over 10 min, as shown in the dot plot. Bar = arithmetic mean; n = 18 for WT and 22 for Δshlp1 lines.
(E) Numbers of oocysts in A. stephensi midguts on day 14 postblood feeding show complete ablation of oocyst development in Δshlp1 parasites compared to WT (Mann-Whitney U test, ∗∗∗p < 0.001).
(F) Hemocoel injection of Δshlp1 ookinetes shows that mutant sporozoites invade the salivary glands, can be successfully transmitted to mice, and are able to establish new infections.
(G) Genetic complementation of Δshlp1 parasites. Mosquitoes were fed on mice infected with WT or Δshlp1 mutant alone or coinfected with Δshlp1 and either a Δmap2 mutant or a Δnek4 mutant. Scale bar, 100 μm (Kruskal-Wallis test, ∗∗∗p < 0.001).
See also Figure S2.
For ookinetes that did fully differentiate, we analyzed their motility (Moon et al., 2009) and found that gliding motility in Δshlp1 ookinetes was indistinguishable from that of WT parasites (Figure 3D; Movies S1 and S2), indicating that their motor function was unimpaired. Parasite motility is governed by an actomyosin motor termed the glideosome, which resides within the pellicle of invasive zoites (Opitz and Soldati, 2002). Motility and midgut invasion by the ookinete involves the secretion of a number of membrane proteins, micronemal proteins such as circumsporozoite and TRAP-related protein (CTRP) (Dessens et al., 1999; Yuda et al., 1999), and secreted ookinete adhesive protein (SOAP) (Dessens et al., 2003), as well as the action of the calcium-dependent protein kinases, CDPK3 (Siden-Kiamos et al., 2006) and CDPK1 (Sebastian et al., 2012).
To extend these findings in vivo, A. stephensi mosquitoes were fed on mice infected with either WT or Δshlp1 parasites to analyze both oocyst development and sporogony. While the WT developed normally, no Δshlp1 oocysts were found on day 14 or 21 postinfection (p.i.) (Figure 3E). Hence, to determine whether the lack of oocyst formation in Δshlp1 mutant parasites was due to a defect in invasion of the midgut epithelium, we bypassed the midgut barrier by injecting ookinetes directly into the hemocoel of A. stephensi mosquitoes and analyzed salivary glands for sporozoites 20 days postinjection (Ecker et al., 2008). Δshlp1 parasites were able to form viable sporozoites, which could migrate to and actively invade the salivary gland (Figure 3F). Parasite transmission experiments using these mosquitoes resulted in infection of mice with both WT and Δshlp1 lines, strongly suggesting that the mutant parasites undergo normal exo- and intraerythrocytic proliferation, but oocyst development in vivo is ablated. Subsequent in vivo invasion assays using A. gambiae L3-5 mosquitoes (Collins et al., 1986) with WT and Δshlp1 parasites showed a slight but nonsignificant reduction in melanized Δshlp1 ookinetes compared to WT (Figure S3A). These data indicate that Δshlp1 ookinetes are able to cross the midgut epithelium but cannot form oocysts.
Figure S3.

Phenotypic Analyses of WT and Δshlp1 Ookinetes, Related to Figure 4
(A) Ookinete invasion assay in A. gambiae L3-5 mosquitoes. Microscopy images show melanized ookinetes (arrows) present in mid-guts of mosquitoes infected with either WT control and Δshlp1 parasites but not with Δctrp parasites. Scale bar = 50 μm. The number of melanized Δshlp1 ookinetes was not significantly reduced compared to the wild-type, but significantly higher compared to the Δctrp mutant (Mann-Whitney U-test, ∗∗∗ = p < 0.001).
(B) Montage of longitudinal sections of the anterior apical end of WT (three left panels) and Δshlp1 ookinetes (seven right panels) emphasizing the marked reduction or absence in apical micronemes, whereas all other structures at the apical end appear normal. Bar = 100 nm.
(C) Normalized wild-type RNA expression of female-specific genes (cith, dozi and nek4), shlp1 and microneme specific genes (cht1, ctrp and soap), relative to arginyl-tRNA synthetase and hsp70 used as endogenous controls. Each point is the mean of three biological replicates consisting of three technical replicates ±SD (normalized using qBasePlus).
(D) Relative RNA expression of female-specific genes (cith, dozi, nek4) and shlp1 (relative to arginyl-tRNA synthetase and hsp70) in Δshlp1 mutant parasites compared to wild-type controls. Each point is the mean of three biological replicates consisting of three technical replicates ±SD (normalized using qBasePlus).
Asexual blood = ASB; Activated gametocytes = AG; Ookinete = OO. ∗ = p < 0.05; ∗∗ = p < 0.01.
SHLP1 Function Is Contributed by the Female Gamete
We next examined whether the defect was sex specific by performing genetic crosses between Δshlp1 parasites and lines deficient in either male (Δmap2) or female (Δnek4) gametes (Tewari et al., 2010). We found that only the Δmap2 mutant was able to rescue the Δshlp1 phenotype and form oocysts in the mosquito midgut (p < 0.001), confirming a strict requirement for a functional female copy of the shlp1 gene. Ookinetes resulting from female gametocytes with this genotype can be complemented by mutant parasites with viable female forms (Figure 3G).
SHLP1 Is Important for Microneme Formation in Ookinetes
To identify whether deletion of shlp1 results in morphological defects in the viable ookinetes at the ultrastructure level, we analyzed Δshlp1 parasites using transmission electron microscopy (TEM). While no defects were found in the apical architecture of Δshlp1 ookinetes (i.e., the plasmalemma, inner membrane complex [IMC], central aperture, and subpellicular microtubules) (Figures 4Ai–4Aiii and S3B), there was a marked reduction in the number of apical micronemes (Figures 4Aii, 4Aiv, 4Av, and S3B). Of the various sections examined, 80%–90% of Δshlp1 ookinetes had either no apical micronemes (Figures 4Aiv, 4Av, and S3B) or had markedly reduced microneme numbers, which were not apically located (Figures 4Aiv and S3B).
Figure 4.

Microneme Reduction and Mislocalization in Δshlp1 Ookinetes
(A) (i) Transmission electron micrograph (TEM) of a longitudinal section through a WT ookinete showing the conical apical end. The cytoplasm contains a number of apically located micronemes (M) and a more posteriorly located nucleus (N). Scale bar, 1 μm. (ii) Longitudinal TEM section through a Δshlp1 ookinete showing substantial reduction of micronemes and mislocalization of remaining micronemes at the posterior end. Bar = 1 μm. (iii) Enlargement of the anterior of a WT ookinete showing the complex nature of the apical end consisting of a conical shaped electron dense collar (C) with a central aperture and an underlying less electron dense ring (R). Micronemes (M) are located within the cytoplasm. Scale bar, 100 nm. (iv and v). Detail of the anterior of Δshlp1 ookinetes showing reduction in micronemes (M). Scale bar, 100 nm.
(B) Indirect immunofluorescence of a number of motor complex and micronemal proteins in WT and Δshlp1 ookinetes. Compared to WT, staining for micronemal proteins SOAP and CTRP was reduced in the Δshlp1 mutant and spread throughout the cell body as opposed to the normal concentration at the apical end (arrow). No difference was observed between WT and mutant ookinetes for glideosomal GAP45 and MTIP.
(C) Western blot analyses of ookinete micronemal proteins CTRP and SOAP. Expression levels were compared between WT and Δshlp1 ookinetes and normalized to the GFP control using ImageJ software. CTRP expression was unaltered in the mutant whereas SOAP expression was reduced to 40%.
(D) Relative RNA expression of microneme specific genes cht1, ctrp, and soap in Δshlp1 mutant parasites compared to WT controls. Each point is the mean of three biological replicates ±SD (normalized using qBasePlus). AG, activated gametocytes; OO, ookinete. ∗p < 0.05; ∗∗p < 0.01.
See also Figure S3.
For confirmation, we performed indirect immunofluorescence and western blotting for proteins associated with either the ookinete motor complex or the micronemes (Figure 4B). While the glideosome-associated protein 45 (GAP45) and myosin A tail domain-interacting protein (MTIP) displayed no obviously abnormal pattern in the Δshlp1 line, micronemal CTRP showed a less distinct apical staining and overall a less intense staining distributed throughout the body of the ookinete relative to that of the WT. Similar differences were observed for microneme-associated SOAP consistent with the marked reduction of micronemes in Δshlp1 ookinetes observed by TEM. However, western blot analysis of CTRP and SOAP revealed that overall levels of CTRP were comparable between mutant and WT, whereas SOAP was reduced in the Δshlp1 ookinete to 40% of WT levels (Figure 4C).
The RNA expression profile for female-specific and micronemal genes was comparable for all genes analyzed in WT parasites with the exception of soap, which appeared to be upregulated by 2-fold in ookinetes compared to blood and gametocyte stages (Figure S3C). We used quantitative RT-PCR (qRT-PCR) to investigate whether the defect in microneme formation in the Δshlp1 ookinete is accompanied by downregulation of expression of genes encoding ookinete micronemal proteins in activated gametocytes and ookinetes. Of the three genes for micronemal proteins assayed, both ctrp and soap were significantly downregulated in Δshlp1 ookinetes (p < 0.05 and p < 0.01, respectively). Expression levels of the invasion-associated chitinase 1 (cht1) were completely unaffected in both stages (Figure 4D). Of the female-specific genes investigated, only nek4 expression was significantly altered in Δshlp1 mutant ookinetes (Figure S3D).
A variety of micronemal proteins have previously been proposed to have a role in ookinete motility and ookinete to oocyst transition. CTRP deletion mutants are nonmotile and thus do not form oocysts (Dessens et al., 1999; Yuda et al., 1999), while membrane attack ookinete protein (MAOP) (Kadota et al., 2004) and SOAP (Dessens et al., 2003) -null mutants are motile but unable or severely impaired in their capability to invade the midgut epithelium, respectively. To date, there is no information on the ultrastructure of these mutant parasites.
It is possible that the absence of micronemes is not due to a lack of cargo but incomplete maturation of the organelles or misdirected trafficking of the proteins as indicated by the diffuse overall staining of CTRP in the Δshlp1 cells. De novo biosynthesis of micronemes begins at the Golgi (Schrevel et al., 2008), which suggests a role for SHLP1 in the regulation of vesicular trafficking and microneme biogenesis. This suggestion is further corroborated by our colocalization and subcellular fractionation data showing SHLP1-GFP in the ER as a peripheral and integral membrane protein. Furthermore, while there is a marked effect on microneme development and ookinete function in the Δshlp1 mutant, there is no obvious developmental effect on the other invasive stages (i.e., sporozoites and merozoites) or on asexual development. However, the micronemes of the ookinete are structurally unique with a thin duct connecting the micronemes to the cell apex, a structure that is lacking in other developmental stages. Future studies are needed to understand the function of micronemal proteins and their secretion in the absence of micronemes.
The ability of ookinetes with no micronemes to invade the midgut epithelium has recently been described for a mutant lacking the paternally inherited MISFIT gene, corroborating our finding that mutants with a defect in microneme organelle biogenesis are capable of midgut invasion. However, our genetic complementation result showed that shlp1 is inherited through the female line, in contrast to MISFIT that showed a paternal effect (Bushell et al., 2009).
In summary, our results indicate that SHLP1 in Plasmodium is essential for parasite transmission via the mosquito and thus a potential target for the development of transmission-blocking drugs. Ookinete-to-oocyst transition represents one of the biggest bottlenecks in the life cycle of the malarial parasite, and we show here that SHLP1 plays a crucial role in this process.
Experimental Procedures
See Extended Experimental Procedures for details.
Extended Experimental Procedures.
Ethics Statement
All animal work has passed an ethical review process and was approved by the United Kingdom Home Office. Work was carried out in accordance with the United Kingdom ‘Animals (Scientific Procedures) Act 1986’ and in compliance with ‘European Directive 86/609/EEC’ for the protection of animals used for experimental purposes. The permit number for the project license is 40/3344.
Bioinformatic Analysis
AtSLP1 and AtSLP2 were used in BLAST searches of diverse prokaryote and eukaryotic proteomes to identify 47 SHLP orthologs and A. thaliana PPP-family phosphatases were added as an outgroup. The sequences were aligned using ClustalW2 and optimized using CLC Genomics Workbench (CLC bio, Cambridge, MA). After identifying the PP domains, the sequences were realigned using the same program and a neighbor-joining bootstrap tree was generated. The phylogenetic tree was drawn using Fig Tree v1.3.1. Targeting prediction was performed using the prediction of apicoplast targeting sequences (PATS) server (http://gecco.org.chemie.uni-frankfurt.de/pats/pats-index.php).
Animals
Tuck-Ordinary (TO) (Harlan) outbred mice were used for all experiments except for mosquito infections where C57/Bl6 mice were used.
Generation of Transgenic Parasites
The targeting vector for shlp1 was constructed using the pBS-DHFR plasmid, which contains polylinker sites flanking a Toxoplasma gondii dhfr/ts expression cassette conveying resistance to pyrimethamine. PCR primers P0171 (5′-CCCCGGGCCCGTTAATATTGAATAACGTTGTCAAC-3′) and P0172 (5′-GGGGAAGCTTCTATAGCTATAATTTTGCCATTCC-3′) were used to generate a 466 bp fragment of 5′ upstream sequence of shlp1 from WT genomic DNA, which was inserted into ApaI and HindIII restriction sites upstream of the dhfr/ts cassette of pBS-DHFR. A 498 bp fragment generated with primers P0173 (5′-CCCCGAATTCCAGGAAAGTCAAACTAATTTGAACCCAG-3′) and P0174 (5′-GGGGTCTAGAGAACAGAATTTGTATGTGTGAATGTGATC-3′) from the 3′ flanking region of shlp1 was then inserted downstream of the dhfr/ts cassette using EcoRI and XbaI restriction sites. For transfection a linear fragment was released using ApaI and XbaI restriction enzymes. Genotyping of transgenic parasites was performed by integration PCR, Southern blotting and PFGE.
For GFP-tagging by single homologous recombination, a 703 bp region of shlp1 starting 383 bp downstream of the ATG start codon and omitting the stop codon was amplified using primers T0801 (5′-CCCCGGTACCTAGGGTAATATTAATAATGGGAAACCACG-3′) and T0802 (5′-CCCCGGGCCCCAAAACTGGGTTCAAATTAGTTTGAC-3′). This was inserted upstream of the gfp sequence in the pOB277 vector using KpnI and ApaI restriction sites. The pOB277 vector contains the human dhfr cassette, also conveying resistance to pyrimethamine. Before transfection, the sequence was linearized using EcoRV and P. berghei ANKA line 2.34 was then transfected by electroporation (Janse et al., 2006). Briefly, electroporated parasites were mixed immediately with 200 μl of reticulocyte-rich blood from a phenylhydrazine (Sigma) treated, naive mouse, incubated at 37°C for 20 min and then injected intraperitoneally into naive mice. From day 1 post infection pyrimethamine (70 μg/ml) (Sigma) was supplied in the drinking water for four days. Mice were monitored for 15 days and drug selection was repeated after passage to a second mouse. Resistant parasites were then used for cloning by limiting dilution and subsequent genotyping.
Genotypic Analysis of Mutants
For the gene knockout parasites, two diagnostic PCR reactions were used as illustrated in Figure S2. Primer 1 (INT P17, 5′-CTATGTTTTACATCAATAATTGCATGG-3′) and primer 2 (ol248, 5′-GATGTGTTATGTGATTAATTCATACAC-3′) were used to determine successful integration of the selectable marker at the targeted locus. Primers 3 (P17 KO1, 5′-GTAAACCCCTATGGAGAATATCATAAAAG-3′) and 4 (P17 KO2, 5′-GGTGTGATGGTTGTCTAGTATGTCCGAC-3′) were used to verify deletion of the gene. Having confirmed integration, genomic DNA from wild-type (WT) and mutant parasites was digested with EcoRV and the fragments were separated on a 0.8% agarose gel, blotted onto a nylon membrane (GE Healthcare), and probed with a PCR fragment homologous to the P. berghei genomic DNA just outside of the targeted region.
Chromosomes of WT and gene knockout parasites were separated by pulsed field gel electrophoresis (PFGE) on a CHEF DR III (BioRad) using a linear ramp of 60–500 s for 72 hr at 4 V/cm. Gels were blotted and hybridized with a probe recognizing both the resistance cassette in the targeting vector and, more weakly, the 3′UTR of the P. berghei dhfr/ts locus on chromosome 7.
For the C-terminal GFP-tagged parasites, one diagnostic PCR reaction was used as illustrated in Figure S2. Primer 1 (INT P17tag, 5′-GCCTAATTAGGTGCAACTTTCGC-3′) and primer 2 (ol492, 5′- ACGCTGAACTTGTGGCCG-3′) were used to determine the correct integration of the gfp sequence into the targeted locus. Having confirmed correct integration, parasites were also visualized on a Zeiss AxioImager M2 (Carl Zeiss, Inc) microscope fitted with an AxioCam ICc1 digital camera (Carl Zeiss, Inc) and analyzed by Western blot to confirm GFP expression as described.
Phenotypic Analysis
Infections for phenotype screens were initiated by the intraperitoneal injection of infected blood containing 5x106 parasites into mice pretreated with 0.2 ml of 6 mg/ml phenylhydrazine in PBS intraperitoneally to induce reticulocytosis. Asexual stages and gametocyte production were monitored on Giemsa-stained blood films.
Exflagellation was examined on days 4-5 post infection. 10 μl of gametocyte-infected blood were obtained from the tail with a heparinized pipette tip and mixed immediately with 40 μl of ookinete culture medium (RPMI1640 containing 25 mM HEPES, 25% fetal bovine serum, 10 mM sodium bicarbonate, 50 μM xanthurenic acid at pH 7.6). The mixture was placed under a Vaseline-coated coverslip and 15 min later exflagellation centers were counted by phase contrast microscopy in 12-15 fields of view using a 63x objective and 10x ocular lens. Ookinete formation was assessed the next day. 10 μl of infected tail blood were obtained as above, mixed immediately with 40 μl ookinete culture medium, and incubated for 2 hr at 20°C to allow completion of gametogenesis and fertilization. Each culture was then diluted with 0.45 ml of ookinete medium and incubated at 20°C for a further 21–24 hr to allow ookinete differentiation. Cultures were pelleted for 2 min at 5000 rpm and then incubating with 50 μl of ookinete medium containing Hoechst 33342 DNA dye to a final concentration of 5 μg/ml and a Cy3-conjugated mouse monoclonal antibody recognizing the P28 protein on the surface of ookinetes (13.1) and any undifferentiated macrogametes or zygotes. P28-positive cells were counted with a Zeiss AxioImager M2 microscope (Carl Zeiss, Inc) fitted with an AxioCam ICc1 digital camera. Ookinete conversion was expressed as the percentage of P28 positive parasites that had differentiated into ookinetes (Liu et al., 2008). Motility assays were performed as previously described (Moon et al., 2009). Briefly, ookinete cultures were mixed with an equal volume of Matrigel on ice, mounted onto a slide, covered and sealed with nail varnish and left to set at room temperature for at least 30 min. Time lapse movies of ookinetes (1 frame every 5 s for 10 min) were taken using a Zeiss AxioImager M2 microscope as described above. Ookinete speed was determined as number of body lengths moved per min.
High resolution live cell imaging was performed using an Olympus-based personal Delta Vision work station using a 63x oil immersion objective. Subsequent off line image preparation was carried out using Applied Precision software and images were finalized using Volocity 3D image analysis software (Perkin Elmer). Images presented are 2D projections of 0.1 μM stepped Z-stacks. For mosquito transmission experiments 20-50 A. stephensi SD500 or A. gambiae L3-5 mosquitoes were allowed to feed for 20 min on anaesthetized infected mice whose asexual parasitaemia had reached ∼7%–23% and were carrying comparable numbers of gametocytes as determined on Giemsa stained blood films. On day 14 post feeding approximately 20 A. stephensi mosquitoes were dissected and oocysts on their midguts were counted. Oocyst formation was examined by Hoechst 33342 staining for 10-15 min and guts were washed and mounted under Vaseline-rimmed coverslips. Images were recorded using a 63x oil immersion objective on a Zeiss AxioImager M2 microscope fitted with an AxioCam ICc1 digital camera. Day 21 post feeding another 20 mosquitoes were dissected and their guts and salivary glands crushed separately in a loosely fitting homogenizer to release sporozoites, which were then quantified using a haemocytometer. Due to day-to-day variations in transmission levels, all data were normalized to a matching number of WT controls analyzed on the same day.
A. gambiae L3-5 guts were analyzed on day 8 p.i. for the presence of melanised ookinetes. Briefly, guts were fixed in 4% formaldehyde/PBS, washed 3 times in PBS and mounted in Vectashield (Vector laboratories).
Genetic Crosses and Haemocoel Injections
Genetic complementation crosses were carried out between different mutant parasite lines as described (Ecker et al., 2008). Briefly, Mice were infected with combinations of different strains and 3-6 days old female A. stephensi mosquitoes were infected by directly feeding on these mice. Mosquitoes were dissected 12-14 days post infection and the presence of oocysts was determined.
For haemocoel injections, A. stephensi mosquitoes were anaesthetised with CO2 and injected into the thorax with approximately 500 ookinetes harvested from standard cultures in 69 μl of complete ookinete medium using a microinjector (Drummond, Nanoject II). 20 days post-injection bite-back infections of mice and salivary gland dissections for quantification of sporozoites were performed.
Purification of Schizonts, Gametocytes, and Ookinetes
Purification of gametocytes was achieved using a protocol modified from (Beetsma et al., 1998). Mice were treated by intraperitoneal injection of 0.2 ml of phenylhydrazine (6 mg/ml) (Sigma) in PBS to encourage reticulocyte formation four days prior to infection with parasites. Day four post infection (p.i.) mice were treated with sulfadiazine (Sigma) at 20 mg/l in their drinking water for two days to eliminate asexual blood stage parasites. On day six p.i. mice were bled by cardiac puncture into heparin and gametocytes separated from uninfected erythrocytes on a 48% v/v NycoDenz gradient (Stock: 27.6% w/v NycoDenz in 5 mM Tris-HCl, pH 7.20, 3 mM KCl, 0.3 mM EDTA) in coelenterazine loading buffer (CLB), containing PBS, 20 mM HEPES, 20 mM Glucose, 4 mM sodium bicarbonate, 1 mM EGTA, 0.1% w/v bovine serum albumin, pH 7.25. Gametocytes were harvested from the interface and washed twice in RPMI 1640 ready for activation of gamete formation. Blood cells from day 5 p.i. mice were placed in culture for 24 hr at 37°C for schizont (with rotation at 100 rpm) and 20°C for ookinete production as described above. Schizonts and ookinetes were purified on a 60% v/v and 63% v/v NycoDenz gradient, respectively and harvested from the interface and washed.
Quantitative RT-PCR
Parasites were purified from blood and the RNA was isolated using Trizol (Sigma) according to the manufacturer’s instructions. Purified RNA was treated with DNase I (QIAGEN) and cDNA was synthesized using the high capacity RNA-to-cDNA kit (Applied Biosystems). Gene expression was quantified from 250 ng of total RNA. qPCR reactions consisted of 2.5 μl of cDNA, 5 μl SYBR green fast master mix (Applied Biosystems), 0.5 μl (250 nM) each of the forward and reverse primers, and 1.5 μl DEPC-treated water. All of the primers were designed either with PerlPrimer (Marshall, 2004) or primer3 (Primer-blast, NCBI) design software and were 18-24 bp in length, containing 40%–60% GC content, amplified a region 70-200 bp long and where possible, annealing within 600 bp of the 3′ end of the gene of interest.
Analysis was conducted using an Applied Biosystems 7500 fast machine with the following cycling conditions: 95°C for 20 s followed by 40 cycles of 95°C for 3 s; 60°C for 30 s. Three biological replicates, consisting of three technical replicates, were performed for each assayed gene. Wild-type and relative expression were determined using qBasePlus v1.3 (Biogazelle) (Hellemans et al., 2007; Vandesompele et al., 2002), with relative quantification in mutant lines normalized against wild-type expression. Both methods used hsp70 (PBANKA_081890) (forward, 5′-GTATTATTAATGAACCCACCGCT-3′; reverse, 5′-GAAACATCAAATGTACCACCTCC-3′) and arginyl-tRNA synthetase (PBANKA_143420) (forward, 5′-TTGATTCATGTTGGATTTGGCT-3′; reverse, 5′-ATCCTTCTTTGCCCTTTCAG-3′) as reference genes. shlp1 primers were: forward, 5′-CATCACACCAAATTGGCTCTT-3′; reverse, 5′-AATTCGGATAAGGCTGACCA-3′. dozi (PBANKA_121770) primers were: forward, 5′- GCAAGAATGTCGCAAACAC-3′; reverse, 5′-TCTGAGGAAACTAAACATCGAC-3′. nek4 (PBANKA_061670) primers were: forward, 5′-CTTCAGATGTATGGGCTATTGG-3′; reverse, 5′- TTCCCTTTGTTGAATGAAATGG-3′. cht1 (PBANKA_080050) primers were: forward, 5′- ACGTTGACTACCCAACAGGA-3′; reverse, 5′-ATCCTGGGCAGCTGTATTCG-3′. cith (PBANKA_130130) primers were: forward, 5′- ATAGTAACATACAGGTAGGAGGA-3′; reverse, 5′-CCGAAAGTATCAACATCTAGCA-3′. ctrp (PBANKA_041290) primers were: forward, 5′- ACCTCCCGTTTTAGATGGATCG-3′; reverse, 5′-ATGTGGTGTCTCTCCTCCATT-3′. soap (PBANKA_103780) primers were: forward, 5′- AAGGCATCTTCACGATCCGA-3′; reverse, 5′-TTAGTCCCTTTGCATGTGCG-3′.
Immunofluorescence Assays
Thin blood films were prepared from ookinete cultures and air-dried. For various antibodies different procedures were followed. For anti-GAP45, anti-MTIP and anti-SOAP staining, slides were fixed for 10 min in 4% paraformaldehyde in MTSB buffer and for CTRP slides were fixed for 5 min in 1% formaldehyde. Concentrations of primary antibodies were: 1:250 for both rabbit antibodies against GAP45 and MTIP (Green et al., 2008), 1:1000 for the mouse monoclonal anti-CTRP antibody (Dessens et al., 1999) and 1:100 for mouse anti-SOAP (Dessens et al., 2003). Secondary antibodies used were Alexa Fluor 488 goat anti-rabbit, Alexa Fluor 488 goat anti-mouse and Alexa Fluor 568 goat anti-mouse (all 1:1000) from Invitrogen.
Subcellular Fractionation
The protocol used for subcellular fractionation was adapted from (Hopp et al., 2012). Briefly, blood aliquots from infected mice (with WT-GFP and SHLP1-GFP parasites) were prepared as described previously (Guttery et al., 2012a). The parasite pellets obtained were resuspended in hypotonic lysis buffer (10mM Tris-HCl pH 8.4, 5mM EDTA) containing protease inhibitors (Roche), freeze-thawed twice, incubated for 1 hr at 4°C and centrifuged at 100,000 g for 30 min. The supernatants were collected as the soluble protein fraction (S). The pellets were then washed, resuspended in carbonate solution (0.1M Na2CO3 pH 11.0) containing protease inhibitors (Roche), incubated for 30 min at 4°C and centrifuged at 100,000 g for 30 min. The supernatants were saved as the peripheral membrane fraction (PM) and the pellets were washed and solubilized in 4% SDS and 0.5% Triton X-100 in PBS, forming the integral membrane fraction (IM). Equal amounts of these three fractions were then analyzed by western blot using anti-GFP antibody. A control using anti-GAP45 antibody was used to confirm the presence of the integral membrane GAP45 in the carbonate insoluble (IM) fraction (Ridzuan et al., 2012).
Western Blotting
Western blot analysis was performed on cell lysates prepared by re-suspending parasite pellets in a 1:1 ratio of PBS containing Protease inhibitor (Roche) and either Laemmli sample buffer (anti-SOAP, anti GAP45 and anti-GFP blots) or non-reducing SDS buffer (CTRP), boiling and separating on either 4%–15% gradient or 12% SDS-polyacrylamide gel. Samples were subsequently transferred to nitrocellulose membranes (Amersham Biosciences) and immunoblotting was performed using the Western Breeze Chemiluminescent Anti-Rabbit and Anti-Mouse kit (both Invitrogen) according to the manufacturer’s instructions. Primary antibody concentrations were: anti-GFP rabbit polyclonal (Invitrogen) 1:1250, anti-GAP45 rabbit polyclonal (Green et al., 2008) 1:5000, mouse monoclonal anti-CTRP antibody (Dessens et al., 1999) 1:10,000 and mouse anti-SOAP (Dessens et al., 2003) 1:2000. Band intensities quantification was done using the ImageJ software (National Institutes of Health).
Phosphatase Activity Assay
Protein phosphatase activity of the immunoprecipitated SHLP1-GFP was assessed using the SensoLyte MFP Protein Phosphatase Assay Kit (AnaSpec) according to manufacturer’s instructions. Briefly, blood aliquots from infected mice (with WT, WT-GFP and SHLP1-GFP parasites) were incubated in ookinete medium for 30 min at 20°C and processed as described previously (Guttery et al., 2012a). The parasite pellets obtained were lysed for 30 min at 4°C in lysis buffer (ChromoTek) supplemented with protease inhibitors (Roche), the resulting lysates were then immunoprecipitated using GFP-TRAP beads (ChromoTek) according to manufacturer’s instructions. The GFP-TRAP beads were resuspended and diluted in phosphatase assay buffer (100 mM Tris-HCl pH 7.5, 4 mM DTT, 0.2 mM EDTA, 0.5 mM MnCl2, 0.4 mg/ml BSA), incubated for 30 min at 37°C in the presence or absence of MFP fluorogenic phosphatase substrate, and centrifuged for 2 min at 2700 g. Supernatants were transferred to a 96-well microplate and the fluorescence generated by the dephosphorylation of MFP was measured using a microplate fluorimeter. A recombinant PP2A enzyme (Cayman Chemical) was used as positive control for the assay.
SHLP1 In Vivo Phosphorylation
As described previously (Guttery et al., 2012a), schizonts and activated gametocytes (purified as described above) were washed in phosphate-free Krebs buffer (118 mM NaCl, 4.7 mM KCl, 4.2 mM NaHCO3, 1.2 mM MgSO4, 11.7 mM glucose, 10 mM HEPES, 1.3 mM CaCl2, pH 7.4) and metabolically labeled with 3-5 MBq 32P-orthophosphate (Perkin Elmer) in phosphate-free Krebs buffer for 30 min at 20°C or 37°C for activated gametocytes and schizonts, respectively. After two washes in phosphate-free Krebs buffer, the labeled parasites were lysed for 30 min at 4°C in lysis buffer (10 mM Tris-HCl pH 7.5, 150 mM NaCl, 0.5 mM EDTA, 0.5% NP-40) supplemented with protease and phosphatase inhibitors (Roche), the resulting lysate was centrifuged at 20,000 g for 5 min and the supernatant collected. GFP tagged proteins were isolated using GFP-TRAP beads (ChromoTek), the immunoprecipitated proteins were then resuspended in Laemmli sample buffer and separated by SDS-PAGE. 32P-labeled proteins were visualized using a phosphorimager (Molecular Dynamics) and GFP-tagged proteins analyzed by western blot as described above.
Ethics Statement
All animal work has passed an ethical review process and was approved by the United Kingdom Home Office. Work was carried out in accordance with the United Kingdom “Animals (Scientific Procedures) Act 1986” and in compliance with ‘European Directive 86/609/EEC’ for the protection of animals used for experimental purposes. The permit number for the project license is 40/3344.
Generation and Genotyping of Transgenic Parasites
To tag the endogenous shlp1 at the C terminus with GFP, a single homologous recombination technique was used (Guttery et al., 2012a). To replace shlp1 by double homologous recombination, a targeting vector was generated using the pBS-DHFR cassette (Tewari et al., 2010). See Extended Experimental Procedures for all oligonucleotide sequences and detailed methods.
Bioinformatic Analysis
Sequence alignments were performed using ClustalW2 and CLC Genomics Workbench (CLC bio, Cambridge, MA). The phylogenetic tree was generated using Fig Tree v1.3.1.
Phenotypic Analysis
Phenotypic screening was performed as previously described (Guttery et al., 2012a; Tewari et al., 2010). Briefly, asexual proliferation and gametocytogenesis were analyzed using blood smears. Gametocyte activation, zygote formation, and ookinete conversion rates were monitored in in vitro cultures using as a marker the surface antigen P28. For mosquito transmission, triplicate sets of 20–60 Anopheles stephensi were used.
Purification of Schizonts, Gametocytes, and Ookinetes
Schizonts, gametocytes, and ookinetes were purified as described previously (Guttery et al., 2012a).
Quantitative RT-PCR
Gene expression was quantified from 250 ng of total RNA as previously described (Guttery et al., 2012a). hsp70 and arginyl-tRNA synthetase were used as reference genes. Primer sequences and cycling conditions are given in Extended Experimental Procedures.
Isolation of SHLP1-GFP Protein, Phosphatase Assay, and Subcellular Fractionation
Blood aliquots from parasite-infected mice were incubated in ookinete medium for 30 min at 20°C and processed for protein purification and phosphatase assay as described previously (Guttery et al., 2012a). Subcellular fractionation was performed using a carbonate method adapted from Hopp et al. (2012). Soluble (hypotonic lysis), peripheral membrane (carbonate soluble), and integral membrane (carbonate insoluble) fractions were analyzed by western blotting.
SHLP1 In Vivo Phosphorylation
Purified activated gametocytes and purified schizonts were metabolically labeled with 3–5 MBq 32P-orthophosphate for 30 min at 20°C and processed for phosphorylation assay as described previously (Guttery et al., 2012a).
Injections of Ookinetes into the Mosquito Hemocoel and Genetic Crosses
Hemocoel injection of ookinetes and genetic complementation crosses were carried out as described previously (Ecker et al., 2008). See Extended Experimental Procedures for detailed methods.
Electron Microscopy, IFA, and ER Staining
Ookinetes cultured and purified as described above were fixed and processed for routine electron microscopy and IFAs were performed on air-dried ookinete slides from ookinete cultures as described recently (Guttery et al., 2012b). To stain for the ER, ER-Tracker Red (Invitrogen) was used at 2 μM. Blood stages were stained in schizont medium for 60 min at 37°C. Following complete conversion, ookinetes were stained in ookinete medium for 90 min at 25°C. Cells were washed once with respective medium and resuspended in PBS containing Hoechst 33342 DNA stain before being mounting for fluorescent microscopy.
Statistical Analyses
All statistical analyses were performed using GraphPad Prism (GraphPad Software). For phenotypic analysis, nonparametric Mann-Whitney U tests were used. For RNA expression levels, an unpaired t test was used. For genetic complementation experiments, Kruskal-Wallis one-way ANOVA with Dunn’s multiple comparison post hoc test was used.
Acknowledgments
We thank Drs. Johannes Dessens and Judith Green for antibodies, Dr. Sara Sandrini, Julie Rodgers, and Jason Irwin for technical assistance, Dr. Robert Moon for support with motility assays, Dr. Emma King and Ian Ward for high resolution imaging, and Professor Liz Sockett for lab facilities. D.S.G. and B.P. were funded by an MRC Investigator Award (G0900109) and E.-M.P. by an MRC grant (G0900278), both to R.T. A.A.H. is funded by the MRC (U117532067 and G0900278) and the EU FP7 grant agreement 242095 (EviMalar). B.S. is funded by Wellcome Trust grants (92383/Z/10/Z and 095831), and D.J.P.F. is funded by a Wellcome Trust Equipment Grant.
Published: February 21, 2013
Footnotes
Supplemental Information includes Extended Experimental Procedures, three figures, one table, and two movies and can be found with this article online at http://dx.doi.org/10.1016/j.celrep.2013.01.032.
This is an open-access article distributed under the terms of the Creative Commons Attribution-NonCommercial-No Derivative Works License, which permits non-commercial use, distribution, and reproduction in any medium, provided the original author and source are credited.
Supplemental Information
The table also contains the known NCBI or GenBank identifiers of the Shelp orthologs used in the analysis. The enzymes’ PP catalytic domains’ similarities to AtSLP1 and AtSLP2 are shown as percentage of identities.
Δshlp1 ookinetes are fully motile and indistinguishable from wild-type ookinetes.
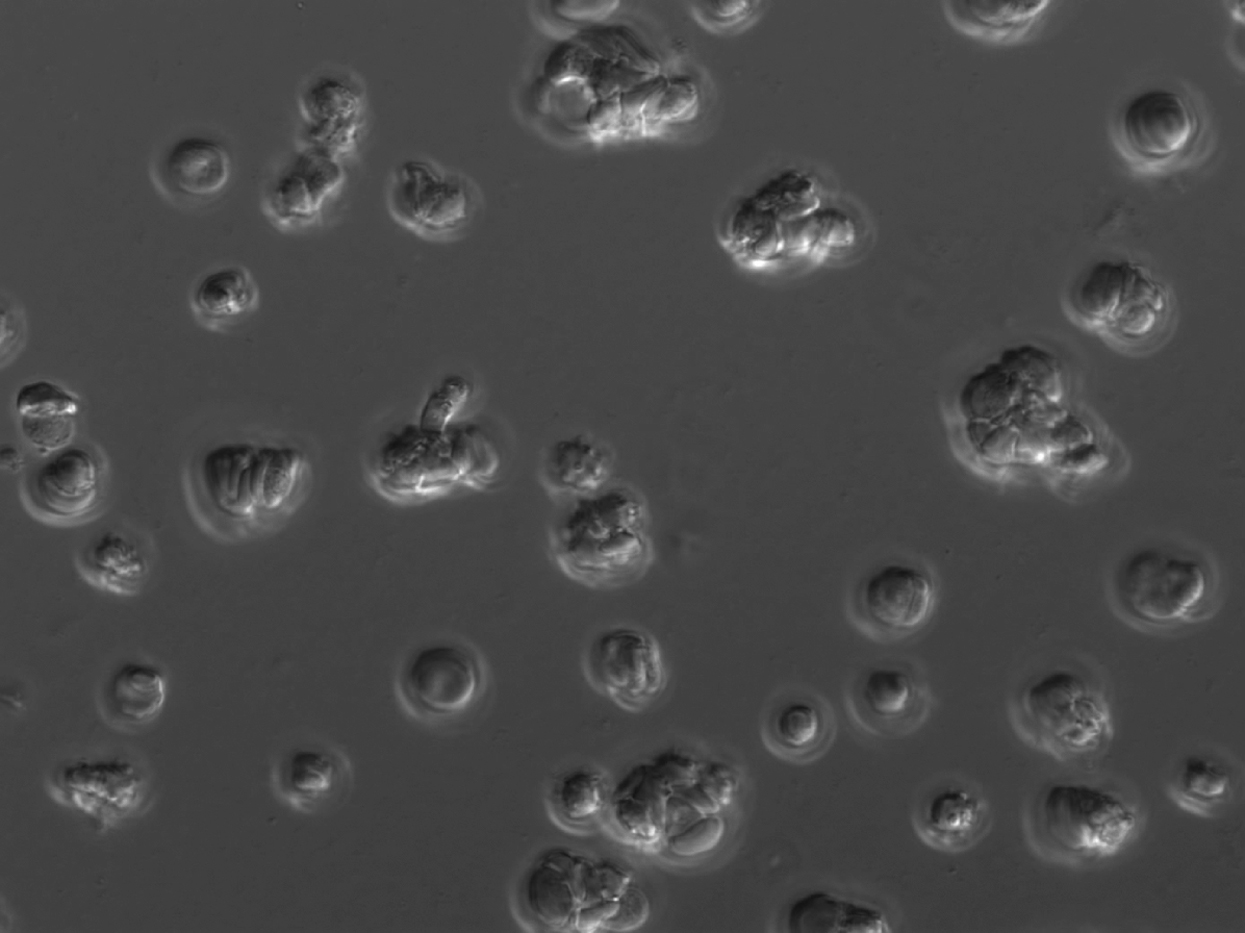{kind=link}
Δshlp1 ookinetes are fully motile and indistinguishable from wild-type ookinetes.
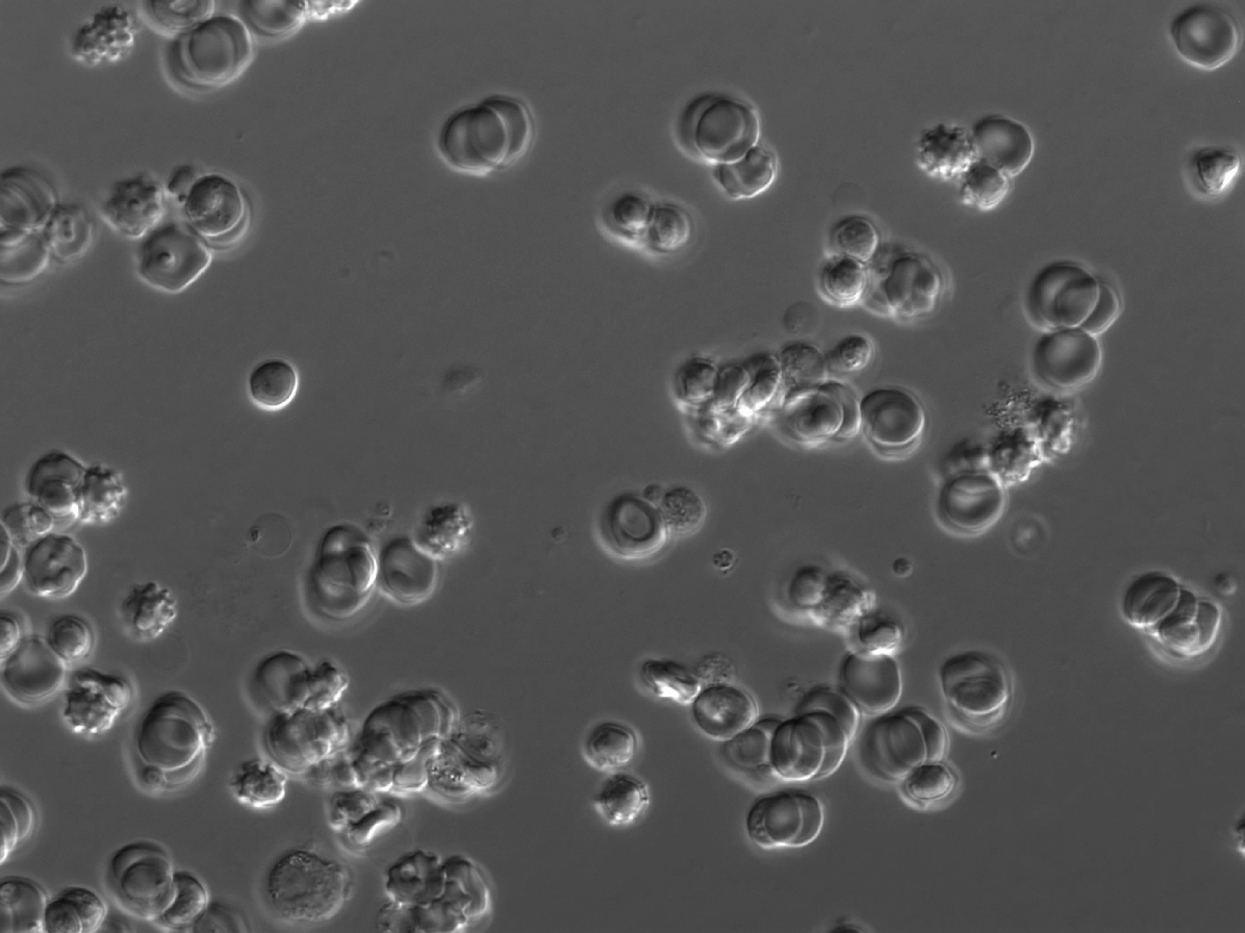{kind=link}
References
- Andreeva A.V., Kutuzov M.A. Widespread presence of “bacterial-like” PPP phosphatases in eukaryotes. BMC Evol. Biol. 2004;4:47. doi: 10.1186/1471-2148-4-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bannister L.H., Sherman I.W. Encyclopedia of Life Sciences (ELS) Wiley; Chichester, UK: 2009. Plasmodium. [Google Scholar]
- Bushell E.S., Ecker A., Schlegelmilch T., Goulding D., Dougan G., Sinden R.E., Christophides G.K., Kafatos F.C., Vlachou D. Paternal effect of the nuclear formin-like protein MISFIT on Plasmodium development in the mosquito vector. PLoS Pathog. 2009;5:e1000539. doi: 10.1371/journal.ppat.1000539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins F.H., Sakai R.K., Vernick K.D., Paskewitz S., Seeley D.C., Miller L.H., Collins W.E., Campbell C.C., Gwadz R.W. Genetic selection of a Plasmodium-refractory strain of the malaria vector Anopheles gambiae. Science. 1986;234:607–610. doi: 10.1126/science.3532325. [DOI] [PubMed] [Google Scholar]
- Dessens J.T., Beetsma A.L., Dimopoulos G., Wengelnik K., Crisanti A., Kafatos F.C., Sinden R.E. CTRP is essential for mosquito infection by malaria ookinetes. EMBO J. 1999;18:6221–6227. doi: 10.1093/emboj/18.22.6221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dessens J.T., Sidén-Kiamos I., Mendoza J., Mahairaki V., Khater E., Vlachou D., Xu X.J., Kafatos F.C., Louis C., Dimopoulos G., Sinden R.E. SOAP, a novel malaria ookinete protein involved in mosquito midgut invasion and oocyst development. Mol. Microbiol. 2003;49:319–329. doi: 10.1046/j.1365-2958.2003.03566.x. [DOI] [PubMed] [Google Scholar]
- Ecker A., Bushell E.S., Tewari R., Sinden R.E. Reverse genetics screen identifies six proteins important for malaria development in the mosquito. Mol. Microbiol. 2008;70:209–220. doi: 10.1111/j.1365-2958.2008.06407.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guttery D.S., Ferguson D.J., Poulin B., Xu Z., Straschil U., Klop O., Solyakov L., Sandrini S.M., Brady D., Nieduszynski C.A. A putative homologue of CDC20/CDH1 in the malaria parasite is essential for male gamete development. PLoS Pathog. 2012;8:e1002554. doi: 10.1371/journal.ppat.1002554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guttery D.S., Poulin B., Ferguson D.J., Szöőr B., Wickstead B., Carroll P.L., Ramakrishnan C., Brady D., Patzewitz E.M., Straschil U. A unique protein phosphatase with kelch-like domains (PPKL) in Plasmodium modulates ookinete differentiation, motility and invasion. PLoS Pathog. 2012;8:e1002948. doi: 10.1371/journal.ppat.1002948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopp C.S., Flueck C., Solyakov L., Tobin A., Baker D.A. Spatiotemporal and functional characterisation of the Plasmodium falciparum cGMP-dependent protein kinase. PLoS ONE. 2012;7:e48206. doi: 10.1371/journal.pone.0048206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu G., Cabrera A., Kono M., Mok S., Chaal B.K., Haase S., Engelberg K., Cheemadan S., Spielmann T., Preiser P.R. Transcriptional profiling of growth perturbations of the human malaria parasite Plasmodium falciparum. Nat. Biotechnol. 2010;28:91–98. doi: 10.1038/nbt.1597. [DOI] [PubMed] [Google Scholar]
- Kadota K., Ishino T., Matsuyama T., Chinzei Y., Yuda M. Essential role of membrane-attack protein in malarial transmission to mosquito host. Proc. Natl. Acad. Sci. USA. 2004;101:16310–16315. doi: 10.1073/pnas.0406187101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kutuzov M.A., Andreeva A.V. Prediction of biological functions of Shewanella-like protein phosphatases (Shelphs) across different domains of life. Funct. Integr. Genomics. 2012;12:11–23. doi: 10.1007/s10142-011-0254-z. [DOI] [PubMed] [Google Scholar]
- Lasonder E., Treeck M., Alam M., Tobin A.B. Insights into the Plasmodium falciparum schizont phospho-proteome. Microbes Infect. 2012;14:811–819. doi: 10.1016/j.micinf.2012.04.008. [DOI] [PubMed] [Google Scholar]
- McConnell J.L., Wadzinski B.E. Targeting protein serine/threonine phosphatases for drug development. Mol. Pharmacol. 2009;75:1249–1261. doi: 10.1124/mol.108.053140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mochida S., Hunt T. Protein phosphatases and their regulation in the control of mitosis. EMBO Rep. 2012;13:197–203. doi: 10.1038/embor.2011.263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moon R.W., Taylor C.J., Bex C., Schepers R., Goulding D., Janse C.J., Waters A.P., Baker D.A., Billker O. A cyclic GMP signalling module that regulates gliding motility in a malaria parasite. PLoS Pathog. 2009;5:e1000599. doi: 10.1371/journal.ppat.1000599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moorhead G.B., Trinkle-Mulcahy L., Ulke-Lemée A. Emerging roles of nuclear protein phosphatases. Nat. Rev. Mol. Cell Biol. 2007;8:234–244. doi: 10.1038/nrm2126. [DOI] [PubMed] [Google Scholar]
- Murray C.J.L., Rosenfeld L.C., Lim S.S., Andrews K.G., Foreman K.J., Haring D., Fullman N., Naghavi M., Lozano R., Lopez A.D. Global malaria mortality between 1980 and 2010: a systematic analysis. Lancet. 2012;379:413–431. doi: 10.1016/S0140-6736(12)60034-8. [DOI] [PubMed] [Google Scholar]
- Opitz C., Soldati D. ‘The glideosome’: a dynamic complex powering gliding motion and host cell invasion by Toxoplasma gondii. Mol. Microbiol. 2002;45:597–604. doi: 10.1046/j.1365-2958.2002.03056.x. [DOI] [PubMed] [Google Scholar]
- Schrevel J., Asfaux-Foucher G., Hopkins J.M., Robert V., Bourgouin C., Prensier G., Bannister L.H. Vesicle trafficking during sporozoite development in Plasmodium berghei: ultrastructural evidence for a novel trafficking mechanism. Parasitology. 2008;135:1–12. doi: 10.1017/S0031182007003629. [DOI] [PubMed] [Google Scholar]
- Sebastian S., Brochet M., Collins M.O., Schwach F., Jones M.L., Goulding D., Rayner J.C., Choudhary J.S., Billker O. A Plasmodium calcium-dependent protein kinase controls zygote development and transmission by translationally activating repressed mRNAs. Cell Host Microbe. 2012;12:9–19. doi: 10.1016/j.chom.2012.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siden-Kiamos I., Ecker A., Nybäck S., Louis C., Sinden R.E., Billker O. Plasmodium berghei calcium-dependent protein kinase 3 is required for ookinete gliding motility and mosquito midgut invasion. Mol. Microbiol. 2006;60:1355–1363. doi: 10.1111/j.1365-2958.2006.05189.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solyakov L., Halbert J., Alam M.M., Semblat J.P., Dorin-Semblat D., Reininger L., Bottrill A.R., Mistry S., Abdi A., Fennell C. Global kinomic and phospho-proteomic analyses of the human malaria parasite Plasmodium falciparum. Nat Commun. 2011;2:565. doi: 10.1038/ncomms1558. [DOI] [PubMed] [Google Scholar]
- Tewari R., Straschil U., Bateman A., Böhme U., Cherevach I., Gong P., Pain A., Billker O. The systematic functional analysis of Plasmodium protein kinases identifies essential regulators of mosquito transmission. Cell Host Microbe. 2010;8:377–387. doi: 10.1016/j.chom.2010.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuruta H., Mikami B., Yamamoto C., Yamagata H. The role of group bulkiness in the catalytic activity of psychrophile cold-active protein tyrosine phosphatase. FEBS J. 2008;275:4317–4328. doi: 10.1111/j.1742-4658.2008.06575.x. [DOI] [PubMed] [Google Scholar]
- Uhrig R.G., Moorhead G.B. Two ancient bacterial-like PPP family phosphatases from Arabidopsis are highly conserved plant proteins that possess unique properties. Plant Physiol. 2011;157:1778–1792. doi: 10.1104/pp.111.182493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward P., Equinet L., Packer J., Doerig C. Protein kinases of the human malaria parasite Plasmodium falciparum: the kinome of a divergent eukaryote. BMC Genomics. 2004;5:79. doi: 10.1186/1471-2164-5-79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkes J.M., Doerig C. The protein-phosphatome of the human malaria parasite Plasmodium falciparum. BMC Genomics. 2008;9:412. doi: 10.1186/1471-2164-9-412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuda M., Sakaida H., Chinzei Y. Targeted disruption of the plasmodium berghei CTRP gene reveals its essential role in malaria infection of the vector mosquito. J. Exp. Med. 1999;190:1711–1716. doi: 10.1084/jem.190.11.1711. [DOI] [PMC free article] [PubMed] [Google Scholar]
Supplemental References
- Beetsma A.L., van de Wiel T.J., Sauerwein R.W., Eling W.M. Plasmodium berghei ANKA: purification of large numbers of infectious gametocytes. Exp. Parasitol. 1998;88:69–72. doi: 10.1006/expr.1998.4203. [DOI] [PubMed] [Google Scholar]
- Green J.L., Rees-Channer R.R., Howell S.A., Martin S.R., Knuepfer E., Taylor H.M., Grainger M., Holder A.A. The motor complex of Plasmodium falciparum: phosphorylation by a calcium-dependent protein kinase. J. Biol. Chem. 2008;283:30980–30989. doi: 10.1074/jbc.M803129200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellemans J., Mortier G., De Paepe A., Speleman F., Vandesompele J. qBase relative quantification framework and software for management and automated analysis of real-time quantitative PCR data. Genome Biol. 2007;8:R19. doi: 10.1186/gb-2007-8-2-r19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janse C.J., Franke-Fayard B., Mair G.R., Ramesar J., Thiel C., Engelmann S., Matuschewski K., van Gemert G.J., Sauerwein R.W., Waters A.P. High efficiency transfection of Plasmodium berghei facilitates novel selection procedures. Mol. Biochem. Parasitol. 2006;145:60–70. doi: 10.1016/j.molbiopara.2005.09.007. [DOI] [PubMed] [Google Scholar]
- Liu Y., Tewari R., Ning J., Blagborough A.M., Garbom S., Pei J., Grishin N.V., Steele R.E., Sinden R.E., Snell W.J., Billker O. The conserved plant sterility gene HAP2 functions after attachment of fusogenic membranes in Chlamydomonas and Plasmodium gametes. Genes Dev. 2008;22:1051–1068. doi: 10.1101/gad.1656508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall O.J. PerlPrimer: cross-platform, graphical primer design for standard, bisulphite and real-time PCR. Bioinformatics. 2004;20:2471–2472. doi: 10.1093/bioinformatics/bth254. [DOI] [PubMed] [Google Scholar]
- Ridzuan M.A., Moon R.W., Knuepfer E., Black S., Holder A.A., Green J.L. Subcellular location, phosphorylation and assembly into the motor complex of GAP45 during Plasmodium falciparum schizont development. PLoS ONE. 2012;7:e33845. doi: 10.1371/journal.pone.0033845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandesompele J., De Preter K., Pattyn F., Poppe B., Van Roy N., De Paepe A., Speleman F. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 2002 doi: 10.1186/gb-2002-3-7-research0034. i, RESEARCH0034. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The table also contains the known NCBI or GenBank identifiers of the Shelp orthologs used in the analysis. The enzymes’ PP catalytic domains’ similarities to AtSLP1 and AtSLP2 are shown as percentage of identities.
Δshlp1 ookinetes are fully motile and indistinguishable from wild-type ookinetes.
Δshlp1 ookinetes are fully motile and indistinguishable from wild-type ookinetes.