

Abstract
Neuregulin 1 (NRG1) is an axon-derived factor that is critical for Schwann cell (SC) development and myelinogenesis in a manner dependent on transmembrane tyrosine kinases ErbB2 and ErbB3. Recent studies suggest that NRG1 signaling plays a role in remyelination of regenerated nerves after injury. In this study, we investigated the role of Erbin, a protein that interacts with ErbB2 in remyelination of injured nerves. We show that Erbin expression increased dramatically in injured nerves. Myelinated axons were fewer, and g-ratios of those that were myelinated were increased in erbin−/− mice, which were impaired in functional recovery from nerve injury. These results indicate a necessary role of Erbin in remyelination of regenerating axons. Erbin ablation had little effect on numbers of BrdU-labeled and TUNEL-labeled SCs, suggesting mechanisms independent of altered proliferation or apoptosis. We demonstrated that Erbin mutant mice were impaired in raising or maintaining the levels of ErbB2 and in producing NRG1 in axons. Together, these observations demonstrate that Erbin is required for remyelination of regenerated axons after injury, probably by regulating ErbB2 and NRG1 levels, identifying a novel player in regulating remyelination.
Introduction
Schwann cells (SCs) are critical for myelin sheath thickness and internodal distance, which are important determinants of nerve conduction velocity (Baumann and Pham-Dinh, 2001). They are also necessary for the recovery from nerve injury (Fu and Gordon, 1997; Suter and Scherer, 2003; Chen et al., 2007; Nave et al., 2007; Fancy et al., 2011). After injury, axon segments distal to the injury site undergo Wallerian degeneration (Dubový, 2011), a complex process characterized by axon disintegration, myelin breakdown, and debris phagocytosis by SCs and macrophages. Concomitantly, normally quiescent SCs dedifferentiate and proliferate to remyelinate axons sprouted from proximal segments. Unlike myelination that has been studied extensively, less is understood about regulatory mechanisms of remyelination.
Neuregulin 1 (NRG1) is a factor that regulates SC development and myelination of axons (Marchionni et al., 1993; Shah et al., 1994; Dong et al., 1995; Grinspan et al., 1996; Mahanthappa et al., 1996; Wolpowitz et al., 2000; Winseck et al., 2002; Michailov et al., 2004; Mei and Xiong, 2008). Of six types of NRG1 (Steinthorsdottir et al., 2004), type III seems to be the isoform implicated in axonal regulation of SC myelination (Michailov et al., 2004; Taveggia et al., 2005; Lemke, 2006; Nave and Salzer, 2006). The function of NRG1 is regulated by BACE1, an extracellular protease, whose mutation impairs myelination (Hu et al., 2006, 2008; Willem et al., 2006). NRG1 binds to ErbB3, which heterodimerizes with and thus activates ErbB2. NRG1 regulation of myelination requires ErbB2 and ErbB3 (Nave and Salzer, 2006; Mei and Xiong, 2008). However, NRG1 and ErbB2 seem to be dispensable for myelin maintenance (Atanasoski et al., 2006; Fricker et al., 2011).
Evidence suggests that NRG1 regulates remyelination after nerve injury. It promotes remyelination of regenerated axons and functional recovery after injury (Chen et al., 1998; Joung et al., 2010). It also increases the survival of terminal SCs after denervation (Trachtenberg and Thompson, 1996) and promotes their process extension (Hayworth et al., 2006). On the other hand, remyelination is delayed in peripheral nerves of BACE1 null mice (Hu et al., 2008). Recently, motoneuron-specific ablation of type III NRG1 impairs remyelination (Fricker et al., 2011), indicating a critical role of NRG1 in remyelination.
In the current study, we investigated how NRG1 signaling is regulated in remyelination of regenerated axons. We found that nerve injury increases the expression of Erbin, a binding protein that specifically interacts with the ErbB2 (Borg et al., 2000; Huang et al., 2001). Our previous work demonstrated that Erbin is expressed in SCs and is critical for myelination and ensheathment of peripheral nerves during development (Tao et al., 2009). To study its role in remyelination, we compared myelination of regenerated axons after nerve injury between wild-type and erbin−/− mice and found that Erbin is necessary for remyelination. We explored possible cellular and molecular mechanisms. Results suggest that Erbin is critical for remyelination by promoting SC differentiation and by maintaining ErbB2 levels. These observations identified a novel player in regulating remyelination.
Materials and Methods
Animals.
The erbin−/− mice were described previously (Tao et al., 2009) and were backcrossed with C57BL/6 mice for multiple generations. Mice were housed in a room with a 12 h light/dark cycle with access to food and water ad libitum. Animal experiments were approved by the Institutional Animal Care and Use Committee of the Georgia Health Sciences University.
Antibodies and chemicals.
Rabbit Erbin antibody was generated by GST-Erbin-PDZ as described previously (Huang et al., 2001) and purified by GST-fusion proteins immobilized on Affi-Gel 10, following the manufacturer's instruction. Primary antibodies were as follows: ErbB2 (1:500, sc-284), ErbB3 (1:500, sc-285), NRG1 (1:500, sc-348), and Integrinβ4 (1:500, sc-9090), all from Santa Cruz Biotechnology; and myelin basic protein (MBP; 1:250, M1891), α-tubulin (1:1000, T5168), β-actin (1:1000, A1978), and BrdU (1:1000, B2531), all from Sigma-Aldrich. Secondary antibodies for immunostaining were as follows: Alexa Fluor 488 anti-rabbit (1:500, A11018; Invitrogen) and Alexa Fluor 594 anti-mouse (1:500, A11015; Invitrogen).
Nerve crush injury and transplant.
Animals (either sex, >3 months old) were anesthetized with ketamine and xylazine (100 and 20 mg/kg, i.p., respectively), and right thighs were sterilized with 2% iodide and 70% alcohol. Right sciatic nerves at the midthigh were exposed. The tips of No. 5 Dumont microforceps (11252-00; F.S.T.), which were precooled with liquid nitrogen, were used to clamp and crush the nerves three times (30 s each) as described previously (Hu et al., 2008). Skins were closed with steel clips (39465012; Leica). Animals were kept warm on a thermostatic rubber pad (37°C), which was connected with a heat therapy pump (GAYMAR) during the entire process to increase the survival rate. In some experiments, segments of sciatic nerves (∼10 mm) were isolated from wild-type mice and transplanted into mutant mice or vice versa under a surgical OPMI microscope (Carl Zeiss). Nerve segments were sutured to recipient nerves in correct proximal–distal orientation with 10-0 nylon black (STREF: 03171; F.S.T.).
Toe-spreading analysis.
Toe-spreading analysis was performed as described previously (Wakatsuki et al., 2009). At the designated time after nerve injury, the animals with palm and toes of the hindpaws painted with ink were allowed to walk on white paper along a 30 × 10 cm corridor. Each mouse was tested three times. The distances between the first and fifth toes were measured as the toe-spreading index.
Electron microscopy.
Electron microscopic (EM) analysis of myelin was performed as described previously (Tao et al., 2009). Mice were anesthetized and cardiac perfused with 4% formaldehyde and 2% glutaraldehyde in 0.1 m sodium cacodylate buffer (NaCac; pH 7.4). Sciatic nerves were removed and fixed overnight at 4°C in the same perfusion fixative for 24 h. They were washed by 0.1 m NaCac and osmicated with 2% osmium tetroxide for 30–60 min at 4°C, washed by 0.1 m NaCac and by deionized H2O for 30–60 min at 4°C, and dehydrated in graded (30–70%) ethanol. Samples were stained with 2% uranyl acetate in 70% ethanol at 4°C for 30 min, followed by dehydration with 70–100% ethanol. Samples were incubated two times with propylene oxide for 10 min and embedded with resins. Ultrathin sections were photographed with a Phillips 400 Transmission electron microscope. EM images were analyzed by Image J (NIH). To minimize the variation in different regions of nerves, cross sections of nerves were divided into 10 regions, and two pictures were randomly taken from each region and analyzed by an individual blind to genotypes or treatments. The g-ratios were calculated by the perimeters of axons (inner) divided by the perimeters of corresponding fibers (outer). Axonal diameters were normalized by perimeters through the following equation: diameter = perimeter/π. This procedure allowed for inclusion of irregularly shaped axons and fibers and helped to eliminate biased measurement of diameters based on circularity.
Immunostaining and BrdU and TUNEL staining.
Immunostaining was performed as described previously with modification (Tao et al., 2009). Sciatic nerves were embedded in OCT, frozen in liquid nitrogen, and cut into 20 μm sections that were mounted on SuperFrost plus slides. Sections and cultured cells on coverslips were fixed in 4% phosphate-buffered paraformaldhyde (PFA) and incubated at 4°C for 30 min with 0.3% Triton X-100 plus 5% goat serum in PBS (to permeabilize cells). They were incubated at 4°C overnight with primary antibodies in PBS containing 5% goat serum. After washing three times with PBS, samples were incubated at room temperature for 1 h with Alexa-488 goat anti-rabbit or Alexa-594 goat anti-mouse secondary antibody and mounted with Vectashield mounting medium (Vector Laboratories). Images were taken by an LSM510 confocal microscope (Zeiss) and analyzed by Image J (NIH).
BrdU labeling was performed as described previously with modification (Xu et al., 2007). BrdU (B5002; Sigma-Aldrich) was injected into mice 5 d after sciatic nerve crush (100 mg/kg, i.p.; three times with a 6 h interval). Two hours after the final injection, mice were cardiac perfused with 4% PFA and 4% sucrose in PB. Sciatic nerves were dissected out and subjected to cryostat section. Longitudinal or cross sections were permeabilized three times with 1% Triton X-100 for 5 min and incubated with 2N HCl for 2 h at room temperature to expose BrdU-incorporated DNA. Samples were neutralized by borate buffer (0.1 m, pH 8.5) for 12 min and washed by PBS three times before immunostaining using anti-BrdU antibody (1:1000 in blocking buffer: PBS containing 5% goat serum).
A TUNEL apoptosis detection kit (T9162-80; US Biological) was used for DNA fragmentation fluorescence staining according to the manufacturer's protocol with modification. Frozen tissue sections (15 d after crush) were fixed with 4% PFA overnight at 4°C. Samples were washed three times for 10 min with PBS and permeabilized with 0.5% Triton X-100 in PBS overnight at 4°C. Samples were incubated with a TUNEL reaction mixture (enzyme solution plus labeling solution) for 60 min at 37°C, washed with PBS, mounted by Vectorshield with DAPI, and examined under a fluorescence microscope.
Western blotting.
Western blotting was performed as described previously (Tao et al., 2009). Proteins were transferred from an SDS-PAGE gel to a nitrocellulose membrane, which was incubated in blocking buffer–PBS with 0.2% Tween 20 and 5% milk for 1 h at room temperature and subsequently incubated with the indicated antibodies in the blocking buffer overnight at 4°C, washed with PBS containing 0.3% Tween 20, and incubated with an HRP-conjugated secondary antibody for 1 h at room temperature (Tao et al., 2009). Immunoreactive bands were visualized using enhanced chemiluminescence (32106; Pierce Chemical). Autoradiographic films were scanned with an Epson 1680 scanner, and the captured images were analyzed with Image J (NIH).
Reverse transcription quantitative PCR.
Total RNA was isolated from dissected sciatic nerves using Trizol (Invitrogen) and purified using the RNeasy mini kit (QIAGEN). Reverse transcription quantitative PCR (RT-qPCR) was performed as described previously (Liu et al., 2011). Equal amounts of total RNAs (1 μg) were reverse transcribed by random hexamer primers using the Maxima Enzyme Mix (Fermentas). Quantitative PCRs were run in a PTC-200 Peltier Thermal Cycler (Bio-Rad MJ Research) using SYBR Green/ROX (Fermentas). For erbB2 transcripts, primers were 5′-CGCGGGTACCCAAGTGTGTA (forward) and 5′-CGTTGTCCAAAGGGTCTCG (reverse), which generated a product of 317 bp. For erbB3 transcripts, primers were 5′-CACACCTGGTCATAGCGGTGAC (forward) and 5′-TGGGTCCAGAGGCTCGATGCTC (reverse), which generated a product of 152 bp. For erbin transcripts, primers were 5′-GCAAGGTTCCTCATGACTG (forward) and 5′CTCGAATCTCTTGCTTTGCC (reverse), which generated a product of 540 bp. For NRG1 type III transcripts, primers were 5′-GGACCCCTGAGGTGAGAACA (forward) and 5′-CAGTCGTGGATGTCGATGTGG (reverse), which generated a product of 102 bp.
Statistical analyses.
Data were analyzed by the paired or unpaired t test. Unless otherwise indicated, data were presented as mean ± SEM. Statistical significance was considered when p < 0.05.
Results
Expression of Erbin is increased in SCs of injured sciatic nerves
To investigate whether Erbin regulates remyelination, we determined expression of Erbin in injured sciatic nerves. Right sciatic nerves were injured by crushes with liquid nitrogen-cooled microforceps (Hu et al., 2008), and distal segments of injured nerves were isolated for Western blot analysis. ErbB2 is known to be increased in injured nerves and is implicated in SC proliferation, differentiation, and remyelination after injury (Carroll et al., 1997; Kwon et al., 1997; Atanasoski et al., 2006; Hu et al., 2008). Accordingly, as shown in Figure 1, ErbB2 levels were increased in distal segments of injured sciatic nerves by 6.7- and 16.3-fold, respectively, at days 5 and 20 after injury. This increase was preceded by a brief reduction at days 1 and 3, probably because of nerve degeneration (Fig. 1A,B). Similar changes were observed for ErbB3 expression after nerve injury, except the increase was less than ErbB2. Interestingly, Erbin levels were also increased in distal segments of injured sciatic nerves (Fig. 1A,B). As a control, Integrin β4, a laminin receptor, was reduced in distal nerves after injury (Fig. 1A,B), confirming a previous report (Einheber et al., 1993), and suggesting that the elevating effect on Erbin after nerve injury was specific. Together, these results indicate that Erbin expression was specifically increased in injured sciatic nerves.
Figure 1.

Increased expression of Erbin and ErbB receptors in injured nerves. A, Western blots showing increased expression of Erbin, ErbB2, and ErbB3 in distal segments of injured nerves. Distal segments at indicated days after injury were homogenized and subjected to Western blotting with indicated antibodies. B, Quantitative data of A. n = 3. **p < 0.01; *p < 0.05, compared with uninjured control. ErbB2 levels were increased in distal segments of injured sciatic nerves to 6.7 ± 1.67- and 16.3 ± 1.02-fold and ErbB3 levels to 1.8 ± 0.07- and 1.4 ± 0.08-fold (n = 3; p < 0.01) on days 5 and 20 after injury, respectively, whereas Erbin levels were increased to 4.7 ± 0.63-, 4.6 ± 0.86-, and 5.4 ± 1.30-fold (n = 3; p < 0.01) on days 3, 5, and 20, respectively. C, Erbin expression was increased in regenerated SCs. Longitudinal sections of injured distal segments on day 20 were costained with anti-Erbin antibody and DAPI. White arrows indicate Erbin staining; white arrowheads indicate hollow space between Erbin-stained myelin; yellow arrows indicate nuclei (elongated in proximal regions but round in distal segments). CTL, Control.
To determine in which cells of sciatic nerves Erbin was increased, we stained longitudinal sections of injured nerves with a purified anti-Erbin antibody that recognizes a single band on Western blot (data not shown). In proximal segments (20 d after injury), Erbin was present in longitudinal tracks with a hallow space in between that is presumably occupied by axons (Fig. 1C, enlarged image), suggesting that Erbin was present in myelin, in agreement with a previous report (Tao et al., 2009). Notice that SC nuclei in this segment were elongated in shape as a result of myelination. However, distal segments (20 d after injury) were filled with proliferating and differentiating SCs (as indicated by DAPI staining). Erbin immunoreactivity was increased (Fig. 1C), in agreement with results from Western blotting (Fig. 1A,B). Moreover, the immunoreactivity was in close proximity with cell nuclei (Fig. 1C), suggesting presence in regenerating SCs. The increase of Erbin in injured nerves started as early as day 3, 2 d ahead of those of ErbB2 and ErbB3 (Fig. 1A,B), suggesting a preceding role of Erbin in the reparative process after nerve injury.
Erbin is required for remyelination after nerve injury
Nerve injury leads to Wallerian degeneration (Dubový, 2011). To determine whether Erbin mutation alters this process or regeneration thereafter, we divided nerve segments into different parts. As shown in Figure 2A, proximal segments of nerves 2 mm from the injury site were designated as P2-3, whereas distal segments of nerves 2 and 4 mm from the injury site were designated as D2-3 and D4-5, respectively. Nerve segments were processed for EM analysis. As shown in Figure 2B, both myelinated and unmyelinated axons of P2-3 segments in wild-type and erbin−/− mice were relatively intact 5 d after injury. Myelin thickness correlated with axonal sizes (Fig. 2C). The g-ratio of myelinated axons, obtained by dividing the axon diameter by the fiber (axon plus myelin) diameter, was higher in erbin−/− P2-3 segments compared with wild-type controls. This result could suggest that Erbin is involved in myelination development, in agreement with our previous report (Tao et al., 2009). In contrast, the segments distal to the injury site underwent Wallerian degeneration. On day 5 after injury, in D2-3 segments, axonal degeneration and myelin breakdown or collapse was apparent, and the debris was phagocytosed by macrophages (Fig. 2B). Degeneration was less severe in D4-5 segments, which were farther away from the injury site: a few intact myelinated axons remained. The degeneration prevented accurate measurement of the g-ratio of axons in distal segments on day 5 after injury. However, there was no difference in Wallerian degeneration between wild-type and erbin−/− mice, suggesting that Erbin ablation did not alter this process.
Figure 2.

Wallerian degeneration of distal segments of injured nerves in both wild-type and Erbin null mice. A, Schematic illustration of the injury site (red) and distal and proximal segments. Segments in yellow were analyzed by electron microscopy. P, Proximal; D, distal. Numbers indicate distance from the crush site in millimeters. B, Electron micrographs of P2-3 (left), D2-3 (middle), and D4-5 (right) of wild-type (top) and erbin−/− (bottom) mice on day 5 after injury. P2-3 shows unmyelinated axons (red arrowhead) and myelinated axons (yellow arrowhead). D2-3 and D4-5 show degenerated axons (star), myelin debris (white arrow), and macrophages with phagosomes (yellow arrow). Scale bar, 5 μm. C, g-ratio of myelinated axons in P2-3 segments on day 5 after injury, plotted against the axon diameter. The averaged g-ratio was presented as means ± SEM. ***p < 0.001; n ≥ 20 images per mouse, three mice per group.
To explore the role of Erbin in nerve regeneration, we studied remyelination of injured sciatic nerves in erbin−/− mice. After nerve injury, axons are regenerated from proximal ends and sprout into distal parts of the injury site within 10–14 d (Fu and Gordon, 1997; Suter and Scherer, 2003; Chen et al., 2007; Nave et al., 2007; Fancy et al., 2011). As shown in Figure 2B, on day 5, myelin sheathes collapsed and axons were degenerated in D2-3 segments of both control and Erbin mutant mice. However, on day 20, axons were detected in these segments in wild-type mice, indicating that axons were regenerated (Fig. 3C). Moreover, these D2-3 axons were myelinated (Fig. 3C). However, fewer axons were detected and fewer axons were myelinated in distal segments in erbin−/− mice compared with wild-type controls (Fig. 3G), indicating an impairment of axonal regeneration and remyelination when Erbin is ablated. This notion is supported by three additional pieces of evidence. First, many large D2-3 axons in erbin−/− mice were not myelinated or were naked on day 20 after injury, in contrast to those in wild-type controls (Fig. 3C). The number of unmyelinated axons with a diameter of 1 μm or larger was dramatically increased in erbin−/− mice (Fig. 3H). Second, of those myelinated axons in erbin−/− D2-3 segments, the g-ratio was significantly larger than that of wild-type controls. Similar myelination defects were observed in D4-5 segments in erbin−/− mice. Third, concomitantly with morphological changes were alterations of MBP, a major myelin component. On day 5 after injury, MBP levels were reduced in distal segments compared with those in proximal segments of both wild-type and erbin−/− mice (Fig. 3I). This time course was consistent with axonal degeneration and myelin collapse in Wallerian degeneration (Carroll et al., 1997) (Fig. 2). On day 20, MBP levels were increased in wild-type distal segments compared with those on day 5 (lanes 7 and 3, respectively; Fig. 3I,J), in agreement with increased myelination on day 20. However, although MBP in erbin−/− distal segments was also elevated (compare lanes 4, 8), levels in distal segments were significantly lower in erbin−/− mice compared with wild-type controls. These results suggest that Erbin ablation attenuates the ability of mutant mice to produce MBP. Together, these morphological and biochemical studies indicate that Erbin is essential for axonal remyelination.
Figure 3.

Deficient remyelination of regenerated axons in erbin−/− mice on day 20 after nerve injury. Shown are representative electron micrographs and g-ratios of respective segments of injured sciatic nerves of indicated genotypes on day 20 after injury. A, B, P2-3 segments. C, D, D2-3 segments. E, F, D4-5 segments. Open arrowheads indicate naked axons with a diameter equal to or larger than 1 μm. Scale bar, 5 μm. The averaged g-ratio was presented as means ± SEM. n ≥ 20 EM images of each mouse, three mice per genotype. ***p < 0.001. G, Fewer axons were myelinated in regenerating nerves of erbin−/− mice. Scored were axons from over 20 EM images per mouse, three mice per genotype in an area of 0.01 mm2. H, Increased number of unmyelinated axons with diameters equal to or larger than 1 μm in injured erbin−/− sciatic nerves. Axons were scored as in G. **p < 0.01. I, Reduced MBP levels in distal segments of injured sciatic nerves in erbin−/− mice. Proximal and distal segments were isolated from injured sciatic nerves of indicated genotypes on days 5 and 20 after injury, and subjected to Western blotting with MBP antibody and α-tubulin (to indicate equal loading). J, Quantitative analyses of data in I. n = 3. **p < 0.01, compared with wild type; ##p < 0.01, compared with data of distal segments on day 5. P, Proximal; D, distal.
We have also examined regenerated nerves on day 40. The g-ratios of both wild-type and erbin−/− regenerated axons on day 40 were reduced (Fig. 4A,B) compared with those on day 20 (Fig. 3C–F), suggesting recovery from nerve injury. However, myelin of regenerated axons in erbin−/− mice remained thinner (with larger g-ratios) than wild-type controls, suggesting again impaired remyelination. Quantitatively, compared with day 20, the number of myelinated axons was increased and that of unmyelinated axons were reduced in wild-type mice (Fig. 4B). Myelinated axons were fewer and unmyelinated axons more in erbin−/− mice than controls (Fig. 4B), supporting continuing impairment in remyelination on day 40. However, the numbers of large (>1 μm in diameter) unmyelinated axons in distal segments were similar between the injured two genotypes, indicating delayed remyelination of large axons in erbin−/− mice. These results corroborate that Erbin is necessary for remyelination of newly generated axons.
Figure 4.

Deficient remyelination of regenerated axons in erbin−/− mice on day 40 after nerve injury. A, Representative electron micrographs and g-ratios of respective segments of injured sciatic nerves of indicated genotypes on day 40 after injury. The averaged g-ratio was presented as means ± SEM. n ≥ 20 EM images per mouse, three mice per genotype. ***p < 0.001. B, Fewer axons were myelinated in regenerating nerves of erbin−/− mice. Scored were axons from over 20 EM images per mouse, three mice per genotype in an area of 0.01 mm2. C, No difference in unmyelinated axons with a diameter of 1 μm or larger between wild-type and erbin−/− mice. Axons were scored as in B. D, Distribution curves of axon size of myelinated axons in D4-5 segments of wild-type and erbin−/− mice on days 20 and 40 after injury.
Notice that axons from proximal segments of injured nerves in both mice remained intact on days 20 and 40 after injury (Fig. 3A, 4A), although axons with a similar size in proximal nerves of erbin−/− had thinner myelin and an increased g-ratio (Figs. 3A,B, 4A,B), the same as previously reported during development (Tao et al., 2009). Intriguingly, the distribution curves of myelinated axon size in D4-5 segments shifted toward the right on both days 20 and 40 (Fig. 4D), suggesting an increased size of regenerating axons in erbin−/− mice. We speculate that this may be attributable to loss of the physical boundary by myelin or a compensatory mechanism.
Functional recovery after nerve injury is impaired in erbin−/− mice
To determine whether impaired remyelination in erbin−/− mice impedes functional recovery, we performed walking footprint analysis. As shown in Figure 5A, on day 5 after injury, the ink mark left by right heels (the side of sciatic nerve injury) was more intense compared with the normal left heels, indicating impaired gastrocnemius innervation (de Medinaceli et al., 1982; Brown et al., 1989; Haastert et al., 2006). This impairment was observed in both wild-type and erbin−/− mice. On day 20, the ink mark intensity of right heels became similar to that of left heels in wild-type mice, indicating functional recovery of injured sciatic nerves. However, marks of right heels of erbin−/− mice remained heavier than those of left heels on day 20, although they became similar by day 40. These results suggest a delay in functional recovery in erbin−/− mice. On the other hand, after contact with a surface, rodents spread the toes of the hind feet, which is a reflex that requires sensory innervation (Contreras et al., 1995). Sciatic nerve injury attenuates the contraction of paw extensor and paw intrinsic muscles, and, consequently, mice have difficulty spreading toes (Fig. 5B). The ability to spread the toes has been used to indicate the recovery of sensory function after sciatic nerve injury (de Medinaceli et al., 1982; Brown et al., 1989; Klapdor et al., 1997; Haastert et al., 2006). As shown in Figure 5C, on day 5 after injury, the distance between the first and fifth toes was significantly smaller in the right paws compared with left paw controls in both wild-type and erbin−/− mice, indicating impaired sensory function. Consistent with the heel preference, the toe distance of right hindpaws recovered by day 20 after injury in wild-type mice (Fig. 5C). In erbin−/− mice, however, the distance remained abnormal even on day 40 after injury (Fig. 5C), suggesting that sensory function was not fully restored in erbin−/− mice 40 d after injury. Together, these observations provide evidence that Erbin ablation inhibits functional recovery from nerve injury, corresponding to the morphological deficiency of remyelination in regenerating nerves of erbin−/− mice.
Figure 5.

Delayed functional recovery of erbin−/− mice after nerve injury. A, Gait patterns of wild-type and erbin−/− mice on indicated days after nerve injury. Paw prints were recorded by painting hindpaws with black ink and having them walk on white paper along a 30 × 10 cm corridor (toward the top). Prints of the right feet (where sciatic nerves were injured) were on the right. B, Representative footprints of left (L) and right (R) hindpaws of wild-type mice on day 5 after injury. The arrowhead indicates heel marks. Toe distance was measured between the first and fifth toes (indicated by arrows). C, Delayed recovery in toe distances in erbin−/− mice. Toe distances were measured as indicated in B on different days after injury. n = 5; **p < 0.01, paired t test.
Erbin is not required for proliferation and survival of regenerated SCs in injured nerves
To investigate mechanisms of Erbin in remyelination, we determined whether proliferation or survival of SCs was altered in injured nerves of erbin−/− mice by BrdU labeling and TUNEL assays, respectively. Mice received injections of BrdU on day 5 after nerve injury, and sciatic nerves were collected 24 h later and stained with anti-BrdU antibody for BrdU incorporation. Few nuclei were labeled by BrdU in proximal segments of injured nerves or in uninjured sciatic nerves (Fig. 6A,C). No difference was observed between proximal segments from wild-type and erbin−/− mice. In contrast, BrdU-incorporated nuclei increased dramatically in distal segments of injured nerves of wild-type mice (Fig. 6A,C), consistent with the concept that SCs undergo proliferation during Wallerian degeneration (Kwon et al., 1997; Adilakshmi et al., 2011). However, a similar increase in BrdU-labeled nuclei was also observed in erbin−/− mice, indicating that Erbin is not required for SC proliferation after nerve injury (Fig. 6A–C).
Figure 6.

No effect by Erbin mutation on SC proliferation and survival in regenerating nerves after injury. A, B, Representative pictures of BrdU staining from intact nerves (CTL) or of different segments from injured nerves of wild-type (A) or erbin−/− (B) mice. C, No difference in the number of BrdU-labeled nuclei between wild-type and erbin−/−. Labeled nuclei in an area of 0.1 mm2 were quantified. n = 3. D, E, Representative pictures of TUNEL staining from intact nerves (CTL) or of different segments from injured nerves of wild-type (D) or erbin−/− (E) mice. F, No difference in the number of TUNEL-positive nuclei between wild-type and erbin−/− mice. TUNEL-positive nuclei were counted in an area of 0.1 mm2 from different regions and normalized with total nuclear numbers. n = 3. P, Proximal; D, distal.
Next, we investigated whether ablation of Erbin affected SC survival in regenerating nerves. To this end, sections from proximal and distal sciatic nerves of injured and uninjured nerves were processed to fluorescence labeling of the terminal uridine nucleotides in apoptotic nuclei. As shown in Figure 6D–F, TUNEL-positive cells were in sections from distal segments of injured nerves in both control and erbin−/− mice, and there was no difference between control and erbin−/− samples. These results suggest that Erbin is not required for SC survival in injured nerves. In agreement with the notion that SCs increase in number in injured nerves, the ratios of TUNEL-positive nuclei over total nuclei were reduced in distal segments compared with uninjured or proximal segments (Fig. 6F). Together, these results demonstrate that Erbin is not required for SC proliferation or survival, suggesting that it may regulate the myelinating ability of regenerated SCs.
Requirement of Erbin for elevated ErbB2 expression in injured nerves
Erbin is implicated in ErbB2 stability in transfected cells and during myelination (Tao et al., 2009). We determined whether nerve injury-induced ErbB2 expression requires Erbin. Wild-type and erbin−/− mice were subjected to nerve injury as described in Figure 1. Proximal and distal segments of injured nerves were isolated for Western blot analysis. Compared with proximal segments in wild-type mice, ErbB2 and ErbB3 levels in distal segments were significantly increased 5 d after injury (Fig. 7A,B). In erbin−/− mice, however, the ErbB2 levels in distal segments reduced significantly to 19.2% and ErbB3 levels reduced to 40.0% of those in wild type (Fig. 7A,B), despite the similar proliferation and survival of SCs in distal segments (Fig. 6). These results indicated that the increase in ErbB2/3 in injured nerves requires Erbin. Notice that ErbB2 were reduced in erbin−/− proximal segments (Fig. 7A,B), which did not exhibit consistent morphological changes after nerve injury (Fig. 2). This observation was in agreement with our previous study (Tao et al., 2009). Despite the reduction at absolute levels, ErbB2 levels were higher in distal segments of erbin−/− injured nerves compared with that in proximal segments, suggesting some mechanisms related to protein production may not be severely impaired by ablation of Erbin. We measured ErbB2 and ErbB3 mRNAs in injured nerves by quantitative RT-PCR. ErbB2 mRNA was increased in distal segments in both wild-type and erbin−/− mice compared with those in proximal segments, although the levels were significantly lower in erbin−/− injured nerves (Fig. 7C), suggesting similar amplification of transcriptional machinery for ErbB2 production. Notably, the increase of ErbB2 at mRNA levels was only 70.1 ± 7.82%, in contrast to 187 ± 24.5% at protein levels in distal segments compared with those in proximal segments of wild-type nerves, implying a requirement for protein-stabilizing mechanisms. Moreover, the reduction of ErbB2 or ErbB3 in injured nerves of erbin−/− mice at mRNA levels was much less than that at protein levels. Together, these observations suggest that Erbin mutation prevents ErbB2 and ErbB3 increase in injured nerves, mainly at protein levels. This mechanism contributes to reduced NRG1/ErbB signaling in SCs, which is necessary for remyelination.
Figure 7.
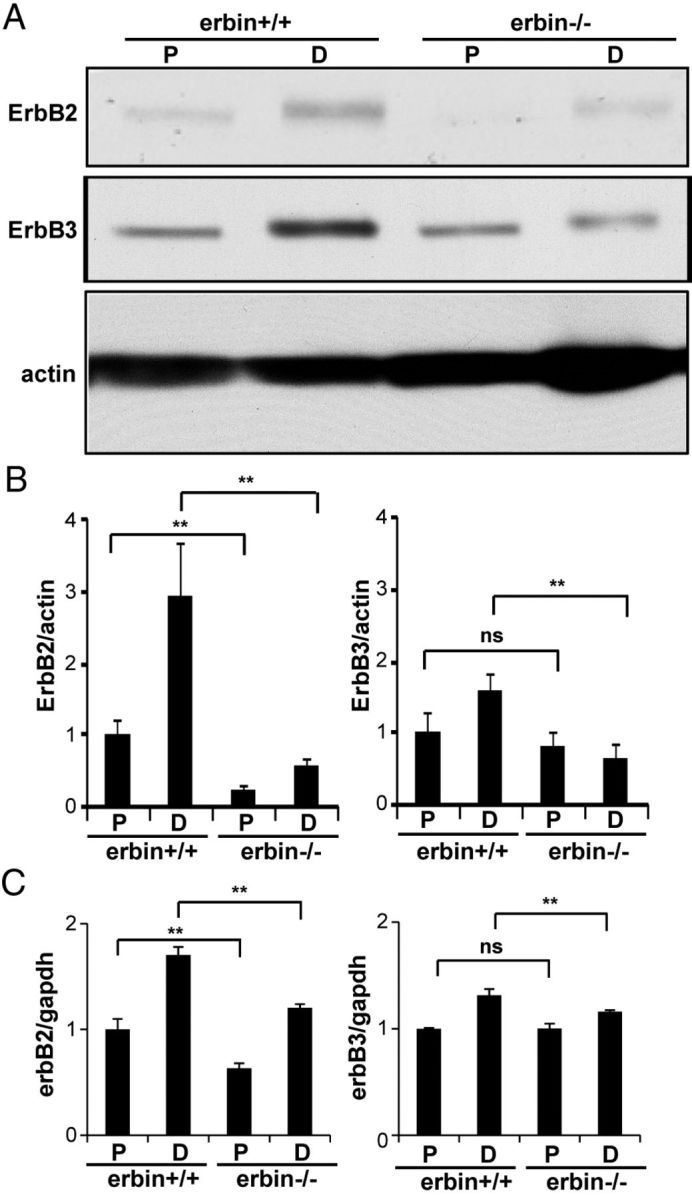
Injury-induced expression of ErbB2 was inhibited in erbin−/− mice. A, Inability of injury to increase ErbB2 expression in erbin−/− distal segments. Five days after injury, proximal (P) and distal (D) segments were isolated and subjected to Western blot analysis for ErbB2 and ErbB3. B, Quantitative analysis of data in A. n = 3; **p < 0.01, compared with wild-type controls. ErbB2 and ErbB3 levels were increased to 2.9 ± 0.25- and 1.6 ± 0.22-fold, respectively, above those in proximal segments in wild-type mice (n = 3; p < 0.01). ErbB2 levels in erbin−/− mice were 2.3 ± 0.46-fold (n = 3; p < 0.05) above those in proximal segments, whereas ErbB3 had no increase. C, mRNA levels of erbB2 and erbB3 in proximal and distal segments of injured sciatic nerves. mRNA was analyzed by quantitative RT-PCR. n = 3; **p < 0.01, compared with wild-type controls.
Erbin regulates remyelination via influence on both axonal and SC factors
Myelination is determined by the interaction between axons and SCs (Jessen and Mirsky, 2005; Nave and Trapp, 2008; Salzer et al., 2008). Since both axonal (type III NRG1) and SC (ErbB2) factors were altered in injured nerves of erbin−/− mice (see below), we have to determine which factor is the key for Erbin to control remyelination. To that end, we performed allograft transplantation, which is transplanting sciatic nerve segments of donor mice to replace segments in recipient mice. Nerve allografts not only provide a scaffold for host nerve regeneration, but also carry SCs of donor mice to host mice. Axon segments in allografts will be degenerated, whereas regenerating axons will grow through the allografts and interact with and eventually are remyelinated by donor SCs. This method has been used to differentiate functions of axons and SCs of different genetic background or after different treatments in nerve regeneration (Aguayo et al., 1976; Haney et al., 1999). As shown in Figure 8, A and B, a sciatic nerve segment (∼10 mm) from a wild-type donor mouse was transplanted to a wild-type recipient mouse (+/+ to +/+). Twenty days after transplantation, 1 mm segments from the middle of transplanted nerves were dissected out and processed to electron microscopy. As shown in Figure 8, C and D, axons were evident in +/+ allografts in +/+ mice and were myelinated with a g-ratio of 0.7529 ± 0.00393. Notice that these axons were newly regenerated because donor axons should have been degenerated within days after surgery.
Figure 8.
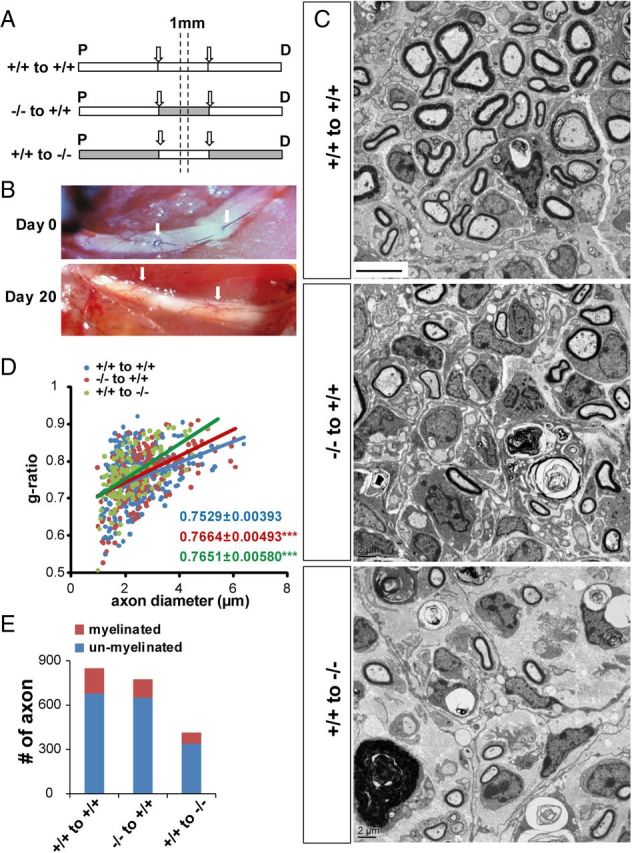
Impaired remyelination in allographs of different genotypes. A, Schematic illustration of allograph transplant experiments between wild-type and erbin−/− sciatic nerves. Arrows mark the allograph segment. Dashed lines mark the central region in allographs for EM examination. B, Sciatic nerves on days 0 and 20 after transplant surgery. Arrows indicate allographs. C, Representative EM images from indicated allograph/transplant groups. Scale bar, 5 μm. D, Impaired myelination of −/− to +/+ and +/+ to −/− allograph axons. The g-ratios of myelinated axons in allographs were plotted against axonal diameters. n ≥ 20 images per mouse, three mice per group. ***p < 0.001, compared with +/+ to +/+. E, Reduced myelinated axons in −/− to +/+ and +/+ to −/− allograph axons. Scored were axons from over 20 EM images per mouse, three mice per genotype in an area of 0.01 mm2. P, Proximal; D, distal.
Interestingly, when erbin−/− allografts were transplanted to wild-type mice (−/− to +/+), myelination of regenerated axons was impaired with g-ratios of 0.7664 ± 0.00493 (Fig. 8D), and the myelinated axon number decreased (Fig. 8E). This result suggests that erbin−/− SCs brought over by allografts were impaired in their ability to myelinate regenerated axons, in support of the hypothesis that Erbin in SCs is critical for remyelination. Unexpectedly, remyelination was also impaired in “+/+ to −/−” allografts (Fig. 8C–E), suggesting a role of Erbin from cells of proximal segments in regulating axonal factors for remyelination. Total axon numbers were reduced in allograft +/+ to −/−, suggesting impaired axon regeneration when axotomized neurons were surrounded with Erbin-deficient supporting cells.
Requirement of Erbin for elevated type III NRG1 expression in injured nerves
NRG1, in particular the type III isoform, is a critical axonal factor for myelination during development and remyelination after injury (Michailov et al., 2004; Taveggia et al., 2005; Hu et al., 2006, 2008; Willem et al., 2006; Fricker et al., 2011). Two bands were detected in sciatic nerves using verified antibody to type III NRG1, representing the full-length Pro-NRG1 (NRG-FL) and the C-terminal fragment (CTF), which was generated during NRG1 maturation (Hu et al., 2006, 2008; Willem et al., 2006; Tao et al., 2009) (Fig. 9A,B). In agreement, we also found increased NRG1 expression (both NRG1-FL and NRG-CTF) in injured nerves of wild-type mice (Fig. 9A,B). The increase at the protein level was readily detectable on days 1, 3, and 20. The reduction of NRG1 in injured nerves on day 5 may be caused by severe axon degeneration in response to nerve injury (Carroll et al., 1997; Wakatsuki et al., 2009). Intriguingly, the levels of NRG1 in distal segments of injured nerves were significantly lower than those in wild-type controls (Fig. 9C,D). Notice that type III NRG1 levels were lower in erbin−/− proximal segments than controls (Fig. 9C,D), implying a possible requirement of Erbin for neuronal NRG1 expression. These results suggest that Erbin mutation also prevents NRG1 increase in injured nerves, suggesting that NRG1/ErbB signaling and/or Erbin activity in SCs may regulate NRG1 production in regenerating axons.
Figure 9.

Requirement of Erbin for elevated type III NRG1 expression in injured nerves. A, Western blotting results showing increased type III NRG1 expression in wild-type mice at indicated times after injury. B, Quantitative analysis of data in A. n = 3. *p < 0.05; **p < 0.01, compared with uninjured controls. C, Reduced type III NRG1 levels in injured distal segments in erbin−/− mice. Proximal (P) and distal (D) segments were isolated 5 d after injury and subjected to Western blot analysis. D, Quantitative analyses of data in C. n = 3; **p < 0.01, compared with wild-type controls. CTL, Control.
Discussion
Major findings of this study include the following. First, Erbin expression increased dramatically in injured nerves. The increase was more in distal segments of the injury site, and the increase was mainly in SCs (Fig. 1). Second, in erbin−/− mice, myelinated axons were fewer, and g-ratios of those that were myelinated were increased (Figs. 3, 4), indicating a necessary role of Erbin in remyelination of regenerating axons. In accordance with morphological myelin deficits, functional recovery from nerve injury was impaired in Erbin mutant mice (Fig. 5). Third, BrdU-labeled cells and TUNEL-labeled cells were not altered by Erbin ablation (Fig. 6), suggesting that impaired remyelination is not caused by reduced proliferation or increased apoptosis of SCs. Fourth, Erbin elevation was ahead of that of ErbB2/3 increase in injured nerves, and Erbin mutation attenuates the elevation of ErbB2/3 and type III NRG1 in injured nerves, suggesting that the mutant mice are impaired in raising or maintaining NRG1/ErbB signaling, which is critical for remyelination (Carroll et al., 1997; Hu et al., 2008; Fricker et al., 2011). Finally, Erbin mutation also prevents type III NRG1 increase in injured nerves, suggesting that NRG1/ErbB signaling and/or Erbin activity in SCs may regulate NRG1 production in regenerating axons. Together, these observations demonstrate that Erbin is required for remyelination of regenerated axons after injury.
Communication between SCs and axons controls myelin repair as well as development (Fu and Gordon, 1997; Jessen and Mirsky, 2005; Taveggia et al., 2010). NRG1 signaling seems to be critical for remyelination of injured nerves, although it may be dispensable for myelin maintenance under physiological conditions (Atanasoski et al., 2006). First, expression of ErbB2 and ErbB3 in SCs and NRG1 (SC-derived type II and axon-derived type III) is increased in injured sciatic nerves (Carroll et al., 1997; Kwon et al., 1997; Atanasoski et al., 2006; Hu et al., 2008). Second, the increase is more in distal segments, where remyelination is active, than in proximal segments (Carroll et al., 1997; Hu et al., 2008). Third, NRG1-treated rodents showed earlier, and more robust, remyelination of regenerated axons after injury and functional recovery (Chen et al., 1998; Joung et al., 2010). Moreover, the addition of NRG1 could increase the survival of terminal Schwann cells at the neuromuscular junction after postnatal denervation (Trachtenberg and Thompson, 1996) and promote their process extension (Hayworth et al., 2006). Fourth, remyelination was delayed in peripheral nerves of BACE1 null mice, probably because of impairment in NRG1 cleavage and subsequent activation of PI3K/Akt, which has been implicated in myelination (Ogata et al., 2004; Chen et al., 2006; Hu et al., 2008). Finally, neuron-specific ablation of type III NRG1 leads to hypomyelination or loss of myelin sheath in mice (Fricker et al., 2011).
During development, NRG1 has been implicated in promoting SC proliferation (Dong et al., 1995). However, no change in BrdU-labeled SCs in erbin−/−-damaged nerves suggests that Erbin is not required for increased SC proliferation after injury. Impaired myelination did not seem to result from an increase in SC apoptosis because TUNEL-labeled SCs were not altered. This notion is in agreement with a report that NRG1/ErbB signaling was not required for SC proliferation or survival (Atanasoski et al., 2006). We propose that impairment in remyelination may be caused by compromised remyelinating ability of regenerated SCs. This notion is supported by the following additional evidence. First, Erbin is expressed mainly in SCs or myelin of the peripheral system under physiological conditions (Tao et al., 2009) as well as after nerve injury (Fig. 1C). Second, erbin−/− SCs in “−/− to +/+” allografts are impaired to remyelinate regenerated axons in wild-type recipient mice.
How does Erbin regulate remyelination? NRG1 signaling necessary for remyelination requires proper levels of ErbB2 for signaling. On the other hand, activation of ErbB kinases leads to endocytic removal of active ligand–receptor complexes from the cell surface and subsequent sorting to proteasomes for degradation or endosomes for recycling (Waterman et al., 1998; Yang et al., 2005; Liu et al., 2007; Mei and Xiong, 2008). In the current study, we found that Erbin increases at protein levels ahead of ErbB2 (Fig. 1) and Erbin mutation prevents injury from elevating ErbB2 to similar levels as control wild-type mice (Fig. 7). Moreover, in contrast to its effects on ErbB2 protein, Erbin mutation has a less effect on mRNA levels of ErbB2, suggesting that a major mechanism of action is to increase the stability of ErbB2 proteins. In agreement, we showed previously that Erbin could prevent both constitutive and ligand-induced ErbB2 endocytosis (Tao et al., 2009). These observations suggest that Erbin regulates remyelination of regenerated axons mainly by stabilizing ErbB2 in SCs.
Intriguingly, remyelination was also impaired in +/+ to −/− allografts (Fig. 8C,D). This result suggests that axonal factors necessary for remyelination may be impaired in erbin−/− mice. In response to nerve injury, axons increase the expression of various factors, including cytokines and growth factors including type III NRG1 (Hammarberg et al., 1996; Kurek et al., 1996; Hu et al., 2008) (Fig. 9). Axonal NRG1 promotes remyelination after nerve injury (Joung et al., 2010; Fricker et al., 2011). Interestingly, injury was unable to increase type III NRG1 to similar levels in erbin−/− mice, compared with controls (Fig. 9), suggesting that Erbin is necessary for NRG1 production in axons after nerve injury. This result provides an additional mechanism for Erbin to regulate remyelination. Type III NRG1 mRNA was not detectable in proximal or distal segments of injured nerves (data not shown); therefore, increased axonal NRG1 in distal segments of wild-type mice (Fig. 9) (Hu et al., 2008), where remyelination is active, is likely transported from soma of axotomized neurons.
Footnotes
The authors declare no competing financial interests.
This work was supported in part by grants from VA Merit Awards and the NIH (L.M. and W.-C.X.). L.M. is the Georgia Research Alliance Eminent Scholar in Neuroscience. We thank R. Smith (Georgia Health Sciences University) for assistance in electron microscopic analysis.
References
- Adilakshmi T, Ness-Myers J, Madrid-Aliste C, Fiser A, Tapinos N. A nuclear variant of ErbB3 receptor tyrosine kinase regulates ezrin distribution and Schwann cell myelination. J Neurosci. 2011;31:5106–5119. doi: 10.1523/JNEUROSCI.5635-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aguayo AJ, Epps J, Charron L, Bray GM. Multipotentiality of Schwann cells in cross-anastomosed and grafted myelinated and unmyelinated nerves: quantitative microscopy and radioautography. Brain Res. 1976;104:1–20. doi: 10.1016/0006-8993(76)90643-0. [DOI] [PubMed] [Google Scholar]
- Atanasoski S, Scherer SS, Sirkowski E, Leone D, Garratt AN, Birchmeier C, Suter U. ErbB2 signaling in Schwann cells is mostly dispensable for maintenance of myelinated peripheral nerves and proliferation of adult Schwann cells after injury. J Neurosci. 2006;26:2124–2131. doi: 10.1523/JNEUROSCI.4594-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumann N, Pham-Dinh D. Biology of oligodendrocyte and myelin in the mammalian central nervous system. Physiol Rev. 2001;81:871–927. doi: 10.1152/physrev.2001.81.2.871. [DOI] [PubMed] [Google Scholar]
- Borg JP, Marchetto S, Le Bivic A, Ollendorff V, Jaulin-Bastard F, Saito H, Fournier E, Adélaïde J, Margolis B, Birnbaum D. ERBIN: a basolateral PDZ protein that interacts with the mammalian ERBB2/HER2 receptor. Nat Cell Biol. 2000;2:407–414. doi: 10.1038/35017038. [DOI] [PubMed] [Google Scholar]
- Brown CJ, Mackinnon SE, Evans PJ, Bain JR, Makino AP, Hunter DA, Hare GM. Self-evaluation of walking-track measurement using a sciatic function index. Microsurgery. 1989;10:226–235. doi: 10.1002/micr.1920100317. [DOI] [PubMed] [Google Scholar]
- Carroll SL, Miller ML, Frohnert PW, Kim SS, Corbett JA. Expression of neuregulins and their putative receptors, ErbB2 and ErbB3, is induced during Wallerian degeneration. J Neurosci. 1997;17:1642–1659. doi: 10.1523/JNEUROSCI.17-05-01642.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen LE, Liu K, Seaber AV, Katragadda S, Kirk C, Urbaniak JR. Recombinant human glial growth factor 2 (rhGGF2) improves functional recovery of crushed peripheral nerve (a double-blind study) Neurochem Int. 1998;33:341–351. doi: 10.1016/s0197-0186(98)00037-0. [DOI] [PubMed] [Google Scholar]
- Chen S, Velardez MO, Warot X, Yu ZX, Miller SJ, Cros D, Corfas G. Neuregulin 1-erbB signaling is necessary for normal myelination and sensory function. J Neurosci. 2006;26:3079–3086. doi: 10.1523/JNEUROSCI.3785-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen ZL, Yu WM, Strickland S. Peripheral regeneration. Annu Rev Neurosci. 2007;30:209–233. doi: 10.1146/annurev.neuro.30.051606.094337. [DOI] [PubMed] [Google Scholar]
- Contreras PC, Steffler C, Yu E, Callison K, Stong D, Vaught JL. Systemic administration of rhIGF-I enhanced regeneration after sciatic nerve crush in mice. J Pharmacol Exp Ther. 1995;274:1443–1449. [PubMed] [Google Scholar]
- de Medinaceli L, Freed WJ, Wyatt RJ. An index of the functional condition of rat sciatic nerve based on measurements made from walking tracks. Exp Neurol. 1982;77:634–643. doi: 10.1016/0014-4886(82)90234-5. [DOI] [PubMed] [Google Scholar]
- Dong Z, Brennan A, Liu N, Yarden Y, Lefkowitz G, Mirsky R, Jessen KR. Neu differentiation factor is a neuron-glia signal and regulate survival, proliferation, and maturation of rat Schwann cell precursors. Neuron. 1995;15:585–596. doi: 10.1016/0896-6273(95)90147-7. [DOI] [PubMed] [Google Scholar]
- Dubový P. Wallerian degeneration and peripheral nerve conditions for both axonal regeneration and neuropathic pain induction. Ann Anat. 2011;193:267–275. doi: 10.1016/j.aanat.2011.02.011. [DOI] [PubMed] [Google Scholar]
- Einheber S, Milner TA, Giancotti F, Salzer JL. Axonal regulation of Schwann cell integrin expression suggests a role for alpha 6 beta 4 in myelination. J Cell Biol. 1993;123:1223–1236. doi: 10.1083/jcb.123.5.1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fancy SP, Chan JR, Baranzini SE, Franklin RJ, Rowitch DH. Myelin regeneration: a recapitulation of development? Annu Rev Neurosci. 2011;34:21–43. doi: 10.1146/annurev-neuro-061010-113629. [DOI] [PubMed] [Google Scholar]
- Fricker FR, Lago N, Balarajah S, Tsantoulas C, Tanna S, Zhu N, Fageiry SK, Jenkins M, Garratt AN, Birchmeier C, Bennett DL. Axonally derived neuregulin-1 is required for remyelination and regeneration after nerve injury in adulthood. J Neurosci. 2011;31:3225–3233. doi: 10.1523/JNEUROSCI.2568-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu SY, Gordon T. The cellular and molecular basis of peripheral nerve regeneration. Mol Neurobiol. 1997;14:67–116. doi: 10.1007/BF02740621. [DOI] [PubMed] [Google Scholar]
- Grinspan JB, Marchionni MA, Reeves M, Coulaloglou M, Scherer SS. Axonal interactions regulate Schwann cell apoptosis in developing peripheral nerve: neuregulin receptors and the role of neuregulins. J Neurosci. 1996;16:6107–6118. doi: 10.1523/JNEUROSCI.16-19-06107.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haastert K, Lipokatic E, Fischer M, Timmer M, Grothe C. Differentially promoted peripheral nerve regeneration by grafted Schwann cells over-expressing different FGF-2 isoforms. Neurobiol Dis. 2006;21:138–153. doi: 10.1016/j.nbd.2005.06.020. [DOI] [PubMed] [Google Scholar]
- Hammarberg H, Piehl F, Cullheim S, Fjell J, Hökfelt T, Fried K. GDNF mRNA in Schwann cells and DRG satellite cells after chronic sciatic nerve injury. Neuroreport. 1996;7:857–860. doi: 10.1097/00001756-199603220-00004. [DOI] [PubMed] [Google Scholar]
- Haney CA, Sahenk Z, Li C, Lemmon VP, Roder J, Trapp BD. Heterophilic binding of L1 on unmyelinated sensory axons mediates Schwann cell adhesion and is required for axonal survival. J Cell Biol. 1999;146:1173–1184. doi: 10.1083/jcb.146.5.1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayworth CR, Moody SE, Chodosh LA, Krieg P, Rimer M, Thompson WJ. Induction of neuregulin signaling in mouse Schwann cells in vivo mimics responses to denervation. J Neurosci. 2006;26:6873–6884. doi: 10.1523/JNEUROSCI.1086-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang YZ, Wang Q, Xiong WC, Mei L. Erbin is a protein concentrated at postsynaptic membranes that interacts with PSD-95. J Biol Chem. 2001;276:19318–19326. doi: 10.1074/jbc.M100494200. [DOI] [PubMed] [Google Scholar]
- Hu X, Hicks CW, He W, Wong P, Macklin WB, Trapp BD, Yan R. Bace1 modulates myelination in the central and peripheral nervous system. Nat Neurosci. 2006;9:1520–1525. doi: 10.1038/nn1797. [DOI] [PubMed] [Google Scholar]
- Hu X, He W, Diaconu C, Tang X, Kidd GJ, Macklin WB, Trapp BD, Yan R. Genetic deletion of BACE1 in mice affects remyelination of sciatic nerves. FASEB J. 2008;22:2970–2980. doi: 10.1096/fj.08-106666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jessen KR, Mirsky R. The origin and development of glial cells in peripheral nerves. Nat Rev Neurosci. 2005;6:671–682. doi: 10.1038/nrn1746. [DOI] [PubMed] [Google Scholar]
- Joung I, Yoo M, Woo JH, Chang CY, Heo H, Kwon YK. Secretion of EGF-like domain of heregulin beta promotes axonal growth and functional recovery of injured sciatic nerve. Mol Cells. 2010;30:477–484. doi: 10.1007/s10059-010-0137-5. [DOI] [PubMed] [Google Scholar]
- Klapdor K, Dulfer BG, Hammann A, Van der Staay FJ. A low-cost method to analyze footprint patterns. J Neurosci Methods. 1997;75:49–54. doi: 10.1016/s0165-0270(97)00042-3. [DOI] [PubMed] [Google Scholar]
- Kurek JB, Austin L, Cheema SS, Bartlett PF, Murphy M. Up-regulation of leukaemia inhibitory factor and interleukin-6 in transected sciatic nerve and muscle following denervation. Neuromuscul Disord. 1996;6:105–114. doi: 10.1016/0960-8966(95)00029-1. [DOI] [PubMed] [Google Scholar]
- Kwon YK, Bhattacharyya A, Alberta JA, Giannobile WV, Cheon K, Stiles CD, Pomeroy SL. Activation of ErbB2 during wallerian degeneration of sciatic nerve. J Neurosci. 1997;17:8293–8299. doi: 10.1523/JNEUROSCI.17-21-08293.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemke G. Neuregulin-1 and myelination. Sci STKE. 2006;2006:pe11. doi: 10.1126/stke.3252006pe11. [DOI] [PubMed] [Google Scholar]
- Liu X, Bates R, Yin DM, Shen C, Wang F, Su N, Kirov SA, Luo Y, Wang JZ, Xiong WC, Mei L. Specific regulation of NRG1 isoform expression by neuronal activity. J Neurosci. 2011;31:8491–8501. doi: 10.1523/JNEUROSCI.5317-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Tao YM, Woo RS, Xiong WC, Mei L. Stimulated ErbB4 internalization is necessary for neuregulin signaling in neurons. Biochem Biophys Res Commun. 2007;354:505–510. doi: 10.1016/j.bbrc.2007.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahanthappa NK, Anton ES, Matthew WD. Glial growth factor 2, a soluble neuregulin, directly increases Schwann cell motility and indirectly promotes neurite outgrowth. J Neurosci. 1996;16:4673–4683. doi: 10.1523/JNEUROSCI.16-15-04673.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchionni MA, Goodearl AD, Chen MS, Bermingham-McDonogh O, Kirk C, Hendricks M, Danehy F, Misumi D, Sudhalter J, Kobayashi K. Glial growth factors are alternatively spliced erbB2 ligands expressed in the nervous system. Nature. 1993;362:312–318. doi: 10.1038/362312a0. [DOI] [PubMed] [Google Scholar]
- Mei L, Xiong WC. Neuregulin 1 in neural development, synaptic plasticity and schizophrenia. Nat Rev Neurosci. 2008;9:437–452. doi: 10.1038/nrn2392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michailov GV, Sereda MW, Brinkmann BG, Fischer TM, Haug B, Birchmeier C, Role L, Lai C, Schwab MH, Nave KA. Axonal neuregulin-1 regulates myelin sheath thickness. Science. 2004;304:700–703. doi: 10.1126/science.1095862. [DOI] [PubMed] [Google Scholar]
- Nave KA, Salzer JL. Axonal regulation of myelination by neuregulin 1. Curr Opin Neurobiol. 2006;16:492–500. doi: 10.1016/j.conb.2006.08.008. [DOI] [PubMed] [Google Scholar]
- Nave KA, Trapp BD. Axon-glial signaling and the glial support of axon function. Annu Rev Neurosci. 2008;31:535–561. doi: 10.1146/annurev.neuro.30.051606.094309. [DOI] [PubMed] [Google Scholar]
- Nave KA, Sereda MW, Ehrenreich H. Mechanisms of disease: inherited demyelinating neuropathies–from basic to clinical research. Nat Clin Pract Neurol. 2007;3:453–464. doi: 10.1038/ncpneuro0583. [DOI] [PubMed] [Google Scholar]
- Ogata T, Iijima S, Hoshikawa S, Miura T, Yamamoto S, Oda H, Nakamura K, Tanaka S. Opposing extracellular signal-regulated kinase and Akt pathways control Schwann cell myelination. J Neurosci. 2004;24:6724–6732. doi: 10.1523/JNEUROSCI.5520-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salzer JL, Brophy PJ, Peles E. Molecular domains of myelinated axons in the peripheral nervous system. Glia. 2008;56:1532–1540. doi: 10.1002/glia.20750. [DOI] [PubMed] [Google Scholar]
- Shah NM, Marchionni MA, Isaacs I, Stroobant P, Anderson DJ. Glial growth factor restricts mammalian neural crest stem cells to a glial fate. Cell. 1994;77:349–360. doi: 10.1016/0092-8674(94)90150-3. [DOI] [PubMed] [Google Scholar]
- Steinthorsdottir V, Stefansson H, Ghosh S, Birgisdottir B, Bjornsdottir S, Fasquel AC, Olafsson O, Stefansson K, Gulcher JR. Multiple novel transcription initiation sites for NRG1. Gene. 2004;342:97–105. doi: 10.1016/j.gene.2004.07.029. [DOI] [PubMed] [Google Scholar]
- Suter U, Scherer SS. Disease mechanisms in inherited neuropathies. Nat Rev Neurosci. 2003;4:714–726. doi: 10.1038/nrn1196. [DOI] [PubMed] [Google Scholar]
- Tao Y, Dai P, Liu Y, Marchetto S, Xiong WC, Borg JP, Mei L. Erbin regulates NRG1 signaling and myelination. Proc Natl Acad Sci U S A. 2009;106:9477–9482. doi: 10.1073/pnas.0901844106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taveggia C, Zanazzi G, Petrylak A, Yano H, Rosenbluth J, Einheber S, Xu X, Esper RM, Loeb JA, Shrager P, Chao MV, Falls DL, Role L, Salzer JL. Neuregulin-1 type III determines the ensheathment fate of axons. Neuron. 2005;47:681–694. doi: 10.1016/j.neuron.2005.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taveggia C, Feltri ML, Wrabetz L. Signals to promote myelin formation and repair. Nat Rev Neurol. 2010;6:276–287. doi: 10.1038/nrneurol.2010.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trachtenberg JT, Thompson WJ. Schwann cell apoptosis at developing neuromuscular junctions is regulated by glial growth factor. Nature. 1996;379:174–177. doi: 10.1038/379174a0. [DOI] [PubMed] [Google Scholar]
- Wakatsuki S, Yumoto N, Komatsu K, Araki T, Sehara-Fujisawa A. Roles of meltrin-beta/ADAM19 in progression of Schwann cell differentiation and myelination during sciatic nerve regeneration. J Biol Chem. 2009;284:2957–2966. doi: 10.1074/jbc.M803191200. [DOI] [PubMed] [Google Scholar]
- Waterman H, Sabanai I, Geiger B, Yarden Y. Alternative intracellular routing of ErbB receptors may determine signaling potency. J Biol Chem. 1998;273:13819–13827. doi: 10.1074/jbc.273.22.13819. [DOI] [PubMed] [Google Scholar]
- Willem M, Garratt AN, Novak B, Citron M, Kaufmann S, Rittger A, DeStrooper B, Saftig P, Birchmeier C, Haass C. Control of peripheral nerve myelination by the beta-secretase BACE1. Science. 2006;314:664–666. doi: 10.1126/science.1132341. [DOI] [PubMed] [Google Scholar]
- Winseck AK, Caldero J, Ciutat D, Prevette D, Scott SA, Wang G, Esquerda JE, Oppenheim RW. In vivo analysis of Schwann cell programmed cell death in the embryonic chick: regulation by axons and glial growth factor. J Neurosci. 2002;22:4509–4521. doi: 10.1523/JNEUROSCI.22-11-04509.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolpowitz D, Mason TB, Dietrich P, Mendelsohn M, Talmage DA, Role LW. Cysteine-rich domain isoforms of the neuregulin-1 gene are required for maintenance of peripheral synapses. Neuron. 2000;25:79–91. doi: 10.1016/s0896-6273(00)80873-9. [DOI] [PubMed] [Google Scholar]
- Xu M, Bruchas MR, Ippolito DL, Gendron L, Chavkin C. Sciatic nerve ligation-induced proliferation of spinal cord astrocytes is mediated by kappa opioid activation of p38 mitogen-activated protein kinase. J Neurosci. 2007;27:2570–2581. doi: 10.1523/JNEUROSCI.3728-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang XL, Huang YZ, Xiong WC, Mei L. Neuregulin-induced expression of the acetylcholine receptor requires endocytosis of ErbB receptors. Mol Cell Neurosci. 2005;28:335–346. doi: 10.1016/j.mcn.2004.10.001. [DOI] [PubMed] [Google Scholar]