

Abstract
(−)-Lobeline (2R,6S,10S; 1a), an antagonist at nicotinic acetylcholine receptors (nAChRs), inhibits the neurochemical and behavioral effects of methamphetamine and inhibits dopamine transporter (DAT) and vesicular monoamine transporter (VMAT2) function. VMAT2 is a target for the development of treatments for methamphetamine abuse. Structural modification of lobeline affords the defunctionalized analogues meso-transdiene (MTD) and lobelane, which have high potency and selectivity for VMAT2. To establish the structure–activity relationships within this novel class of VMAT2 ligands, specific stereochemical forms of MTD, lobelane, and other structurally related analogues have been synthesized. These compounds have been evaluated for inhibition of [3H]nicotine ([3H]NIC) binding (α4β2* nAChR), [3H]methyllycaconitine ([3H]MLA) binding (α7* nAChR), and [3H]dihydrotetrabenazine ([3H]DTBZ) binding (VMAT2). Generally, all of these analogues had lower affinities at α4β2* and α7* nAChRs compared to lobeline, thereby increasing selectivity for VMAT2. The following structural modifications resulted in only modest changes in affinity for VMAT2, affording analogues that were less potent than the lead compound, lobelane: (1) altering the stereochemistry at the C-2 and C-6 positions of the piperidino ring, (2) varying unsaturation in the piperidino C-2 and C-6 substituents, (3) introducing unsaturation into the piperidine ring, (4) ring-opening or eliminating the piperidine ring, and (5) removing the piperidino N-methyl group. Furthermore, incorporating a quaternary ammonium group into defunctionalized lobeline molecules in the cis-series resulted in significant loss of affinity for VMAT2, whereas only a modest change in affinity was obtained in the trans-series. The most potent (Ki = 630 nM) and VMAT2-selective compound evaluated was the N-methyl-2,6-cis-bis(naphthaleneethyl)piperidine analogue 28b (1-NAP-lobelane), in which the phenyl groups of lobelane were replaced with 1-naphthyl moieties. Thus, initial structure–activity relationship studies reveal that the most promising structural changes to the lobeline molecule that lead to enhancement of VMAT2 affinity and selectivity are defunctionalization, affording lobelane and MTD, and replacement of the phenyl rings of lobelane with other aromatic moieties that have a π-extended structure.
Introduction
Psychostimulant-induced behavioral activation and reinforcement are mediated, at least in part, via interaction with neurotransmitter transporters, which regulate synaptic dopamine (DA) concentrations.1,2 Recent studies have demonstrated that psychostimulants alter vesicular monoamine transporter (VMAT2) function.3 Methamphetamine decreases VMAT2 function4,5 and is also a substrate for the DA transporter (DAT).6,7 VMAT2 heterologous knockout mice exhibit reduced amphetamine conditioned reward,8 enhanced amphetamine locomotion,8 and enhanced sensitivity to cocaine and amphetamine,9 indicating that VMAT2 plays an important role in mediating the behavioral effects of psychostimulants. These results support the idea that VMAT2 should be considered as a target for the development of pharmacotherapies to treat psychostimulant abuse. Other evidence supporting the role of VMAT2 in psychostimulant pharmacology is the finding that benzoquinolizine derivatives, such as tetrabenazine, have high affinity for VMAT2, decrease locomotor activity and aggressiveness in monkeys, and, moreover, depress methamphetamine-induced hyperactivity in rats and mice.13
(−)-Lobeline (2R,6S,10S; 1a) (Figure 1), a weakly basic lipophilic alkaloid from Lobelia inflata, interacts with VMAT2 and DAT via a novel mechanism of action.14 Lobeline potently inhibits [3H]dihydroxytetrabenazine ([3H]DTBZ) binding to VMAT2 with an IC50 of 0.90 μM and inhibits [3H]DA uptake into rat striatal vesicle preparations with an IC50 of 0.88 μM.15,16 Furthermore, lobeline also inhibits (IC50 = 80 μM) [3H]DA uptake into rat striatal synaptosomes via DAT, but with 100-fold lower affinity.15 Importantly, lobeline has been shown to inhibit both the neurochemical and behavioral effects of amphetamine in rodents.17–19 Specifically, methamphetamine-evoked DA release from rat striatal slices is inhibited by lobeline.17 Furthermore, lobeline attenuates d-amphetamine-, methamphetamine-, and nicotine-induced hyperactivity in rodents.17,20 Importantly, lobeline is not self-administered but decreases methamphetamine self-administration in rats.18,19 The lobeline-induced decrease in methamphetamine self-administration was not surmounted by increasing unit doses of methamphetamine.18,19 Taken together, these results suggest that lobeline lacks abuse liability, while decreasing the stimulant and rewarding effects of methamphetamine via a noncompetitive interaction with VMAT2. Consistent with the observation that lobeline is not self-administered,19 lobeline does not evoke DA release but stimulates dihydroxyphenylacetic acid overflow,15 which likely results from alterations in presynaptic DA storage via an interaction with VMAT2.14 Furthermore, the observation that lobeline inhibits d-amphetamine- and methamphetamine-evoked DA release from superfused rat striatal slices17 is consistent with its inhibition of methamphetamine self-administration.18 Thus, VMAT2 appears to be a novel target for development of therapeutic candidates to treat methamphetamine abuse.
Figure 1.

In addition to its activity at VMAT2, lobeline acts as an antagonist at nicotinic acetylcholine receptors (nAChRs).21 Lobeline inhibits nicotine-evoked [3H]DA overflow from rat striatal slices with an IC50 of 1 μM, suggesting that lobeline acts as an antagonist at α6β2β3* nAChRs that mediate DA release.21 Lobeline also inhibits nicotine-evoked 86Rb+ efflux from rat thalamic synaptosomes with an IC50 of 0.7 μM and inhibits [3H]nicotine ([3H]NIC) binding to rat striatal membranes with a Ki of 4.7 nM, indicating that lobeline is also an antagonist at α4β2* nAChRs.21 Moreover, lobeline also inhibits [3H]methyllycaconitine ([3H]MLA) binding to rat brain membranes with a Ki of 6.26 μM, indicating an interaction with the α7* nAChR subtype.22 Lobeline has also been reported to be an antagonist at human α7* nAChRs expressed in Xenopus oocytes with an IC50 of 8.5 μM.23
Due to the multiple pharmacological actions of lobeline, it is important to examine structural analogues of lobeline to begin to define the receptor pharmacophores and develop compounds with enhanced selectivity. Our preliminary studies22,24 show that structural changes to the lobeline molecule, such as removal of one or both of the oxygen functionalities, result in a decrease in α4β2* nAChR affinity, which is consistent with the findings of others.25 Thus, it is possible that analogues of lobeline can be developed with selectivity for VMAT2. Drug discovery selectively targeting VMAT2 may provide a unique opportunity to further probe the underlying neurochemical mechanisms responsible for psychostimulant abuse and may provide a novel approach for its treatment. To date, there are very few VMAT2 ligands reported in the literature; these include low affinity ligands, such as 3-amino-2-phenylpropene derivatives (2),26 and high affinity tetrabenazine analogues, such as dihydrotetrabenazine (3) and Ro4-1284 (4)27,28 (Figure 1). Thus, lobeline analogues with selectivity for VMAT2 provide a novel structural class of VMAT2 ligand as potential leads for therapeutic development.
Selective targeting of VMAT2 by systematic structural modification of lobeline provided two non-oxygen containing lobeline analogues: N-methyl-2,6-bis(cis-phenylethenyl)piperidine (meso-transdiene, MTD, 5a) and N-methyl-2,6-bis(cis-phenylethyl)piperidine (lobelane, 6a) (Scheme 1). The latter two analogues showed good affinity for VMAT2 and DAT, with negligible affinity for α4β2* and α7* nAChRs.22 Of note, both 5a and 6a are optically inactive meso-compounds. These interesting results prompted us to carry out a more detailed investigation of the structure–activity of the various isomeric forms of 5a and 6a, as well as structurally related defunctionalized analogues, to identify selective ligands for VMAT2 that have potential as therapeutic interventions in psychostimulant abuse.
Scheme 1a.

a Reagents and conditions: (a) K2CO3, MeOH; (b) NaBH4, EtOH, rt; (c) 85% H3PO4, 60 °C; (d) H2, 10% Pd/C, MeOH, 45 psi; (e) Zn/Hg, HCl (5%), reflux; (f) CrO3, H2SO4, acetone, 0 °C; (g) H2, 10% Pd/C, 10% HOAc/MeOH, 45 psi.
Chemistry
The synthetic routes to the defunctionalized analogues 5a–5c, 6a–6c, and 13a–13d all emanate from lobeline (1a) and are illustrated in Scheme 1. C-2 epimerization of lobeline (1a) in K2CO3/methanol for 48 h at room temperature afforded an equal mixture of the two C-2 epimers, 1a and 1b,29,30 which were transformed into compound 7 via reduction with NaBH4 in absolute ethanol. Utilizing previously described dehydration methodologies,25,31 compound 7 was treated with 85% phosphoric acid to yield a mixture of the cis-and trans-distyryl compounds 5a31 (MTD) and 5b31 ((−)-TTD), which were obtained in their pure isomeric forms via silica gel column chromatographic separation. Catalytic hydrogenation of 5a afforded lobelane (6a),25,32 and similar treatment of 5b afforded (−)-2S,6S trans-lobelane [(−)-trans-lobelane, 6b]. The unsaturated lobeline analogue, 8, which was prepared by Clemmensen reduction of lobeline (1a),25 was converted into a mixture of the C-6 epimers of 9 through Jones oxidation, followed by treatment with K2CO3/methanol. The mixture of the epimers of 9 was reduced with NaBH4/EtOH to afford 10, which was a mixture of the four possible diastereomers. Treatment of this isomeric mixture with 85% H3PO4 afforded epimers 5a (MTD) and 5c [(+)-TTD], which were separated by silca gel chromatography. Subsequently, catalytic hydrogenation of 5c gave (+)-2R,6R trans-lobelane [(+)-trans-lobelane, 6c] (Scheme 1a).
Dehydration of 1a with 85% H3PO4, followed by epimerization at C-2, afforded 11 as a mixture of C-2 epimers. Concomitant reduction of the olefinic bond and the carbonyl group in 11 was achieved by catalytic hydrogenation to afford 12, which was a mixture of all four possible diastereomers. Dehydration of 12 with H3-PO4 afforded 13a and 13b, which were obtained in isomerically pure form by silica gel chromatography. Compound 13c and 13d were obtained from 9 by utilizing the same procedure for the synthesis of 13a and 13b from 11 (vide supra) (Scheme 1b).
The synthesis of compounds 17 and 19 is illustrated in Scheme 2. Compound 15 was prepared by condensation of 2,6-lutidine with benzaldehyde.32 Hydrogenation and then methylation of 15 afforded the quaternary ammonium compound 16,32 which upon reduction with NaBH4/EtOH afforded compound 17. A similar procedure was applied to the synthesis of compound 19 from 15. Both 17 and 19 can be transformed into lobelane (6a) under Pd–C catalytic hydrogenation conditions, which indicates that the C2, C6 stereochemistry in 17 and 19 is cis. The COSY NMR spectrum of 19 showed correlations of H-6 (δ 3.00) with H-5 (δ 2.22) and H-7 (δ 6.21) and correlations of H-5′ (δ 2.04) with H-4 (δ 5.82) and H-5 (δ 2.22). Thus, as a result of these data, the structure of 19 was assigned as shown in Scheme 2. Norlobelane (20) (Scheme 2) was prepared by catalytic hydrogenation of compound 15 by utilizing PtO2 as the catalyst.32 The preparation of compound 22a, the N-demethylated analogue of MTD (5a), is illustrated in Scheme 3. A mixture of cis- and trans-norlobelanine (21) was prepared.31 Utilizing a similar procedure for the preparation of 5a and 5b from the mixture of lobeline isomers 1a and 1b, the meso-compound 22a was prepared from 21 and obtained in an isomerically pure form; the racemic compound 22b/c was also isolated from this reaction.
Scheme 2a.

a Reagents and conditions: (a) H2, 10% Pd/C, MeOH, 45 psi; (b) p-C6H4SO3CH3, 170–180 °C; (c) NaBH4, EtOH, 0 °C; (d) H2, PtO2, HOAc, 45 psi.
Scheme 3a.
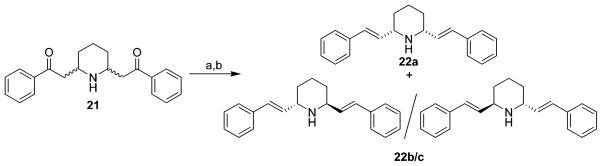
a Reagents and conditions: (a) NaBH4, EtOH, rt; (b) 85% H3PO4, 60 °C.
The quaternary ammonium compounds 23a, 23b, 24a, and 24b (Figure 2) were prepared by treatment of 5a, 5b, 6a, and 6b, respectively, with excess iodomethane in acetone. Ring-opened compounds 25a and 25b (Figure 2) were obtained during the hydrogenation of 5b to 6b and of 5c to 6c, respectively, as byproducts. Acyclic compound 2633 and 2734 (Figure 2) were prepared as previously reported. The nor-1-naphthyl and nor-2-naphthyl compounds 28a and 29a were prepared according to the procedure utilized to prepare norlobelane (20),32 except that 1- or 2-naphthaldehyde was used instead of benzaldehyde (Scheme 4). N-Methylation was then carried out using NaCNBH3/(CH2O)n to afford the corresponding N-methyl compounds, 28b (1-NAP-lobelane) and 29b (2-NAP-lobelane).
Figure 2.

Scheme 4a.
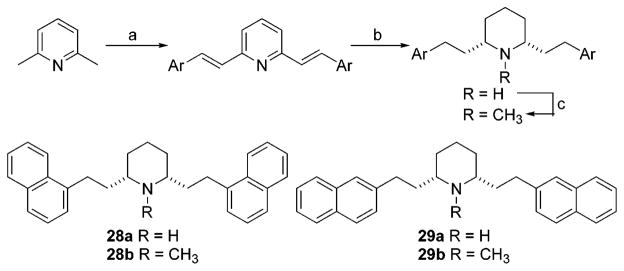
a Reagents and conditions: (a) 1- or 2-naphthaldehyde, Ac2O, reflux; (b) H2, PtO2, 10% HOAc/MeOH, 45 psi; (c) NaCNBH3, (CH2O)n, MeOH, rt.
Results and Discussion
Table 1 shows that lobeline (1a) potently inhibited [3H]NIC binding to rat striatal membranes with a Ki value of 4 nM, had low affinity (Ki = 6.26 μM) for α7* nAChRs, and, importantly, inhibited [3H]DTBZ binding at VMAT2 (Ki = 2.76 μM). Partially defunctionalized lobeline analogues, retaining a single oxygen functionality in the molecule, have low affinity at α4β2* 22,25 but are equipotent or exhibit higher affinity at α7*.22 More importantly, these compounds retained affinity at VMAT2, comparable with lobeline.22 Further structural modification of the lobeline molecule revealed that the fully deoxygenated, unsaturated meso-compound, MTD (5a), had no affinity at either α4β2* or α7* nAChRs but had comparable but slightly lower affinity (Ki = 9.88 μM) compared with lobeline at VMAT2. Thus, MTD was more selective, but slightly less potent at VMAT2 than lobeline. Reduction of the trans double bonds in MTD afforded lobelane (6a, Ki value = 0.97 μM at VMAT2), which was more selective and 3-fold more potent than lobeline at VMAT2.
Table 1.
Ki Values for Lobeline Analogue Inhibition of [3H]NIC (α4β2* nAChR) and [3H]MLA (α7* nAChR) Binding to Rat Brain Membranes and Inhibition of [3H]DTBZ Binding (VMAT2) to Rat Vesicle Membranes
| compd |
Ki, μM, (SEM)a
|
Ki ratio
|
|||
|---|---|---|---|---|---|
| [3H]NIC binding (α4α2*) | [3H]MLA binding (α7*) | [3H]DTBZ binding (VMAT2) | α4α2*/VMAT2 | α7*/VMAT2 | |
| 1a | 0.004 ± 0.000 | 6.26 ± 1.30 | 2.76 ± 0.64 | 0.0014/1 | 2.3/1 |
| 5a | 11.6 ± 2.01 | >100 | 9.88 ± 2.22 | 1.2/1 | >10.1/1 |
| 5b | >100 | >100 | 19.4 ± 1.25 | >5.2/1 | >5.2/1 |
| 5c | >100 | >100 | 7.09 ± 2.42 | >14.1/1 | >14.1/1 |
| 6a | 14.9 ± 1.67 | 26.0 ± 6.57 | 0.97 ± 0.19 | 15.4/1 | 26.8/1 |
| 6b | >100 | 25.3 ± 4.27 | 5.32 ± 0.45 | >18.8/1 | 4.8/1 |
| 6c | >100 | 40.0 ± 14.1 | 6.46 ± 1.70 | >15.5/1 | 6.2/1 |
| 13a | >100 | >100 | 2.50 ± 0.23 | >40.0/1 | >40.0/1 |
| 13b | >100 | 55.1 ± 13.3 | 5.27 ± 1.32 | >19.0/1 | 10.5/1 |
| 13c | >100 | >100 | 2.67 ± 0.56 | >37.4/1 | >37.4/1 |
| 13d | >100 | 25.2 ± 3.00 | > 100 | – | <0.2/1 |
| 17 | >100 | >100 | 3.02 ± 0.41 | >33.1/1 | >33.1/1 |
| 19 | >100 | >100 | 3.84 ± 1.20 | >26.0/1 | >26.0/1 |
| 20 | >100 | >100 | 2.31 ± 0.21 | >43.3/1 | >43.3/1 |
| 22a | >100 | >100 | 2.08 ± 0.08 | >48.1/1 | >48.1/1 |
| 22b/c | >100 | >100 | 5.19 ± 2.45 | >19.3/1 | >19.3/1 |
| 23a | >100 | 33.8 ± 9.79 | >100 | – | <0.3/1 |
| 23b | >100 | 15.1 ± 4.31 | 9.24 ± 2.14 | >10.8/1 | 1.6/1 |
| 24a | 24.3 ± 7.20 | 9.63 ± 0.81 | 16.5 ± 9.26 | 1.5/1 | 0.6/1 |
| 24b | 16.2 ± 2.05 | 2.39 ± 0.16 | 24.6 ± 14.0 | 0.7/1 | 0.1/1 |
| 25a | >100 | >100 | 5.21 ± 1.23 | >19.2/1 | >19.2/1 |
| 25b | >100 | >100 | 3.96 ± 0.80 | >25.2/1 | >25.2/1 |
| 26 | >100 | >100 | 2.37 ± 0.55 | >42.2/1 | >42.2/1 |
| 27 | >100 | >100 | 3.07 ± 1.72 | >32.3/1 | >32.3/1 |
| 28a | >100 | >100 | 4.68 ± 0.70 | >21.4/1 | >21.4/1 |
| 28b | >100 | >100 | 0.63 ± 0.16 | >158.7/1 | >158.7/1 |
| 29a | >100 | >100 | >100 | -- | -- |
| 29b | >100 | >100 | 2.03 ± 0.45 | >49.3/1 | >49.3/1 |
Each Ki value represents data from four independent experiments, each performed in duplicate.
The trans analogues of MTD, 5b and 5c, and the trans analogues of lobelane, 6b and 6c, were synthesized to assess the importance of the C-2 and C-6 stereochemistry of the piperidine ring on VMAT2 affinity and selectivity. Enantiomers 5b and 5c had no affinity at either α4β2* or α7* nAChRs. Interestingly, 5c was equipotent with its meso-isomer, MTD, but 5b was slightly less potent (2–3-fold) than MTD. Surprisingly, only a small difference in affinity between these two trans enantiomers was observed at VMAT2. These results indicate a surprising lack of stereochemical sensitivity at the ligand recognition site at VMAT2, in that a change in the stereochemistry of the piperidino ring at the C2 and C6 positions from cis to trans within the MTD series (i.e., from 5a to 5b and 5c) does not affect interaction with VMAT2. More surprisingly, the observation that there was little difference in affinity for VMAT2 for the trans enantiomers 5b and 5c suggests that the specific configuration at C2 and C6 in these compounds is not recognized by the VMAT2 binding site. Within the lobelane series of compounds (i.e., 6a, 6b, and 6c), a change of C2, C6 stereochemistry from cis to trans afforded a modest reduction (5–6-fold) in affinity at VMAT2. Again, the trans enantiomers 6b and 6c exhibited comparable affinities at VMAT2. Taken together, these data indicate that the VMAT2 binding site does not recognize major stereochemical changes to the MTD and lobelane molecules at the C2 and C6 piperidino ring carbons.
Reduction of only one double bond in MTD affords enantiomers 13a (cis-lob-7E-ene) and 13c (cis-lob-9E-ene), both of which had no affinity at either α4β2* or α7* nAChRs and, importantly, had significantly higher (4-fold) affinity than MTD at VMAT2, indicating that reduction of just one of the double bonds in MTD results in an increase in affinity and selectivity for VMAT2. Again, it is surprising that there are no differences in affinity between these enantiomers at VMAT2. Similarly, the enantiomers 13b (trans-lob-7E-ene) and 13d (trans-lob-9E-ene) showed no affinity at either α4β2* or α7* nAChRs but interestingly did show very different affinities for VMAT2. Specifically, compound 13b had similar affinity to MTD at VMAT2, whereas 13d exhibited no affinity for VMAT2. These results suggest a somewhat complex structure–activity relationship within these structurally related analogues, in that the VMAT2 recognition site appears to accommodate three trans-lobelene isomers, with different stereochemistry at C2 and C6, but does not recognized one specific stereochemical form 13d, which has the 2R,6R stereochemical configuration.
Increasing conformational rigidity in the piperidine ring by introducing a C-3, C-4 double-bond afforded two racemic cis-analogues, 17 and 19. Compound 19 is a mixture of enantiomers that are structural analogues of the chirally pure compounds 13a and 13c. The racemic compound 17 is structurally related to the meso-compound lobelane (6a). As expected, these defunctionalized analogues [(±)-17 and (±)-19] exhibited no affinity at α4β2* and α7* nAChRs, were equipotent with compounds 13a and 13c at VMAT2, and exhibited 4-fold lower affinity than lobelane (6a) at VMAT2. Although 17 and 19 are racemic, making the structure–activity relationships more difficult to elucidate, it would appear that introduction of unsaturation into the piperidine ring does not markedly affect affinity at VMAT2, since only a modest decrease in affinity is observed in these compounds when compared with the corresponding saturated piperidino ring analogues.
Two conformationally flexible, ring-opened compounds, 25a and 25b, were evaluated. Compound 25a and 25b exhibited a 4–5-fold lower affinity compared to lobelane (6a) at VMAT2. Additionally, the two acyclic compounds, 26 and 27 (Ki = 2.37 and 3.07 μM, respectively), exhibited lower affinity for VMAT2 compared to lobelane but were comparable to 25a and 25b. Thus, ring-opening or complete removal of the piperidine ring results in only a modest reduction in affinity at VMAT2 compared to lobelane (6a).
The effect of removing the piperidine N-methyl substituent of lobelane and its analogues was also investigated. Accordingly, compounds 20, 22a, and 22b/c, which are the corresponding nor-compounds of lobelane (6a), 5a, and 5b/5c, respectively, were prepared and evaluated. None of these nor-compounds exhibited affinity at either α4β2* or α7* nAChRs. With respect to VMAT2, removal of the N-methyl group did not have a dramatic effect when compared to the corresponding parent N-methyl compound. These results suggest that the presence of the N-methyl group is not critical for interaction with VMAT2.
1-Methyl-4-phenylpyridinium (MPP+), a quaternary ammonium compound, is a well-known ligand for VMAT2.35–38 Thus, incorporating a quaternary ammonium group into the above lobeline analogues was considered a structural modification worthy of investigation with regard to enhancing VMAT2 affinity. Four quaternary ammonium analogues of MTD and lobelane, i.e., 23a, 23b, 24a, and 24b, were prepared and evaluated. All of these analogues had negligible affinity for α4β2* nAChRs, although 24a and 24b exhibited low affinity (Ki = 2.4–9.6 μM) at α7* nAChRs. Interestingly, compound 23a exhibited no affinity for VMAT2, indicating that N-quaternization of MTD eliminates the interaction with this transporter. Similarly, compound 24a, the N-quaternized form of lobelane, exhibited a near 20-fold decrease in affinity for VMAT2, compared with lobelane (6a). In the trans-MTD and trans-lobelane series, N-quaternization to afford 23b and 24b afforded only modest changes in affinity for VMAT2, when compared to the corresponding nonquaternized parent compounds 5b and 6b, respectively. In summary, incorporating a quaternary ammonium group into defunctionalized lobeline molecules in the cis-series resulted in significant loss of affinity for VMAT2, whereas only a modest effect was obtained in the trans-series.
Four analogues of lobelane (compounds 28a, 28b, 29a, and 29b), in which the phenyl moieties were replaced with 1-naphthyl or 2-naphthyl moieties to determine the relative contribution of π∠π or hydrophobic interactions at the VMAT2 binding site, were prepared. The 1-naph-thyl (28a, 1-NAP-norlobelane, Ki = 4.68 μM) and 2-naphthyl (29a, 2-NAP-norlobelane, Ki > 100 μM) nor-compounds were both less potent than lobelane; however, a dramatic difference in VMAT2 affinity was observed between these meso-analogues. The corresponding N-methyl analogues (28b, 1-NAP-lobelane, Ki = 0.63 μM; 29b, 2-NAP-lobelane, Ki = 2.03 μM) exhibited a more modest difference in VMAT2 affinity. More importantly, 1-NAP-lobelane (28b), which incorporates aromatic moieties with a greater π-surface area, was found to be the most potent and selective lobelane analogue evaluated in this series, suggesting that structural modification of the aromatic moieties of lobelane, and specifically those that increase π∠πinteractions and/or hydrophobic interactions at the recognition site of VMAT2, are worth pursuing further.
Summary
In summary, based upon structural modification of the lobeline molecule, a novel class of VMAT2-selective ligands has been identified. Initial structure–activity relationship studies reveal that the most promising structural changes that enhance VMAT2 affinity and selectivity are defunctionalization, affording lobelane and MTD, and replacement of the phenyl rings with other aromatic moieties that have a π-extended structure, i.e., a 1-naphthyl moiety.
Experimental Section
Chemistry
All purchased reagents were used without further purification. Flash column chromatography was carried out using ICN SilicTech 32–63, 60 Å silica gel. TLC analysis was carried out on EMD Chemicals Inc. glass plates precoated with 250 μm silica gel 60 F254. Melting points were determined on a Fisher Scientific melting point apparatus and are uncorrected. NMR spectra were recorded in CDCl3 on a Varian 300 MHz instrument and are reported in ppm relative to TMS as internal standard. Mass spectra were recorded on a JEOL JMS-700T MStation or on a Bruker Autoflex MALDI-TOF MS. GC–mass spectra were recorded on an Agilent 6890 GC incorporating an Agilent 7683 autosampler and an Agilent 5973 MSD. Optical rotation data were obtained on a Perkin-Elmer 241 polarimeter. All of the final amine compounds were converted to their hydrochloride salts with 2 N HCl in Et2O. Elemental analyses were carried out on a COSTECH elemental combustion system and are within ±0.4% of theory. Compounds 20,32 26,33 and 2734 were prepared according to previously reported methods.
N-Methyl-2,6-cis-di-(E)-styrylpiperidine (5a) and N-Methyl-2S,6S-trans-di-(E)-styryl piperidine (5b)
(−)-Lobeline semisulfate salt (1.85 g) was dissolved in methanol (100 mL) and treated with excess K2CO3 for 48 h at room temperature, concentrated, brought into water, extracted with CHCl3, dried over Na2SO4, filtered, and concentrated to afford a mixture of 2R,6S- and 2S,6S-lobeline free base in nearly equal ratio (determined by NMR) as a white solid (1.35 g, 4.00 mmol). This product was suspended in absolute ethanol (40 mL), and NaBH4 (300 mg, 8.00 mmol) was added at room temperature. The mixture was stirred for 1 h and then quenched with acetone. The solvents were evaporated under reduced pressure, and the residue was suspended in water (60 mL) and extracted with CHCl3 (50 mL × 3). The combined organic extract was dried (Na2SO4), filtered, and concentrated to give a mixture of lobelanidine and its isomers (7) as a white solid, which was used directly in the next reaction. The crude product 7 was dissolved in 85% H3PO4 (40 mL) and the solution allowed to stir at 60 °C for 24 h. The reaction mixture was cooled to room temperature, diluted with water (150 mL), and basified with solid K2CO3 and then NaOH (pH ~10). The aqueous solution was then extracted with EtOAc (80 mL × 3). The combined organic extracts were dried (Na2SO4), filtered, and concentrated. The crude product was purified by silica gel column chromatography (10:1 to 1:1 hexanes:EtOAc gradient) to give title compounds 5a (390 mg, 32%) and 5b (341 mg, 28%), each as white solids. Compound 5a: mp 149–150 °C (lit.31 mp 149–150 °C); 1H NMR δ 1.42–1.88 (m, 6H), 2.25 (s, 3H), 2.63 (m, 2H), 6.21 (dd, J = 15.9, 9.0 Hz, 2H), 6.51 (d, J = 15.9 Hz, 2H), 7.19–7.41 (m, 10H) ppm; 13C NMR δ 24.1, 33.9, 42.6, 68.6, 126.3, 127.5, 128.7, 130.5, 134.0, 137.2 ppm; MS m/z 303 (M+). Anal. (C22H25N·HCl·0.5H2O) C, H, N. Compound 5b: [α]25D −180.2 (c 1.0, CHCl3); mp 93–94 °C (lit.31 mp 96–97 °C); 1H NMR δ 1.60–1.75 (m, 4H), 1.82–1.97 (m, 2H), 2.28 (s, 3H), 3.38 (m, 2H), 6.38 (dd, J = 15.9, 8.7 Hz, 2H), 6.52 (d, J = 15.9 Hz, 2H), 7.17–7.42 (m, 10H) ppm; 13C NMR δ 19.6, 32.9, 42.0, 62.3, 126.4, 127.5, 128.7, 130.5, 131.8, 137.2 ppm; MS m/z 303 (M+). Anal. (C22H25N·HCl·1/3H2O) C, H, N.
N-Methyl-2,6-cis-bis(2-phenethyl)piperidine (6a)
Compound 5a (420 mg, 1.38 mmol) was dissolved in methanol (50 mL) and 10% Pd/C (40 mg) was added. The mixture was hydrogenated on a Parr hydrogenation apparatus (45 psi) for 18 h. The catalyst was removed by filtration through a Celite pad. The filter cake was rinsed with methanol, and the combined organic portions were concentrated under reduced pressure. The resulting residue was purified by silica gel column chromatography (CHCl3:MeOH 30:1) to afford 6a (223 mg, 53%) as a white solid: mp 171–172 °C; 1H NMR δ 1.30–1.42 (m, 4H), 1.60–1.85 (m, 6H), 2.19 (s, 3H), 2.32–2.44 (m, 2H), 2.60–2.77 (m, 4H), 7.12–7.35 (m, 10H) ppm; 13C NMR δ 25.4, 27.1, 31.0, 32.5, 36.6, 62.7, 125.7, 128.4, 128.5, 143.0 ppm; MS m/z 307 (M+). Anal. (C22H29N·HCl·0.2H2O) C, H, N.
N-Methyl-2S,6S-trans-bis(2-phenethyl)piperidine (6b) and N-Methyl-1,9-diphenyl-3S-nonanamine (25a)
Compound 6b was prepared by utilizing a similar procedure to that described for 6a, except that the starting material was 5b (3.0 g, 9.90 mmol), to give 1.32 g (43%) of product as a colorless oil, along with the ring opened product 25a (1.35 g, 44%) as a colorless oil. Compound 6b: [α]25D −35.4 (c 0.5, CHCl3); mp 152–153 °C (HCl salt); 1H NMR δ 1.37–1.52 (m, 2H), 1.55–1.75 (m, 6H), 1.81–1.95 (m, 2H), 2.36 (s, 3H), 2.55–2.67 (m, 4H), 2.67–2.78 (m, 2H), 7.14–7.35 (m, 10H) ppm; 13C NMR δ 20.1, 26.6, 32.4, 33.1, 37.9, 57.6, 125.8, 128.4, 128.5, 142.9 ppm; MS m/z 307 (M+). Anal. (C22H29N·HCl·0.2H2O) C, H, N. Compound 25a: [α]25D −4.4 (c 1.0, CHCl3); 1H NMR δ 1.24–1.45 (m, 6H), 1.53–1.71 (m, 4H), 1.94 (m, 2H), 2.52 (s, 3H), 2.59 (t, J = 7.5 Hz, 2H), 2.66–2.84 (m, 3H), 6.51 (brs, 1H), 7.09–7.37 (m, 10H) ppm; 13C NMR δ 25.5, 29.3, 29.7, 31.3, 31.5, 31.6, 31.8, 33.2, 36.2, 59.1, 125.7, 126.2, 128.3, 128.5 (2C), 128.6, 141.1, 142.8 ppm; MS m/z 309 (M+). Anal. (C22H31N·HCl) C, H, N.
N-Methyl-2R,6R-trans-di-(E)-styrylpiperidine (5c)
Jones reagent (prepared by dissolving 26.72 g of CrO3 in 23 mL of concentrated H2SO4, followed by dilution with water to 100 mL) was added dropwise to a solution of compound 825 (1.22 g, 3.80 mmol) in acetone (60 mL) at 0 °C until an orange-colored reaction mixture was formed. The mixture was stirred for another 30 min, and then methanol was added to quench the reaction. The mixture was then filtered and washed with acetone. The filter cake was dissolved in water (50 mL) and extracted with CHCl3 (25 mL × 3). The filtrate was concentrated under vacuum and diluted with water (30 mL). The aqueous solution was extracted with CHCl3 (25 mL × 3). The combined organic extracts were dried over anhydrous Na2SO4, filtered, and concentrated to afford a yellow solid. Following a similar procedure previously described for the preparation of compound 5a and 5b from LOB sulfate, compound 5c was prepared to give 3.45 g (23%) as a white solid, along with compound 5a (3.22 g, 22%): [α]25D 182.8 (c 1.0, CHCl3); mp 92–93 °C; 1H NMR δ 1.62–1.75 (m, 4H), 1.80–1.96 (m, 2H), 2.28 (s, 3H), 3.38 (m, 2H), 6.38 (dd, J = 15.9, 8.7 Hz, 2H), 6.52 (d, J = 15.9 Hz, 2H), 7.20–7.42 (m, 10H) ppm; 13C NMR δ 19.6, 32.9, 41.9, 62.3, 126.4, 127.5, 128.7, 130.5, 131.7, 137.2 ppm; MS m/z 303 (M+). Anal. (C22H25N·HCl·H2O) C, H, N.
N-Methyl-2R,6R-trans-bis(2-phenethyl)piperidine (6c) and N-Methyl-1,9-diphenyl-3R-nonanamine (25b)
Compound 6c was prepared utilizing a similar procedure to that described for 6a, except the starting material was 5c (2.20 g, 7.26 mmol) to give 1.06 g (48%) of product as a colorless oil, along with the ring-opened product 25b (990 mg, 44%) as a colorless oil. Compound 6c: [α]25D 36.0 (c 1.0, CHCl3); mp 151–152 °C (HCl salt); 1H NMR δ 1.39–1.55 (m, 2H), 1.50–1.75 (m, 6H), 1.81–1.95 (m, 2H), 2.39 (s, 3H), 2.58–2.70 (m, 4H), 2.63–2.78 (m, 2H), 7.15–7.38 (m, 10H) ppm; 13C NMR δ 20.0, 26.6, 32.3, 33.0, 38.0, 57.8, 125.6, 128.5, 128.6, 142.8 ppm; MS m/z 307 (M+). Anal. (C22H29N·HCl·0.2H2O) C, H, N. Compound 25b: [α]25D 4.3 (c 1.0, CHCl3); 1H NMR δ 1.24–1.50 (m, 8H), 1.55–1.77 (m, 4H), 2.39 (s, 3H), 2.47 (m, 1H), 2.56–2.68 (m, 4H), 7.10–7.34 (m, 10H) ppm; 13C NMR δ 25.8, 29.5, 30.1, 31.7, 32.3, 33.5, 33.6, 35.4, 36.2, 58.9, 125.7, 125.8, 128.3, 128.4 (2C), 128.5, 142.7, 142.9 ppm; MS m/z 309 (M+). Anal. (C22H31N·HCl·0.5H2O) C, H, N.
N-Methyl-2R-(E)-styryl-6S-(2-phenethyl)piperidine (13a) and N-Methyl-2S-(E)-styryl-6S-(2-phenethyl)piperidine (13b)
Compound 11 (1.20 g, 3.76 mmol), which was prepared by dehydration of 1a and then epimerization in methanol, was dissolved in 10% HOAc/CH3OH (50 mL), and 10% Pd/C (120 mg) was added. The mixture was hydrogenated on a Parr hydrogenation apparatus (45 psi) for 24 h. The catalyst was removed by filtration through a Celite pad. The filter cake was rinsed with methanol, and the combined organic portion was concentrated under reduced pressure. The crude product 12 was dissolved in 85% H3PO4 (30 mL) and allowed to stir at 60 °C for 24 h. Work up was as describe for 5a and 5b above. The dehydration product was purified by column chromatography (4:1 to 1:1 hexanes:EtOAc gradient) to give the title compound 13a (188 mg, 16%), as a white solid, and 13b (167 mg, 15%), as a colorless oil. Compound 13a: [α]25D 53.8 (c 1.0, CHCl3); mp 75–76 °C; 1H NMR δ 1.30–1.89 (m, 7H), 1.92–2.09 (m, 2H), 2.27 (s, 3H), 2.51–2.69 (m, 2H), 2.70–2.83 (m, 1H), 6.21 (dd, J = 15.9, 8.4 Hz, 1H), 6.47 (d, J = 15.9 Hz, 1H), 7.15–7.40 (m, 10H) ppm; 13C NMR δ 24.5, 31.3, 31.9, 33.7, 36.2, 40.3, 63.9, 68.9, 125.8, 126.3, 127.4, 128.4, 128.5, 128.7, 130.2, 134.9, 137.3, 142.9 ppm; MS m/z 305 (M+). Anal. (C22H27N·HCl·1/3H2O) C, H, N. Compound 13b: [α]25D −103.6 (c 1.0, CHCl3); mp 106–107 °C (HCl salt); 1H NMR δ 1.50–1.82 (m, 7H), 1.84–1.98 (m, 1H), 2.32 (s, 3H), 2.51 (ddd, J = 13.5, 10.8, 5.7 Hz, 1H), 2.68 (ddd, J = 13.5, 10.8, 5.4 Hz, 1H), 2.83 (m, 1H), 3.23 (m, 1H), 6.28 (dd, J = 15.9, 8.4 Hz, 1H), 6.47 (d, J = 15.9 Hz, 1H), 7.13–7.39 (m, 10H) ppm; 13C NMR δ 19.4, 28.9, 29.1, 32.2, 33.2, 40.7, 58.6, 61.8, 125.9, 126.4, 127.4, 128.4, 128.5, 128.7, 131.0, 132.0, 137.3, 142.7 ppm; MS m/z 305 (M+). Anal. (C22H25N·HCl) C, H, N.
N-Methyl-2R-(2-phenylethyl)-6S-(E)-styrylpiperidine (13c) and N-Methyl-2R-(2-phenylethyl)-6R-(E)-styrylpi-peridine (13d)
Compounds 13c and 13d were prepared by utilizing a similar procedure to that described above for compounds 13a and 13b, except the starting material was compound 8 (1.20 g 3.74 mmol), to give the title compound 13c (160 mg, 14%), as a white solid, and 13d (113 mg, 10%), as a colorless oil. Compound 13c: [α]25D −51.5 (c 1.0, CHCl3); mp 74–75 °C; 1H NMR δ 1.30–1.84 (m, 7H), 1.92–2.09 (m, 2H), 2.27 (s, 3H), 2.51–2.69 (m, 2H), 2.70–2.83 (m, 1H), 6.21 (dd, J = 15.9, 8.4 Hz, 1H), 6.47 (d, J = 15.9 Hz, 1H), 7.15–7.40 (m, 10H) ppm; 13C NMR δ 24.5, 31.2, 31.9, 33.7, 36.2, 40.2, 63.9, 68.9, 125.8, 126.3, 127.4, 128.4, 128.5, 128.7, 130.2, 134.8, 137.3, 142.9 ppm; MS m/z 305 (M+). Anal. (C22H27N·HCl) C, H, N. Compound 13d: [α]25D 103.5 (c 1.0, CHCl3); mp 105–106 °C (HCl salt); 1H NMR δ 1.52–1.85 (m, 7H), 1.85–1.90 (m, 1H), 2.32 (s, 3H), 2.51 (ddd, J = 13.5, 10.8, 5.7 Hz, 1H), 2.68 (ddd, J = 13.5, 10.8, 5.7 Hz, 1H), 2.85 (m, 1H), 3.25 (m, 1H), 6.28 (dd, J = 15.9, 8.4 Hz, 1H), 6.47 (d, J = 15.9 Hz, 1H), 7.11–7.41 (m, 10H) ppm; 13C NMR δ 19.2, 28.7, 29.0, 32.0, 33.1, 40.5, 58.6, 61.9, 125.9, 126.3, 127.5, 128.3, 128.5, 128.6, 131.3, 131.4, 137.1, 142.5 ppm; MS m/z 305 (M+). Anal. (C22H25N·HCl) C, H, N.
(−)-N-Methyl-2,6-cis-diphenethyl-1,2,3,6-tetrahydro-pyridine (17)
Compound 15 (colorless crystals recrystallized from benzene, mp 167–168 °C) was synthesized from the condensation reaction of 2,6-lutidine with benzaldehyde utilizing the reported procedure.32 The double bonds of the 2,6-side chains of compound 15 were then reduced by Pd–C catalytic hydrogenation, and the resulting product was treated with methyl p-toluenesulfonate following the reported procedure,32 to afford the N-methylated compound 16 (yellow crystals recrystallized from methanol, mp 238–239 °C). NaBH4 (160 mg, 4.22 mmol) was added to a suspension of compound 16(2.00 g, 4.22 mmol) in absolute ethanol (100 mL) at 0 °C. The mixture was stirred for 2 h and then quenched with acetone. Solvent was removed under reduced pressure and the residue was taken up in water (50 mL) and extracted with CHCl3 (50 mL × 3). The combined organic extracts were dried over Na2SO4, filtered, and concentrated. The crude product was purified by silica gel column chromatography (1:1 hexanes: EtOAc) to give the title compound 17 (245 mg, 19%) as a white solid: mp 162–163 °C (HCl salt); 1H NMR δ 1.62–1.97 (m, 4H), 1.99 (m, 2H), 2.19 (s, 3H), 2.60–2.81 (m, 5H), 3.09 (m, 1H), 5.52 (ddd, J = 10.2, 3.6, 1.8 Hz, 1H), 5.78 (ddd, J = 10.2, 3.9, 3.0 Hz, 1H), 7.13–7.32 (m, 10H) ppm; 13C NMR δ 28.4, 32.2, 32.3, 32.9, 35.6, 36.0, 58.5, 61.2, 125.6, 125.8, 128.4, 128.5, 128.6, 128.8, 142.76, 142.82 ppm; MS m/z 305 (M+). Anal. (C22H27N·HCl·0.5H2O) C, H, N.
(±)-N-Methyl-2,6-cis-2-phenethyl-6-(E)-styryl-1,2,5,6-tetrahydropyridine (19)
Compound 19 was prepared by utilizing a procedure similar to that described above for 17, except the starting material was compound 1832 (1.08 g, 2.30 mmol), to give title compound 19 (265 mg, 38%) as a white solid. mp 90–91 °C; 1H NMR δ 1.85–1.96 (m, 2H), 2.04 (m, 1H), 2.22 (m, 1H), 2.29 (s, 3H), 2.59 (m, 1H), 2.79 (m, 1H), 2.85 (m, 1H), 3.00 (m, 1H), 5.64 (d, J = 10.2 Hz, 1H), 5.82 (m, 1H), 6.21 (dd, J = 15.9, 8.4 Hz, 1H), 6.51 (d, J = 15.9 Hz, 1H), 7.13–7.41 (m, 10H) ppm; 13C NMR δ 31.2, 33.1, 35.6, 40.3, 62.6, 64.2, 124.6, 125.7, 126.4, 127.5, 128.4, 128.7, 129.2, 130.6, 133.7, 137.1, 142.8 ppm; MS m/z 303 (M+). Anal. (C22H25N·HCl) C, H, N.
2,6-cis-Diphenethylpiperidine (20)
mp 199–200 °C (HCl salt) (lit.32 mp 192–194 °C); 1H NMR δ 1.07 (ddd, J = 24.0, 13.2, 3.9 Hz, 2H), 1.32 (ddt, J = 26.4, 12.9, 3.9 Hz, 1H), 1.62–1.74 (m, 6H), 1.78 (dq, J = 13.2, 3.0 Hz, 1H), 2.50 (m, 2H), 2.63 (t, J = 8.1 Hz, 4H), 7.12–7.33 (m, 10H) ppm; 13C NMR δ 25.0, 32.7, 32.9, 39.2, 56.9, 125.9, 128.4, 128.5, 142.3 ppm; MS m/z 293 (M+). Anal. (C21H27N·HCl) C, H, N.
2,6-cis-Di-(E)-styrylpiperidine (22a) and (±)-2,6-trans-Di-(E)-styrylpiperidine (22b/c)
Norlobelanine (21) was prepared according to a reported method.31 Utilizing a similar procedure to that described for the preparation of 5a and 5b from lobeline (1a/1b), compounds 22a (18%) and 22b (13%) were obtained in pure form from norlobelanine (21). Compound 22a: mp 226–227 °C (HCl salt); 1H NMR δ 1.23–1.62 (m, 3H), 1.75 (dd, J = 12.3, 3.0 Hz, 2H), 1.91 (m, 1H), 3.37 (t, J = 8.1 Hz, 2H), 6.26 (dd, J = 15.9, 7.2 Hz, 2H), 6.54 (d, J = 15.9 Hz, 2H), 7.17–7.40 (m, 10H) ppm; 13C NMR δ 24.7, 32.4, 59.7, 126.4, 127.4, 128.6, 129.6, 133.4, 137.2 ppm; MS m/z 289 (M+). Anal. (C21H23N·HCl) C, H, N. Compound 22b/c: mp 194–195 °C (HCl salt); 1H NMR δ 1.53–1.64 (m, 2H), 1.64–1.78 (m, 2H), 1.78–1.92 (m, 2H), 3.78 (m, 2H), 6.41 (dd, J = 15.9, 6.3 Hz, 2H), 6.52 (d, J = 15.9 Hz, 2H), 7.19–7.43 (m, 10H) ppm; 13C NMR δ 20.3, 31.5, 53.8, 126.4, 127.5, 128.7, 129.9, 132.8, 137.3 ppm; MS m/z 289 (M+). Anal. (C21H23N·HCl·0.25H2O) C, H, N.
N,N-Dimethyl-2,6-cis-di-(E)-styrylpiperidine Iodide (23a)
Iodomethane (490 mg, 3.45 mmol) was added to a stirred solution of compound 5a (350 mg, 1.15 mmol) in acetone (8 mL), and the stirring was continued for 24 h. The solvent was removed under reduced pressure, and the residue was washed thoroughly with Et2O to afford the title compound 23a as a white solid (460 mg, 90%): mp 223–224 °C; 1H NMR δ 1.89–2.07 (m, 3H), 2.09–2.25 (m, 3H), 2.93 (s, 3H), 3.34 (s, 3H), 5.36 (brt, J = 9.0 Hz, 2H), 6.17 (dd, J = 15.9, 9.6 Hz, 2H), 7.25 (d, J = 15.9 Hz, 2H), 7.30–7.39 (m, 6H), 7.44–7.52 (m, 4H) ppm; 13C NMR δ 21.7, 27.7, 38.6, 50.5, 73.9, 118.9, 127.4, 128.9, 129.6, 134.6, 142.0 ppm; MS m/z maldi 318 (M − 127)+. Anal. (C23H28NI) C, H, N.
N,N-Dimethyl-2S,6S-trans-di-(E)-styrylpiperidine Iodide (23b)
Compound 23b was prepared by utilizing a similar procedure to that described above for 23a, except using the starting material 5b (50 mg, 0.16 mmol), to give 69 mg (95%) of product as a white solid: [α]25D −182.8 (c 0.5, CHCl3); mp 126–127 °C; 1H NMR δ 1.65–2.30 (m, 6H), 3.28 (s, 6H), 4.71 (m, 2H), 6.44 (dd, J = 15.6, 9.9 Hz, 2H), 7.21 (d, J = 15.6 Hz, 2H), 7.26–7.40 (m, 6H), 7.51–59 (m, 4H) ppm; 13C NMR δ 17.5, 26.8, 48.7, 71.8, 118.3, 127.6, 129.0, 129.6, 134.7, 141.7 ppm; MS (maldi) m/z 318 (M − 127)+. Anal. (C23H28NI) C, H, N.
N,N-Dimethyl-2,6-cis-bis(2-phenethyl)piperidine Iodide (24a)
Compound 24a was prepared by utilizing a similar procedure to that described above for 23a, except using the starting material 6a (116 mg, 0.38 mmol) to give 166 mg (97%), to produce a white solid: mp 241–242 °C; 1H NMR δ 1.69–1.95 (m, 6H), 2.04–2.18 (m, 2H), 2.28–2.43 (m, 2H), 2.67–2.86 (m, 4H), 2.85 (s, 3H), 3.21 (s, 3H), 3.73 (m, 2H), 7.19–7.35 (m, 10H) ppm; 13C NMR δ 22.0, 26.8, 32.3, 33.0, 38.4, 50.1, 74.5, 126.9, 128.6, 128.9, 139.5 ppm; MS (maldi) m/z 322 (M − 127)+. Anal. (C23H32NI·1/3H2O) C, H, N.
N,N-Dimethyl-2S,6S-trans-bis(2-phenethyl)piperidine Iodide (24b)
Compound 24b was prepared by utilizing a similar procedure to that described above for 23a, except using the starting material 6b (58 mg, 0.19 mmol), to give 82 mg (96%) of product as a white solid: [α]25D −37.1 (c 0.3, CHCl3); mp 222–223 °C; 1H NMR δ 1.70–2.00 (m, 6H), 2.08–2.32 (m, 4H), 2.67–2.82 (m, 4H), 3.25 (s, 6H), 3.73 (m, 2H), 7.18–7.36 (m, 10H) ppm; 13C NMR δ 16.8, 24.1, 29.4, 32.6, 49.3, 70.0, 126.9, 128.6, 128.9, 139.5 ppm; MS (maldi) m/z 322 (M − 127)+. Anal. (C23H32NI·0.5H2O) C, H, N.
2,6-cis-Bis(1-naphthalenethyl)piperidine (28a)
Utilizing a similar procedure32 to that described for compound 20, compound 28a was prepared from 2,6-lutidine and 1-naph-thaldehyde: mp 218–219 °C (HCl salt); 1H NMR δ 1.16 (ddd, J = 23.4, 13.2, 3.3 Hz, 2H), 1.39 (ddt, J = 26.4, 13.2, 3.3 Hz, 1H), 1.72–1.92 (m, 7H), 2.63 (m, 2H), 3.09 (dd, J = 9.0, 6.3 Hz, 4H), 7.29–7.42 (m, 4H), 7.42–7.53 (m, 4H), 7.70 (d, J = 7.8 Hz, 2H), 7.84 (dd, J = 7.2, 2.4 Hz, 2H), 8.04 (d, J = 7.5 Hz, 2H) ppm; 13C NMR δ 25.1, 29.9, 32.9, 38.6, 57.4, 124.0, 125.6, 125.7, 125.9, 126.0, 126.7, 128.9, 131.9, 134.0, 138.5 ppm; MS (maldi) m/z 394 (M + 1)+. Anal. (C29H31N·HCl) C, H, N.
N-Methyl-2,6-cis-bis(1-naphthalenethyl)piperidine(28b)
NaBH3CN (344 mg, 5.50 mmol) was added to a mixture of 28a (434 mg, 1.10 mmol), paraformaldehyde (330 mg, 11.00 mmol), and methanol (10 mL). The mixture was stirred at room temperature overnight. The solvent was then evaporated under reduced pressure. The residue was dissolved in water (30 mL) and the aqueous solution was extracted with CHCl3 (20 mL × 3). The combined organic extracts were dried over Na2SO4, filtered, and concentrated. The crude product was purified by silica gel column chromatography (30:1 CHCl3: MeOH) to afford 425 mg (95%) of 28b as a white solid. mp 217–218 °C (HCl salt); 1H NMR δ 1.40–1.64 (m, 4H), 1.68–1.90 (m, 4H), 2.03 (m, 2H), 2.24 (s, 3H), 2.61 (m, 2H), 3.09 (dd, J = 9.0, 6.3 Hz, 4H), 3.18 (t, J = 8.1 Hz, 4H), 7.34–7.41 (m, 4H), 7.42–7.54 (m, 4H), 7.71 (dd, J = 6.3, 2.7 Hz, 2H), 7.85 (dd, J = 7.5, 1.5 Hz, 2H), 8.11 (d, J = 8.1 Hz, 2H) ppm; 13C NMR δ 25.3, 26.7, 29.6, 30.5, 35.8, 63.1, 124.0, 125.5, 125.7, 125.9, 126.1, 126.6, 128.9, 132.0, 134.0, 139.1 ppm; MS (maldi) m/z 408 (M + 1)+. Anal. (C30H33N·HCl·0.75H2O) C, H, N.
2,6-cis-Bis(2-naphthalenethyl)piperidine (29a)
By utilizing a similar procedure to that described for the preparation of compound 20,32 compound 29a was prepared from 2,6-lutidine and 2-naphthaldehyde: mp 222–223 °C (HCl salt); 1H NMR δ 1.12 (ddd, J = 23.7, 12.9, 3.0 Hz, 2H), 1.39 (ddt, J = 25.8, 12.9, 3.6 Hz, 1H), 1.60–1.86 (m, 7H), 2.52 (m, 2H), 2.77 (t, J = 7.8 Hz, 4H), 7.30 (dd, J = 8.7, 1.2 Hz, 2H), 7.36–7.47 (m, 4H), 7.58 (brs, 2H), 7.71–7.82 (m, 6H) ppm; 13C NMR δ 25.0, 32.8, 32.9, 39.0, 56.9, 125.2, 126.0, 126.4, 127.4, 127.5, 127.7, 128.0, 132.1, 133.7, 139.8 ppm; MS (maldi) m/z 394 (M + 1)+. Anal. (C29H31N·HCl·0.5H2O) C, H, N.
N-Methyl-2,6-cis-bis(2-naphthalenethyl)piperidine(29b)
Utilizing a similar procedure to that described for the preparation of compound 28b, compound 29b was prepared from 29a: mp 230–231 °C (HCl salt); 1H NMR δ 1.33–1.45 (m, 4H), 1.70–1.83 (m, 4H), 2.02 (m, 2H), 2.23 (s, 3H), 2.48 (m, 2H), 2.85 (m, 4H), 7.35 (dd, J = 6.6, 1.5 Hz, 2H), 7.38–7.46 (m, 4H), 7.65 (brs, 2H), 7.74–7.82 (m, 6H) ppm; 13C NMR δ 25.2, 29.7, 30.1, 32.9, 36.5, 64.1, 125.9, 126.1, 126.5, 127.6, 127.7, 127.8, 128.2, 131.9, 133.6, 139.7 ppm. MS (maldi) m/z 408 (M + 1)+. Anal. (C30H33N·HCl·0.75H2O) C, H, N.
[3H]NIC and [3H]MLA Binding Assay
Rat striatal and whole brain (excluding cerebellum) membranes were used for the [3H]NIC and [3H]MLA binding assays, respectively. Membrane suspensions (150–200 μg/100 μL) were added to assay tubes containing analogue (7–9 concentrations, 1 nM–1 mM) and 3 nM [3H]NIC or [3H]MLA for a final assay volume of 200–250 μL. For the [3H]NIC and [3H]MLA binding assays, tubes were incubated for 90 and 120 min, respectively. Reactions were terminated by addition of ice-cold buffer and rapid filtration onto either Whatman GF/B glass fiber filters presoaked in 0.5% polyethylenimine using a Brandel Harvester (Biomedical Research and Development Laboratory, Inc., Gaithersburg, MD) or onto Unifilter-96 GF/B 96-well filter plates also presoaked in 0.5% polyethylenimine using a Packard Filter Mate Harvester (Packard BioScience Co., Meriden, CT). Bound radioactivity was determined via liquid scintillation spectrometry. Nonspecific binding was determined in the presence of 10 μM NIC for the [3H]NIC binding assay and in the presence of 10 μM MLA or 1 mM NIC for the [3H]MLA binding assays.
Preparation of Rat Brain Synaptic Vesicles
Synaptic vesicles were prepared as previously described.15 Briefly, fresh whole brain (excluding cerebellum and brain stem) was homogenized in 20 vol of ice-cold 0.32 M sucrose using a glass homogenizer (seven strokes of a Teflon pestle, clearance = 0.003 in). Homogenates were centrifuged at 1000g for 12 min at 4 °C. Resulting supernatants (S1) were centrifuged at 22 000g for 10 min. The resulting pellets (P2), containing the synaptosomes, were resuspended in 18 mL of ice-cold Milli-Q water for 5 min with seven strokes of the Teflon pestle homogenizer. Osmolarity was restored by immediate addition of 2 mL of 25 mM HEPES and 100 mM K2-tartrate buffer (pH 7.5). Samples were centrifuged at 20 000g for 20 min. MgSO4 (final concentration, 1 mM) was added to the resulting supernatants (S3). Final centrifugations were performed at 100 000g for 45 min. Pellets (P4) were resuspended immediately in ice-cold buffer (see below) providing ~15 μg of protein/100 μL.
[3H]DHTBZ Binding Assay
[3H]DHTBZ binding to synaptic vesicles was performed according to previously described procedures.16 Briefly, 100 μL of vesicles suspension was incubated in assay buffer (in 25 mM HEPES, 100 mM K2 tartrate, 5 mM MgSO4, 0.1 mM EDTA, and 0.05 mM EGTA, pH 7.5, 25 °C) in the presence of 5 nM [3H]DHTBZ and 1 nM–1 mM lobeline analogues (final concentrations) for 30 min at room temperature. Nonspecific binding was determined in the presence of 20 μM TBZ. Assays were performed in duplicate using the Unifilter-96 96-well GF/B filter plates (presoaked in 0.5% polyethylenimine) plates and terminated by harvesting using the FilterMate harvester. After washing five times with 350 μL of the ice-cold wash buffer (in 25 mM HEPES, 100 mM K2 tartrate, 5 mM MgSO4, and 10 mM NaCl, pH 7.5), filter plates were dried, bottoms were sealed, and each well was filled with 40 μL of Packard’s MicroScint 20 cocktail. Bound [3H]DHTBZ was measured using a Packard TopCount NXT scintillation counter and a Packard Windows NT-based operating system.
Supplementary Material
Acknowledgments
This research was supported by NIH grants DA 00399 and DA 13519. The authors also gratefully acknowledge the generous gift of [3H]DTBZ from Dr. Michael R. Kilbourn (supported by NIH grant MH 47611). The authors also thank Mr. Robert King of Environmental Research and Training Laboratory, University of Kentucky, for the elemental analyses. For purposes of full disclosure, the University of Kentucky holds patents on lobeline and lobeline analogues that have been licensed by Yaupon Therapeutics Inc. (Lexington, KY). A potential royalty stream to L.P.D. and P.A.C. may occur consistent with University of Kentucky policy. Both L.P.D. and P.A.C. are founders of and have financial interest in Yaupon Therapeutics Inc.
Footnotes
Supporting Information Available: Elemental analyses data. This information is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Wise RA, Bozarth MA. A psychomotor stimulant theory of addiction. Psychol Rev. 1987;94:469–492. [PubMed] [Google Scholar]
- 2.Koob GF. Neural mechanisms of drug reinforcement. Ann NY Acad Sci. 1992;654:171–191. doi: 10.1111/j.1749-6632.1992.tb25966.x. [DOI] [PubMed] [Google Scholar]
- 3.Fleckenstein AE, Hanson GR. Impact of psychostimulants on vesicular monoamine transporter function. Eur J Pharmacol. 2003;479:283–289. doi: 10.1016/j.ejphar.2003.08.077. [DOI] [PubMed] [Google Scholar]
- 4.Brown JM, Hanson GR, Fleckenstein AE. Methamphetamine rapidly decrease vesicular dopamine uptake. J Neurochem. 2000;74:2221–2223. doi: 10.1046/j.1471-4159.2000.0742221.x. [DOI] [PubMed] [Google Scholar]
- 5.Brown JM, Hanson GR, Fleckenstein AE. Regulation of the vesicular monoamine transporter-2: A novel mechanism for cocaine and other psychostimulants. J Pharmacol Exp Ther. 2001;296:762–767. [PubMed] [Google Scholar]
- 6.Sulzer D, Chen TK, Lau YY, Kristensen H, Rayport S, Ewing A. Amphetamine redistributes dopamine from synaptic vesicles to the cytosol and promotes reverse transport. J Neurosci. 1995;15:4102–4108. doi: 10.1523/JNEUROSCI.15-05-04102.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Johnson RA, Eshleman AJ, Meyers T, Neve KA, Janowsky A. [3H]Substrate- and cell-specific effects of uptake inhibitors on human dopamine and serotonin transporter-mediated efflux. Synapse. 1998;30:97–106. doi: 10.1002/(SICI)1098-2396(199809)30:1<97::AID-SYN12>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- 8.Takahashi N, Miner LL, Sora I, Ujike H, Revay RS, Kostic V, Jackson-Lewis V, Prezedborski S, Uhl GR. VMAT2 knockout mice: Heterozygotes display reduced amphetamine conditioned reward enhanced amphetamine locomotion enhanced MPTP toxicity. Proc Natl Acad Sci USA. 1997;94:9938–9943. doi: 10.1073/pnas.94.18.9938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang Y, Gainetdinov RR, Fumagalli F, Xu F, Jones SR, Bock CB, Miller GW, Wightman RM, Caron MG. Knockout of the vesicular monoamine transporter 2 gene results in neonatal death and supersensitivity to cocaine and amphetamine. Neuron. 1997;19:1285–1296. doi: 10.1016/s0896-6273(00)80419-5. [DOI] [PubMed] [Google Scholar]
- 10.Fuente-Fernandez RDL, Furtado S, Guttman M, Furukawa Y, Lee CS, Calne DB, Ruth TJ, Stoessl AJ. VMAT2 binding is elevated in dopa-responsive dystonia: Visualizing empty vesicles by PET. Synapse. 2003;49:20–28. doi: 10.1002/syn.10199. and references therein. [DOI] [PubMed] [Google Scholar]
- 11.Rocha BA, Fumagalli F, Gainetdinov RR, Jones SR, Ator R, Giros B, Miller GW, Caron MG. Cocaine self-administration in dopamine transporter knockout mice. Nat Neurosci. 1998;1:132–137. doi: 10.1038/381. [DOI] [PubMed] [Google Scholar]
- 12.Sora I, Wichems C, Takahashi N, Li XF, Zeng Z, Revay R, Lesch KP, Murphy DL, Uhl GR. Cocaine reward models: Conditioned place preference can be established in dopamine- and in serotonin-transporter knock out mice. Proc Natl Acad Sci US A. 1998;95:7699–7704. doi: 10.1073/pnas.95.13.7699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pletscher A, Brossi A, Gey KF. Benzoquinolizine derivatives: A new class of monoamine decreasing drugs with psychotropic action. Int Rev Neurobil. 1962;4:275–306. [Google Scholar]
- 14.Dwoskin LP, Crooks PA. A novel mechanism and potential use for lobeline as a treatment for psychostimulant abuse. Biochem Pharmacol. 2002;63:89–98. doi: 10.1016/s0006-2952(01)00899-1. [DOI] [PubMed] [Google Scholar]
- 15.Teng L, Crooks PA, Sonsalla PK, Dwoskin LP. Lobeline and nicotine evoke [3H]overflow from rat striatal slices preloaded with [3H]dopamine: Differential inhibition of synaptosomal and vesicular [3H]dopamine uptake. J Pharmacol Exp Ther. 1997;280:1432–1444. [PubMed] [Google Scholar]
- 16.Teng L, Crooks PA, Dwoskin LP. Lobeline displaces [3H]dihydroxytetrabenazine binding and releases [3H]dopamine from rat striatal synaptic vesicles: Comparison with d-amphetamine. J Neurochem. 1998;71:258–265. doi: 10.1046/j.1471-4159.1998.71010258.x. [DOI] [PubMed] [Google Scholar]
- 17.Miller DK, Crooks PA, Teng L, Witkin JM, Munzar P, Goldberg SR, Acri JB, Dwoskin LP. Lobeline inhibits the neurochemical and behavioral effects of amphetamine. J Pharmacol Exp Ther. 2001;296:1023–1034. [PubMed] [Google Scholar]
- 18.Harrod SB, Dwoskin LP, Crooks PA, Klebaur JE, Bardo MT. Lobeline attenuates d-methamphetamine self-administration in rats. J Pharmacol Exp Ther. 2001;298:172–179. [PubMed] [Google Scholar]
- 19.Harrod SB, Dwoskin LP, Green TA, Gehrke BJ, Bardo MT. Lobeline does not serve as a reinforcer in rats. Psychopharmacology. 2003;165:397–404. doi: 10.1007/s00213-002-1289-6. [DOI] [PubMed] [Google Scholar]
- 20.Miller DK, Harrod SB, Green TA, Wong MY, Bardo MT, Dwoskin LP. Lobeline attenuates the locomotor stimulation induced by repeated nicotine administration in rats. Pharmacol Biochem Behav. 2002;74:279–286. doi: 10.1016/s0091-3057(02)00996-6. [DOI] [PubMed] [Google Scholar]
- 21.Miller DK, Crooks PA, Dwoskin LP. Lobeline inhibits nicotine-evoked [3H]dopamine overflow from rat striatal slices and nicotine-evoked 86Rb+ efflux from thalamic synaptosomes. Neuropharmacology. 2000;39:2654–2662. doi: 10.1016/s0028-3908(00)00140-4. [DOI] [PubMed] [Google Scholar]
- 22.Miller DK, Crooks PA, Zheng G, Grinevich VP, Norrholm S, Dwoskin LP. Lobeline analogues with enhanced affinity and selectivity for plasmalemma and vesicular mono-amine transporters and diminished affinity at α4β2* and α7* nAChRs. J Pharmacol Exp Ther. 2004;310:1035–1045. doi: 10.1124/jpet.104.068098. [DOI] [PubMed] [Google Scholar]
- 23.Briggs CA, McKenna DG. Activation and inhibition of the human α7 nicotinic acetylcholine receptor by agonist binding affinity. Neuropharmacology. 1998;37:1095–1102. doi: 10.1016/s0028-3908(98)00110-5. [DOI] [PubMed] [Google Scholar]
- 24.Crooks PA, Jones MD, Chesnut MD, Jaromczyk AM, Dwoskin LP. Stereochemically defined lobeline analogues: Inhibition of [3H]dopamine uptake and [3H]nicotine binding in rat striatum. College Problems Drug Dependence. 1999;61:29. [Google Scholar]
- 25.Flammia D, Malgorzata D, Damaj MI, Martin B, Glennon RA. Lobeline: Structure-affinity investigation of nicotinic acetylcholinergic receptor binding. J Med Chem. 1999;42:3726–3731. doi: 10.1021/jm990286m. [DOI] [PubMed] [Google Scholar]
- 26.Perera RP, Wimalasena DS, Wimalasena K. Characterization of a series of 3-amino-2-phenylpropene derivatives as novel bovine chromaffin vesicular monoamine transporter inhibitors. J Med Chem. 2003;46:2599–2605. doi: 10.1021/jm030004p. [DOI] [PubMed] [Google Scholar]
- 27.Canney DJ, Kung M, Kung HF. Amino- and amidotetrabenazine derivatives: Synthesis and evaluation as potential ligands for the vesicular monoamine transporter. Nucl Med Biol. 1995;22:527–535. doi: 10.1016/0969-8051(94)00118-4. [DOI] [PubMed] [Google Scholar]
- 28.Lee LC, Vander BT, Sherman PS, Frey KA, Kilbourn MR. In vitro and in vivo studies of benzisoquinoline ligands for the brain synaptic vesicle monoamine transporter. J Med Chem. 1996;39:191–196. doi: 10.1021/jm950117b. [DOI] [PubMed] [Google Scholar]
- 29.Compere D, Marazano C, Das BC. Enantioselective access to lobelia alkaloids. J Org Chem. 1999;64:4528–4532. [Google Scholar]
- 30.Zheng G, Dwoskin LP, Crooks PA. Indirect trapping of the retro-conjugate addition reaction intermediate involved in the epimerization of lobeline: Application to the synthesis of (−)-sedamine. J Org Chem. 2004;69:8514–8517. doi: 10.1021/jo048848j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ebnöther A. 48. Über die Mutarotation des Lobelins. Cis-trans-Isomere in der Reihe der Lobelia-alkaloide. Helv Chim Acta. 1958;41:386–396. [Google Scholar]
- 32.Lee J, Freudenberg W. Piperidine derivatives. Part 1. Lobelan and related compounds. J Org Chem. 1944;9:537–546. [Google Scholar]
- 33.Blickle FF, Zienty FB. Antispasmodics. IV. J Am Chem Soc. 1939;61:774–776. [Google Scholar]
- 34.Stuhmer W, Elbrachter E. N-Substituierte bis(3,3′-phenylpropyl)amine. Arch Pharmacol. 1954;287:139–142. doi: 10.1002/ardp.19542870308. [DOI] [PubMed] [Google Scholar]
- 35.Daniels AJ, Reinhard JF., Jr Energy-driven uptake of the neurotoxin 1-methyl-4-phenylpyridine into chromaffin granules via the catecholamine transporter. J Biol Chem. 1988;263:5034–5036. [PubMed] [Google Scholar]
- 36.Darchen F, Scherman D, Henry JP. Characteristics of the transport of quaternary ammonium 1-methyl-4-phenylpyridine by chromaffin granules. Biochem Pharmacol. 1988;37:4381–4387. doi: 10.1016/0006-2952(88)90621-1. [DOI] [PubMed] [Google Scholar]
- 37.Del Zompo M, Piccardi MP, Quartu SRM, Gessa GL, Vaccari A. Selective uptake into synaptic dopamine vesicles: Possible involvement in MPTP neurotoxicity. Br J Pharm. 1993;109:411–414. doi: 10.1111/j.1476-5381.1993.tb13584.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Moriyama Y, Amakatsu K, Futai M. Uptake of the neurotoxin, 4-methylphenylpyridinium, into chromaffin granules and synaptic vesicles: A proton gradient drives its uptake through monoamine transporter. Arch Biochem Biophys. 1993;305:271–277. doi: 10.1006/abbi.1993.1422. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.