

Abstract
Deubiquitination has emerged as an essential regulatory mechanism of a number of cellular processes. An in-depth understanding of deubiquitinating enzyme (DUB) catalysis, particularly the mode of ubiquitin binding and the individual steps in the DUB catalytic turnover, is imperative for exploiting DUBs for therapeutic intervention. In this work we present a transient kinetic study of USP2 in hydrolyzing a model substrate Ub-AMC and a physiological substrate K48-linked diubiquitin. We carried out stopped-flow fluorescence analyses of the binding of mono- and diubiquitin to an inactive USP2 mutant and unveiled interesting differences in the binding kinetics between the two substrates. While a simple one-step binding of monoubiquitin to USP2 was observed, a biphasic binding was evident for diubiquitin. We further followed the deubiquitination reaction of Ub-AMC and K48-linked IQF-diubiquitin by USP2 using stopped-flow florescence under a single turnover condition. Global fitting of the reaction traces revealed differences in the microscopic rate constants between Ub-AMC and the physiological diubiquitin substrate. Our binding and single-turnover data support a conformational rearrangement of the diubiquitin substrate in USP2-catalyzed deubiquitination. This finding is of significance given the recent finding that the K48-linked diubiquitin is dynamic in its conformation. Our results provide useful insights into the mechanism of how USP recognizes ubiquitin moieties in a chain structure, which bears importance for understanding the USP catalysis and developing inhibitors against USPs.
INTRODUCTION
Ubiquitination has now been established as an essential posttranslational modification required for numerous cellular processes, including DNA repair, cell cycle regulation, transcription, and proteasome-mediated protein degradation (1, 2). While ubiquitination has been extensively characterized, the opposing process of deubiquitination remains less well understood. There are approximately 100 deubiquitinating enzymes (DUBs) encoded by the human genome (3, 4). DUBs can be grouped into at least five families, including ubiquitin-specific protease (USP), ubiquitin C-terminal hydrolase (UCH), the ovarian tumor (OTU) domain DUB, the Machado-Joseph (MJD) domain DUB, and the Jab1/MPN metalloenzyme (JAMM) domain DUB (5, 6). The ubiquitin-specific proteases represent the largest of the five DUB families.
The human ubiquitin-specific protease 2 (USP2) has recently gained much attention due to its association with numerous protein factors implicated in cancer (7). The rat homologue of USP2 was originally identified to be upregulated when prostate cancer cell lines were exposed to dihydrotestosterone (8). In humans USP2 was found to be overexpressed in 44% of all prostate tumors (8). Moreover, USP2 itself was determined to be a bona fide oncogene because of its ability to transform NIH-3T3 cells (8). Since its discovery USP2 has been found to stabilize a number of proteins, including fatty acid synthase (FAS), MDM2, MDMX, Aurora-A, epithelial Na+ channel (ENaC), and KCNQ1, by processing the K48-linked polyubiquitin chain (8-14). USP2 is known to contain an N-terminal extension that was suggested to determine the target specificity (9). To date two different USP2 isoforms, Usp2-45 (Usp2b) and Usp2-69 (Usp2a), were identified and have been found in different tissues (15). USP2’s function has also been associated with different target proteins (8-14). The variable N-terminal domains of the USP2 isoforms likely affect cellular localization and target specificity of USP2.
Many different forms of ubiquitination are observed in cells. Proteins can be monoubiquitinated where a single ubiquitin moiety is conjugated to the target protein through an isopeptide bond formed between the C-terminal carboxylate of ubiquitin and the side-chain amino group of a lysine residue on the target proteins. Another common ubiquitin modification is polyubiquitination where substrates are tagged with a ubiquitin chain linked through an isopeptide bond between the C-terminal carboxylate in one ubiquitin and a lysine residue in a second ubiquitin. Notably all seven lysine residues in ubiquitin are found to form ubiquitin chains. Available evidences suggest that the structure and dynamics of ubiquitin chains of different linkage vary largely. K48-linked ubiquitin chain, which typically triggers proteasome-mediated protein degradation, has been extensively characterized structurally and dynamically amongst the different ubiquitin chain structures. Initial X-ray crystallography revealed that K48-linked diubiquitin exists in a highly compact, closed structure where the major site of the interaction exists between the Ile44 residues of the distal and proximal ubiquitin moieties (16). Interestingly, this conformation is sterically unfavorable for ubiquitin’s interaction with USPs, which raises the important question of how USPs bind diubiquitin. More recent NMR analysis revealed that a dynamic conformational equilibrium of K48-linked diubiquitin exists in solution. The K48-linked diubiquitin exists in either a closed conformation or an open conformation (17-19). This open conformation of diubiquitin is sterically favorable for its interaction with USPs. Thus a conformational isomerization step is likely critical for the binding and recognition of K48-linked diubiquitin by USPs. To date details on the ubiquitin binding and conformational rearrangements that occur during USP catalysis remain scarce.
Most USPs contain a cysteine-catalytic triad. During catalysis the active site cysteine residue nucleophilically attacks the isopeptide bond that connects ubiquitin to a target protein, resulting in a thioester intermediate that engages the C-terminal carboxylate of ubiquitin. This thioester intermediate then undergoes hydrolysis to liberate the ubiquitin moiety. Although the general mechanism of the ubiquitin-specific proteases (USPs) is predicted to follow the above-described steps, kinetic details of the catalytic cycle remain undefined. Here we report pre-steady-state kinetic analyses of the USP2-catalyzed hydrolysis of a model DUB substrate, Ub-AMC and a physiological substrate, K48-linked diubiquitin. Using a kinetic approach we were able to elucidate the catalytic mechanism of USP2 and obtained the individual microscopic rate constants in a full catalytic cycle. Our results support a mechanism of USP2-catalyzed hydrolysis of the K48-linked diubiquitin in which diubiquitin undergoes a conformational rearrangement prior to catalysis.
EXPERIMENTAL PROCEDURES
Plasmid construction and gene cloning
The human USP2 catalytic core (amino acid 259 to 605) was amplified by PCR using an Addgene plasmid (#22577, Flag-HA-USP2) (20) as a template. The amplified gene product was cloned into the E. coli expression vector pET-28a using the restriction sites SalI and XhoI for expression of USP2 with an N-terminal 6xHis tag. The C276A mutation was introduced to USP2 gene by QuikChange mutagenesis.
Protein expression and purification
Wild-type USP2 catalytic core and the active site mutant C276A USP2 catalytic core were expressed in Rosetta(DE3) cells (Novagen). Cells were cultured at 37 °C until the OD600 reached 0.8. The cell culture was induced with 0.4 mM IPTG and was cultured for an additional 15 hours at 17 °C following the induction. Cells were then harvested and sonicated in lysis buffer [50 mM NaH2PO4 (pH 8.0), 500 mM NaCl, 5% glycerol, 1 mM β-mercaptoethanol, and 10 mM imidazole]. Cell free extract was bound to Ni-NTA resin (Invitrogen) and washed extensively with lysis buffer. USP2 was eluted with lysis buffer containing 80 mM imidazole. The eluted USP2 was dialyzed against a buffer containing 50 mM Tris (pH 8.0), 100 mM NaCl, 5% glycerol, and 1 mM DTT. The purity of USP2 catalytic core was analyzed using SDS-PAGE and Coomassie Blue staining. We also generated an inactive USP2 mutant in which the catalytic residue Cys276 was replaced by an alanine. This mutant has been previously shown to be void of deubiquitinating activity (21). The mutant C276A USP2 catalytic core was purified following the same protocol as described for the wild-type USP2.
In order to generate Ub-AMC (ubiquitin-7-amino-4-methylcoumarin), human ubiquitin (amino acids 1-75) was expressed in BL21(DE3) cells using the pTYB1 expression plasmid following a reported protocol (22). Specifically cells were cultured at 37 °C until OD600 reached 0.8. The cell culture was induced with 0.25 mM IPTG and was cultured for an additional 15 hours at 17 °C following the induction. Cells were then harvested and sonicated in lysis buffer containing 20 mM Tris (pH 7.5), 100 mM NaCl, 2 mM EDTA, 5% glycerol, 1 mM PMSF. Ubiquitin1-75 - intein fusion protein was bound to chitin affinity beads for 3 hours at 4 °C and washed with 300 ml high salt buffer [20 mM Tris (pH 7.5), 500 mM NaCl, 2 mM EDTA, 5% glycerol]. The bound fusion protein was then washed with 80 ml low salt buffer [20 mM HEPES (pH 6.5), 100 mM NaCl]. Ubiquitin1-75 was cleaved from intein using the low salt buffer containing 75 mM sodium 2-mercaptoethane-sulfonate (MESNA). Cleavage was allowed to proceed overnight at room temperature. The eluted Ub1-75-MESNA (4 mg/ml) was then incubated with glycine-AMC (10 mg/ml) in low salt buffer containing 20% DMSO in the presence of N-hydroxysuccinimide (5 mg/ml) and collidine (20 mg/ml). The reaction was allowed to proceed for 30 hours at room temperature. Ub-AMC was purified by HPLC using a Phenomenex Jupiter C18 column (300 Å, 10 μm, 250 × 10 mm) with an acetonitrile gradient. Ub-AMC eluted at approximately 35% acetonitrile. HPLC fractions containing pure Ub-AMC were lyophilized, resuspended in DMSO, and stored at −80 °C. The native K48-linked diubiquitin was either purchased from Enzo Life Sciences or prepared as previously described (23).
Steady-state kinetic analysis of USP2-catalyzed deubiquitination
The steady-state kinetics of USP2-catalyzed deubiquitination was assayed using the fluorogenic substrate Ub-AMC as previously described (24).
We also determined the steady-state kinetics of USP2-catalyzed hydrolysis of K48-linked IQF diubiquitin substrate DiUb48-6 (LifeSensors). The proximal and distal ubiquitin contain an internally quenched fluorescence (IQF) pair. DUB hydrolysis results in separation of the quencher from the fluorophore TAMRA, resulting in an increase in fluorescence. The K48-linked IQF diubiquitin substrate was incubated in a reaction buffer [50 mM HEPES (pH 7.8), 1 mM DTT, 0.1 mg/ml BSA] at 25 °C. Following addition of enzyme, fluorescence emission at 580 nm was recorded with an excitation at 540 nm using a Fluoromax-4 fluorescence spectrophotometer (Horiba, CA). The initial rate in nM/s was calculated by creating a standard calibration curve of the fluorescence signal versus the fully turned over K48-linked IQF diubiquitin substrate concentration. The initial rate was then plotted against substrate concentrations ranging from 0.25-7.5 μM. Steady-state rate constants were determined by fitting the initial rates to the Michaelis-Menten equation V = (Vmax [S]) / ([S] + Km) using GraphPad Prism (GraphPad Software, CA). kcat is equal to Vmax/[E].
Steady-state kinetic measurement of USP2-catalyzed hydrolysis of a native K48-linked diubiquitin (Enzo Life Sciences) was carried out in a reaction buffer containing 50 mM HEPES (pH 7.8), 1 mM DTT, 0.1 mg/ml BSA at 37° C for up to 20 minutes. The concentration of K48-linked diubiquitin was varied from 0.5-20 μM while USP2 concentration was held constant at 20 nM. The reaction was quenched by the addition of SDS loading solution. The reaction products were separated on a 20% SDS-PAGE gel and visualized by Coomassie Blue staining. The intensity of the individual monoubiquitin and diubiquitin gel bands were quantified using Quantity One software (Bio-Rad). The initial rate was then plotted against substrate concentration and the steady-state rate constants were determined by fitting the initial rates to the Michaelis-Menten equation V = (Vmax [S]) / ([S] + Km) using GraphPad Prism (GraphPad Software, CA). kcat is equal toVmax /[E].
Apparent dissociation constant of Ub-AMC binding to USP2
In order to determine the apparent dissociation constant of Ub-AMC binding to USP2, increasing concentrations of C276A USP2 (0 - 25 μM) were incubated with Ub-AMC (0.5 μM) in a buffer containing 50 mM HEPES (pH 7.8), 1 mM DTT, and 0.1 mg/ml BSA at 25 °C. Binding was monitored by following the decrease of Ub-AMC fluorescence using a Fluoromax-4 fluorescence spectrophotometer (Horiba, CA). The excitation wavelength was set to 350 nm while emission was monitored at 395 nm. Using GraphPad Prism the dissociation constant was determined through nonlinear curve fitting of the plot of the change in fluorescence (Y) against the concentration of USP2 (X) using equation 1.
| (eq.1) |
Y designates the fluorescence change and is equal to (Yinitial – Yobs)/Yinitial, X is the concentration of C276A USP2, B is equal to (Yinitial – Ymin) / Yinitial, [L] stands for the concentration of Ub-AMC, and Kd stands for the apparent dissociation constant (25, 26).
Apparent dissociation constant of K48-linked IQF diubiquitin binding to USP2
In order to determine the apparent dissociation constant of K48-linked IQF diubiquitin binding to USP2, increasing concentrations of C276A USP2 (0-8 μM) were incubated with K48-linked IQF diubiquitin (25 nM) in a buffer containing 50 mM HEPES (pH 7.8), 1 mM DTT, and 0.1 mg/ml BSA at 25 °C. Binding was monitored by following the increase in TAMRA fluorescence in the IQF diubiquitin substrate using a Fluoromax-4 fluorescence spectrophotometer (Horiba, CA). The excitation wavelength was set to 550 nm while emission was monitored at 575 nm. Using GraphPad Prism the dissociation constant was determined through nonlinear curve fitting of the plot of the fluorescence change (Y) against the concentration of USP2 (X) using equation 1, where Y is equal to (Yobs – Yinitial) / Yinitial, X is the concentration of C276A USP2, B is equal to (Ymax – Yinitial) / Yinitial, [L] stands for the concentration of K48-linked IQF diubiquitin, and Kd stands for the apparent dissociation constant (25, 26).
Transient kinetics of the binding of monoubiquitin to USP2
An Applied Photophysics SX.18MV-R stopped-flow spectrophotometer was used to monitor the binding of native ubiquitin to the USP2 catalytic core. Reactions were carried out in a buffer containing 50 mM HEPES (pH 7.8), 0.1 mg/ml BSA, and 1 mM DTT at 25 °C. Ubiquitin binding to USP2 was monitored through the quenching of the native tryptophan fluorescence of the wild-type USP2 catalytic core and an inactive C276A USP2 mutant. Fluorescence emission was recorded using a 320 nm cutoff filter with a 280 nm excitation. Ubiquitin concentration was varied from 4 to 70 μM while USP2 concentration was held constant at 1 μM. At least 3 stopped-flow traces were averaged for each data set. Averaged data sets were fit to a single exponential equation. The observed rates (Y) were plotted against the concentration of ubiquitin (X). Binding constants were determined using the equation Y = kon • X + koff and Kd = koff / kon.
Ub-AMC binding to USP2 was monitored by following the change of AMC fluorescence. Fluorescence emission was recorded using a 420 nm cutoff filter with a 350 nm excitation. An inactive C276A USP2 mutant was used for the Ub-AMC binding assay. The C276A USP2 concentration was varied from 3 to 12 μM while Ub-AMC concentration was held constant at 0.8 μM. At least 3 stopped-flow traces were averaged for each data set. Averaged data sets were fit to a single exponential equation. The observed rates (Y) were plotted against the concentration of C276A USP2 (X). Binding constants were determined using the equation Y = kon • X + koff and Kd = koff / kon.
Transient kinetics of the binding of diubiquitin to USP2
Binding of K48-linked IQF diubiquitin to C276A USP2 was monitored by following TAMRA fluorescence change. Fluorescence emission was recorded using a 570 nm cutoff filter with a 540 nm excitation. C276A USP2 concentration was varied from 1-9 μM while K48-linked IQF diubiquitin concentration was held constant at 0.4 μM. At least 3 stopped-flow traces were averaged for each data set. Averaged data sets were fit to a double exponential equation. The observed rates were plotted against C276A USP2 concentration and analyzed using GraphPad Prism. Individual microscopic rate constants were determined using equations 2-7 as detailed in Results. These equations are approximations of the full solutions which were previously published (27).
| (eq.2) |
| (eq.3) |
| (eq.4) |
| (eq.5) |
| (eq.6) |
| (eq.7) |
where kobs,1 and kobs,2 are the observed rate constants of the fast and slow phases of K48-linked IQF diubiquitin binding to USP2 respectively. k1, k−1, k2, and k−2 are the individual forward and reverse rate constants of the fast and slow phases respectively. is the maximal rate of kobs,2. K1 and K2 are the association constants and Kd,app is the apparent dissociation constant.
Binding of native K48-linked diubiquitin to C276A USP2 was monitored through the quenching of the USP2 tryptophan fluorescence. Fluorescence emission was recorded using a 320 nm cutoff filter with a 280 nm excitation. Diubiquitin concentration was varied from 2-32 μM while USP2 concentration was held constant at 1 μM. At least 3 stopped-flow traces were averaged for each data set. Averaged data sets were fit to a double exponential equation. The observed rates were plotted against diubiquitin concentration and analyzed using GraphPad Prism. Individual microscopic rate constants were determined in a manner similar to IQF K48-linked diubiquitin.
Pre-steady-state kinetic analysis of USP2-catalyzed Ub-AMC hydrolysis
The USP2-catalyzed hydrolysis of Ub-AMC was followed under a single turnover condition. An Applied Photophysics SX.18MV-R stopped-flow spectrophotometer was used to monitor changes in fluorescence. All reactions were carried out in a buffer containing 50 mM HEPES (pH 7.8), 0.1 mg/ml BSA, and 1 mM DTT at 25 °C. The USP2 concentration was varied from 1.25-5 μM while Ub-AMC concentration was held constant at 0.2 μM. Fluorescence emission was recorded using a 420 nm cutoff filter with excitation at 350 nm. At least 3 time traces were averaged for each data set. Data sets were globally fit using the program KinTek Explorer (28, 29). The burst kinetics of USP2-catalyzed hydrolysis of Ub-AMC was followed under multiple turnover conditions. All reactions were carried out in a buffer containing 50 mM HEPES (pH 7.8), 0.1 mg/ml BSA, and 1 mM DTT at 25 °C. The USP2 concentration was varied from 0.4 to 2 μM while Ub-AMC concentration was held constant at 12 μM. Fluorescence emission was recorded using a 420 nm cutoff filter with excitation at 350 nm. At least 3 time traces were averaged for each data set. The data sets were nonlinearly fit to equation 8 using GraphPad Prism,
| (eq.8) |
where Y is fluorescence, t is time, A stands for amplitude, λ defines the observed rate constant of the exponential phase, kss is the linear steady-state rate.
Pre-steady-state kinetic analysis of USP2-catalyzed K48-linked IQF diubiquitin hydrolysis
Single-turnover experiments were performed for USP2-catalyzed hydrolysis of K48-linked IQF diubiquitin. An Applied Photophysics SX.18MV-R stopped-flow spectrophotometer was used to monitor changes in fluorescence during each time trace. All reactions were carried out in a buffer containing 50 mM HEPES (pH 7.8), 0.1 mg/ml BSA, and 1 mM DTT at 25 °C. The USP2 concentration was varied from 1.25 to 4 μM while diubiquitin concentration was held constant at 0.2 μM. Fluorescence emission was recorded using a 570 nm cutoff filter with an excitation at 540 nm. At least 3 time traces were averaged for each data set. Data sets were globally fit using the program KinTek Explorer (28, 29).
RESULTS
Steady-state kinetic analysis of USP2 hydrolyzing Ub-AMC
USP2 hydrolyzing Ub-AMC under the steady-state condition was monitored by fluorescence emission associated with substrate hydrolysis. The initial velocity was plotted against substrate concentration (Figure 1A), and the steady-state rate constants were determined using nonlinear regression curve fitting to the Michaelis-Menten equation (Table 1). USP2 was efficient in hydrolyzing Ub-AMC with a kcat of 0.35 ± 0.03 s−1, a Km of 2.4 ± 0.2 μM, and a kcat/Km of 1.5 × 105 M−1 s−1. These values are consistent with two previous studies, where a kcat/Km of 2.5 × 105 M−1 s−1 was reported (21, 30).
Figure 1.

Steady-state kinetic analysis of USP2-catalyzed hydrolysis of Ub-AMC (A), K48-linked IQF diubiquitin (B), and K48-linked native diubiquitin (C). The initial rates are plotted against the substrate concentration and fit to the Michaelis-Menten equation.
Table 1.
Kinetic rate constants for USP2
| Substrate | kcat (s−1) | Km (μM) | kcat/KM (M−1s−1) | k1 (μM−1s−1) | k−1 (s−1) | k2 (s−1) | k−2 (s−1) | Kd,app (μM) |
|---|---|---|---|---|---|---|---|---|
| Ubiquitin | N/A | N/A | N/A | 0.40 ± 0.01 | 2.64 ± 0.56 | N/A | N/A | ND |
| Ub-AMC | 0.35 ± 0.03 | 2.4 ± 0.2 | 1.5 × 105 | 0.33 ± 0.02* | 1.82 ± 0.17* | N/A | N/A | 2.9 ± 0.6 * |
| IQF-diubiquitin (K48) | 0.10 ± 0.01 | 1.2 ± 0.1 | 8.3 × 104 | 0.14 ± 0.01* | 0.06 * | 0.21 * | 0.36 * | 0.27 ± 0.08 * |
| Native diubiquitin (K48) | 0.19 ± 0.01 | 2.8 ± 0.3 | 6.8 × 104 | 0.23 ± 0.01 * | 0.06* | ND | ND | ND |
Indicates the constant was derived using the USP2 C276A mutant. ND stands for not determined and N/A stands for not applicable
Steady-state kinetic analysis of USP2 hydrolyzing K48-linked IQF diubiquitin
In order to facilitate USP2 steady-state kinetic analysis, a physiologically relevant diubiquitin substrate was used to characterize USP2 catalysis under the steady-state condition. The K48-linked internally quenched fluorescence (IQF) diubiquitin substrate was derivatized with a TAMRA fluorophore donor in the proximal ubiquitin and a quencher in the distal ubiquitin. The internally quenched fluorescence pair in diubiquitin is separated upon hydrolysis, leading to an increase in donor fluorescence (Figure 2). The initial velocity was plotted against substrate concentration (Figure 1B), and the steady-state rate constants were determined using nonlinear regression curve fitting to the Michaelis-Menten equation (Table 1). A kcat of 0.10 ± 0.01 s−1, a Km of 1.2 ± 0.1 μM, and a kcat/Km of 8.3 × 104 M−1 s−1 were determined using K48-linked IQF-diubiquitin as a substrate. The kcat and Km values are 3.5 and 2-fold lower than those of Ub-AMC as a substrate respectively. Overall a similar catalytic efficiency was observed for the K48-linked IQF diubiquitin and Ub-AMC in USP2-catalyzed hydrolysis.
Figure 2.

Diagram of the fluorescence quenching of the K48-linked IQF diubiquitin and the release of quenching upon hydrolysis. The fluorescence quencher (pink) is positioned on the distal ubiquitin (blue) and the TAMRA fluorophore (yellow) on the proximal ubiquitin (green). When the C-terminal carboxylate of the distal ubiquitin is covalently bonded to the side-chain amino group of the lysine 48 in the proximal ubiquitin, the TAMRA fluorescence is quenched. After USP2-catalyzed hydrolysis, the proximal ubiquitin is liberated from the distal ubiquitin and the quencher loses its ability to quench the TAMRA fluorescence (pink glow).
We also determined the kinetic constants of USP2-catalyzed hydrolysis of native K48-linked diubiquitin in order to corroborate the results obtained with the K48-linked IQF diubiquitin substrate under the steady-state condition. Native K48-linked diubiquitin hydrolysis was performed under the same reaction condition as the IQF K48-linked diubiquitin substrate. The reaction product was analyzed on a 20% denaturing SDS-PAGE gel. After Coomassie Blue staining, the mono- and diubiquitin bands were quantified to determine the rate of diubiquitin cleavage at each substrate concentration as described previously (31). The initial rate was plotted against diubiquitin concentration (Figure 1C) and the steady-state rate constants were determined through nonlinear curve fitting to the Michaelis-Menten equation (Table 1). A kcat of 0.19 ± 0.01 s−1, a Km of 2.8 ± 0.3 μM, and a kcat/Km of 6.8 × 104 M−1 s−1 were determined using native K48-linked diubiquitin as a substrate. The rate constants are close to those determined for USP2-catalyzed hydrolysis of K48-linked IQF diubiquitin. Thus the fluorophore-quencher pair on diubiquitin had no adverse effect on the interaction of the diubiquitin substrate with the USP2 catalytic core.
Kinetics of monoubiquitin binding to USP2
To date there has been no report of the transient kinetics of ubiquitin binding to USP. Here we developed two methods to follow the binding of monoubiquitin to USP2 catalytic core. First we exploited the fluorescence quenching of tryptophan residues in the USP2 catalytic core upon binding of ubiquitin (ubiquitin contains no tryptophan). Using stopped-flow fluorescence spectroscopy, binding of ubiquitin was monitored by following the decrease of tryptophan fluorescence intensity of the wild type USP2 (Figure 3A). The data can be fit to a single exponential function with satisfaction. An observed rate constant (kobs) was obtained for each ubiquitin concentration. Linear regression analysis of the plot of kobs versus ubiquitin concentration (Figure 3A inset) was used to determine the binding constants kon = 0.40 ± 0.01 μM−1 s−1 and koff = 2.64 ± 0.56 s−1 as described in the Experimental Procedures. An equilibrium binding constant (Kd) of 6.6 μM was calculated from the fitted kon (k1) and koff (k−1) (see Table 1) and is in accord with previously reported dissociation constant of ubiquitin for other USPs determined using ITC and SPR (32, 33). The koff of 2.64 s−1 is relatively slow, which can be understood in view of the numerous interactions that exist between ubiquitin and the USP2 catalytic core as shown in the cocrystal structure of USP2 and ubiquitin (30). The calculated Kd of 6.6 μM is approximately three times higher than the Km determined for USP2-catalyzed hydrolysis of Ub-AMC. Thus it is not reliable to approximate Km to Kd for USP2-catalyzed Ub-AMC hydrolysis.
Figure 3.
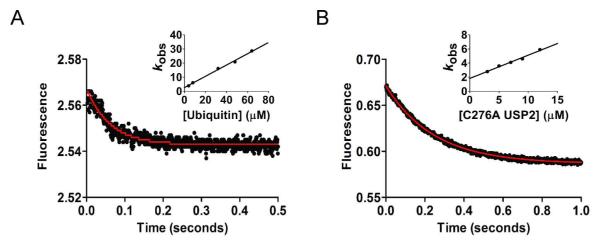
Stopped-flow fluorescence traces of ubiquitin and Ub-AMC binding to the USP2 catalytic core. (A) Stopped-flow fluorescence traces of ubiquitin binding to USP2 obtained by USP2 tryptophan fluorescence quenching. (B) Stopped-flow fluorescence traces of Ub-AMC binding to C276A USP2. The data were fit to a single exponential function (solid red line) using GraphPad Prism (GraphPad Software, CA). The fitted rates were plotted against the concentration of either ubiquitin (inset in panel A) or USP2 (inset in panel B). The binding and dissociation constants, kon and koff, were determined through linear regression (described in the Experimental Procedures section).
In addition to the native tryptophan fluorescence quenching method, we also followed Ub-AMC binding to an inactive USP2 by monitoring the fluorescence quenching of the fluorophore AMC that is conjugated to the C-terminus of ubiquitin (Figure 3B). In order to monitor USP2 binding of Ub-AMC without hydrolysis, we generated a catalytically inactive mutant, C276A USP2. This mutant has been previously shown to be completely void of deubiquitinating activity (21). We observed a strong quenching of AMC fluorescence upon its binding to the USP2 catalytic core (Figure 3B). The fluorescence data was fit to a single exponential binding equation with satisfaction. Linear regression analysis of the plot of kobs versus C276A USP2 concentration (Figure 3B inset) was used to determine the kinetic binding constants, kon = 0.33 ± 0.02 μM−1 s−1 and koff = 1.82 ± 0.17 s−1. A Kd of 5.5 μM was calculated based on the kon and koff (Table 1). The kinetic constants are similar to those determined for native monoubiquitin, suggesting that the conjugation of an AMC group to the C-terminus of ubiquitin does not affect the kinetic behavior of ubiquitin binding to USP2. Notably, both native ubiquitin and Ub-AMC bind to USP2 with monophasic kinetics. This suggests that ubiquitin binds to USP2 in a single step despite that USP2 catalytic core interacts with ubiquitin at multiple sites as revealed by the cocrystal structure of USP2-ubiquitin complex (30).
In order to probe the effect of C276A mutation on the interaction of USP2 with ubiquitin, we characterized the binding of C276A USP2 to ubiquitin as described for the wild-type USP2 (Figure S1). A kon of 0.48 ± 0.05 μM−1s−1 and a koff of 7.54 ± 2.60 s−1 were determined for ubiquitin binding to C276A USP2. These values are close to those determined for ubiquitin binding to the wild-type USP2 (kon = 0.40 ± 0.01 μM−1s−1 and koff = 2.64 ± 0.56 s−1). Therefore the C276A mutation has no adverse effect on the interaction between USP2 and ubiquitin.
Binding of Ub-AMC to USP2 under an equilibrium condition
We also determined the equilibrium binding constant of Ub-AMC binding to USP2 by measuring the decrease in Ub-AMC fluorescence in the presence of increasing concentrations of C276A USP2. The change in fluorescence was plotted against USP2 concentration (Figure 4A). Nonlinear curve fitting to equation 1 yielded an apparent dissociation constant Kd,app of 2.9 ± 0.6 μM. The determined apparent dissociation constant is close to the Kd calculated from the kon and koff determined by the stopped-flow binding experiments (Figure 3, Table 1).
Figure 4.
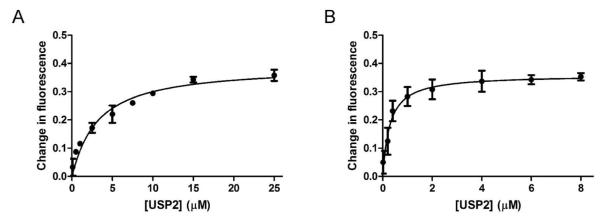
Steady-state fluorescence change of Ub-AMC and K48-linked IQF diubiquitin binding to C276A USP2. (A) The fluorescence change of 0.5 μM Ub-AMC was plotted against increasing concentrations of USP2 (0-25 μM). (B) The fluorescence change of 25 nM K48-linked IQF diubiquitin was recorded with increasing concentrations of USP2 (0-8 μM). The apparent dissociation constant was determined through nonlinear curve fitting to equation 1 using GraphPad Prism.
Binding of K48-linked IQF diubiquitin to USP2 under an equilibrium condition
In order to define the equilibrium binding constant (Kd,app) of diubiquitin binding to USP2, we measured the K48-linked IQF diubiquitin fluorescence in the presence of increasing concentrations of C276A USP2. Binding was monitored by following the enhancement of TAMRA fluorescence upon the binding of K48-linked IQF diubiquitin substrate (25 nM) to USP2 (0-8 μM). The increase in fluorescence was plotted against USP2 concentration (Figure 4B). Nonlinear curve fitting to equation 1 yielded an apparent dissociation constant (Kd,app) of 0.27 ± 0.08 μM. This apparent dissociation constant was later used in calculating the individual microscopic binding rate constants in a proposed two-step process of diubiquitin binding to USP2 (vide infra). Notably USP2 has a significantly higher affinity for K48-linked diubiquitin compared to monoubiquitin.
Transient kinetics of K48-linked diubiquitin binding to USP2
Stopped-flow fluorescence analysis was used to follow K48-linked IQF diubiquitin binding to USP2 under a pre-steady-state condition. Changes in fluorescence over time were monitored by following the increase of TAMRA fluorescence observed upon mixing K48-linked IQF diubiquitin and the inactive C276A USP2 mutant (see Figure 5A). The binding traces were fit to both single and double exponential functions for comparison. As shown in Figure 5, the binding of K48-linked IQF diubiquitin to USP2 core was clearly biphasic, with a fast increase in TAMRA fluorescence followed by a slower phase of fluorescence increase. The observed rates were plotted against USP2 concentration (Figure 6A and 6B). The observed rate of the first phase (kobs,1) was linearly dependent on USP2 concentration and can be fit by linear regression analysis (Figure 6A). The second phase (kobs,2) was nonlinear (Figure 6B) and was fit to a hyperbola as previously described (27). Based on the regression analysis of the plots of kobs,1 and kobs,2, using equations 2-7 we were able to calculate each microscopic binding rate constant for a two-step process describing the binding of diubiquitin to USP2. Specifically, from the linear regression analysis of kobs,1 against USP2 concentration (see Figure 6A), the slope (k1) was equal to 0.14 ± 0.01 μM−1 s−1. The rate of k−1 (0.06 s−1) was calculated by taking the difference between the y-intercept of the linear regression analysis of kobs,1 (0.63 ± 0.04 s−1) and the maximum of kobs,2 (0.57 ± 0.03 s−1 ) as defined by the hyperbola. The equilibrium constant of the second slower step (K2 = 0.59) was calculated using equation 5 from the determined Kd(app) and K1 (2.33 μM−1). The rate constants of the forward and reverse directions of the second binding step, k2 and k−2, were calculated to be 0.21 s−1and 0.36 s−1 respectively using equations 4 and 7. Together this analysis suggests a two-step binding mechanism with binding occurring in the first step and a possible conformational rearrangement occurring in the second step (34, 35). It should be noted that the increase in TAMRA fluorescence upon binding to USP2 is most likely caused by the separation of the two ubiquitin moieties that releases quenching of the donor TAMRA fluorescence. This suggests that the diubiquitin adopts a more open conformation in the binary complex upon binding to the USP2 catalytic core. Notably, if the dissociation constant was calculated from fitting the fluorescence data to a single exponential function, the value was significantly higher (Kd = 2.42 μM) compared to the experimentally determined value of 0.27 μM. This provides further support for a two-step binding mechanism for K48-linked IQF diubiquitin.
Figure 5.
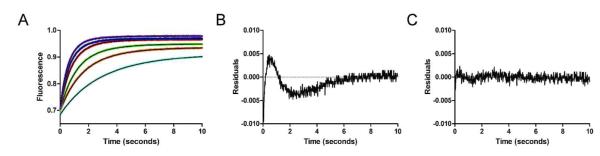
Stopped-flow fluorescence traces of K48-linked IQF diubiquitin binding to the USP2 catalytic core. (A) Fluorescence enhancement associated with the TAMRA fluorophore in the IQF diubiquitin substrate was monitored over 10 seconds to follow the binding of K48-linked IQF diubiquitin to USP2. USP2 concentration was varied (1, 2, 3, 5, 7, and 9 μM) while K48-linked IQF diubiquitin concentration was held constant at 0.4 μM. The data set was fit to a single or double exponential function for a comparison. The double exponential fitting results are shown as black lines. Residuals for the single (B) and double (C) exponential fitting to a representative binding trace for 9 μM USP2 and 0.4 μM K48-linked IQF diubiquitin are shown. Analyses of the residuals reveal that a double exponential function provides a significantly better fit to the data.
Figure 6.
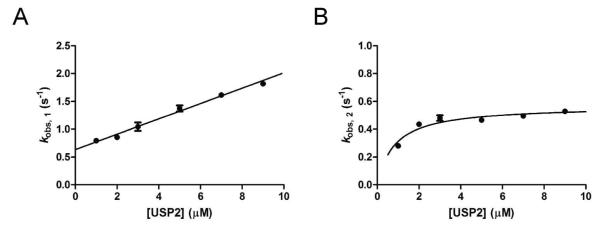
Concentration dependence of the observed rate constants of K48-linked IQF diubiquitin binding to USP2. In A and B the rate constants kobs, 1 and kobs, 2 determined by fitting the fluorescence data reporting the K48-linked IQF diubiquitin binding to USP2 (see figure 5) were plotted against the concentration of USP2. Individual microscopic binding constants were derived from linear regression analysis of the plot of kobs, 1 (A) and nonlinear curve fitting to a hyperbola of the plot of kobs, 2 (B).
The binding interaction between USP2 and native K48-linked diubiquitin was also characterized using stopped-flow fluorescence analysis under the pre-steady-state condition. Binding was monitored by following the C276A USP2 tryptophan fluorescence quenching. The binding of native K48-linked diubiquitin to USP2 was again clearly biphasic (Figure S2). The fitted rate constants were plotted against diubiquitin concentration (Figure S3). Similar to IQF, the observed rate constant for the first phase (kobs,1) was linearly dependent on diubiquitin concentration and was fit by linear regression analysis (Figure S3A), while the second phase (kobs,2) was nonlinear (Figure S3B) and was fit to a hyperbola as described above for K48-linked IQF diubiquitin. The binding constants of native K48-linked diubiquitin (k1 = 0.23 μM−1 s−1, k−1 = 0.06 s−1, and the maximum value of kobs,2 = 0.80 s−1 ) were determined to be very similar in value to the rate constants determined for K48-linked IQF diubiquitin (k1 = 0.14 μM−1 s−1, k−1 = 0.06 s−1, and the maximum value of kobs,2 = 0.57 s−1). Due to an extremely week fluorescence signal we were unable to determine the apparent dissociation constant of native K48-linked diubiquitin binding to C276A USP2 under an equilibrium condition, which prevented us from calculating the k2 and k−2 of native diubiquitin binding to USP2. Nonetheless, the good agreement in the measured k1, k−1 for the native K48-linked diubiquitin and the K48-linked IQF diubiquitin support that the fluorophores on K48-linked IQF diubiquitin do not adversely affect USP2’s interaction with diubiquitin.
Transient kinetic analysis of USP2-catalyzed hydrolysis of Ub-AMC
Stopped-flow fluorescence traces were recorded for USP2-catalyzed hydrolysis of Ub-AMC under a single turnover condition. Upon rapid mixing of Ub-AMC with the USP2 catalytic core, a rapid increase in fluorescence was observed. Fluorescence emission was recorded using a 420 nm cutoff filter with 350 nm excitation. The USP2 concentration was varied from 1.25 to 5 μM while the Ub-AMC concentration was held constant at 0.2 μM. The fluorescence changes were recorded over a period of 20 seconds (Figure 7A). The pre-steady-state data was fit to a kinetic model as illustrated in Scheme 1 using KinTek Explorer (28, 29) and plotted as red traces in Figure 7A.
Figure 7.
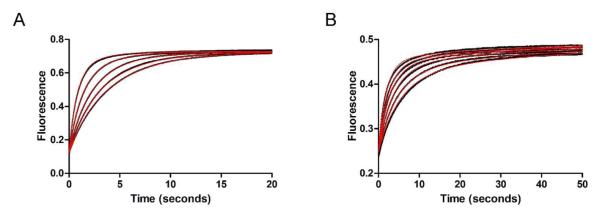
Stopped-flow fluorescence kinetic traces of USP2-catalyzed hydrolysis of Ub-AMC and K48-linked IQF diubiquitin under single-turnover conditions. (A) USP2-catalyzed hydrolysis of Ub-AMC. Fluorescence changes (black dots) were monitored over 20 seconds. USP2 concentration was varied (1.25, 1.5, 2.0, 3.0, and 5.0 μM) and shown in ascending order while Ub-AMC concentration was held constant at 0.2 μM. The data was then globally fit (red line) to reaction Scheme 1. (B) USP2-catalyzed hydrolysis of K48-linked IQF diubiquitin. Fluorescence changes (black dots) were monitored over 50 seconds. USP2 concentration was varied (1.25, 1.5, 2.0, 2.5, 3.0, and 4.0 μM) and shown in ascending order while K48-linked IQF-diubiquitin concentration was held constant at 0.2 μM. The data was then globally fit (red line) to reaction Scheme 2 which represents a two-step binding mechanism.
Scheme 1.

Kinetic scheme of USP2-catalyzed hydrolysis of Ub-AMC.
Shown in Scheme 1 is the proposed kinetic mechanism of USP2-catalyzed hydrolysis of Ub-AMC. USP2•Ub-AMC is the Michaelis complex of USP2 and Ub-AMC; USP2-Ub is an acyl-enzyme intermediate where ubiquitin is covalently bound to the USP2 active site cysteine through a thioester bond. USP2•Ub is USP2 bound with ubiquitin following the deacylation reaction. The k1, k−1, k4, and k−4 were determined from the binding studies of Ub-AMC and ubiquitin respectively (Table 1). These values were fixed during the global fitting. The fluorescence data in Figure 7A was then globally fit to the reaction mechanism shown in Scheme 1 and the obtained rate constants are summarized in Table 2. The fitting was satisfactory with a χ2/ndf value of 38. Also importantly the fitted values were well constrained (see Table 2). Our fitting results indicate that the acylation step (k2 = 4.40 ± 0.04 s−1) was significantly faster than the deacylation step (k3 =0.51 ± 0.01 s−1). The rate constant of the deacylation step is similar to the steady-state rate constant (kcat = 0.35 ± 0.03 s−1) determined for the USP2-catalyzed hydrolysis of Ub-AMC.
Table 2.
Pre-steady-state kinetic analysis for USP2-catalyzed hydrolysis of Ub-AMC
| rate | best fit | lower limit | upper limit |
|---|---|---|---|
| k 1 a | 0.33 μM−1s−1 | n/a | n/a |
| k −1 a | 1.82 s−1 | n/a | n/a |
| k 2 | 4.40 ± 0.04 s−1 | 4.02 s−1 | 5.14 s−1 |
| k −2 | 1.74 ± 0.08 s−1 | 0.89 s−1 | 6.65 s−1 |
| k 3 | 0.51 ± 0.01 s−1 | 0.33 s−1 | 1.24 s−1 |
| k 4 a | 2.64 s−1 | n/a | n/a |
| k −4 a | 0.40 μM−1s−1 | n/a | n/a |
| F(Ub-AMC) a | 0.61 | n/a | n/a |
| F(USP2•Ub-AMC) | 2.58 ± 0.03 | 2.32 | 2.71 |
| F(AMC) | 3.64 ± 0.01 | 3.63 | 3.65 |
| χ2/ndf | 38 | n/a | n/a |
Indicates the value was fixed during global fitting. The values for k1, k−1, k4, and k−4, were derived as previously explained for the USP2/Ub-AMC and the USP2/ubiquitin stopped-flow binding data (Figure 3 and Table 1). Fluorescence output expression factors (F) for Ub-AMC, USP2•Ub-AMC, and AMC are also listed within the table. The fluorescence factor for Ub-AMC was determined based on the observed fluorescence signal and the known concentration. The factor for Ub-AMC was fixed during the global fitting. The remaining rates and fluorescence factors were solved by fitting the fluorescence data in Figure 7A to reaction Scheme 1 using the KinTek Explorer software. The upper and lower limits were determined using the FitSpace Editor function of the KinTek Explorer program and the overall goodness of the fit was judged by Chi2/degrees of freedom (χ2/ndf).
Our global fitting of USP2-catalyzed hydrolysis of Ub-AMC utilized a minimal kinetic scheme that has been used in a previous study of the serine protease subtilisin (36). We also evaluated the fitting of the transient kinetic data to a more complex kinetic scheme (Figure S4). This scheme included a tetrahedral intermediate, followed by a chemical step accounting for the collapse of the intermediate to generate an acyl-enzyme intermediate and then the dissociation of AMC. The fluorescence data of USP2-catalyzed hydrolysis of Ub-AMC can be fit to the model. However, with the inclusion of additional reaction steps and intermediates, the microscopic rate constants were no longer constrained (Figure S4).
Transient kinetic analysis of USP2-catalyzed hydrolysis of K48-linked IQF diubiquitin
Single-turnover experiments were performed to determine the transient kinetics of USP2-catalyzed hydrolysis of K48-linked IQF diubiquitin. Upon rapid mixing of K48-linked IQF diubiquitin and USP2, a rapid increase in fluorescence was observed when using a 570 nm cutoff filter (Figure 7B). In the K48-linked IQF diubiquitin substrate, the C-terminal carboxylate of the distal ubiquitin (red) is covalently linked to the side-chain amino group of Lys48 on the proximal ubiquitin (grey) by an isopeptide bond (see Figure 8). During USP2-catalyzed hydrolysis, the C-terminal carboxylate of the distal ubiquitin forms a covalent adduct with the USP2 (blue) active site cysteine residue forming a thioester intermediate, which is subsequently hydrolyzed in a deacylation step. The internally quenched fluorescence (IQF) FRET pair in diubiquitin is separated following the USP2-catalyzed cleavage of the isopeptide bond, thus producing the TAMRA fluorescence signal.
Figure 8.
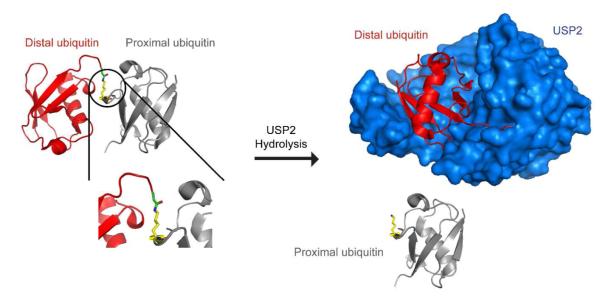
Illustration of the proximal and distal ubiquitin in USP2-catalyzed deubiquitination. Before USP2-catalyzed hydrolysis of diubiquitin, the C-terminal carboxylate of the distal ubiquitin (red) is covalently linked to the side-chain amino group of a lysine 48 residue of the proximal ubiquitin (grey) by an isopeptide bond. The C-terminal carboxylate of the distal ubiquitin becomes covalently bound to the USP2 (blue) active site cysteine residue forming a thioester intermediate. Then a deacylation step occurs followed by release of distal ubiquitin. This illustration was generated using the X-ray crystal structure of a Lys48-linked diubiquitin with an open conformation (17) and the cocrystal structure of USP2 in complex with ubiquitin (30).
For USP2-catalyzed hydrolysis of K48-linked IQF diubiquitin, USP2 concentration was varied from 1.25 μM to 4 μM while diubiquitin concentration was held constant at 0.2 μM. The pre-steady-state fluorescence traces were then globally fit (red line) to reaction Scheme 2 using KinTek Explorer (Figure 7B). In Scheme 2, USP2•DiUbA represents the initial binary complex between diubiquitin and USP2. USP2•DiUbB represents a new conformation in the binary complex. USP2-Ub is the acyl-enzyme intermediate. Ubproximal is the released proximal ubiquitin. USP2•Ub is USP2 bound with the distal ubiquitin, and Ubdistal is the released free distal ubiquitin. The rate constants k1, k−1, k2, k−2, k5, and k−5 were determined from the stopped-flow binding studies of K48-linked IQF diubiquitin and ubiquitin (see Table 1, Figure 3 and 5). These values were fixed during the global fitting. Because the deacylation step in USP2-catalyzed hydrolysis of diubiquitin and Ub-AMC is identical with the assumption that the leaving groups are released before hydrolysis of the acyl-enzyme intermediate, we fixed the rate constant of deacylation to 0.51 s−1. The fluorescence data in Figure 7B was globally fit to the reaction mechanism shown in Scheme 2 and the obtained rate constants are summarized in Table 3. The data globally fit with satisfaction.
Scheme 2.

Kinetic scheme of USP2-catalyzed hydrolysis of K48-linked IQF diubiquitin.
Table 3.
Pre-steady-state kinetic analysis for USP2-catalyzed hydrolysis of K48-linked IQF diubiquitin
| rate | best fit | lower limit | upper limit |
|---|---|---|---|
| k 1 a | 0.14 μM−1s−1 | n/a | n/a |
| k −1 a | 0.06 s−1 | n/a | n/a |
| k 2 a | 0.21 s−1 | n/a | n/a |
| k −2 a | 0.36 s−1 | n/a | n/a |
| k 3 | 0.24 ± 0.03 s−1 | 0.11 s−1 | 2.40 s−1 |
| k −3 | 17 ± 3 s−1 | 4.54 s−1 | 208 s−1 |
| k 4 a | 0.51 s−1 | n/a | n/a |
| k 5 a | 2.64 s−1 | n/a | n/a |
| k −5 a | 0.40 μM−1 s−1 | n/a | n/a |
| F(DiUb)a | 1.24 | n/a | n/a |
| F(DiUb•USP2A) | 2.27 ± 0.01 | 2.26 | 2.27 |
| F(DiUb•USP2B) | 2.44 ± 0.01 | 2.43 | 2.53 |
| F(Ubprox)a | 2.50 | n/a | n/a |
| χ2/ndf | 43 | n/a | n/a |
Indicates the value was fixed during global fitting. The values for k1 , k−1, k2 , k−2, k5, and k−5, were derived as explained for the USP2/K48-linked IQF diubiquitin and the USP2/ubiquitin stopped-flow binding data (Figure 3,5 and Table 1). Since the deacylation step is identical for both Ub-AMC and diubiquitin hydrolysis, the rate of deacylation that was previously determined from the global fitting of USP2-catalyzed hydrolysis ofUb-AMC was fixed for the rate of deacylation in the global fitting of diubiquitin hydrolysis. Fluorescence output expression factors (F) for DiUb, DiUb•USP2A, DiUb•USP2B, and Ubprox are also provided within the table. The fluorescence factor for DiUb and fully hydrolyzed Ubprox were determined based on the observed fluorescence signal and the known concentrations of the species. The factors for these two species were fixed during the global fitting. The remaining rates and fluorescence factors were globally fit for the fluorescence data in Figure 7B to reaction Scheme 2 using the KinTek Explorer software. The upper and lower limits were determined using the FitSpace Editor function ofthe KinTek Explorer program and the overall goodness of fit was judged by Chi2/degrees of freedom (χ2/ndt).
Burst kinetics for USP2-catalyzed hydrolysis of Ub-AMC
The USP2 single turnover experiments indicate that deacylation is rate limiting for Ub-AMC hydrolysis. In accord with this observation, a burst phase was observed for USP2-catalyzed hydrolysis of Ub-AMC under pre-steady-state multiple turnover conditions (Figure 9). We fit the individual traces to equation 8 that describes burst phase kinetics. The rate constants of the burst phase were determined to be in the range of 3.6-4.1s−1 and were largely concentration independent. We noted that the burst phase rate constants are in close agreement with the k2 of 4.4 s−1 that describes an acylation step (Scheme 1 and Table 2). Our attempt to globally fit these traces to both the simplified mechanism (Scheme 1) and to the more complex reaction scheme (Figure S4) was hampered because of the difficulty in constraining the individual rate constants.
Figure 9.

Burst kinetics of USP2-catalyzed hydrolysis of Ub-AMC. The burst kinetics of USP2-catalyzed hydrolysis of Ub-AMC was characterized under multiple turnover conditions. Fluorescence changes were monitored over 2 seconds. USP2 concentration was varied (0.4, 1, 1.5, and 2 μM) and shown in ascending order while Ub-AMC concentration was held constant at 12 μM. The fluorescence data was nonlinearly fit (black line) to equation 8 using GraphPad Prism (GraphPad Software, CA).
DISCUSSION
Deubiquitinating enzymes (DUBs) are an important family of enzymes that regulate the ubiquitination state of a large group of cellular proteins in eukaryotes. There are approximately 100 DUBs encoded by the human genome (3, 4). How DUBs recognize the various physiological ubiquitin substrates and catalyze the hydrolysis of the isopeptide bond between ubiquitin and the lysine residue on a target protein are important questions that remain to be addressed. Here we report a pre-steady-state kinetic analysis of the binding and hydrolysis of both mono- and diubiquitin substrates by a prototypical ubiquitin-specific protease, USP2, in order to understand the mode of substrate recognition and the mechanism of hydrolysis. Using a kinetic approach we obtained the individual microscopic rate constants in a full catalytic cycle of USP2 using both a model substrate Ub-AMC and a physiological substrate K48-linked diubiquitin.
We first determined the binding kinetics of monoubiquitin (ubiquitin and Ub-AMC) and K48-linked diubiquitin to the USP2 catalytic core using stopped-flow fluorescence. Despite the addition of the fluorophore AMC to the C-terminus of ubiquitin, the binding kinetics of Ub-AMC to an inactive USP2 is similar to that of the native ubiquitin binding to the wild-type USP2 (Table 1), indicating that the AMC group does not disrupt the interaction of ubiquitin with the USP2 catalytic core. Different from monoubiquitin, we observed biphasic kinetics for the binding of diubiquitin to USP2. For monoubiquitin, the kinetic constants kon and koff were used to calculate the equilibrium binding constant. Ubiquitin itself has a modest affinity for USP2 (Kd = 6.6 μM, Table 1). This value is in accord with the measured equilibrium dissociation constant of Ub-AMC to USP2 and the previously reported dissociation constants of ubiquitin for other USPs (32, 33). We found that the K48-linked diubiquitin has a significantly higher affinity for USP2 compared to monoubiquitin with an apparent Kd of 0.27 ± 0.08 μM. Although a cocrystal structure of USP2 and K48-linked diubiquitin is not available, our binding data suggests that extra binding interactions between the proximal ubiquitin and the USP2 catalytic core likely exist, thus contributing to the higher binding affinity.
Our stopped-flow data for diubiquitin revealed that the binding of K48-linked IQF diubiquitin to USP2 is biphasic. The apparent rate of the first step is linearly dependent on USP2 concentration while the second step is best fit by a hyperbola. This observation suggests a possible conformational rearrangement following the initial binding step. Our global fitting of the stopped-flow fluorescence traces of USP2-catalyzed hydrolysis of K48-linked IQF diubiquitin agrees well with a two-step binding mechanism and indicates that the two-step binding mechanism is on the kinetic pathway, where the second step of binding is partially rate-limiting.
The early X-ray crystal structure of K48-linked diubiquitin revealed a highly compact, closed structure in which the major site of the interaction defined by Ile44 on both the distal and proximal ubiquitin moieties are buried (16). Based on the USP2 structure bound with a distal ubiquitin, this closed conformation of diubiquitin is sterically unfavorable for its interaction with USP2 (see Figure 10). Recently, another K48-linked diubiquitin crystal structure was solved and revealed that K48-linked diubiquitin can also exist in an open conformation (17). Based on our modeling this open conformation of diubiquitin allows an unhindered interaction of diubiquitin with the USP2 catalytic core (see Figure 10B). Recent NMR analysis revealed an equilibrium between the compact and the open diubiquitin conformations in solution (17-19). Moreover NMR analyses determined that the conformational rearrangement in K48-linked diubiquitin occurs within 10 ns (18). Thus this conformation change step is beyond the detection by stopped-flow fluorescence technique. Therefore, the biphasic binding kinetics observed for K48-linked diubiquitin can be best interpreted by the binding of the diubiquitin to USP2, followed by an additional conformational rearrangement in the USP2-diubiquitin complex to position diubiquitin in a productive conformation for catalysis.
Figure 10.
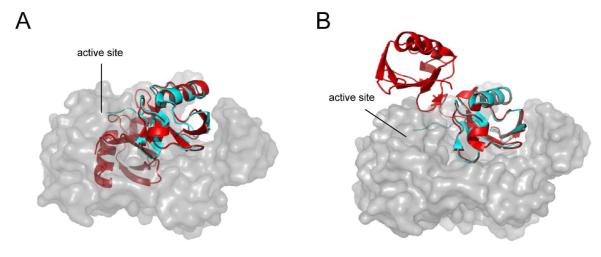
Binding of K48-linked diubiquitin to the USP2 catalytic core requires an open diubiquitin conformation. The X-ray cocrystal structure of USP2 (grey) and ubiquitin (cyan) (30) are overlaid with the X-ray structure of a closed (A) or open (B) K48-linked diubiquitin (red) (16, 17). Due to steric constraints USP2 requires an open K48-linked diubiquitin conformation for binding to USP2.
Our global fitting analysis revealed a conformational rearrangement step following the binding of the diubiquitin to USP2 catalytic core. Although a previous cocrystal structure study of the USP catalytic core has showed a small conformational rearrangement in the finger and thumb domains of USP upon the binding of the distal ubiquitin (37), this conformation change is less likely in what we observed in the stopped-flow analysis. As a support of this notion, the binding of monoubiquitin to the USP2 catalytic core is monophasic and lacks a conformational change step following the bimolecular binding step. Thus we conclude that the observed conformational change step most likely originates from the diubiquitin moiety, which is reported by the IQF pair on the K48-linked diubiquitin.
By globally fitting the stopped-flow fluorescence traces, we obtained the individual microscopic rate constants of the complete USP2 catalytic cycle in hydrolyzing Ub-AMC. Pre-steady-state kinetic characterization of hydrolysis of Ub-AMC clearly showed that the rate of acylation (4.40 ± 0.04 s−1) is 9-fold faster than the rate of deacylation (0.51 ± 0.01 s−1). This notion was corroborated by burst kinetic analyses of USP2-catalyzed hydrolysis of Ub-AMC.
A recent report compared USP2-catalyzed hydrolysis of Ub-AMC, a synthetic K48-linked diubiquitin substrate, and Ub-V77 that contains an additional valine residue C-terminal to Gly76 (21). A LC-MS based assay was used to quantify the hydrolysis of the diubiquitin and the Ub-V77 substrates. It was found that an oxyanion hole mutant (N271A/D575A) was more sensitive in activity when hydrolyzing diubiquitin and Ub-V77 than Ub-AMC. Asn271 and Asp575 are believed to stabilize the oxyanion intermediate formed during the acylation step of deubiquitination. Because elimination of the oxyanion hole did not affect Ub-AMC hydrolysis, the authors suspected deacylation is the rate-limiting step for USP2 catalyzed hydrolysis of Ub-AMC, but not the hydrolysis of the K48-linked diubiquitin and Ub-V77. Here we provide direct evidence that deacylation is rate limiting in USP2-catalyzed Ub-AMC hydrolysis.
Deacylation as a rate-limiting step was also observed for other cysteine and serine proteases when peptidyl-AMC was used as a substrate. Specifically, deacylation is the rate-limiting step for cathepsin C catalyzed hydrolysis of a dipeptide-AMC substrate (28-fold slower than the rate of acylation) (38). Likewise, deacylation is the rate-limiting step for the subtilisin superfamily protease Kex2 in cleaving peptidyl-AMC substrates (39). Interestingly we observed a change in several microscopic rate constants for hydrolysis of K48-linked IQF diubiquitin by USP2. Further study will be needed to determine whether the rate-limiting step in USP2-catalyzed K48-diubiquitin hydrolysis is different from that of Ub-AMC as a substrate. It should be noted that the K48-linked IQF diubiquitin is a better mimic of the polyubiquitin chain linked by K48 on ubiquitin. The kinetic mechanisms unveiled using this substrate should provide further information for our understanding of the ubiquitin-specific protease catalyzed deubiquitination reaction.
Supplementary Material
ACKNOWLEDGMENTS
We thank Wade Harper for the USP2 plasmid. We also thank Progenra, Inc for the K48-linked IQF diUb reagent.
This work was supported by a grant from the US National Institutes of Health to Z. Zhuang (R01GM097468).
1Abbreviations used are
- DUB
Deubiquitinating enzyme
- USP
ubiquitin-specific protease
- Ub-AMC
ubiquitin-7-amino-4-methylcoumarin
- IQF
internally quenched fluorescence
- diUb
diubiquitin
- TAMRA
carboxytetramethylrhodamine
Footnotes
SUPPORTING INFORMATION Figures S1–S4. This material is available free of charge via the internet at http://pubs.acs.org.
REFERENCES
- 1.Chen ZJ, Sun LJ. Nonproteolytic functions of ubiquitin in cell signaling. Mol Cell. 2009;33:275–286. doi: 10.1016/j.molcel.2009.01.014. [DOI] [PubMed] [Google Scholar]
- 2.Ulrich HD, Walden H. Ubiquitin signalling in DNA replication and repair. Nat Rev Mol Cell Biol. 2010;11:479–489. doi: 10.1038/nrm2921. [DOI] [PubMed] [Google Scholar]
- 3.Komander D, Clague MJ, Urbe S. Breaking the chains: structure and function of the deubiquitinases. Nat Rev Mol Cell Biol. 2009;10:550–563. doi: 10.1038/nrm2731. [DOI] [PubMed] [Google Scholar]
- 4.Nijman SM, Luna-Vargas MP, Velds A, Brummelkamp TR, Dirac AM, Sixma TK, Bernards R. A genomic and functional inventory of deubiquitinating enzymes. Cell. 2005;123:773–786. doi: 10.1016/j.cell.2005.11.007. [DOI] [PubMed] [Google Scholar]
- 5.Amerik AY, Hochstrasser M. Mechanism and function of deubiquitinating enzymes. Biochim Biophys Acta. 2004;1695:189–207. doi: 10.1016/j.bbamcr.2004.10.003. [DOI] [PubMed] [Google Scholar]
- 6.Reyes-Turcu FE, Ventii KH, Wilkinson KD. Regulation and cellular roles of ubiquitin-specific deubiquitinating enzymes. Annu Rev Biochem. 2009;78:363–397. doi: 10.1146/annurev.biochem.78.082307.091526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sacco JJ, Coulson JM, Clague MJ, Urbe S. Emerging roles of deubiquitinases in cancer-associated pathways. IUBMB Life. 2010;62:140–157. doi: 10.1002/iub.300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Graner E, Tang D, Rossi S, Baron A, Migita T, Weinstein LJ, Lechpammer M, Huesken D, Zimmermann J, Signoretti S, Loda M. The isopeptidase USP2a regulates the stability of fatty acid synthase in prostate cancer. Cancer Cell. 2004;5:253–261. doi: 10.1016/s1535-6108(04)00055-8. [DOI] [PubMed] [Google Scholar]
- 9.Oberfeld B, Ruffieux-Daidie D, Vitagliano JJ, Pos KM, Verrey F, Staub O. Ubiquitin-specific protease 2-45 (Usp2-45) binds to epithelial Na+ channel (ENaC)-ubiquitylating enzyme Nedd4-2. Am J Physiol Renal Physiol. 2011;301:F189–196. doi: 10.1152/ajprenal.00487.2010. Page 30 of 55. [DOI] [PubMed] [Google Scholar]
- 10.Fakitsas P, Adam G, Daidie D, van Bemmelen MX, Fouladkou F, Patrignani A, Wagner U, Warth R, Camargo SM, Staub O, Verrey F. Early aldosterone-induced gene product regulates the epithelial sodium channel by deubiquitylation. J Am Soc Nephrol. 2007;18:1084–1092. doi: 10.1681/ASN.2006080902. [DOI] [PubMed] [Google Scholar]
- 11.Krzystanek K, Rasmussen HB, Grunnet M, Staub O, Olesen SP, Abriel H, Jespersen T. Deubiquitylating enzyme USP2 counteracts Nedd4-2-mediated downregulation of KCNQ1 potassium channels. Heart Rhythm. 2012;9:440–448. doi: 10.1016/j.hrthm.2011.10.026. [DOI] [PubMed] [Google Scholar]
- 12.Shi Y, Solomon LR, Pereda-Lopez A, Giranda VL, Luo Y, Johnson EF, Shoemaker AR, Leverson J, Liu X. Ubiquitin-specific cysteine protease 2a (USP2a) regulates the stability of Aurora-A. J Biol Chem. 2011;286:38960–38968. doi: 10.1074/jbc.M111.231498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stevenson LF, Sparks A, Allende-Vega N, Xirodimas DP, Lane DP, Saville MK. The deubiquitinating enzyme USP2a regulates the p53 pathway by targeting Mdm2. EMBO J. 2007;26:976–986. doi: 10.1038/sj.emboj.7601567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Verrey F, Fakitsas P, Adam G, Staub O. Early transcriptional control of ENaC (de)ubiquitylation by aldosterone. Kidney Int. 2008;73:691–696. doi: 10.1038/sj.ki.5002737. [DOI] [PubMed] [Google Scholar]
- 15.Gousseva N, Baker RT. Gene structure, alternate splicing, tissue distribution, cellular localization, and developmental expression pattern of mouse deubiquitinating enzyme isoforms Usp2-45 and Usp2-69. Gene Expr. 2003;11:163–179. doi: 10.3727/000000003108749053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cook WJ, Jeffrey LC, Carson M, Chen Z, Pickart CM. Structure of a diubiquitin conjugate and a model for interaction with ubiquitin conjugating enzyme (E2) J Biol Chem. 1992;267:16467–16471. doi: 10.2210/pdb1aar/pdb. [DOI] [PubMed] [Google Scholar]
- 17.Hirano T, Serve O, Yagi-Utsumi M, Takemoto E, Hiromoto T, Satoh T, Mizushima T, Kato K. Conformational dynamics of wild-type Lys-48-linked diubiquitin in solution. J Biol Chem. 2011;286:37496–37502. doi: 10.1074/jbc.M111.256354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ryabov Y, Fushman D. Interdomain mobility in di-ubiquitin revealed by NMR. Proteins. 2006;63:787–796. doi: 10.1002/prot.20917. [DOI] [PubMed] [Google Scholar]
- 19.Varadan R, Walker O, Pickart C, Fushman D. Structural properties of polyubiquitin chains in solution. J Mol Biol. 2002;324:637–647. doi: 10.1016/s0022-2836(02)01198-1. [DOI] [PubMed] [Google Scholar]
- 20.Sowa ME, Bennett EJ, Gygi SP, Harper JW. Defining the human deubiquitinating enzyme interaction landscape. Cell. 2009;138:389–403. doi: 10.1016/j.cell.2009.04.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang W, Sulea T, Tao L, Cui Q, Purisima EO, Vongsamphanh R, Lachance P, Lytvyn V, Qi H, Li Y, Menard R. Contribution of active site residues to substrate hydrolysis by USP2: insights into catalysis by ubiquitin specific proteases. Biochemistry. 2011;50:4775–4785. doi: 10.1021/bi101958h. [DOI] [PubMed] [Google Scholar]
- 22.Chen J, Ai Y, Wang J, Haracska L, Zhuang Z. Chemically ubiquitylated PCNA as a probe for eukaryotic translesion DNA synthesis. Nat Chem Biol. 2010;6:270–272. doi: 10.1038/nchembio.316. [DOI] [PubMed] [Google Scholar]
- 23.Piotrowski J, Beal R, Hoffman L, Wilkinson KD, Cohen RE, Pickart CM. Inhibition of the 26 S proteasome by polyubiquitin chains synthesized to have defined lengths. The Journal of biological chemistry. 1997;272:23712–23721. doi: 10.1074/jbc.272.38.23712. [DOI] [PubMed] [Google Scholar]
- 24.Bozza WP, Zhuang Z. Biochemical characterization of a multidomain deubiquitinating enzyme Ubp15 and the regulatory role of its terminal domains. Biochemistry. 2011;50:6423–6432. doi: 10.1021/bi200529z. [DOI] [PubMed] [Google Scholar]
- 25.Yang G, Liu RQ, Taylor KL, Xiang H, Price J, Dunaway-Mariano D. Identification of active site residues essential to 4-chlorobenzoyl-coenzyme A dehalogenase catalysis by chemical modification and site directed mutagenesis. Biochemistry. 1996;35:10879–10885. doi: 10.1021/bi9609533. [DOI] [PubMed] [Google Scholar]
- 26.Anderson KS, Sikorski JA, Johnson KA. Evaluation of 5-enolpyruvoylshikimate-3-phosphate synthase substrate and inhibitor binding by stopped-flow and equilibrium fluorescence measurements. Biochemistry. 1988;27:1604–1610. doi: 10.1021/bi00405a032. [DOI] [PubMed] [Google Scholar]
- 27.Johnson KA. Rapid kinetic analysis of mechanochemical adenosinetriphosphatases. Methods Enzymol. 1986;134:677–705. doi: 10.1016/0076-6879(86)34129-6. [DOI] [PubMed] [Google Scholar]
- 28.Johnson KA, Simpson ZB, Blom T. FitSpace explorer: an algorithm to evaluate multidimensional parameter space in fitting kinetic data. Anal Biochem. 2009;387:30–41. doi: 10.1016/j.ab.2008.12.025. [DOI] [PubMed] [Google Scholar]
- 29.Johnson KA, Simpson ZB, Blom T. Global kinetic explorer: a new computer program for dynamic simulation and fitting of kinetic data. Anal Biochem. 2009;387:20–29. doi: 10.1016/j.ab.2008.12.024. [DOI] [PubMed] [Google Scholar]
- 30.Renatus M, Parrado SG, D’Arcy A, Eidhoff U, Gerhartz B, Hassiepen U, Pierrat B, Riedl R, Vinzenz D, Worpenberg S, Kroemer M. Structural basis of ubiquitin recognition by the deubiquitinating protease USP2. Structure. 2006;14:1293–1302. doi: 10.1016/j.str.2006.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen J, Dexheimer TS, Ai Y, Liang Q, Villamil MA, Inglese J, Maloney DJ, Jadhav A, Simeonov A, Zhuang Z. Selective and Cell-Active Inhibitors of the USP1/ UAF1 Deubiquitinase Complex Reverse Cisplatin Resistance in Non-small Cell Lung Cancer Cells. Chemistry & biology. 2011;18:1390–1400. doi: 10.1016/j.chembiol.2011.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Luna-Vargas MP, Faesen AC, van Dijk WJ, Rape M, Fish A, Sixma TK. Ubiquitin-specific protease 4 is inhibited by its ubiquitin-like domain. EMBO Rep. 2011;12:365–372. doi: 10.1038/embor.2011.33. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 33.Faesen AC, Dirac AM, Shanmugham A, Ovaa H, Perrakis A, Sixma TK. Mechanism of USP7/HAUSP activation by its C-terminal ubiquitin-like domain and allosteric regulation by GMP-synthetase. Mol Cell. 2011;44:147–159. doi: 10.1016/j.molcel.2011.06.034. [DOI] [PubMed] [Google Scholar]
- 34.Hsieh J, Fierke CA. Conformational change in the Bacillus subtilis RNase P holoenzyme--pre-tRNA complex enhances substrate affinity and limits cleavage rate. RNA. 2009;15:1565–1577. doi: 10.1261/rna.1639409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wands AM, Wang N, Lum JK, Hsieh J, Fierke CA, Mapp AK. Transient-state kinetic analysis of transcriptional activator.DNA complexes interacting with a key coactivator. J Biol Chem. 2011;286:16238–16245. doi: 10.1074/jbc.M110.207589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Strausberg SL, Ruan B, Fisher KE, Alexander PA, Bryan PN. Directed coevolution of stability and catalytic activity in calcium-free subtilisin. Biochemistry. 2005;44:3272–3279. doi: 10.1021/bi047806m. [DOI] [PubMed] [Google Scholar]
- 37.Hu M, Li P, Li M, Li W, Yao T, Wu JW, Gu W, Cohen RE, Shi Y. Crystal structure of a UBP-family deubiquitinating enzyme in isolation and in complex with ubiquitin aldehyde. Cell. 2002;111:1041–1054. doi: 10.1016/s0092-8674(02)01199-6. [DOI] [PubMed] [Google Scholar]
- 38.Schneck JL, Villa JP, McDevitt P, McQueney MS, Thrall SH, Meek TD. Chemical mechanism of a cysteine protease, cathepsin C, as revealed by integration of both steady-state and pre-steady-state solvent kinetic isotope effects. Biochemistry. 2008;47:8697–8710. doi: 10.1021/bi8007627. [DOI] [PubMed] [Google Scholar]
- 39.Rockwell NC, Fuller RS. Direct measurement of acylenzyme hydrolysis demonstrates rate-limiting deacylation in cleavage of physiological sequences by the processing protease Kex2. Biochemistry. 2001;40:3657–3665. doi: 10.1021/bi0020877. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.