

Abstract
Opioids increase dopamine release in the brain through inhibition of GABA-A IPSCs onto dopamine cells. Immunolabeling indicates that GABA neurons in the rostromedial tegmental nucleus (RMTg), also known as the tail of the ventral tegmental area, send a dense projection to midbrain dopamine neurons stain for μ-opioid receptors. There is however, little functional evidence that these neurons play a role in the opioid-dependent increase in dopamine neuron activity. The present study used retrograde tracers injected into the ventral tegmental area and substantia nigra (VTA/SN) to identify RMTg neurons that project to the VTA/SN. Whole-cell current-clamp and cell-attached recordings from labeled RMTg neurons were performed in sagittal slices from rat. The rhythmic spontaneous firing rate of RMTg neurons was decreased and the membrane potential was hyperpolarized in response to application of μ-opioid agonist DAMGO. Agonists that act at κ- and δ-opioid receptors (U69593 and DPDPE) failed to hyperpolarize RMTg neurons. Whole-cell recordings made in dopamine neurons revealed rhythmic, large amplitude spontaneous IPSCs that had a similar frequency, pattern and opioid sensitivity to the firing of RMTg neurons. In addition, electrical and channelrhodopsin-2 stimulation within the RMTg evoked GABA-A IPSCs in dopamine neurons that were inhibited by μ-opioid agonists DAMGO, but not κ- and δ-opioid agonists. Thus, this study demonstrates functional connection from the RMTg to the VTA/SN mediated by a dense, opioid-sensitive GABA innervation, and that the RMTg is a key structure in the μ-opioid receptor-dependent regulation of dopamine neurons.
Introduction
Many drugs of abuse, including opioids, increase the activity of dopamine neurons resulting in increased dopamine release in the mesocorticolimbic system. Dopamine release specifically in the nucleus accumbens is thought to be essential for reward learning and goal-oriented behavior (Di Chiara and Imperato, 1988; Nicola and Malenka, 1997; Thomas et al., 2001). Thus, the synaptic regulation of dopamine neurons is a key initial step in reward circuits leading to drug addiction (Wise, 1996; Di Chiara et al., 2004). Drugs of abuse also induce adaptive changes that alter the synaptic strength of inputs to dopamine neurons. For example, the probability of GABA release was increased during withdrawal in slices from animals treated chronically with morphine (Bonci and Williams, 1997). In addition, glutamate synaptic transmission was enhanced following treatment of animals with many drugs of abuse, including opioids (Saal et al., 2003). Characterizing the synaptic mechanisms by which opioids alter the excitability of dopamine neurons is a necessary first step in understanding the resulting adaptive changes.
It is well known that μ-opioid agonists inhibit GABA input onto dopamine neurons (Johnson and North, 1992a; Shoji et al., 1999; Ford et al., 2006; Madhavan et al., 2010). The disinhibition results in increased firing of dopamine neurons. The opioid-sensitive cells that underlie this disinhibition were thought to be GABA-interneurons within the ventral tegmental area and substantia nigra (VTA/SN; Johnson and North, 1992a). There is, however, no direct evidence of a functional connection between GABA interneurons and dopamine neurons. Recently, the rostromedial tegmental nucleus (RMTg), also known as tail of VTA, has been recognized as the major GABA afferent to dopamine neurons (Jhou et al., 2009b; Kaufling et al., 2009). The RMTg was originally defined as an area that was caudal and medial to the VTA that was marked by a robust increase in early response genes such as cFOS and Δ-FOS expression following an injection of cocaine or amphetamine (Perrotti et al., 2005; Geisler et al., 2008; Kaufling et al., 2010a,b). Antero- and retrograde tracing studies revealed that RMTg neurons receive a strong glutamate input from lateral habenula and send a dense GABA projection to dopamine neurons in the VTA/SN (Jhou et al., 2009b; Kaufling et al., 2009; Balcita-Pedicino et al., 2011). Stimulation within the lateral habenula in vivo inhibited dopamine neurons, an action that was mediated by GABA-A receptors (Ji and Shepard, 2007). Aversive stimuli increased the activity of lateral habenula neurons and decreased the activity of dopamine neurons. Finally, the observation that neurons in the medial RMTg were immunopositive for μ-opioid receptors suggested that this GABAergic pathway could be another opioid-sensitive pathway that mediates the disinhibition of dopamine cell activity. This study indicates that the RMTg is another opioid-sensitive inhibitory relay to dopamine neurons.
Materials and Methods
All procedures were performed in accordance with the guidelines of the Oregon Health & Science University and the Animal Care and Use Committees approved all of the experimental procedures.
Intracerebral microinjections.
Male and female Sprague Dawley rats (postnatal day 20–23, Charles River Laboratories) were used in all experiments. Rats were anesthetized with isoflurane and immobilized in a Stereotaxic Alignment System (Kopf Instruments). A solution of fluorescent microspheres (100 nl; 0.04 μm diameter; excitation/emission = 488/560; Invitrogen) or cholera toxin subunit B conjugated to Alexa Fluor 488 (200 nl of 0.1% in PBS; Invitrogen) was injected bilaterally over 5–10 min into VTA [from bregma (in mm); −4.35 anteroposterior, ±0.8 lateral, −7.5 ventral]. Successful recordings were made from 30 of 66 injected rats. Channelrhodopsin-2 (ChR2) was expressed using adeno-associated virus constructs prepared by the Vollum Institute Viral Core (Portland, OR). The vector AAV-CAG-ChR2-Venus (100 nl; 5 × 1012 genomes/ml) was injected bilaterally using a Nanoject II (Drummond Scientific Company) into RMTg [from bregma (in mm); −5.7 anteroposterior, ±0.8 lateral, −7.25 ventral]. Successful ChR2 stimulation experiments were made from 21 of 29 injected rats. After 7–10 d acute brain slices were prepared. Animals were excluded from the study if injections missed the target.
Slice preparation and recording.
Adult rats (150–300 g) were anesthetized with isoflurane and killed. Brains were quickly removed and placed in a vibratome (Leica). An additional 36 untreated rats were used for recordings of spontaneous IPSCs and electrical stimulation experiments. Midbrain sagittal slices (220 μm) were prepared in ice-cold physiological solution containing the following (in mm): 126 NaCl, 2.5 KCl, 1.2 MgCl2, 2.4 CaCl2, 1.4 NaH2PO4, 25 NaHCO3, 11 d-glucose, and 0.005 MK-801. Slices were incubated in warm (35°C) 95%O2/5%CO2 oxygenated saline containing MK-801 (10 μm) for at least 30 min. Slices containing midbrain were then transferred to the recording chamber that was constantly perfused with 35°C 95%O2/5%CO2 oxygenated saline solution at the rate of 1.5–2 ml/min.
RMTg and midbrain dopamine neurons were visualized with a 40 × water-immersion objective on an upright fluorescent microscope (BX51WI, Olympus USA) equipped with gradient contrast infrared optics. Wide field activation of ChR2 was achieved with collimated light from an LED (470 nm; Thorlabs). A 100- to 200-μm-diameter focal laser spot (473 nm laser, IkeCool) was used to activate a ChR2 within RMTg nucleus using 4× objective. Physiological identification of dopamine neurons was based on the presence of D2 receptor-mediated GIRK currents and the rate of spontaneous action potential activity (1–5 Hz) with spike widths ≥1.2 ms (Ungless et al., 2004; Ford et al., 2006; Chieng et al., 2011).
Whole-cell and cell-attached recordings were made from RMTg or dopamine neurons using an Axopatch-1D amplifier (Molecular Devices). All recording were digitized at 10 kHz and filtered at 5 kHz. GABA-A IPSCs were recorded with patch pipettes (2–2.5 MΩ) filled with an internal solution containing the following (in mm): 115 KCl, 20 NaCl, 1.5 MgCl2, 5 HEPES (K), 10 BAPTA, 2 ATP, 0.2 GTP, and 10 phosphocreatine, pH 7.4, 280 mOsm. Cells were voltage-clamped at −60 mV. For the recordings from RMTg, KCl was replaced with K-methylsulfate. Series resistance was monitored throughout the experiment (range; 3–15 MΩ), and compensated by 80%. A monopolar saline-filled glass electrode (3–6 MΩ) was used to evoke synaptic release. GABA-A IPSCs were evoked with electric stimulation or presynaptic depolarization induced by ChR2 activation (2 stimuli at 20 Hz; every 20 s). GABA-A IPSCs were pharmacologically isolated with DNQX (10 μm).
Immunohistochemistry.
Identification of the RMTg was based on the stimulation of cFOS activity using d-amphetamine (4 mg/kg, subcutaneous) 2 h before killing the animals and removing the brain (10 animals including saline controls). Brains were postfixed in 2% paraformaldehyde in PBS overnight at 4°C. Sagittal brain sections (100 μm) that included the RMTg, VTA/SN were cut in PBS. Free-floating sections were washed three times for 20 min in PBS and blocked with 0.3% Triton-X with 5% normal goat serum for 4 h. Sections were incubated with rabbit anti-cFOS (1:2000; Santa Cruz Biotechnology) and mouse anti-TH (1:1000; IncStar) overnight. Sections were washed three times in PBS (20 min each) and incubated in Alexa-647 anti-rabbit and Alexa-555 anti-mouse secondary antibody (1:1000; Invitrogen) for 2 h. Sections were washed with PBS and mounted with anti-fade gel mount (Biomeda), and visualized with a laser scanning confocal microscope (LSM710, Zeiss).
Data analysis.
Data were collected on a Macintosh computer using AxoGraphX (Axograph Scientific), and stored for offline analysis. Statistical significance was assessed with Student's t tests or one-way ANOVA (Bonferroni's post hoc analysis) to determine significance; *p < 0.05, ***p < 0.001. Data are presented as mean ± SEM.
Materials.
Drugs were applied by bath perfusion. The solution containing [Met]5enkephalin (ME) included the peptidase inhibitors, bestatin hydrochloride (10 μm) and thiorphan (1 μm). [D-Pen2, 5]-enkephalin (DPDPE) was obtained from Bachem. ME, [D-Ala2, N-Me-Phe4, Gly5-ol]-enkephalin (DAMGO), (+)-(5α,7α,8β)-N-methyl-N-[7-(1-pyrrolidinyl)-1-oxaspiro[4.5]dec-8-yl]-benzeneacetamide (U69593), naloxone, picrotoxin, and tetrodotoxin (TTX) were obtained from Sigma.
Results
Identification of retrograde labeled neurons within the RMTg in sagittal slices
The RMTg was located as previously reported using the increase in cFOS expression following an injection of d-amphetamine (Perrotti et al., 2005; Jhou et al., 2009b; Kaufling et al., 2009). A dense cluster of cFOS immunopositive cells was found in an area 1–1.5 mm caudal to VTA, 0.5–0.8 mm lateral from midline and 0.1 mm ventral to the decussation of the superior cerebellar peduncle (Fig. 1A). The retrograde tracer, cholera toxin B (CTX), injected into VTA resulted in the colocalization with cFOS-positive neurons (Fig. 1A, inset). Because VTA and substantia nigra are in close proximity, retrograde tracers included both areas. Although not all cFOS neurons colocalized with retrograde tracer, ∼80% of retrogradely labeled neurons were cFOS-positive within the RMTg nucleus where dense cFOS immunoreactivity was found. Thus neurons in the RMTg that projected to the VTA/SN could be identified for whole-cell and cell-attached electrophysiological recordings in living slices.
Figure 1.

Hyperpolarization of identified RMTg neurons mediated by μ-opioid agonists. A, Confocal mosaic image of a sagittal slice containing VTA/SN and RMTg. RMTg neurons were identified with elevated cFOS (red) activity 2 h after an injection of amphetamine. The retrograde tracer, cholera toxin subunit B (green), was injected into VTA. cFOS-positive neurons frequently colocalized with retrograde tracer (inset). ml, medial lemniscus; 3n, oculomotor nerve. B, Whole-cell current-clamp recording from retrograde tracer-positive neuron. ME (10 μm) caused hyperpolarization and decreased spontaneous firing. C, Retrogradely labeled neurons in the RMTg were hyperpolarized through an activation of μ-opioid receptors DAMGO. Application of ME and DAMGO but not κ- or δ-opioid agonists (U69593 or DPDPE) significantly induced a hyperpolarization of the membrane potential from the baseline potentials. Number of cells indicated in or above the bars. Error bars indicate SEM. *p < 0.05, ***p < 0.001.
Opioids hyperpolarize RMTg neurons
Retrograde transport of fluorescent microspheres was used to identify neurons in the RMTg that projected to the VTA/SN. This method was preferred over the use of CTX because the microspheres were bright and less susceptible to photobleaching as was found with CTX. Whole-cell current-clamp recordings were made from retrogradely labeled neurons to measure membrane potential and firing rate. Application of ME (1–10 μm) resulted in hyperpolarization and a decrease in the firing rate (Fig. 1B). Only cells that were responsive to ME and recovered to baseline following washout were considered to be opioid-sensitive (36%; 42/118 neurons). Application of ME (1 μm) and the μ-opioid selective agonist, DAMGO (1 μm) significantly hyperpolarized the retrograde tracer-positive neurons (−6.3 ± 0.67 mV; n = 8; t7 = 9.4; p < 0.001, and −5.5 ± 1.67 mV; n = 6; t5 = 3.3; p < 0.05, respectively; Student's t test single group comparison to 0 mV), whereas the κ-opioid agonist, U69593 (1 μm) and the δ-opioid agonist, DPDPE (1 μm) had no significant effect (−0.3 ± 0.30 mV; n = 4; t3 = 1.1; p = 0.36, and −0.9 ± 0.42 mV; n = 4; t3 = 2.1; p = 0.12, respectively, Fig. 1C).
Opioids inhibit spontaneous activity of RMTg neurons
Cell-attached recordings were made to monitor the spontaneous firing of retrogradely labeled neurons in the RMTg (Fig. 2A). Approximately half of these neurons fired spontaneously in a rhythmic pattern at 6.7 ± 0.49 Hz (12/26 neurons; Fig. 2B). The rate of firing was slower than that found in vivo (20.2 Hz, 17.8 Hz, 18.2 Hz, and 15.5 Hz; Jhou et al., 2009a; Hong et al., 2011; Jalabert et al., 2011; Lecca et al., 2011), but slightly higher than reported using whole-cell current-clamp recording (4.7 Hz; Lecca et al., 2011). The firing rate of RMTg neurons was decreased to 1.7 ± 0.55 Hz by ME (1 μm) and DAMGO (1 μm) within 2–3 min (Fig. 2B; n = 12; F(2,33) = 33.1; p < 0.001 one-way ANOVA Bonferroni). In some cases, opioids completely inhibited the firing of RMTg neurons within 1–2 min (n = 4). Following the washout of ME or superfusion with naloxone (1 μm) the firing rate gradually returned to the baseline frequency at 6.9 ± 0.52 Hz within 5 min (Fig. 2B). Thus, the spontaneous activity of retrogradely labeled RMTg neurons was sensitive to μ-opioid agonists.
Figure 2.

The firing rate of retrogradely labeled RMTg neurons and rate of spontaneous GABA-A IPSCs were inhibited by μ-opioid agonists. A, Cell-attached recording from a RMTg neuron. Application of μ-opioid agonist DAMGO (1 μm) gradually inhibited the firing of the neuron. This inhibition was reversed by application of opioid antagonist naloxone (1 μm). B, Summary plot of the firing rate of RMTg neurons in control, during application of ME (•; 1 μm) or DAMGO (○; 1 μm), and washout ME or superfusion of naloxone after DAMGO (n = 12). Application of ME or DAMGO significantly reduced frequency of spontaneous firing from control and washout. Bar graphs represent the average firing frequency of shown data. C, Whole-cell voltage-clamp recording from a VTA dopamine neuron. Rhythmic spontaneous GABA-A IPSCs were examined. The μ-opioid agonist DAMGO (1 μm) decreased large amplitude sIPSCs, but not miniature IPSCs and the inhibition was reversed by naloxone (1 μm). D, Summary plot of the frequency of the large amplitude sIPSCs in control, during ME (•; 1 μm) or DAMGO (○; 1 μm), and washout of ME or superfusion of naloxone after DAMGO. Application of ME or DAMGO significantly reduced frequency of sIPSC from control and washout. Bar graphs represent the average sIPSC frequency of shown values. In B and D, lines connecting circles indicate recordings made from the same cell. Error bars indicate SEM. ***p < 0.001.
In whole-cell voltage-clamp mode, current–voltage plots constructed before and after ME or DAMGO revealed an inwardly rectifying current indicating the activation of an inwardly rectifying potassium channel (GIRK) conductance (data not shown). DAMGO (1 μm) only induced a small outward current (18.0 ± 5.3 pA; n = 6), but this was enough to decrease spontaneous firing of the RMTg neurons because of the high input resistance (552 ± 84.6 MΩ). Therefore, the mechanism by which μ-opioid agonists decreased the spontaneous firing of RMTg neurons was from a hyperpolarization through the activation of GIRK.
Opioids inhibit spontaneous GABA-A IPSCs in DA neurons
Whole-cell voltage-clamp recordings were made from dopamine neurons in the VTA/SN and spontaneous GABA-A IPSCs (sIPSCs) were examined. A high chloride internal was used such that the reversal potential of GABA-A IPSC was 0 mV. Large rhythmic sIPSCs were occasionally observed in recordings from dopamine neurons located in the caudal portion of the VTA/SN (n = 7; Fig. 2C). The average frequency of the rhythmic sIPSCs was 7.1 ± 0.69 Hz and amplitude of 248 ± 44.1 pA. These sIPSCs were eliminated by bath application of TTX (300 nm) indicating the dependence on action potentials and the presence of cell body and axonal projection in the brain slice. ME (1 μm) and DAMGO (1 μm) decreased the frequency of the sIPSCs (1.7 ± 0.70 Hz; F(2,18) = 14.0; p < 0.001 one-way ANOVA Bonferroni). The reduction in sIPSC frequency was selective for μ-opioid receptors because the κ- and δ-opioid agonists U69593 (1 μm) and DPDPE (1 μm) did not significantly affect the sIPSC frequency (87 ± 14.4%; n = 9; F(2,24) = 0.50; p = 0.61, and 81 ± 8.5%; n = 4; F(2,9) = 0.20; p = 0.83, respectively, one-way ANOVA Bonferroni). The amplitude of the sIPSC was also not changed by the κ- and δ-opioid agonists (95 ± 10.3%; n = 9; F(2,24) = 0.80; p = 0.46 and 95 ± 10.6%; n = 4; F(2,9) = 0.70; p = 0.52, respectively, one-way ANOVA Bonferroni). The frequency and opioid sensitivity of sIPSCs on dopamine neurons was remarkably similar to spontaneous firing of RMTg neurons. Stimulation within the RMTg was next used to determine whether the pathway from RMTg to the dopamine cells was intact.
Presynaptic GABA transmission from RMTg to dopamine neurons
Electrical stimulation
Electrical stimulation with electrodes placed within RMTg-evoked GABA-A IPSCs (eIPSCs) in dopamine neurons (Fig. 3A). Stimulating electrodes were placed ∼0.3–1 mm away from the dopamine cells (Fig. 3B). Successful activation of eIPSCs was only possible in certain ‘hot’ spots, a small region (50–100 μm) in the pathway between VTA/SN and RMTg where electrical stimulation successfully activated IPSCs on DA neurons. If the stimulating pipette was moved ±100 μm away from that spot, the same stimulus failed to evoke IPSCs. The delay from the stimulus to the onset of the eIPSC was between 2.2 and 10.7 ms (5.3 ± 0.32 ms) depending on the distance between the stimulating pipette and the dopamine neuron. The GABA projection from RMTg to the VTA is mainly unmyelinated (Balcita-Pedicino et al., 2011), therefore, the long latencies of eIPSC following stimulation within the RMTg was due to the slow conduction velocity of unmyelinated axons. Application of the μ-opioid receptor agonist, DAMGO (1 μm) decreased eIPSCs amplitude to 31 ± 3.6% of control (n = 40; t39 = −19.5; p < 0.0001) and superfusion of the opioid antagonist naloxone (1 μm) reversed this inhibition (96 ± 5.5%, n = 40; t39 = −0.65; p = 0.52; Student's t test single group comparison to 100% control; Fig. 3C). However, κ- and δ-opioid agonists (U69653 1 μm, and DPDPE 1 μm) did not decrease eIPSC amplitude significantly (85 ± 7.5%, n = 20; t19 = −2.1; p = 0.053 and 95 ± 3.3%, n = 10, t9 = −1.6; p = 0.13 respectively; Fig. 3C).
Figure 3.
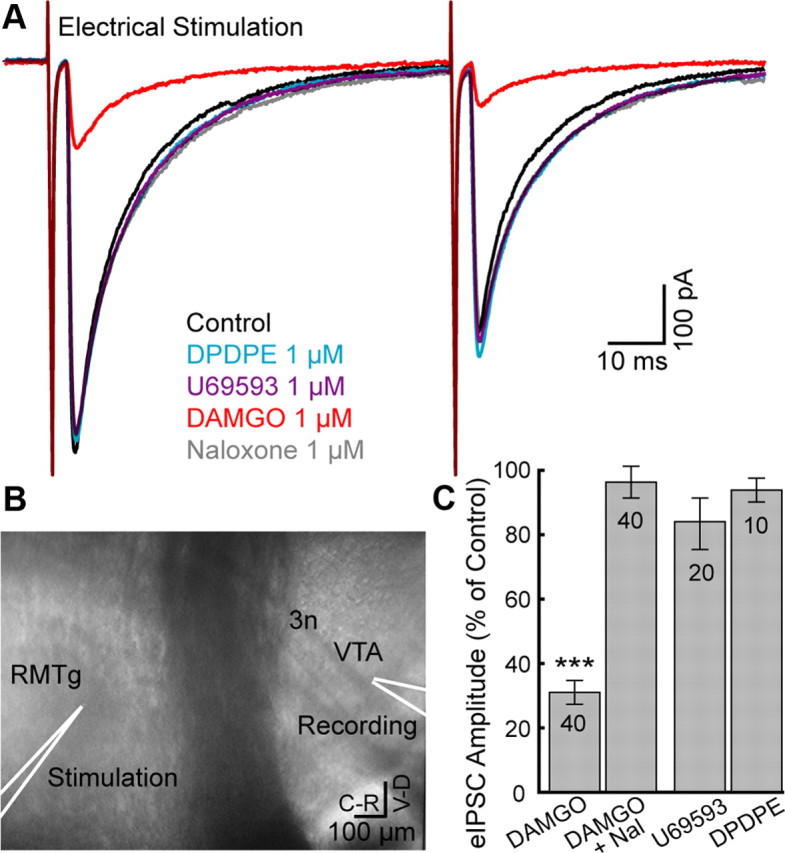
Electrical stimulation in the RMTg evoked μ-opioid-sensitive IPSCs in dopamine neurons. A, Superimposed traces of evoked IPSCs in a dopamine neuron (Vm = −60 mV, average of 10). Application of DAMGO, but not U69593 or DPDPE, decreased the amplitude of eIPSCs. B, Representative image indicating the location of an electrical stimulation and site of recording in the sagittal plane. C, Summary graph of eIPSC amplitude (% of control) during μ-opioid agonist (DAMGO), reversal by opioid antagonist (Naloxone), and during κ- or δ-opioid agonists (U69563 or DPDPE). DAMGO significantly reduced eIPSC amplitude compared with 100% control. Number of cells indicated in bars. Error bars indicate SEM. ***p < 0.001.
ChR2 stimulation
The expression of ChR2 in the RMTg allowed selective stimulation of cell bodies within the RMTg and terminals from the RMTg that projected to the VTA/SN. Bilateral injections of AAV-ChR2-Venus constructs into the RMTg were used to express ChR2. Light stimulation was used to activate ChR2 expressed on the terminals of RMTg neurons and resulted in the release of GABA on to dopamine cells (Fig. 4A). The first experiment used wide field stimulation in the VTA/SN. Fluorescent fibers extended from the RMTg toward VTA/SN (Fig. 4B). Whole-cell voltage-clamp recordings (Vm = −60 mV) were made from dopamine neurons in VTA/SN and flashes of light (0.3 ms) were applied at the dopamine cell body (latency 2.8 ± 0.12 ms). A paired light flash (50 ms separation) evoked GABA-A IPSCs (fIPSCs) in most of the caudal portion of VTA/SN. Light stimuli did not induce a direct current in the dopamine neurons indicating that ChR2 was not expressed in the cell that was being recorded (data not shown). The amplitude of fIPSCs varied between 2 nA and 50 pA depending upon the expression level of ChR2, the area that was illuminated and a duration of the light flash. The fIPSCs were completely blocked by TTX (300 nm; 4.6 ± 2.6% of control, n = 5) confirming that the GABA release was action potential dependent. In addition, the GABA-A receptor antagonist picrotoxin (100 μm) blocked fIPSC (3.5 ± 0.5% of control, n = 12).
Figure 4.

DAMGO inhibited ChR2-evoked IPSCs on dopamine neurons. A, Superimposed traces of fIPSCs (0.3 ms; 470 nm) in a dopamine neuron (Vm = −60 mV, average of 10). DAMGO decreased the amplitude of fIPSCs in a concentration-dependent manner. The GABA-A receptor antagonist picrotoxin (100 μm) blocked fIPSCs. B, Confocal image stained for tyrosine hydroxylase (TH; red) and ChR2 conjugated with Venus (green) in VTA. ChR2-expressing axons were densely innervated in the VTA/SN 7 d after AAV viral expression of ChR2 in the RMTg. C, Concentration-response curve of the inhibition of fIPSC by DAMGO. Error bars indicate SEM.
To investigate the action of opioids, stimulation was adjusted to evoke ∼600 pA GABA-A IPSC (627 ± 75.9 pA, n = 34 neurons). Application of ME or DAMGO inhibited the amplitude of fIPSC in all cells tested, the inhibition was graded and reversed by naloxone (1 μm; Fig. 4A). The inhibition by DAMGO was concentration dependent having an EC50 of 64 ± 10 nm (Fig. 4C). The paired-pulse ratio (PPR, fIPSC2/fIPSC1) was significantly increased from 0.79 ± 0.08 in control to 1.91 ± 0.52 after ME application (1 μm; n = 13; t24 = −2.1; p = 0.04). The PPR was also increased from 0.83 ± 0.05 in control to 1.26 ± 0.15 after application of μ-opioid specific agonist DAMGO (300 nm or 1 μm; n = 16; t30 = −2.7; p = 0.01; Student's t tests unpaired data). The increased PPR suggested that opioid inhibition of fIPSCs was mediated by a presynaptic mechanism. Thus, in addition to reducing the firing rate of RMTg neurons, μ-opioid agonists acted directly at presynaptic terminals to inhibit GABA release onto dopamine neurons.
Light stimulation within the RMTg in slices expressing ChR2 was used to demonstrate the connection between the RMTg and dopamine neurons. For these experiments a focal laser flash (100–200 μm diameter) was applied ∼0.6–2 mm away from the dopamine neuron in the area of the RMTg (Fig. 5B). This focal stimulation evoked GABA-A IPSCs with an average amplitude of 338 ± 109 pA (Fig. 5A). When the focal laser stimulation was moved by ±100 μm the flash failed to evoke an IPSC. The latency between the flash and the initial rise of the GABA-A IPSC was 5.4 ± 0.50 ms, similar to that found with electrical stimulation in the RMTg. Both ME (1 μm) and DAMGO (1 μm) significantly inhibited the IPSC amplitude to 27 ± 5.7% (n = 13; t12 = −12.8; p < 0.001) and 17 ± 5.1% of control (n = 10; t9 = −16.3; p < 0.001), respectively (Fig. 5C). This inhibition reversed upon washout of ME or application of naloxone (96 ± 4.0%; n = 13; t12 = −1.1; p = 0.29, and 97 ± 6.7%; n = 10; t9 = −0.47; p = 0.65, of control, respectively, Student's t tests single group comparison to 100% control). Thus, electrical or ChR2 stimulation within RMTg evoked GABA-A IPSCs in VTA/SN that were sensitive to inhibition by μ-opioid receptors.
Figure 5.

Activation of ChR2 in the RMTg-evoked IPSCs in dopamine neurons that were inhibited by μ-opioid agonists. A, Superimposed traces of IPSCs in a dopamine neuron evoked by laser flashes (5 ms; 473 nm, average of 5) in the RMTg. DAMGO (1 μm) and picrotoxin (100 μm) inhibited amplitude of IPSCs. Notice the latency between laser stimulation and the onset of the IPSC (5.4 ± 0.5 ms). B, Wide-field mosaic confocal image stained for tyrosine hydroxylase (TH; red) and ChR2 conjugated with Venus (green) in sagittal plane. The star indicates the location of laser stimulation in RMTg. C, Summary graph of flash-evoked IPSC amplitude. ME (1 μm) and DAMGO (1 μm) significantly decreased IPSC amplitude compared with 100% control. The inhibitions were reversed by washout of ME or by naloxone (1 μm) after DAMGO that were not significantly different from 100% control. Number of cells indicated in the bars. Error bars indicate SEM. ***p < 0.001.
Discussion
This study shows that the dense GABA afferent connection from RMTg to the dopamine neurons is another GABAergic pathway that is sensitive to inhibition by μ-opioid agonists. Application of μ-opioid agonists inhibited the spontaneous firing of RMTg neurons and spontaneous GABA-A IPSCs recorded in dopamine neurons. Electric stimulation in the RMTg triggered GABA-A IPSCs that were blocked by μ-opioid agonists. Expression of ChR2 in the RMTg resulted in light-sensitive terminals in the VTA/SN that upon stimulation resulted in GABA-A IPSCs on dopamine neurons. These GABA-A IPSCs were potently inhibited by μ-opioid agonists. Finally, in slices where ChR2 was expressed in the RMTg/tVTA, focal light stimulation within the RMTg resulted in opioid-sensitive GABA-A IPSCs on dopamine neurons. The results indicate that the neurons in the RMTg synapse onto dopamine neurons in VTA/SN and the strength of that connection is reduced upon μ-opioid receptor activation.
GABA interneurons in the VTA/SN are believed to play a major role in opioid disinhibition on dopamine neurons (Johnson and North, 1992a). This hypothesis was based on the observation that spontaneous opioid-sensitive GABA-A IPSPs were present in horizontal brain slices. The fact that that these spontaneous IPSCs were sensitive to TTX indicated that the cell bodies were located within the slice. There have also been multiple publications demonstrating that non-dopamine neurons in the VTA/SN are sensitive to inhibition by opioids (Johnson and North, 1992a,b; Cameron et al., 1997; Chieng et al., 2011). There is, however, no direct functional evidence that the non-dopamine neurons synapse onto dopamine neurons in VTA/SN. It may be that interneurons and RMTg neurons both synaptically regulate dopamine neurons. Previous reports indicated that 35–50% of VTA and 20% of substantia nigra neurons are GABA neurons (Margolis et al., 2006; Nair-Roberts et al., 2008; Balcita-Pedicino et al., 2011; Chieng et al., 2011) although the density of synapses onto a dopamine neuron arising from interneurons is not known. In addition, VTA/SN neurons receive GABA inputs from striatum, nucleus accumbens, external globus pallidus. However, the relative role of opioid action on those pathways is not known. With nonselective electrical stimulation within the VTA/SN opioids inhibited GABA-A IPSCs by a maximum that varied between 40 and 75% (Johnson and North, 1992a; Ford et al., 2006; Madhavan et al., 2010). With selective stimulation of the RMTg used in this study DAMGO (1 μm) inhibited the amplitude of IPSCs by an average of 69% using electrical stimulation and 83% using ChR2 stimulation. In addition, in 5 of 10 experiments using ChR2 stimulation in the RMTg, the amplitude of the IPSC was decreased by >90%. This observation indicates that the IPSCs from the RMTg are more sensitive to inhibition by opioids than when nonselective stimulation is used to evoke IPSCs. Whole-cell recordings from RMTg neurons showed that not all RMTg neurons were inhibited by opioids. Yet, stimulation in the RMTg resulted in GABA-A IPSCs that were always sensitive to opioids. This observation suggests that there may be a heterogeneous population of RMTg neurons but that most neurons that project to the VTA/SN were sensitive to opioids. Given the density of GABA innervation from the RMTg to dopamine neurons, it is possible that this pathway is another major contributor in the opioid-sensitive GABA input onto dopamine neurons (Jhou et al., 2009b; Kaufling et al., 2009; Balcita-Pedicino et al., 2011; Jalabert et al., 2011; Lecca et al., 2011). This suggestion is supported by a recent study in vivo demonstrating that morphine acts in the RMTg to increase the firing of dopamine neurons (Jalabert et al., 2011).
Electrophysiological characteristics of RMTg/tVTA
Neurons in the RMTg receive a strong glutamate input from lateral habenula (Hong et al., 2011) and fire action potentials at ∼10–20 Hz in vivo (Jhou et al., 2009a; Hong et al., 2011; Jalabert et al., 2011; Lecca et al., 2011). The spontaneous activity of RMTg neurons in the present study and previous publications using brain slices was 4.7–10.5 Hz. This activity was considered to be intrinsic because the glutamate pathway from the lateral habenula was severed (Lecca et al., 2011). Thus, the difference in frequency between in vivo and in vitro preparations most likely results from the removal of afferent excitatory drive in brain slices.
The frequency and sensitivity to opioids of sIPSCs recorded in dopamine neurons was also similar to the firing rate of RMTg neurons. The all-or-none waveform and rhythmic pattern of sIPSCs probably originates from the firing of a single neuron. As previously reported, opioids gradually decreased the frequency of sIPSC without affecting the amplitude (Johnson and North, 1992b). This all-or-none inhibition suggests that the spontaneously firing of presynaptic GABA neurons in the slice was decreased by opioids. The potential mechanism for the inhibition of sIPSCs is mainly through a hyperpolarization of the presynaptic membrane potential as was observed in the RMTg.
Opioid agonists also inhibit transmitter release through an inhibition of voltage-dependent calcium channels as well as a direct action of βγ-subunits on the release machinery SNAP-25 (Blackmer et al., 2005; Gerachshenko et al., 2005). Unlike the all-or-none inhibition of sIPSCs by opioids, a graded inhibition of IPSCs by opioids was observed in experiments where axons and terminals within the VTA/SN were activated using ChR2 (Fig. 4). This observation suggests that there are multiple opioid-sensitive release sites that synapse on a single dopamine neuron that arise from the RMTg. This also suggests that the underlying mechanism for the inhibition of GABA release from terminals may differ from the activation of GIRK conductance measured in RMTg somata.
The activity of lateral habenula neurons is increased following the absence of an expected reward or an aversive stimulus. This increased activity preceded a pause in dopamine cell firing (Matsumoto and Hikosaka, 2007; Hong et al., 2011). Although the RMTg might receive inputs from reward related brain regions other than lateral habenula, some RMTg neurons were activated in response to a negative reward cue and this activation was followed by an inhibition of dopamine neurons (Hong et al., 2011). Thus, RMTg neurons are an inhibitory relay structure to dopamine neurons.
The relative role of RMTg in opioid actions within the VTA/SN is not known. FOS expression in the RMTg increased during acute withdrawal from several psychostimulants, however, morphine applied acutely did not elevate FOS expression (Kaufling et al., 2010b). The lack of FOS expression might be due to the inhibition of RMTg neurons by opioids. During the withdrawal from a 1-week exposure to morphine, the probability of GABA release was increased in the VTA and substantia nigra (Bonci and Williams, 1996, 1997). This may suggest that during withdrawal from morphine the firing of dopamine neurons is suppressed through an increased GABA input from the RMTg. The role of the RMTg in this observation remains to be determined given that there are multiple GABA inputs onto dopamine neurons.
In conclusion, the GABA neurons in the RMTg send a dense projection to dopamine neurons in the VTA/SN. This GABA input is potently inhibited by the activation of μ-, but not κ-or δ-opioid receptors. Given the density of the GABA innervation and the potency of the opioid inhibition of GABA release from these cells it appears that the neurons of the RMTg are a key site of opioid action in the mesolimbic reward pathway.
Footnotes
This work was supported by NIH Grants DA08163 and DA04523 to J.T.W.
References
- Balcita-Pedicino JJ, Omelchenko N, Bell R, Sesack SR. The inhibitory influence of the lateral habenula on midbrain dopamine cells: ultrastructural evidence for indirect mediation via the rostromedial mesopontine tegmental nucleus. J Comp Neurol. 2011;519:1143–1164. doi: 10.1002/cne.22561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackmer T, Larsen EC, Bartleson C, Kowalchyk JA, Yoon EJ, Preininger AM, Alford S, Hamm HE, Martin TF. G protein betagamma directly regulates SNARE protein fusion machinery for secretory granule exocytosis. Nat Neurosci. 2005;8:421–425. doi: 10.1038/nn1423. [DOI] [PubMed] [Google Scholar]
- Bonci A, Williams JT. A common mechanism mediates long-term changes in synaptic transmission after chronic cocaine and morphine. Neuron. 1996;16:631–639. doi: 10.1016/s0896-6273(00)80082-3. [DOI] [PubMed] [Google Scholar]
- Bonci A, Williams JT. Increased probability of GABA release during withdrawal from morphine. J Neurosci. 1997;17:796–803. doi: 10.1523/JNEUROSCI.17-02-00796.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cameron DL, Wessendorf MW, Williams JT. A subset of ventral tegmental area neurons is inhibited by dopamine, 5-hydroxytryptamine and opioids. Neuroscience. 1997;77:155–166. doi: 10.1016/s0306-4522(96)00444-7. [DOI] [PubMed] [Google Scholar]
- Chieng B, Azriel Y, Mohammadi S, Christie MJ. Distinct cellular properties of identified dopaminergic and GABAergic neurons in the mouse ventral tegmental area. J Physiol. 2011;589:3775–3787. doi: 10.1113/jphysiol.2011.210807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Chiara G, Imperato A. Drugs abused by humans preferentially increase synaptic dopamine concentrations in the mesolimbic system of freely moving rats. Proc Natl Acad Sci U S A. 1988;85:5274–5278. doi: 10.1073/pnas.85.14.5274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Chiara G, Bassareo V, Fenu S, De Luca MA, Spina L, Cadoni C, Acquas E, Carboni E, Valentini V, Lecca D. Dopamine and drug addiction: the nucleus accumbens shell connection. Neuropharmacology. 2004;47:227–241. doi: 10.1016/j.neuropharm.2004.06.032. [DOI] [PubMed] [Google Scholar]
- Ford CP, Mark GP, Williams JT. Properties and opioid inhibition of mesolimbic dopamine neurons vary according to target location. J Neurosci. 2006;26:2788–2797. doi: 10.1523/JNEUROSCI.4331-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geisler S, Marinelli M, Degarmo B, Becker ML, Freiman AJ, Beales M, Meredith GE, Zahm DS. Prominent activation of brainstem and pallidal afferents of the ventral tegmental area by cocaine. Neuropsychopharmacology. 2008;33:2688–2700. doi: 10.1038/sj.npp.1301650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerachshenko T, Blackmer T, Yoon EJ, Bartleson C, Hamm HE, Alford S. Gbetagamma acts at the C terminus of SNAP-25 to mediate presynaptic inhibition. Nat Neurosci. 2005;8:597–605. doi: 10.1038/nn1439. [DOI] [PubMed] [Google Scholar]
- Hong S, Jhou TC, Smith M, Saleem KS, Hikosaka O. Negative reward signals from the lateral habenula to dopamine neurons are mediated by rostromedial tegmental nucleus in primates. J Neurosci. 2011;31:11457–11471. doi: 10.1523/JNEUROSCI.1384-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jalabert M, Bourdy R, Courtin J, Veinante P, Manzoni OJ, Barrot M, Georges F. Neuronal circuits underlying acute morphine action on dopamine neurons. Proc Natl Acad Sci U S A. 2011;108:16446–16450. doi: 10.1073/pnas.1105418108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jhou TC, Fields HL, Baxter MG, Saper CB, Holland PC. The rostromedial tegmental nucleus (RMTg), a GABAergic afferent to midbrain dopamine neurons, encodes aversive stimuli and inhibits motor responses. Neuron. 2009a;61:786–800. doi: 10.1016/j.neuron.2009.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jhou TC, Geisler S, Marinelli M, Degarmo BA, Zahm DS. The mesopontine rostromedial tegmental nucleus: a structure targeted by the lateral habenula that projects to the ventral tegmental area of Tsai and substantia nigra compacta. J Comp Neurol. 2009b;513:566–596. doi: 10.1002/cne.21891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji H, Shepard PD. Lateral habenula stimulation inhibits rat midbrain dopamine neurons through a GABA(A) receptor-mediated mechanism. J Neurosci. 2007;27:6923–6930. doi: 10.1523/JNEUROSCI.0958-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson SW, North RA. Opioids excite dopamine neurons by hyperpolarization of local interneurons. J Neurosci. 1992a;12:483–488. doi: 10.1523/JNEUROSCI.12-02-00483.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson SW, North RA. Two types of neurone in the rat ventral tegmental area and their synaptic inputs. J Physiol. 1992b;450:455–468. doi: 10.1113/jphysiol.1992.sp019136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufling J, Veinante P, Pawlowski SA, Freund-Mercier MJ, Barrot M. Afferents to the GABAergic tail of the ventral tegmental area in the rat. J Comp Neurol. 2009;513:597–621. doi: 10.1002/cne.21983. [DOI] [PubMed] [Google Scholar]
- Kaufling J, Veinante P, Pawlowski SA, Freund-Mercier MJ, Barrot M. gamma-Aminobutyric acid cells with cocaine-induced DeltaFosB in the ventral tegmental area innervate mesolimbic neurons. Biol Psychiatry. 2010a;67:88–92. doi: 10.1016/j.biopsych.2009.08.001. [DOI] [PubMed] [Google Scholar]
- Kaufling J, Waltisperger E, Bourdy R, Valera A, Veinante P, Freund-Mercier MJ, Barrot M. Pharmacological recruitment of the GABAergic tail of the ventral tegmental area by acute drug exposure. Br J Pharmacol. 2010b;161:1677–1691. doi: 10.1111/j.1476-5381.2010.00984.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lecca S, Melis M, Luchicchi A, Ennas MG, Castelli MP, Muntoni AL, Pistis M. Effects of drugs of abuse on putative rostromedial tegmental neurons, inhibitory afferents to midbrain dopamine cells. Neuropsychopharmacology. 2011;36:589–602. doi: 10.1038/npp.2010.190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madhavan A, Bonci A, Whistler JL. Opioid-induced GABA potentiation after chronic morphine attenuates the rewarding effects of opioids in the ventral tegmental area. J Neurosci. 2010;30:14029–14035. doi: 10.1523/JNEUROSCI.3366-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margolis EB, Lock H, Hjelmstad GO, Fields HL. The ventral tegmental area revisited: is there an electrophysiological marker for dopaminergic neurons? J Physiol. 2006;577:907–924. doi: 10.1113/jphysiol.2006.117069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumoto M, Hikosaka O. Lateral habenula as a source of negative reward signals in dopamine neurons. Nature. 2007;447:1111–1115. doi: 10.1038/nature05860. [DOI] [PubMed] [Google Scholar]
- Nair-Roberts RG, Chatelain-Badie SD, Benson E, White-Cooper H, Bolam JP, Ungless MA. Stereological estimates of dopaminergic, GABAergic and glutamatergic neurons in the ventral tegmental area, substantia nigra and retrorubral field in the rat. Neuroscience. 2008;152:1024–1031. doi: 10.1016/j.neuroscience.2008.01.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicola SM, Malenka RC. Dopamine depresses excitatory and inhibitory synaptic transmission by distinct mechanisms in the nucleus accumbens. J Neurosci. 1997;17:5697–5710. doi: 10.1523/JNEUROSCI.17-15-05697.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perrotti LI, Bolaños CA, Choi KH, Russo SJ, Edwards S, Ulery PG, Wallace DL, Self DW, Nestler EJ, Barrot M. DeltaFosB accumulates in a GABAergic cell population in the posterior tail of the ventral tegmental area after psychostimulant treatment. Eur J Neurosci. 2005;21:2817–2824. doi: 10.1111/j.1460-9568.2005.04110.x. [DOI] [PubMed] [Google Scholar]
- Saal D, Dong Y, Bonci A, Malenka RC. Drugs of abuse and stress trigger a common synaptic adaptation in dopamine neurons. Neuron. 2003;37:577–582. doi: 10.1016/s0896-6273(03)00021-7. [DOI] [PubMed] [Google Scholar]
- Shoji Y, Delfs J, Williams JT. Presynaptic inhibition of GABA(B)-mediated synaptic potentials in the ventral tegmental area during morphine withdrawal. J Neurosci. 1999;19:2347–2355. doi: 10.1523/JNEUROSCI.19-06-02347.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas MJ, Beurrier C, Bonci A, Malenka RC. Long-term depression in the nucleus accumbens: a neural correlate of behavioral sensitization to cocaine. Nat Neurosci. 2001;4:1217–1223. doi: 10.1038/nn757. [DOI] [PubMed] [Google Scholar]
- Ungless MA, Magill PJ, Bolam JP. Uniform inhibition of dopamine neurons in the ventral tegmental area by aversive stimuli. Science. 2004;303:2040–2042. doi: 10.1126/science.1093360. [DOI] [PubMed] [Google Scholar]
- Wise RA. Neurobiology of addiction. Curr Opin Neurobiol. 1996;6:243–251. doi: 10.1016/s0959-4388(96)80079-1. [DOI] [PubMed] [Google Scholar]