

Abstract
Pdx-1 is a hox-like transcription factor that is responsible for the activation of the insulin gene. Prior studies have demonstrated the interaction in vitro of Pdx-1 with short (20-40 nucleotide) DNA fragments corresponding to “A boxes” of the insulin promoter. Precisely how Pdx-1 binds to DNA in the complex milieu of chromatin, however, has never been studied. Here, we explored how Pdx-1-DNA interactions might be influenced by chromatin accessibility at the insulin gene in β cells (βTC3) vs. pancreatic ductal cells (mPAC). We demonstrate that Pdx-1 occupies the endogenous insulin promoter in βTC3 cells but not in mPAC cells, a finding that is independent of the intracellular Pdx-1 protein concentration. Based on micrococcal nuclease protection assays, the difference in promoter binding between the two cell types appears to be secondary to chromatin accessibility at predicted Pdx-1 binding sites between bp −126 to −296 (relative to the transcriptional start site) of the insulin promoter. Binding studies using pruified Pdx-1 and reconsituted chromatin in vitro suggest that the positioning of a nucleosome(s) within this crucial region of the promoter might account for differences in chromatin accessibility. Consistent with these observations, fluorescence colocalization studies show that Pdx-1 does not occupy regions of compacted, nucleosome-rich chromatin within the nucleus. Our findings suggest a model whereby insulin transcription in the β cell is at least partially facilitated by enahanced chromatin accessibility within a crucial regulatory region between bp −126 to −296, thereby permitting occupancy by transactivators such as Pdx-1.
INTRODUCTION
Type 1 diabetes mellitus results from the autoimmune destruction of the β cells within the pancreatic islets of Langerhans. The catastrophic metabolic consequences of Type 1 diabetes is primarily due to the absence of insulin secretion from these destroyed β cells. Thus, an attractive approach to reversing diabetes in such individuals is to engineer other cell types to produce and secrete insulin. A major hurdle in this engineering process is to induce transcription of the gene encoding preproinsulin (the “insulin gene”) in heterologous cell types. Strikingly, transcription of the insulin gene is nearly exclusive to β cells, and only about 300-400 base pairs (bp) of DNA 5’ of the transcriptional start site (the “insulin promoter”) are sufficient to confer β cell specificity (1-3). Our laboratory has been interested in the mechanism by which the pancreatic Hox-like transcription factor Pdx-1 mediates insulin gene activation. In recent studies, we have demonstrated that Pdx-1 is necessary for the methylation of Lys4 of histone H3 within the promoter and coding regions of the insulin gene, and that this covalent modification is closely linked to the conversion of RNA polymerase II to its elongation-specific isoform (4, 5). Pdx-1 achieves these crucial activation events through cooperation with cofactors in the β cell, particularly Set9 and p300 (5-7). In this transcriptional paradigm, the first step in Pdx-1 action is its binding to key A/T-rich DNA sequences within the “A boxes.” Several prior studies in vitro have shown the interaction of Pdx-1 with short (20-40 nucleotide) DNA fragments corresponding to A boxes of the insulin promoter (8-10). This robust interaction in vitro, however, does not explain why the presence of Pdx-1 in certain non-β cell types, such as pancreatic duct cells, fails to lead to discernable insulin expression (11-13). In this regard, is unclear whether binding studies involving “naked” DNA fragments reliably predicts the specificity or magnitude of Pdx-1/DNA interactions on more relevant chromatin templates.
The basic unit of chromatin is the nucleosome, which consists of 146 base pairs (bp) of DNA wrapped around an octamer of histones (two each of histones H2A, H2B, H3, and H4). Based upon crystal structure analysis, DNA contacts the histone octamer at approximately 120 points within the nucleosome complex, thereby leading to a tight globular core that potentially inhibits transcription factor binding (14). By contrast, DNA binding sites located between nucleosomes or at the very end of nucleosomes would be predicted to be more accessible to their cognate transcription factors. In the absence of ATP-dependent SWI/SNF-type nucleosome repositioning (15), transcription factors may gain variable access to DNA through one or a combination of several mechanisms, including: (a) conformational changes within the nucleosomal core that allow for transient release of histone-DNA contacts, thereby opening binding sites (16), (b) intrinsic histone binding capacities of some transcription factors (e.g. HNF-3) that may lead to changes in higher order chromatin structure (17), and (c) acetylation of the N-terminal tails of the core histones leading to increases in the equilibrium accessibility (18). Taken together, the relative positioning and structural compaction of the nucleosomal complex can account for the degree of transcription factor binding site accessibility, and hence the overall transcriptional activity of the gene (19, 20).
We and others have proposed that the insulin promoter in islet β cell lines (βTC3 and MIN6) is embedded within euchromatin (“open” or “loose” chromatin), whereas the promoter in non-β cell lines (where the gene is inactive) is located within relative heterochromatin (“closed” or “compacted” chromatin). This conclusion was derived primarily from chromatin immunoprecipitation (ChIP) studies demonstrating the enrichment of histone H3 and H4 Lys acetylation and histone H3 Lys4 methylation at the insulin gene in β cell lines (7, 21, 22). However, no studies have directly addressed the physical accessibility of the insulin gene in β cells vs. non-β cells. In this study, we explored how Pdx-1-DNA interactions might be influenced by the chromatin structure of the insulin gene in β cells (βTC3) vs. pancreatic ductal cells (mPAC). We demonstrate that the chromatin structure of the insulin gene in mPAC cells appears to be more restrictive to Pdx-1 binding, a finding that is likely secondary to the accessibility of chromatin relative to predicted Pdx-1 binding sites in this cell type. Binding studies using purified Pdx-1 and nucleosome-reconstituted DNA in vitro demonstrate that Pdx-1 does not contain intrinsic nucleosome repositioning capacity, but rather binds to nucleosomal DNA only at very high (supra-physiologic) concentrations or when its cognate binding sites are located at the very ends of the nucleosome. We propose a model whereby insulin transcription in the β cell is at least at least partially facilitated by enhanced accessibility of chromatin within a crucial regulatory region.
RESULTS
Insulin promoter occupancy by Pdx-1 is cell-type dependent
To determine the physical accessibility of the insulin promoter in different cellular contexts, we initially studied proximal insulin promoter occupancy by Pdx-1 in primary mouse islets and in two pancreatic cell types that express this protein, but have divergent insulin gene expression characteristics: βTC3 cells (a β cell-derived line with robust insulin expression, ref. (23)) and mPAC cells (a pancreatic ductal cell-derived line with no insulin expression, refs. (8, 24)). Immunoblot analysis (inset to Fig. 1A, lanes 1-3) demonstrates that mouse islets, βTC3 cells and mPAC cells all express Pdx-1 protein, but the levels of Pdx-1 protein are substantially lower in mPAC cells compared to islets and βTC3 cells. Overexpression of a hamster Pdx-1 transgene in mPAC cells (mPAC/Pdx) results in Pdx-1 protein levels comparable to that of βTC3 cells (inset to Fig. 1A, lane 4). ChIP analysis shown in Fig. 1A demonstrates that Pdx-1 nearly equivalently occupies the proximal insulin promoter1 in mouse islets and βTC3 cells. Strikingly, however, promoter occupancy by Pdx-1 was only minimally detectable over background in mPAC and mPAC/Pdx cells. These results raise the possibility that Pdx-1 in mPAC cells is either unable to bind to DNA (owing to absent post-translational modifications or interacting factors) or that the structure of chromatin in mPAC cells physically prohibits DNA binding. Notably, insulin mRNA, as measured by real-time RT-PCR, was undetectable in both mPAC cells and mPAC/Pdx cells (data not shown).
Figure 1. Association of Pdx-1 with the insulin promoter in vivo and in vitro.

A, extracts from mouse islets, βTC3 cells, mPAC cells, and mPAC cells overexpressing hamster Pdx-1 (mPAC/Pdx) were subject to ChIP using polyclonal Pdx-1 antiserum, and the recovery of the proximal insulin promoter (-126 to -296 bp relative to the transcriptional start site) was assessed by quantitative real-time PCR. Data are expressed as the percent of input DNA recovered following ChIP. Data represent the mean ± S.E. of at least three independent ChIP experiments; inset is an immunoblot showing Pdx-1 protein levels in islets, βTC3, mPAC, and mPAC/Pdx cells. B, representative EMSA using nuclear extracts isolated from βTC3, mPAC, and mPAC/Pdx cells and a 32P-labeled oligonucleotide probe containing the mouse insulin 1 A4/A3 element; lower arrows indicate positions of the Pdx-1 complex and the upper arrows indicate positions of the supershifted complex upon addition of Pdx-1 antiserum; PI, preimmune serum; Pdx Ab, polyclonal Pdx-1 antiserum; SS, supershift. C, representative EMSA using nuclear extract from mPAC/Pdx cells, a 32P-labeled oligonucleotide containing the insulin 1 A4/A3 element, and increasing concentrations (100- or 1000-fold excess) of unlabeled oligonucleotide (WT) or unlabeled oligonucleotide containing a mutation in the A4/A3 element (MUT), as indicated in Materials and Methods; upper arrow indicates positions of the unknown (UK) protein complex, and lower arrow indicates position of the Pdx-1 complex.
To address the ability of Pdx-1 to bind to DNA in mPAC cells, we next performed electrophoretic mobility shift assays (EMSAs) using nuclear extracts and a 32P-labeled oligonucleotide probe corresponding to the insulin A4/A3 enhancer element. Fig. 1B (lane 2) demonstrates that Pdx-1 protein in βTC3 nuclear extract produces a distinct, shifted complex with the A4/A3 probe, and that this complex is super-shifted upon addition of Pdx-1 antiserum (Fig. 1B, lane 3). mPAC cell nuclear extract produces similar shifted and supershifted complexes with the A3/A4 probe (Fig. 1B, lanes 4 and 5); the weaker intensity of these complexes relative to βTC3 nuclear extract is consistent with the lower expression of Pdx-1 protein in mPAC cells. The intensity of the complex corresponding to Pdx-1 is enhanced in mPAC/Pdx cells to a level even exceeding that of βTC3 cells (Fig. 1B, lanes 6 and 7). These results are consistent with the ability of Pdx-1 in mPAC cells to bind to DNA. Notably, a complex of unknown (UK) identity is observed just above the Pdx-1 complex in mPAC cells (lanes 4-7). To determine if the UK complex competes with Pdx-1 for binding to the A4/A3 element in mPAC cells, we performed a competition assay shown in Fig. 1C. Whereas unlabeled wild-type (WT) competitor effectively competed both the Pdx-1 and UK complexes (Fig. 1C, lanes 2-3), a competitor containing a mutation in the A4/A3 element (MUT) was effective in competing the UK complex, but not the Pdx-1 complex (Fig. 1C, lanes 4-5). These results suggest that the UK complex in mPAC cells appears to bind to a DNA element distinct from the A4/A3. These results, however, do not entirely rule out the possibility that the UK complex might interfere to some extent with the binding of Pdx-1 to the A4/A3 element within cells.
Nucleosome positioning can inhibit DNA binding and transcriptional activation by Pdx-1
To address whether the chromatin structure of the insulin gene in mPAC cells might inhibit Pdx-1/DNA interactions, we first asked how chromatin structure could influence the affinity of Pdx-1 for DNA. We performed EMSA experiments using bacterially-purified Pdx-1 protein and 32P-labeled, ~170 bp DNA fragments of the insulin 1 promoter containing the A4/A3 enhancer (see Fig. 2A). The 32P-labeled probes were either complexed with HeLa cell-purified histone octamers (“chromatinized”)2 or uncomplexed (“naked”). First, to test the integrity of our purified Pdx-1 protein, we performed a competition EMSA using the protein and one of these probes (Probe 1). Fig. 2B demonstrates that Pdx-1 binding to Probe 1 is inhibited by increasing concentrations of an unlabeled competitor A4/A3 oligonucleotide, whereas a competitor containing a mutation in the A4/A3 element does not inhibit Pdx-1 binding. These studies demonstrate that purified Pdx-1 binds specifically to the A4/A3 element of the insulin promoter.
Figure 2. Binding of Pdx-1 to the insulin A4/A3 element and chromatin reconstitution of insulin gene fragments.

A, schematic representation of the two insulin probes used in chromatin reconstitution studies. The location of the A4/A3 element is indicated by the black box, and the numbers indicate positions of the sequences relative to the transcriptional start site of the insulin 1 gene. B, EMSA was performed using 20 ng bacterially-purified His6-Pdx-1 and approximately 32P-labeled Probe 1. Approximate molar excess of unlabeled competitor oligonucleotide is indicated. The mutant (MUT) competitor contains single bp mutations that disrupt the TAAT consensus sequences, as indicated in Materials and Methods. C, 32P-labeled Probe 1 was reconstituted as chromatin using a salt dilution technique as indicated in Materials and Methods. Naked (N) and chromatinized (C) probes were incubated with MNase for varying times (0-10 min), then treated with proteinase K to digest protein, then subjected to electrophoresis on a 5% polyacrylamide gel and to autoradiography.
We next studied the interaction of purified Pdx-1 with naked and chromatinized probes corresponding to 170 bp fragments of the proximal rat insulin 1 gene containing the A4/A3 element. In Probe 1 (corresponding to bp −126 to −296 relative to the transcriptional start site), the A4/A3 element is located in the middle of the probe, whereas in Probe 2 (corresponding to bp −188 to −368 relative to the transcriptional start site) the A4/A3 element is located at the very end (see Fig 2B). The differential location of the A4/A3 element between the two probes allowed us to assess how Pdx-1 binding is affected by the positioning of the element relative to the core histones (see below). To demonstrate that the chromatinized insulin 1 probes formed true nucleosomes, we subjected 32P-labeled chromatinized and naked probes to micrococcal nuclease (MNase) digestion, followed by polyacrylamide gel electrophoresis. MNase efficiently and non-specifically digests only DNA that is not complexed with histones (25). As shown in Fig. 2C, naked Probe 1 is rapidly digested by MNase, whereas the chromatinized Probe 1 is protected from digestion (identical data were observed for Probe 2, data not shown).
Fig. 3A shows a representative EMSA demonstrating that Pdx-1 forms a complex with the naked Probe 1 at 3 nM concentration; complexes of successively higher molecular weight are observed when increasing concentrations of Pdx-1 are added in these EMSAs, consistent with the binding of Pdx-1 to the A4/A3 and other lower affinity A/T-rich elements contained within the probe (Fig. 3A). Based on quantitative phosphorimager analysis of these data, we estimate Pdx-1 binds to naked Probe 1 with an apparent dissociation constant (KD) of ~30 nM. When this Probe 1 is chromatinized, however, the apparent KD of Pdx-1 increases to ≥180 nM (Fig. 3B). These data suggest that the presence of the histone octamer inhibits the binding of Pdx-1 to DNA, but they do not distinguish whether this inhibition is simply secondary to the presence octamer itself or to the positioning of the A4/A3 element relative to the octameric core.
Figure 3. Interaction of Pdx-1 with naked and chromatinized insulin promoter probes.

EMSAs were performed using different concentrations of His6-Pdx-1 and 32P-labeled naked or chromatinized Probes 1 and 2, as detailed in Materials and Methods. Arrows to the right of each panel indicate positions of Pdx-1 complexes formed with each probe, and the positions of the unbound naked probe (NP) and chromatinized probe (CP) is indicated to the left of each panel. A, binding of increasing concentrations of Pdx-1 to naked Probe 1; B, binding of increasing concentrations of Pdx-1 to chromatinized Probe 1; C, binding of increasing concentrations of Pdx-1 to naked Probe 2; D, binding of increasing concentrations of Pdx-1 to chromatinized Probe 2.
To address whether the positioning of the A4/A3 element might impact Pdx-1 binding, we generated a second probe (Probe 2) in which the A4/A3 binding site is located at the end of the DNA fragment and, consequently, would be expected to be situated at the end of the nucleosome complex (see Fig. 2B). In EMSA studies, Pdx-1 exhibited an apparent KD for naked Probe 2 that was similar to that for naked Probe 1 (~30 nM, see Fig. 3C). Upon chromatinization of Probe 2, the estimated apparent KD of Pdx-1 increased to only ~60 nM (see Fig. 3D), a value that is intermediate between that of the naked probes and chromatinized Probe 1. Taken together, the data in Fig. 3 suggest that the affinity of Pdx-1 for the insulin promoter in the setting of chromatin is strongly influenced by the positioning of the nucleosome relative to putative binding sites.
To determine whether nucleosomes also inhibit transcriptional activation by Pdx-1, we performed transcription in vitro using naked and chromatinized DNA templates. To enhance transcript detection, we generated a reporter construct in which 5 copies of the A4/A3 enhancer element were placed upstream of a minimal promoter driving the luciferase gene (see Fig. 4, top) in plasmid pIC2085S (26). This plasmid contains tandem copies of the sea urchin 5S ribosomal nucleosome positioning elements that flank either side of the enhancer/minimal promoter cassette, as shown in Fig. 4. As a negative control, we also generated a reporter in which the A4/A3 element was mutated, as described previously (9, 27). To initiate transcription, we added purified Pdx-1 protein and HeLa cell nuclear extract, and measured transcriptional initiation at the luciferase gene using a real-time PCR-based 5’-RACE protocol (5). As shown in Fig. 4, addition of Pdx-1 to naked A4/A3 reporter resulted in an 4-fold enrichment of transcription. However, when the reporter was reconstituted as chromatin, Pdx-1-induced transcriptional enrichment was diminished to 1.2-fold; these findings are consistent with the overall silencing effect of nucleosomal DNA on binding and transactivation by Pdx-1. No activation of the control reporter was observed with Pdx-1 (Fig. 4). Because our reporter construct represents only an element of the insulin gene, we note that these results should only be interpreted with regard to the effect of chromatin on the transactivation function of Pdx-1, and not as the effect of chromatin on insulin gene regulation. Reconstitution of these reporter constructs as chromatin was verified by MNase digestion, as described previously (5).
Figure 4. Chromatin inhibits transcriptional activation by Pdx-1.
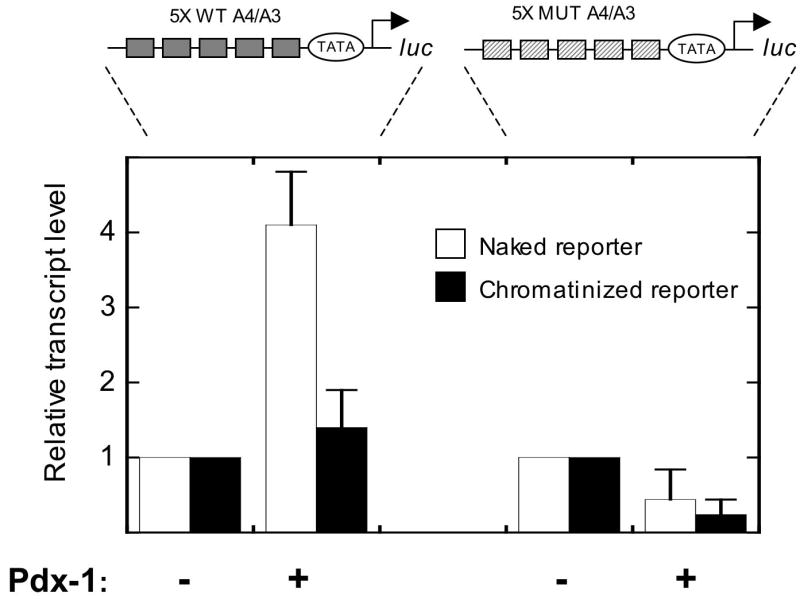
Schematic representations of the insulin reporters used are shown at the top. The reporters contain tandem insulin 1 A4/A3 elements (or the same elements containing mutations in the Pdx-1 binding sequences) placed upstream of a minimal promoter driving the luciferase gene. The reporters were either used directly (naked reporter) or reconstituted as chromatin with histone octamers (chromatinized reporter) and subjected to transcription reactions in vitro using HeLa nuclear extract with or without 100 ng Pdx-1. To ensure that transcriptional initiation was measured, reactions were subjected to 5’ RACE, followed by quantitation using real-time PCR. To highlight the differences in transcription observed after adding Pdx-1, data are expressed as transcript levels relative to a reaction containing only HeLa nuclear extract (defined as 1). Data represent the mean ± S.E. of three independent in vitro transcription reactions.
Chromatin structure may impair insulin promoter accessibility in pancreatic ductal cells
To determine if chromatin structural differences at insulin promoter might account for the differential capacity of Pdx-1 to bind to the insulin gene in mPAC vs. βTC3 cells, we performed chromatin accessibility by real-time PCR (ChART PCR) assays (28). In this assay, nuclei from cells are subject to limited digestion with MNase, which only cleaves DNA that is not tightly bound by nucleosomes (25). Fig. 5A demonstrates that MNase digestion of chromatin from βTC3 and mPAC nuclei yields a ladder of DNA fragments consistent with the average cleavage of chromatin between distinct nucleosomes (~150 bp). Subsequently, we subjected the digestions to real-time PCR to quantitate the fraction of insulin promoter that remained. This assay yields results comparable to traditional endonuclease/southern blot assays, and provides an estimate of the physical accessibility of DNA across regions of the gene (29). Most interestingly, analysis of the ChART PCR assays for accessibility of a crucial transcriptional control region of the insulin promoter containing the A4/A3 element (bp −126 to −296 relative to the transcriptional start site) (1) led to strikingly different results in the two cell types. As shown in Fig. 5B, increasing amounts of MNase in each reaction led to successively greater degradation of this region of the insulin promoter in βTC3 cells compared to mPAC cells. At the highest MNase concentration (2 units/reaction), this fragment of the insulin promoter was degraded almost 10-fold more in βTC3 cells compared to mPAC cells (Fig. 5B). These findings imply that the transcriptional control region of the insulin promoter containing the A4/A3 element is substantially more accessible and possibly less likely occupied by a nucleosome in βTC3 cells. Importantly, an adjacent region of the insulin promoter that is not as crucial for β cell-specific gene transcription (bp −297 to −460 relative to the transcriptional start site) (1) showed comparable accessibility in both cell types, as did the coding region of the β-actin gene (see Fig. 5B); these latter findings confirm that overall digestion by MNase in ChART PCR assays was equivalent in mPAC and βTC3 cells.
Figure 5. Chromatin accessibility by real-time PCR (ChART PCR).

Nuclei (which in total contained 100 ng of DNA) were freshly-isolated from βTC3 and mPAC cells as described in Materials and Methods, and subjected to digestion for 5 min. with varying amounts of MNase. A, 1% agarose gel analysis of total DNA from βTC3 and mPAC cells following MNase digestion with the concentations of MNase (units/reaction) indicated. B, quantitative real-time PCR analysis of residual insulin and β actin gene fragments following MNase digestion with the concentrations of MNase indicated. Locations of the amplified insulin promoter fragments are indicated as base pairs (bp) relative to the transcriptional start site. All data in panel B represent the mean ± S.E. of digestions from nuclei isolated on three separate occasions. “*” indicates that the values for the βTC3 cells shown are statistically different (p<0.05) than the corresponding values for mPAC cells.
Histone acetylation enhances insulin gene occupancy and transcription by Pdx-1 in β cells
Hyperacetylation of histones has been demonstrated to increase the physical accessibility of genes (18, 30). Prior studies demonstrated that treatment of β cell lines with histone deacetylase inhibitors increases insulin transcription (22). To determine whether this increase in transcription might correlate with enhanced accessibility at the insulin gene, we treated βTC3 and mPAC/Pdx cells with the non-specific class 1/2 histone deacetylase inhibitor sodium butyrate (NaBu) (31) to augment histone acetylation. Subsequently, we examined insulin transcription by RT-PCR, as well as histone acetylation and Pdx-1 occupancy at the insulin gene by ChIP. Consistent with other reports (22), Fig. 6A demonstrates that insulin mRNA levels increased almost 5-fold in βTC3 cells at 24 h after treatment with 2.5 mM NaBu, and decrease at 48 h. This increase in message levels was likely secondary to increased gene transcription rather than diminished mRNA breakdown, because levels of insulin pre-mRNA (containing intron 2) were elevated at 24 h (Fig. 6A) as was the recruitment of RNA polymerase II to the promoter (as determined by ChIP, data not shown). Fig. 6B demonstrates that treatment of βTC3 cells with 2.5 mM NaBu for 24 h resulted in approximately 2-3-fold increases in acetylation of histones H3 and H4. Accompanying this increase in acetylation, we observed a 50% increase in Pdx-1 occupancy at the insulin promoter (Fig. 6C). Immunoblot analysis (inset to Fig. 6C) demonstrated that this increase in Pdx-1 occupancy in βTC3 cells was not secondary to an increase in Pdx-1 protein. To determine if histone hyperacetylation might have led to an increase in chromatin accessibility at the insulin promoter, we performed ChART PCR assays using nuclei from control and NaBu-treated βTC3 cells. Fig. 6D shows that NaBu treatment enhanced the sensitivity of the insulin promoter to digestion by MNase, a finding consistent with increased chromatin accessibility. Taken together, these studies suggest that increasing chromatin accessibility may contribute to increasing insulin transcription.
Figure 6. NaBu treatment of βTC3 cells enhances transcription, histone acetylation, and accessibility at the insulin gene.

A, insulin mRNA (closed circles) and pre-mRNA (closed triangles) were measured by real-time RT-PCR at various time points following treatment of βTC3 cells with either 2.5 mM NaBu or vehicle (control). Data represent the mean ± S.E. of three independent experiments, and were normalized to β actin mRNA levels. B, Recovery of the proximal insulin promoter (-126 to -296 bp relative to the transcriptional start site) following quantitative ChIP using antibodies to acetylated H3 or acetylated H4; βTC3 cells were treated with vehicle (control) or NaBu for 24 h. Data represent the mean ± S.E. of three independent ChIP experiments. C, recovery of the proximal insulin promoter following quantitative ChIP using Pdx-1 antibody; βTC3 cells were treated with vehicle (control) or NaBu for 24 h. Data represent the mean ± S.E. of three independent ChIP experiments; inset is an immunoblot showing Pdx-1 protein levels in control- and NaBu-treated βTC3 cells. D, percent residual proximal insulin promoter after digestion of control- and NaBu-treated βTC3 nuclei (100 ng total DNA) with 2 units MNase for 5 min. Data represent the mean ± S.E. of three digestions from nuclei isolated on three independent occasions.
We next performed identical studies in mPAC cells expressing a Pdx-1 transgene (mPAC/Pdx cells). Importantly, treatment of these cells with 2.5 mM NaBu did not detectably enhance insulin transcription (data not shown) or Pdx-1 occupancy at the insulin gene (see Fig. 7A). Although NaBu treatment gave rise to ~3-fold increases in H3 and H4 acetylation at the insulin gene (Fig. 7B), it is noteworthy that the absolute levels of H3 acetylation were nearly 10-fold lower in mPAC cells compared to βTC3 cells. The lower level of H3 acetylation in mPAC cells is consistent with our prior report (21), which suggested that H3 hyperacetylation is a key feature of cells that express the insulin gene. These results suggest that the effect of NaBu in opening chromatin and enhancing transcription at the insulin gene may be dependent upon the pre-existing state histone acetylation.
Figure 7. NaBu treatment of mPAC/Pdx cells enhances histone acetylation, but not accessibility to Pdx-1 at the insulin gene.

A, Recovery of the proximal insulin promoter (-126 to -296 bp relative to the transcriptional start site) following quantitative ChIP using Pdx-1 antibody; mPAC/Pdx cells were treated with vehicle (control) or 2.5 mM NaBu for 24 h. B, recovery of the proximal insulin promoter following ChIP using antibodies to acetylated H3 and acetylated H4; mPAC/Pdx cells were treated with vehicle (control) or NaBu for 24 h. All data represent the mean ± S.E. of three independent ChIP experiments
Pdx-1 localizes to intranuclear regions of euchromatin, but not heterochromatin
Thus far, our data suggest that gene binding and activation by Pdx-1 is likely restricted to euchromatic genetic loci. To determine if the intranuclear localization of Pdx-1 positively correlates with regions of euchromatin, we performed immunofluroescence staining studies in mPAC/Pdx and βTC3 cells. Fig. 8A shows that Pdx-1 (green color) colocalizes with markers of euchromatin (acetylated histone H3, red color) with a nuclear Pearson overlap coefficient (32) of 0.62 in mPAC/Pdx cells and 0.66 in βTC3 cells. By contrast, Pdx-1 shows more disparate localization with a marker of heterochromatin (dimethylated histone H3-Lys9) (Fig. 8B), with significantly lower Pearson overlap coefficients of 0.31 in mPAC/Pdx cells and 0.24 in βTC3 cells. These findings emphasize that gene occupancy by Pdx-1 appears to be preferentially localized to regions of open chromatin.
Figure 8. Pdx-1 in mPAC/Pdx and βTC3 cells localizes to regions of euchromatin.

mPAC and βTC3 cells were fixed and permeabilized, then treated with a mouse monoclonal antibody against Pdx-1 and either rabbit polyclonal antibodies against acetylated H3 (to visualize euchromatin) or dimethylated H3-Lys9 (to visualize heterochromatin), then secondarily treated with antibodies fused to Cy2 (green) and Cy3 (red) recognizing mouse and rabbit Fc portions, respectively. A, an mPAC/Pdx cell (top panels) and a βTC3 cell (bottom panels) were imaged in the green channel to visualize Pdx-1 and the red channel to visualize areas of acetylated H3, then the images were merged. Three-dimensional voxel intensity analysis of the merged image resulted in the Pearson overlap coefficients (r) as indicated in the merged panels. B, an mPAC/Pdx cell and a βTC3 cell were imaged in the green channel to visualize Pdx-1 and the red channel to visualize areas of dimethylated H3-Lys4, then the images were merged. Three-dimensional voxel intensity analysis of the merged image resulted in the Pearson overlap coefficients (r) as indicated in the merged panels.
DISCUSSION
The prevailing paradigm of insulin gene transcription is based on the binding of cell type-specific and ubiquitous transcription factors to individual elements of the insulin promoter, thereby leading to the recruitment and/or activation of basal transcriptional machinery (2, 3, 33). Pdx-1 is perhaps the most extensively-studied of these β cell-specific transcription factors. To the extent that much of the literature regarding the mechanism of gene activation by Pdx-1 is based on reporter gene analysis and EMSA studies, it is not entirely clear if and how such studies may relate to Pdx-1 action on more relevant chromatin templates. Recent studies indicate that chromatin is much more than a mechanism to effect compaction of DNA within the eukaryotic nucleus; among other functions, it serves as a means to control gene expression in a manner independent of the prevailing cell type-specific transcription factors (34). This latter function might explain why, in some cases, the ectopic expression/overexpression of insulin gene transactivators (e.g. Pdx-1, NeuroD1, or MafA) in non-β cell types is insufficient to effect robust gene expression. In this report, we present the first evidence that insulin promoter binding and transactivation by Pdx-1 is dependent upon the nature of chromatin, and that insulin chromatin in pancreatic ductal cells appears closed and inhibitory compared to β cells.
At its most basic level, chromatin is made up of nucleosomes (histone octamers and DNA) that occur at variable intervals (35). Our in vitro EMSA studies demonstrate the Pdx-1 exhibits a dramatic reduction in DNA binding affinity when a nucleosome is positioned over the insulin A4/A3 element. The presence of the nucleosome itself is not the inhibitory factor, however, because placement of the A4/A3 element at the very end of the nucleosome led to almost complete recovery of DNA binding affinity. These results emphasize that relatively subtle positioning of the nucleosome can have a dramatic impact on DNA binding by Pdx-1. We should point out that our studies do not address higher order chromatin structure (i.e. interactions between nucleosomes caused by linker histones and other protein-protein interactions), although it is expected that the presence of such structure might only inhibit DNA accessibility further (35). Whether a transcription factor can bind or not to a specific DNA sequence embedded within a nucleosome appears to be dependent upon several variables. First, some transcription factors, such as HNF3, have the intrinsic capacity to bind to histone proteins and thereby disrupt the compaction of the chromatin (17). Our EMSA studies using mononucleosomes suggest that Pdx-1 has little intrinsic capacity to bind to histones or effect nucleosomal repositioning. Consistent with this observation, we observed Pdx-1 to localize primarily to regions of euchromatin in the nucleus, and not to regions of nucleosome-rich heterochromatin.
A second mechanism that may allow for transcription factor binding in the presence of chromatin is a transitory “release” of DNA from the core histones (16). It has been demonstrated that regions of chromatin that are hyperacetylated at the core histones display this more dynamic picture (18, 30). We hypothesized that the hyperacetylation of histones that is observed at the proximal insulin promoter in β cells (21, 22) may allow for functionally reduced nucleosome occupancy and thus permit DNA binding by Pdx-1 in this cell type. Our ChART PCR assays demonstrate that the region of the promoter (bp -126 to -296 relative to the transcriptional start site) encompassing the A4/A3 element is more accessible in βTC3 cells compared to mPAC cells. As acetylation of the insulin promoter in βTC3 cells is enhanced by treatment with NaBu, we observed enhanced Pdx-1 occupancy and substantially greater chromatin accessibility. By contrast, in mPAC cells we observed an increase in H3 and H4 acetylation, but no increase in insulin gene transcription. Importantly, however, basal and NaBu-stimulated H3 acetylation in mPAC cells was nearly 10-fold lower compared to βTC3 cells. This observation should be considered in the context of our prior report in 2003 (21), which demonstrated that insulin gene activation is marked by H3 hyperacetylation, but not H4 hyperacetylation. Thus, we believe that the inability of NaBu to substantially increase both Pdx-1 occupancy and insulin gene activity in mPAC cells may result from the relative paucity of H3 acetylation. We also note that our ChIP and MNase assays may not be sensitive enough to detect very small increases in accessibility upon addition of NaBu.
Interestingly, a more upstream region of the promoter (bp -297 to -460) is equivalently accessible to MNase digestion in both cell types, emphasizing that chromatin accessibility can vary substantially along the regulatory regions of genes (20). We recognize that our studies in cell lines do not address specific placement of nucleosomes as a mechanism for the variability in accessibility across the insulin gene locus; however, our binding studies using Pdx-1 and reconstituted chromatin in vitro do suggest that chromatin accessibility could be influenced by the precise positioning of nucleosomes.
A third mechanism that may enhance the binding of transcription factors to chromatin is the recruitment of chromatin-modifying enzymes to specific chromatin loci. In this regard, we have recently demonstrated that Pdx-1 recruits the histone H3-Lys4 methyltransferase Set9 to the insulin gene (5), and others have demonstrated recruitment of the histone acetyltransferase p300 by Pdx-1 (7). It would be expected that these recruitments lead to specific covalent modifications of histones (H3-Lys4 methylation and H3/H4 acetylation, respectively) that ultimately increase chromatin accessibility; at the insulin locus, this would facilitate DNA binding by not only Pdx-1, but also by other transcription factors, which themselves may recruit other still other chromatin modifying enzymes. Thus, a chain reaction of this nature during cellular differentiation may lead to the ultimate structure of chromatin that is observed in β cells. This structure is characterized by hyperacetylation of histones H3 and H4, hypermethylation of H3-Lys4, hypomethylation of H3-Lys9, and a relatively accessible proximal promoter region (refs. (5, 21, 22) and this report). Importantly, we did not observe Pdx-1 occupancy at the insulin gene in ductal mPAC cells even after overexpression of Pdx-1 protein; this finding is consistent with the observation that the insulin promoter in mPAC cells is hypoacetylated at H3, hypomethylated at H3-Lys4, hypermethylated at H3-Lys9, and exhibits reduced accessibility (ref. (21) and this report). Because ductal cells express only a subset of histone modifying enzymes and transcription factors found in the β cell (5, 11, 12, 21), we believe that our results emphasize the necessity of having the full complement of such proteins in order to achieve the most favorable chromatin conformation for gene expression.
Taken together, our data in this study suggest that optimal engineering of new β cells may require at least two considerations: (a) starting with an appropriate cell type that harbors histone modifications and overall chromatin accessibility (i.e. a “chromotype”) at key β cell genes that are similar to that of a mature β cell, and (b) co-expression of chromatin-modifying factors that are either enriched in the islet (e.g. the methyltransferase Set9, ref. (5)) or have catalytic activities that modify nucleosome positioning (e.g. ATP-dependent chromatin modulators, ref. (15)). We should emphasize that because our studies utilized cell lines, our conclusions may or may not apply to native islet β cells. In future studies, we propose to investigate how chromatin reorganization may enhance the β cell-like characteristics of transdifferentiated cells.
MATERIALS AND METHODS
Antibodies and vectors
Rabbit polyclonal antiserum against Pdx-1 was a gift from Dr. M. German (University of California, San Francisco). Mouse monoclonal anti-Pdx-1 antiserum was purchased from R&D Systems. Rabbit polyclonal antisera against acetylated histones H3 and H4, and H3-dimethylK9 were purchased from Upstate Biotechnology. RNA Polymerase II (N-20) antiserum was purchased from Santa Cruz Biotechnology. Cy2-conjugated donkey anti-mouse and Cy3-conjugated goat anti-rabbit antibodies were purchased from Jackson ImmunoResearch.
Vector pET.Pdx1 (for generation of His6-tagged Pdx-1 protein in E. coli) was constructed by PCR amplification of the hamster Pdx-1 cDNA from vector pBAT12.Pdx1, and subsequently subcloning it into the NdeI and XhoI sites of vector pET15b. To generate CMV promoter-driven expression constructs for live cell imaging, cDNAs encoding Pdx1 and Nkx6.1 were subcloned in-frame with the cDNA encoding the monomeric variant green fluorescent protein (GFP) in vector pEGFP (Clontech). The 5S ribosomal array fragment containing tandem mutant A4/A3 elements for nucleosome reconstitutions was generated by excising the mutant element cassette (5mEF1) from vector pFoxLucRIP.5mEF1 and subcloning it into the positioning vector pIC-2085S (26) to generate pIC-5mEF1. For reconstitutions, the ~2500 bp array insert was excised from pIC-5mEF1 using enzymes PvuI and ClaI and purified by preparative agarose gel electrophoresis. The array fragment containing the wild-type A4/A3 element was excised from vector pIC-5FF as described previously (5).
Cell culture and transfections
The mouse insulinoma cell line βTC3 and the mouse ductal cell line mPAC L20 were maintained in Dulbecco’s modified Eagle’s medium as previously reported (8, 24). The human cervical carcinoma cell line HeLa was maintained in Dulbecco’s modified Eagle’s medium supplemented with 10% newborn calf serum, 1% sodium pyruvate, and 1% non-essential amino acids (Invitrogen). Where indicated, cells were cultured for 24 hours in medium containing 2.5 mM NaBu. Pancreatic islets were hand-picked from collagenase-digested 6-8-week old CD-1 mouse pancreas (36) using a protocol approved by the Institutional Animal Care and Use Committee. Islets were cultured in RPMI medium containing 25 mM glucose overnight, then used in ChIP experiments.
Quantitative chromatin immunoprecipitation (ChIP)
ChIP assays, including quantitation of coimmunoprecipitated DNA fragments by real-time PCR using the threshold cycle methodology, were performed as previously described (8, 21). ChIP assays were performed on at least 3 independent occasions; for each ChIP assay, recovery of promoter samples were quantitated in triplicate. Forward and reverse primer sequences, respectively, used for PCR amplification of the proximal insulin promoter (-126 to -296) were: 5’-TCAGCCAAAGATGAAGAAGGTCTC-3’ and 5’-TCCAAACACTTGCCTGGTGC-3’.
Chromatin reconstitution, RT-PCR, and in vitro transcription
Chromatin reconstitutions of mouse insulin I promoter fragments and 5S ribosomal array fragments were performed using HeLa-purified histone octamers and a sequential salt dilution procedure as previously described (26). Real time, SYBR Green I-based reverse transcriptase (RT)-PCR was performed using forward and reverse primers to quantitate total insulin and β-actin mRNAs as we detailed previously (4). PCRs were cycled 40 times using the following conditions: 95°C for 15s, 64°C for 1 min. Samples containing no reverse transcriptase were run in parallel reactions to assay for genomic DNA contamination. In all instances, samples without reverse transcriptase exhibited threshold cycle numbers greater than 40, indicating negligible amounts of genomic DNA contamination. In vitro transcription reactions were performed and quantitated for luciferase transcription as we detailed previously (5). To ensure that transcriptional initiation is being measured in these assays, this procedure involves real-time PCR quantitation of luciferase transcript following a modified 5’ rapid amplification of cDNA ends (5’ RACE) protocol.
Protein purification
Pdx-1 protein was prepared as N-terminal, His6-tagged protein from E. coli BL21(DE3)/pLysS cells transformed with vector pET.Pdx1. Protein was purified using nickel-nitrolotriacetic acid agarose (Qiagen) as directed by the manufacturer. Protein exhibited >90% purity as assessed by electrophoresis on a 12% polyacrylamide gel stained with Coomassie Blue. Protein concentration was determined by the method of Bradford (37).
Nuclear extracts and immunoblot analysis
Nuclear extracts were prepared from single, confluent 10-cm plates of cells according to methods described previously (38). 5 μg of nuclear extract were subject to immunoblot analysis after electrophoresis on a 15% SDS-polyacrylamide gel using polyclonal anti-Pdx-1 antiserum. Western blots were visualized using the ECL-Plus® system (Amersham Pharmacia Biotech).
Electrophoretic mobility shift assays
EMSA reactions were performed as described previously in 20 μl volumes at room temperature, followed by analysis on 5% polyacrylamide gels (9, 39). Short oligonucleotide probes were generated by 5’ end-labeling single-stranded oligonucleotides with T4 polynucleotide kinase and γ32P-ATP, and then annealing to an excess of unlabeled complementary strand. The following oligonucleotides were used (top strands shown): wild-type mouse insulin 1 promoter (containing the A4/A3 element): 5’-CTTATTAAGACTATAATAACCCTAAGACTA-3’; mutant mouse insulin 1 promoter (mutations underlined): 5’-CTTACTAAGACTATAGTAACCCTAAGACTA-3’.
To generate the insulin 1 promoter probe for EMSA and mononucleosome studies, two fragments of the mouse insulin 1 5’ regulatory region were amplified by PCR from plasmid DNA in the presence of α-32P-ATP. Probe 1 (-126 to -296 bp relative to the transcriptional start site) was generated using the forward and reverse primers identified above in the ChIP assays section.: 5’-TCAGCCAAAGATGAAGAAGGTCTC-3’ and 5’-TCCAAACACTTGCCTGGTGC-3’. Probe 2 (-188 to -368 bp relative to the transcriptional start site) was generated using the following forward and reverse primers: 5’-CTATCAATGGGAACTGTGAAAC-3’ and 5’-GTTATTATAGTCTTAATAAGGGAC-3’. Radiolabeled DNA fragments or mononucleosomes were incubated with varying concentrations of bacterially-purified Pdx-1 protein for 15 min at room temperature in EMSA buffer (10 mM HEPES pH 7.9, 75 mM KCl, 2.5 mM MgCl2, 0.1 mM EDTA, 1 mM DTT, 3% Ficoll, and 50 ng/μl poly dI-dC). Binding reactions were analyzed by 4% polyacrylamide (60:1 acrylamide:bisacrylamide) gel electrophoresis and subjected to autoradiography. Apparent KDs were estimated by quantitating free and bound probes using a Typhoon phosphorimager (Molecular Dynamics), as described previously (39).
Chromatin accessibility by real-time PCR (ChART PCR)
ChART PCR assays were performed similar to previously published studies (28), but with some modifications. βTC3 and mPAC monolayers were cultured in the presence or absence of 2.5 mM NaBu for 24 hours prior to harvesting. Nuclei from cells cultured in the presence of NaBu were isolated as previously described (40) in buffers containing 2.5 mM NaBu. Nuclei (containing 100 ng of total DNA, as measured by UV absorbance at 260 nm after lysis of an aliquot with 1% SDS) were aliquoted into 75 μl of MNase buffer (10 mM Tris pH 7.5, 4 mM MgCl2, 1 mM CaCl2, 0.32 M sucrose) and digested with 0.1, 0.5, 1, or 2 units of micrococcal nuclease (Sigma) for 5 minutes at room temperature. The reaction was stopped by addition of 100 μl stop solution (20 mM EDTA, 2% SDS, 0.7 mg/ml proteinase K, 0.45 mg/ml glycogen, 0.45 ng/μl pCMVβGal plasmid) and incubated at 37°C for 2 hours to completely digest all protein. DNA was purified by extraction with phenol:chloroform:isoamyl alcohol and ethanol-precipitated. The DNA pellet was resuspended in 100 μl of TE and subjected to either analysis by 1% agarose gel electrophoresis (Fig. 5A) real time PCR amplification for insulin and β actin genes (Fig. 5B). The following primers were used: mouse insulin 1 distal region (-297 to −460): TACCTTGCTGCCTGAGTTCTGC-3’ and 5’-GCATTTTCCACATCATTCCCC-3; mouse insulin 1 proximal region (-126 to -296): same as for Probe 1 above; mouse β actin: 5’-AGGTCATCACTATTGGCAACGA-3’ and 5’-CACTTCATGATGGAATTGAATGTAGTT-3’. Because undigested genomic DNA displayed poor recovery during this procedure, digestion using a limiting amount of MNase (0.1 U) was defined as “100%” in the data in Figs. 5 and 6. To control for small variations in DNA recovery during this procedure, data were normalized to the recovery of the pCMVβGal plasmid, as assessed by real-time PCR using primers specific for the LacZ coding region: 5’-TCAATCCGCCGTTTGTTCCCAC-3’ and 5’-TCCAGATAACTGCCGTCACTCCAAC-3’.
Immunocytochemistry
Immunocytochemical analysis of Pdx1, acetylated histone H3 and dimethylated H3-Lys9 in βTC3 and mPAC cells was performed essentially as described (41). The following concentrations of primary antibodies were used: 1 μg/ml anti-human/mouse Pdx-1 monoclonal antibody, and 5 μg/ml anti-acetylated-histone H3 and anti-dimethylated H3-Lys9 antibodies. Secondary antibodies (Cy2-conjugated anti-mouse and Cy3-conjugated anti-rabbit) were used at 1:200 dilution. Images were acquired with a Zeiss LSM 510 confocal microscope using narrow bandpass filters 450-515 for Cy2 and 546-590 nm for Cy3, respectively. Volocity® software (Improvision) was used for image analysis.
Acknowledgments
We gratefully acknowledge Drs. R. Day and I. Demarco for assistance with the fluorescence microscopy experiments. We also thank Dr. M. German for provision of the polyclonal Pdx-1 antiserum.
This work was supported by grants R01 DK60581 (to R.G.M), T32 GM007055 (to D.A.B and T.G.D), and T32 DK0732025 (to C.E.M.) from the National Institutes of Health, and by a Thomas R Lee Career Development Award from the American Diabetes Association and a grant from the Goldman Philanthropic Partnerships and Meade Family (both to R.G.M.)
ABBREVIATIONS
- 5’ RACE
5’ rapid amplification of cDNA ends
- bp
base pairs
- ChIP
chromatin immunoprecipitation
- ChART PCR
chromatin accessibility by real-time PCR
- EMSA
electrophoretic mobility shift assay
- GFP
green fluorescent protein
- MNase
micrococcal nuclease
- NaBu
sodium butyrate
- RT
reverse transcriptase
Footnotes
Disclosure Statement: The authors have nothing to disclose
Note: the PCR primers used in these studies equally and efficiently amplified the mouse insulin 1 and insulin 2 genes. Unless otherwise stated, the reference to the insulin gene throughout the text refers to both mouse genes.
The histone octamers used in these chromatin reconstitutions were largely unacetylated, based on reactivity to antiacetylated H3 and H4 antiserum in immunoblots (data not shown).
References
- 1.Karlsson O, Edlund T, Moss JB, Rutter WJ, Walker MD. A mutational analysis of the insulin gene transcription control region: expression in beta cells is dependent on two related sequences within the enhancer. Proc Natl Acad Sci U S A. 1987;84:8819–8823. doi: 10.1073/pnas.84.24.8819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Melloul D, Marshak S, Cerasi E. Regulation of insulin gene transcription. Diabetologia. 2002;45:309–326. doi: 10.1007/s00125-001-0728-y. [DOI] [PubMed] [Google Scholar]
- 3.Ohneda K, Ee H, German M. Regulation of insulin gene transcription. Semin Cell Dev Biol. 2000;11:227–233. doi: 10.1006/scdb.2000.0171. [DOI] [PubMed] [Google Scholar]
- 4.Iype T, Francis J, Garmey JC, Schisler JC, Nesher R, Weir GC, Becker TC, Newgard CB, Griffen SC, Mirmira RG. Mechanism of iinsulin gene regulation by the pancreatic transcription factor Pdx-1: application of pre-mRNA analysis and chromatin immunoprecipitation to assess formation of functional transcriptional complexes. J Biol Chem. 2005;280:16798–16807. doi: 10.1074/jbc.M414381200. [DOI] [PubMed] [Google Scholar]
- 5.Francis J, Chakrabarti SK, Garmey JC, Mirmira RG. Pdx-1 links histone H3-Lys-4 methylation to RNA polymerase II Eeongation during activation of insulin transcription. J Biol Chem. 2005;280:36244–36253. doi: 10.1074/jbc.M505741200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Qiu Y, Guo M, Huang S, Stein R. Insulin Gene Transcription Is Mediated by Interactions between the p300 Coactivator and PDX-1, BETA2, and E47. Mol Cell Biol. 2002;22:412–20. doi: 10.1128/MCB.22.2.412-420.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mosley AL, Corbett JA, Ozcan S. Glucose regulation of insulin gene expression requires the recruitment of p300 by the beta-cell-specific transcription factor Pdx-1. Mol Endocrinol. 2004;18:2279–2290. doi: 10.1210/me.2003-0463. [DOI] [PubMed] [Google Scholar]
- 8.Chakrabarti SK, James JC, Mirmira RG. Quantitative assessment of gene targeting in vitro and in vivo by the pancreatic transcription factor, Pdx1. Importance of chromatin structure in directing promoter binding. J Biol Chem. 2002;277:13286–13293. doi: 10.1074/jbc.M111857200. [DOI] [PubMed] [Google Scholar]
- 9.Ohneda K, Mirmira RG, Wang J, Johnson JD, German MS. The homeodomain of PDX-1 mediates multiple protein-protein interactions in the formation of a transcriptional activation complex on the insulin promoter. Mol Cell Biol. 2000;20:900–911. doi: 10.1128/mcb.20.3.900-911.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Peshavaria M, Gamer L, Henderson E, Teitelman G, Wright CV, Stein R. XIHbox 8, an endoderm-specific Xenopus homeodomain protein, is closely related to a mammalian insulin gene transcription factor. Mol Endocrinol. 1994;8:806–16. doi: 10.1210/mend.8.6.7935494. [DOI] [PubMed] [Google Scholar]
- 11.Gasa R, Mrejen C, Leachman N, Otten M, Barnes M, Wang J, Chakrabarti S, Mirmira R, German M. Proendocrine genes coordinate the pancreatic islet differentiation program in vitro. Proc Natl Acad Sci U S A. 2004;101:13245–13250. doi: 10.1073/pnas.0405301101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Heremans Y, Van De Casteele M, in’t Veld P, Gradwohl G, Serup P, Madsen O, Pipeleers D, Heimberg H. Recapitulation of embryonic neuroendocrine differentiation in adult human pancreatic duct cells expressing neurogenin 3. J Cell Biol. 2002;159:303–312. doi: 10.1083/jcb.200203074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hui H, Wright C, Perfetti R. Glucagon-like peptide 1 induces differentiation of islet duodenal homeobox-1-positive pancreatic ductal cells into insulin-secreting cells. Diabetes. 2001;50:785–96. doi: 10.2337/diabetes.50.4.785. [DOI] [PubMed] [Google Scholar]
- 14.Muthurajan UM, Bao Y, Forsberg LJ, Edayathumangalam RS, Dyer PN, White CL, Luger K. Crystal structures of histone Sin mutant nucleosomes reveal altered protein-DNA interactions. EMBO J. 2004;23:260–271. doi: 10.1038/sj.emboj.7600046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vignali M, Hassan AH, Neely KE, Workman JL. ATP-dependent chromatin-remodeling complexes. Mol Cell Biol. 2000;20:1899–1910. doi: 10.1128/mcb.20.6.1899-1910.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Anderson JD, Thastrom A, Widom J. Spontaneous access of proteins to buried nucleosomal DNA target sites occurs via a mechanism that is distinct from nucleosome translocation. Mol Cell Biol. 2002;22:7147–7157. doi: 10.1128/MCB.22.20.7147-7157.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cirillo LA, Lin FR, Cuesta I, Friedman D, Jarnik M, Zaret KS. Opening of compacted chromatin by early developmental transcription factors HNF3 (FoxA) and GATA-4. Mol Cell. 2002;9:279–289. doi: 10.1016/s1097-2765(02)00459-8. [DOI] [PubMed] [Google Scholar]
- 18.Anderson JD, Lowary PT, Widom J. Effects of histone acetylation on the equilibrium accessibility of nucleosomal DNA target sites. J Mol Biol. 2001;307:977–85. doi: 10.1006/jmbi.2001.4528. [DOI] [PubMed] [Google Scholar]
- 19.Lieb JD, Clarke ND. Control of transcription through intragenic patterns of nucleosome composition. Cell. 2005;123:1187–1190. doi: 10.1016/j.cell.2005.12.010. [DOI] [PubMed] [Google Scholar]
- 20.Yuan GC, Liu YJ, Dion MF, Slack MD, Wu LF, Altschuler SJ, Rando OJ. Genome-scale identification of nucleosome positions in S. cerevisiae Science. 2005;309:626–630. doi: 10.1126/science.1112178. [DOI] [PubMed] [Google Scholar]
- 21.Chakrabarti SK, Francis J, Ziesmann SM, Garmey JC, Mirmira RG. Covalent histone modifications underlie the developmental regulation of insulin gene transcription in pancreatic beta cells. J Biol Chem. 2003;278:23617–23623. doi: 10.1074/jbc.M303423200. [DOI] [PubMed] [Google Scholar]
- 22.Mosley AL, Ozcan S. Glucose regulates insulin gene transcription by hyperacetylation of histone H4. J Biol Chem. 2003;278:19660–19666. doi: 10.1074/jbc.M212375200. [DOI] [PubMed] [Google Scholar]
- 23.Efrat S, Linde S, Kofod H, Spector D, Delannoy M, Grant S, Hanahan D, Baekkeskov S. Beta-cell lines derived from transgenic mice expressing a hybrid insulin gene-oncogene. Proc Natl Acad Sci U S A. 1988;85:9037–9041. doi: 10.1073/pnas.85.23.9037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yoshida T, Hanahan D. Murine pancreatic ductal adenocarcinoma produced by in vitro transduction of polyoma middle T oncogene into the islets of Langerhans. Am J Pathol. 1994;145:671–684. [PMC free article] [PubMed] [Google Scholar]
- 25.Levinger LF, Carter CW., Jr Superstructural differences between chromatin in nuclei and in solution are revealed by kinetics of micrococcal nuclease digestion. J Biol Chem. 1979;254:9477–9487. [PubMed] [Google Scholar]
- 26.Steger DJ, Eberharter A, John S, Grant PA, Workman JL. Purified histone acetyltransferase complexes stimulate HIV-1 transcription from preassembled nucleosomal arrays. Proc Natl Acad Sci U S A. 1998;95:12924–12929. doi: 10.1073/pnas.95.22.12924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.German MS, Wang J, Chadwick RB, Rutter WJ. Synergistic activation of the insulin gene by a LIM-homeo domain protein and a basic helix-loop-helix protein: building a functional insulin minienhancer complex. Genes Dev. 1992;6:2165–276. doi: 10.1101/gad.6.11.2165. [DOI] [PubMed] [Google Scholar]
- 28.Rao S, Procko E, Shannon MF. Chromatin remodeling, measured by a novel real-time polymerase chain reaction assay, across the proximal promoter region of the IL-2 gene. J Immunol. 2001;167:4494–4503. doi: 10.4049/jimmunol.167.8.4494. [DOI] [PubMed] [Google Scholar]
- 29.Liang MD, Zhang Y, McDevit D, Marecki S, Nikolajczyk BS. The IL-1beta gene is transcribed from a poised promoter architecture in monocytes. J Biol Chem. 2006;281:9227–9237. doi: 10.1074/jbc.M510700200. [DOI] [PubMed] [Google Scholar]
- 30.Gui CY, Dean A. Acetylation of a specific promoter nucleosome accompanies activation of the epsilon-globin gene by beta-globin locus control region HS2. Mol Cell Biol. 2001;21:1155–1163. doi: 10.1128/MCB.21.4.1155-1163.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Thiagalingam S, Cheng KH, Lee HJ, Mineva N, Thiagalingam A, Ponte JF. Histone deacetylases: unique players in shaping the epigenetic histone code. Ann N Y Acad Sci. 2003;983:84–100. doi: 10.1111/j.1749-6632.2003.tb05964.x. [DOI] [PubMed] [Google Scholar]
- 32.Manders EMM, Verbeek FJ, Aten JA. Measurement of co-localization of objects in dual-colour confocal images. J Microsc. 1993;169:375–382. doi: 10.1111/j.1365-2818.1993.tb03313.x. [DOI] [PubMed] [Google Scholar]
- 33.Chakrabarti SK, Mirmira RG. Transcription factors direct the development and function of pancreatic beta cells. Trends Endocrinol Metab. 2003;14:78–84. doi: 10.1016/s1043-2760(02)00039-5. [DOI] [PubMed] [Google Scholar]
- 34.Fischle W, Wang Y, Allis CD. Histone and chromatin cross-talk. Curr Opin Cell Biol. 2003;15:172–183. doi: 10.1016/s0955-0674(03)00013-9. [DOI] [PubMed] [Google Scholar]
- 35.Wolffe AP, Guschin D. Review: chromatin structural features and targets that regulate transcription. J Struct Biol. 2000;129:102–22. doi: 10.1006/jsbi.2000.4217. [DOI] [PubMed] [Google Scholar]
- 36.Yang Z, Chen M, Ellett JD, Fialkow LB, Carter JD, Nadler JL. The novel anti-inflammatory agent lisofylline prevents autoimmune diabetic recurrence after islet transplantation. Transplantation. 2004;77:55–60. doi: 10.1097/01.TP.0000104844.48064.81. [DOI] [PubMed] [Google Scholar]
- 37.Bradford MM. A Rapid and Sensitive Method for the Quantitation of Microgram Qantities of Protein Utilizing the Priciple of Protein-Dye Binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 38.Sadowski HB, Gilman MZ. Cell-free activation of a DNA-binding protein by epidermal growth factor. Nature. 1993;362:79–83. doi: 10.1038/362079a0. [DOI] [PubMed] [Google Scholar]
- 39.Taylor DG, Babu D, Mirmira RG. The C-terminal domain of the beta cell homeodomain factor Nkx6.1 enhances sequence-selective DNA binding at the insulin promoter. Biochemistry. 2005;44:11269–11278. doi: 10.1021/bi050821m. [DOI] [PubMed] [Google Scholar]
- 40.Lefebvre B, Brand C, Lefebvre P, Ozato K. Chromosomal integration of retinoic acid response elements prevents cooperative transcriptional activation by retinoic acid receptor and retinoid X receptor. Mol Cell Biol. 2002;22:1446–1459. doi: 10.1128/mcb.22.5.1446-1459.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Voss TC, Demarco IA, Booker CF, Day RN. Quantitative methods to analyze subnuclear protein organization in cell populations with varying degrees of protein expression. J Biomed Opt. 2005;10:024011. doi: 10.1117/1.1891085. [DOI] [PMC free article] [PubMed] [Google Scholar]