

Key Points
The Abs to the human fVIII A2 domain in a murine hemophilia A model inhibit fVIIIa and activation of fVIII
Epitopes targeted by hemophilia A mouse Abs cover nearly the entire surface of the human fVIII A2 domain
Abstract
Approximately 30% of patients with severe hemophilia A develop inhibitory anti–factor VIII (fVIII) antibodies (Abs). We characterized 29 anti-human A2 monoclonal Abs (mAbs) produced in a murine hemophilia A model. A basis set of nonoverlapping mAbs was defined by competition enzyme-linked immunosorbent assay, producing 5 major groups. The overlapping epitopes covered nearly the entire A2 surface when mapped by homolog-scanning mutagenesis. Most group A mAbs recognized a previously described epitope bounded by Arg484-Ile508 in the N-terminal A2 subdomain, resulting in binding to activated fVIII and noncompetitive inhibition of the intrinsic fXase complex. Group B and C mAbs displayed little or no inhibitory activity. Group D and E mAbs recognized epitopes in the C-terminal A2 subdomain. A subset of group D mAbs inhibited the activation of fVIII by interfering with thrombin-catalyzed cleavage at Arg372 at the A1-A2 domain junction. Other group D mAbs displayed indeterminate or no inhibitory activity despite inhibiting cleavage at Arg740 at the A2-B domain junction. Group E mAbs inhibited fVIII light-chain cleavage at Arg1689. Inhibition of cleavages at Arg372 and Arg1689 represent novel mechanisms of inhibitor function and, along with the extensive epitope spectrum identified in this study, reveal hitherto unrecognized complexity in the immune response to fVIII.
Introduction
The standard of care for patients with congenital hemophilia A is the infusion of recombinant or plasma-derived factor VIII (fVIII). Approximately 30% of patients with severe hemophilia A develop detectable inhibitory anti-fVIII antibodies (Abs) (inhibitors) in response to these infusions,1-3 which is the most serious complication in the treatment of hemophilia A. In addition, nonhemophiliacs can break tolerance to their native fVIII protein. This results in acquired hemophilia A, which can produce life- or limb-threatening bleeding.
fVIII is a glycoprotein that contains an A1-A2-B-ap-A3-C1-C2 domain sequence. The A domains are homologous to corresponding triplicated domains in factor V (fV) and ceruloplasmin. Each A domain contains 2 ∼20-kDa cupredoxin-like subdomains.4 Intracellular cleavages within the B domain of fVIII or at the A2-B junction prior to its secretion produce A1-A2-B or A1-A2 heavy-chain species and an ap-A3-C1-C2 light-chain. fVIII circulates predominantly as an A1-A2-B/ap-A3-C1-C2 heterodimer tightly bound to von Willebrand factor (VWF). fVIII is activated by thrombin-catalyzed heavy-chain cleavages at Arg372 at the A1-A2 junction and Arg740 at the A2-B junction and by light-chain cleavage at Arg1689 at the ap-A3 junction, which results in dissociation of fVIIIa from VWF.5 The product is a 160-kDa A1/A2/A3-C1-C2 fVIIIa heterotrimer. fVIIIa is a cofactor for factor IXa (fIXa) in the activation of factor X (fX) in the intrinsic pathway of blood coagulation. The A2 subunit participates in the binding of fX to the intrinsic fXase complex.6 Additionally, it has been reported that fIXa interacts with the A2 subunit.7 Following thrombin activation, the A2 domain spontaneously dissociates resulting in loss of cofactor activity.8
fVIII inhibitors in congenital and acquired hemophilia A are polyclonal immunoglobulin G (IgG) populations that typically recognize both the A2 and C2 domains.9 Scandella et al10 identified an immunodominant A2 epitope using a murine monoclonal Ab (mAb), designated 413. 413 competes with human inhibitor plasmas for binding to fVIII, indicating that the Arg484-Ile508 epitope is clinically relevant. The 413 epitope was mapped to a linear segment bounded by residues Arg484-Ile508.11413 interferes with fX binding and is a noncompetitive inhibitor of the intrinsic fXase complex.6 We have produced a large panel of murine anti-human fVIII hybridomas by immunizing mice with human fVIII under conditions that mimic therapeutic use of fVIII.12 Anti-A2 mAbs were identified that do not overlap the 413 epitope. In this study, 29 murine anti-human A2 mAbs were characterized to develop a comprehensive epitope map and to identify additional inhibitory properties of A2 inhibitors.
Methods
Materials
Mouse studies were done in accordance with the Emory University Institutional Animal Care and Use Committee (under protocol no. 2001085, “Low Immunogenicity Factor VIII”; approved July 8, 2011, will expire July 8, 2014). mAb 413 was a gift from the American Red Cross (Rockville, MD). CLB-CAg 9 was a gift from Dr Jan Voorberg (Sanquin-AMC Landsteiner Laboratory, Amsterdam, The Netherlands). GMA-012 (originally described as R8B1213) was purchased from Green Mountain Antibodies (Burlington, VT). Enzyme-linked immunosorbent assay (ELISA) plates were purchased from Thermo Fisher Scientific (Norcross, GA). Pooled citrated normal plasma and fVIII-deficient plasma were purchased from George King Biomedical (Overland Park, KS). Phosphatidylcholine/phosphatidylserine (PCPS) (75/25, w/w) vesicles were prepared as described previously.14 Human B domain–deleted (BDD) fVIII and BDD human/porcine hybrid (HP) constructs were prepared as described previously.11,15 Recombinant full-length human fVIII (Helixate) was a gift from Hemophilia of Georgia. Human VWF was purified and characterized as described previously.5 Molar VWF concentrations are reported in terms of the 265-kDa monomeric subunit.
Anti-human fVIII A2 domain mAbs
Anti-human fVIII A2 domain IgG mAbs were purified from supernatants of splenic B-cell hybridomas that were produced following intravenous immunization of mice with adjuvant-free human fVIII as described previously.12 IgG concentrations were calculated using an extinction coefficient at 280 nm of 1.37 (mg/mL)−1 cm−1. IgG isotypes and subclasses were determined by ELISA using an alkaline-phosphatase isotype and subclass-specific Abs from SouthernBiotech (Birmingham, AL) as previously described.12
Identification of overlapping and nonoverlapping A2 epitopes by competition ELISA
A competition sandwich ELISA was performed using immobilized anti-fVIII A2 domain primary mAb, human fVIII, biotinylated anti-A2 secondary mAb, and streptavidin–alkaline-phosphatase conjugate for detection as previously described.12 In this assay, nonoverlapping epitopes of a mAb pair results in binding of secondary mAb, which was defined as an absorbance at 405 nm >3 times background.
Epitope mapping by homolog-scanning mutagenesis
Hybrid human/porcine constructs (1 U/mL) were captured on microtiter wells using an immobilized anti-C2 domain mAb, I14. Biotinylated anti-A2 test mAbs were added to a final concentration of 1 μg/mL and incubated for 30 minutes at room temperature. Bound mAb was detected using streptavidin–alkaline-phosphatase conjugate as described previously.12 Pymol was used to study the x-ray structure of BDD fVIII (PDB 2R7E) and identify the epitope-containing regions of the A2 domain of fVIII.
FVIII inhibitor assay
FVIII inhibitor titers were measured using the Bethesda assay16 using previously described modifications.17 Pooled citrated normal human plasma was used as the source of fVIII activity. One Bethesda unit (BU) per milliliter is defined as the dilution of inhibitor that produces 50% inhibition of fVIII activity. Inhibition curves were fitted by nonlinear least-squares analysis using the 4-parameter logistic equation to estimate the concentration of mAb producing 50% inhibition.
Intrinsic fXase assay
BDD human fVIII (50nM) was incubated with increasing concentrations of anti-A2 mAbs for 30 minutes at 37°C. Human thrombin (10 nM) was added and samples were removed 30 seconds later and placed into sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) sample buffer. One minute after addition of thrombin, the sample was diluted 100-fold into a solution containing 1.5nM fIXa/20 μM PCPS phospholipid vesicles. fX (300 nM) was added 15 seconds later, followed by removal of aliquots 15, 30, 45, and 60 seconds thereafter and addition to 0.05M EDTA to stop fX activation. fXa activity was measured chromogenically using Spectrozyme Xa as described previously.14
Effect of anti-A2 mAbs on thrombin-catalyzed proteolytic cleavage of fVIII
Full-length or BDD fVIII (100 nM) were incubated for 10 minutes at 37°C with varying concentrations of mAb. In some experiments, fVIII was incubated with VWF (1000 nM) for 10 minutes at 37°C before addition of the Abs. Thrombin (0.5 nM) was added and the incubation was continued for an additional 10 minutes. The reaction was stopped with the addition of 10 µL of nonreducing SDS-PAGE sample buffer (Pierce Biotechnology, Rockford, IL) followed by heating at 98°C for 5 minutes. Samples were analyzed by 10% SDS-PAGE (Ready Gel; Bio-Rad Life Science, Hercules, CA) and were stained overnight with GelCode Blue (Pierce). Gels were destained with water and imaged with an Odyssey Imaging System (Licor Biosciences, Lincoln, NE).
Results
The diversity of epitopes recognized by anti-human A2 mAbs
A competition ELISA was used to determine the overlap pattern of 29 murine anti-human A2 mAbs. Full-length fVIII was bound to an immobilized primary (capture) anti-A2 mAb, followed by addition of biotinylated secondary anti-A2 mAb and detection of the secondary mAb using streptavidin–alkaline phosphatase. Binding of the secondary mAb indicates that the Ab pair recognizes nonoverlapping epitopes.
Each mAb was used in both primary and secondary configurations producing an overlap matrix (Figure 1A). Noncompeting and competing mAbs are depicted as color (yellow) or noncolor (white) elements, respectively. As expected, the diagonal elements are white, indicating that each mAb competed with itself for binding. The matrix is symmetrical with respect to the diagonal element with the exception of biotinylated G6 and G4 and nonbiotinylated B99 and G74, which displayed no binding. Lack of binding of G6 and G4 may have resulted from biotinylation of the critical residues in the Ab paratope. Conversely, lack of binding of B99 and G74 may have resulted from insufficient immobilization of the Ab to the microtiter plate. In total, the competition matrix produced 16 unique epitopes.
Figure 1.
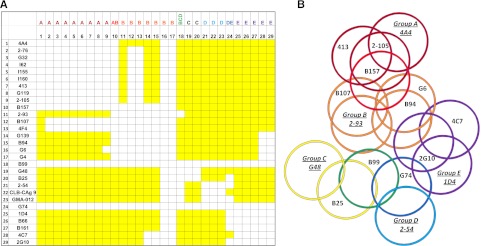
ELISA competition matrix and overlap pattern of anti-A2 mAbs. (A) Competition matrix compiling the results of competition ELISA among 29 anti-A2 mAbs. Rows represent primary mAbs and columns represent biotinylated secondary mAbs. Yellow and white squares represent binding and no binding (no overlap and overlap), respectively, of secondary mAbs. (B) Venn diagram representing overlaps produced by the competition matrix. Each mAb was assigned a group based on its overlap with a basis set of 5 mAbs 4A4—2-93, G48, 2-54, and 1D4—that are underlined and italicized. Colors depict the 8 groups of mAbs defined by their overlap with basis set mAbs: dark red, group A; red, group AB; orange, group B; yellow, group C; green, group BCD; light blue, group D; blue, group DE; and purple, group E.
The results of this analysis yielded a basis set of 5 mAbs: 4A4, 2-93, G48, 2-54, and 1D4. The basis set is defined as a set of mAbs that do not compete with each other for binding to fVIII but as a group compete with all of the remaining mAbs. The members of the basis set in turn defined 5 Ab groups, designated A, B, C, D, and E. Group A consists of 4A4 and mAbs that compete only with 4A4. Group A mAbs represented the largest group, consisting of mAbs derived from 5 different mice, and include the previously characterized mAb 413.10,11 mAbs 2-76, G32, I62, I155, I160, and G119 displayed an overlap pattern identical to 413. Group A mAb 2-105 differs because it overlaps G6 while 413 does not. Group B consists of 2-93 and mAbs that compete only with 2-93. Group C consists of G48 and mAbs that only compete with G48. Group D consists of 2-54 and mAbs that compete only with 2-54. Group E consists of 1D4 and mAbs that compete only with 1D4. Three additional groups were identified based on partial overlaps with the basis set mAbs. Group AB consists of mAbs that compete with 4A4 and 2-93. Group BCD consists of mAbs that compete with 2-93, G48, and 2-54, and group DE mAbs compete with 2-54 and 1D4. The individual group members are identified as a row header in Figure 1A and in Table 1. By comparing all pairwise overlaps and nonoverlaps, a Venn diagram consisting of 16 unique epitopes was produced (Figure 1B).
Table 1.
Properties of anti-A2 mAbs
| mAbs | BU/mg IgG | Type | Isotype | Group | Epitope | |
|---|---|---|---|---|---|---|
| 1 | 4A4 | 40 000 | I | IgG2aκ | A | Asp403-His444 |
| 2 | 2-76 | 38 000 | I | IgG2aκ | A | Arg484-Ile508 |
| 3 | G32 | 3 000 | I | IgG2aκ | A | His444-Ile508 |
| 4 | I62 | 15 000 | I | IgG2aκ | A | Arg484-Ile508 |
| 5 | I155 | 40 000 | I | IgG1κ | A | Arg484-Ile508 |
| 6 | I160 | 20 000 | I | IgG2aκ | A | Arg484-Ile508 |
| 7 | 413 | 21 000 | I | IgG1κ | A | Arg484-Ile508 |
| 8 | G119 | 10 000 | I | IgG1κ | A | Arg484-Ile508 |
| 9 | 2-105 | 40 000 | I | IgG1κ | A | Arg484-Ile508 |
| 10 | B157 | <1 | N/A* | IgG1κ | AB | Indeterminate |
| 11 | 2-93 | 4 | II | IgG1κ | B | Arg541-Glu604 |
| 12 | B107 | Indeterminate† | II | IgG2aκ | B | Arg541-Glu604 |
| 13 | 4F4 | 330 | I | IgG2aκ | B | Indeterminate |
| 14 | G139 | <1 | N/A* | IgG1κ | B | Arg541-Glu604 |
| 15 | B94 | Indeterminate† | II | IgG1κ | B | Arg541-Glu604 |
| 16 | G6 | NI | N/A* | IgG2aκ | B | His444-Ile508 |
| 17 | G4 | 9 | I | IgG1κ | B | Arg541-Glu604 |
| 18 | B99 | 11 | I | IgG1κ | BCD | Indeterminate |
| 19 | G48 | 5 | II | IgG2aκ | C | His444-Arg541 |
| 20 | B25 | 100 | I | IgG1κ | C | His444-Gln468 |
| 21 | 2-54 | 34 000 | II | IgG1κ | D | Glu604-Arg740 |
| 22 | CLB-CAg 9 | Indeterminate† | II | IgG1κ | D | Glu604-Arg740 |
| 23 | GMA-012 | <1 | N/A* | IgG1κ | D | Glu604-Arg740 |
| 24 | G74 | 1 500 | I | IgG1κ | DE | Glu604-Arg740 |
| 25 | 1D4 | 7 000 | I | IgG2aκ | E | Glu604-Arg740 |
| 26 | B66 | 4 000 | I | IgG2aκ | E | Glu604-Arg740 |
| 27 | B161 | 150 | I | IgG2aκ | E | Glu604-Arg740 |
| 28 | 4C7 | <1 | N/A* | IgG2aƙ | E | Indeterminate |
| 29 | 2G10 | Indeterminate† | II | IgG2aƙ | E | Indeterminate |
*Not applicable to noninhibitory mAbs.
†Residual fVIII activity at saturating concentrations of Ab was >50%.
Epitope mapping by homolog-scanning mutagenesis
Most murine anti-human fVIII mAbs cross-react poorly with porcine fVIII.12 Thus, loss of antigenicity of hybrid human/porcine fVIII constructs created by replacement of human A2 regions with the homologous regions of porcine fVIII (Figure 2A) can be used to map regions that contain major epitope determinants.18 Because human/porcine fVIII molecules have normal procoagulant activity, this method has the advantage of using conformationally intact fVIII molecules, avoiding the possibility of artifactual loss of antigenicity due to protein misfolding. Using this approach, an epitope-containing region was found for 24 of the 29 mAbs (Table 1). ELISA results from representative mAbs from groups A through E are shown in Figure 2B and results for the remaining mAb set are shown in supplemental Figure 1 (available on the Blood website). For group A mAb 413, HP9 was the construct that contained the smallest porcine substitution associated with loss of antigenicity. This confirms Arg484-Ile508 as an epitope recognized by this mAb, consistent with earlier results.11 For group B mAb 2-93, loss of antigenicity associated with HP2 (porcine substitution at Ala387-Glu604), but not HP4 (porcine substitution at Ala387-Arg541) placed a major determinant of the epitope between residues Arg541 and Glu604. For group C mAb G48, loss of antigenicity associated with HP5 (porcine substitution at Ala387-Ile508), but not HP6 (porcine substitution at Ala387-His444) placed a major determinant of the epitope between residues Glu445 and Ile508. For group D and E mAbs, loss of antigenicity associated with HP1 (porcine substitution at Ala387-Arg740), but not HP2 (porcine substitution Ala387-Glu604) or other HP constructs place a major determinant of the epitope between residues Asp605 and Arg740 in the C-terminal cupredoxin-like A2 subdomain. The amino acid sequences that contribute to Ab binding identified by homolog-scanning mutagenesis corresponded closely to the overlap groups. For example, 8 of the 9 group A mAbs mapped to epitopes that include the Arg484-Ile508 segment. The exception was group A mAb 4A4, which mapped to residues Asp403-His444 yet competed with all other group A mAbs in the competition ELISA. Regions of Asp403-His444 and Arg484-Ile508 are in close proximity in the x-ray structure of fVIII (Figure 3A). In homolog-scanning mutagenesis, amino acids that constitute major determinants of a mAb’s epitope cannot be identified if they are identical in the porcine and human sequences. Therefore, our overlap data suggest that 4A4 and mAbs that map to Arg484-Ile508 recognize part of a shared discontinuous epitope.
Figure 2.


Epitope mapping of anti-A2 mAbs by homolog-scanning mutagenesis. (A) BDD hybrid human/porcine fVIII constructs. Shaded areas represent porcine fVIII substitutions. (B) Binding of anti-A2 mAbs to human/porcine fVIII. fVIII was captured on a microtiter plate using immobilized anti-C2 mAb, followed by addition of biotinylated anti-A2 mAb and detection using streptavidin–alkaline phosphatase. Bars represent the range of duplicate determinations.
Figure 3.

Map of overlapping mAb epitopes onto the A2 surface. (A) X-ray structure of BDD fVIII (PDB 2R7E) showing regions recognized by group A mAbs (Asp403-His444 and Arg484-Ile508) and group E mAbs (Glu604-Cys711). A2 residues Asp712-Arg740 were not identified in the structure. (B) Regions with the A2 polypeptide chain identified by homolog-scanning mutagenesis were used to anchor the Venn diagram onto the A2 surface.
Mapping the anti-A2 mAb Venn diagram onto the fVIII surface
Low-resolution (3.7-4.0 Å) x-ray structures of BDD human fVIII are available (Figure 3A).19,20 The polypeptide segments recognized by anti-A2 mAbs obtained from homolog-scanning mutagenesis (Table 1) were used to map the Venn diagram (Figure 1B) onto the A2 surface of these models (Figure 3B). Because the Venn diagram delineates only a logical relationship between overlapping epitopes, its rings do not correspond to a physical dimension. However, simultaneous localization of group A, C, and E mAb Venn rings to their corresponding cognate polypeptide segments restricts the size of the Venn structure such that the each ring approximates the footprint associated with its associated mAb. Using this approach, it is evident that nearly the entire A2 surface is targeted by the mAbs used in this study.
Inhibitory properties of anti-A2 mAbs
The anticoagulant properties of the anti-A2 mAbs were investigated using the Bethesda assay. Anti-fVIII Abs are classified as type I or type II inhibitors based on whether they completely (>90%) or incompletely inhibit fVIII at saturating concentrations.21 A representative assay is shown for mAbs from groups A, B, C, D, and E in Figure 4A and the results for the entire mAb set are shown in Table 1. All of the group A mAbs were type I inhibitors and except for G32, had high specific activity. In contrast to most of the other group A mAbs, the epitope of G32 included but was not restricted to Arg484-Ile508. Group B and C mAbs displayed little or no inhibition. Group D and group E mAbs displayed a wide range of inhibitory activity.
Figure 4.

Anticoagulant properties of anti-A2 mAbs. (A) Residual fVIII activity was measured by 1-stage coagulation assay following incubation of normal human plasma with varying concentrations of mAbs for 2 hours at 37°C. Data represent sample means and sample standard deviations. The curves represent least-squares fits to the data. The inhibitor titer in Bethesda units per milliter was obtained by determining the dilution of mAb producing 50% inhibition and converted to Bethesda units per milligram using the mAb concentration. (B) BDD human fVIII (50 nM) was incubated with the indicated concentrations of anti-A2 mAbs for 30 minutes and then thrombin for 60 seconds, followed by sample dilution into fIXa/PCPS phospholipid vesicles, addition of fX, and measurement of fXa as described in “Intrinsic fXase assay.” Results are presented as the percentage of fXa formed in the absence of mAb.
Additionally, the inhibition of fVIIIa by anti-A2 mAbs was analyzed in a plasma-free intrinsic fXase assay using purified components. Figure 4B shows results for representative group A, D, and E mAbs that displayed inhibition in the Bethesda assay. Group A mAb 413 and group E mAb 1D4 completely inhibited fXa formation. Group A mAbs 4A4 and G32 and group E mAb B66 also produced near complete inhibition of fXa formation (data not shown). In contrast, group D mAb 2-54 displayed partial inhibition at saturating concentrations. Thus, the inhibitory properties of the mAbs in the purified intrinsic fXase system are consistent with their anticoagulant properties in the Bethesda assay.
Effect of anti-A2 mAbs on thrombin-catalyzed proteolytic cleavage of fVIII
Because fVIII was allowed to bind Ab before addition of thrombin in the experiment described in Figure 4B, inhibition of fXa formation could be due either to direct inhibition of fVIIIa in the intrinsic fXase complex or inhibition of proteolytic cleavages leading to fVIIIa formation. To evaluate the latter possibility, the effect of anti-A2 mAbs on thrombin-catalyzed cleavage of fVIII was evaluated by SDS-PAGE.
The activation of fVIII by thrombin is initiated by rapid heavy-chain cleavage at Arg740, which releases the B domain and produces the A1-A2 subunit (Figure 5).22 This is followed by Arg1689 cleavage at the ap-A3 light-chain junction producing the thrombin-cleaved light chain (LCIIa), and Arg372 cleavage at the A1-A2 junction, which produces the A1 and A2 subunits of fVIIIa.
Figure 5.

Inhibition of thrombin-catalyzed activation of fVIII by group D and E mAbs. The activation of full-length fVIII (A1-A2-B/ap-A3-C1-C2) is associated with proteolytic cleavages catalyzed by thrombin. The fastest cleavage occurs at Arg740 in the A1-A2-B heavy chain, producing the A1-A2 fragment and the ap-A2-C1-C2 light chain. Cleavage then occurs at Arg372 or Arg1689. Cleavage at Arg372 is necessary for the development of fIXa cofactor activity of fVIIIa, whereas cleavage at Arg1689 is necessary for the dissociation of fVIII from VWF. Cleavage at Arg740 is not necessary for either fVIIIa formation or the dissociation of fVIII from VWF.
Most of the group D and E mAbs inhibited thrombin-catalyzed proteolysis of full-length fVIII (Figure 6). Group D mAb 2-54 and group DE mAb G74 inhibited production of the A1 and A2 subunits, but not the A1-A2 subunit, indicating inhibition of cleavage at Arg372. Additionally, 2-54 and G74 inhibited light-chain cleavage. Analysis of samples taken during the intrinsic fXase experiment described in Figure 4B showed partial inhibition of BDD fVIII heavy-chain and light-chain cleavage at saturating concentrations of 2-54 (supplemental Figure 3), consistent with the partial inhibition of fXa formation.
Figure 6.

Effect of group D and E anti-A2 mAbs on thrombin-catalyzed proteolytic cleavage of full-length fVIII. fVIII (100 nM) was incubated with 0.5 nM thrombin for 10 minutes at 37°C in the presence of the indicated mAb concentrations. Proteolytic cleavages were analyzed by SDS-PAGE as described in “Effect of anti-A2 mAbs on thrombin-catalyzed proteolytic cleavage of fVIII.” Ctrl, fVIII control not exposed to thrombin or anti-A2 mAbs; Stds, Molecular weight standards are 250, 150, 100, 75, 50, 37, and 25 kDa.
Group D mAbs CLB-CAg 9 and GMA-012 also inhibited light-chain cleavage, but in contrast to 2-54 and G74, inhibited production of the A2 subunit, but not the A1 subunit. This result is consistent with normal cleavage at Arg372 and inhibition of cleavage at Arg740. Additionally, the inhibition pattern due to CLB-CAg 9 and GMA-012 was associated with the appearance of a novel 60-kDa band (Figure 6 asterisks). The presence of this band is consistent with a secondary thrombin cleavage within the B domain.
Group E mAbs B161, B66, and 1D4 inhibited light-chain cleavage, but not heavy-chain cleavage (Figure 6). Analysis of samples taken during the intrinsic fXase experiment described in Figure 4B showed complete inhibition of fVIII light-chain cleavage at saturating concentrations of 1D4 (supplemental Figure 3).
In contrast, the noninhibitory group E mAbs 4C7 and 2G10 did not inhibit cleavage of fVIII by thrombin. Group A mAb 413 did not affect cleavage of full-length fVIII (supplemental Figure 2), consistent with its known function as an inhibitor of fVIIIa, not fVIII.6 The weakly inhibitory group B mAb 4F4 and group C mAb B25 also did not affect cleavage of full-length fVIII (supplemental Figure 2).
fVIII circulates bound to VWF and dissociates following cleavage of the light chain.5 Cleavage of the fVIII light chain by thrombin is accelerated severalfold when VWF is bound to either porcine22 or human fVIII (supplemental Figure 4). Therefore, the effect of VWF on the inhibition of light-chain cleavage by group D and group E mAbs was examined (Figure 7). VWF alleviated inhibition of light-chain cleavage produced by group D and group DE mAbs 2-54, CLB-CAg 9, GMA-012, and G74, but did not affect inhibition of heavy-chain cleavages. In contrast, inhibition of light-chain cleavage in the presence of group E mAbs B161, B66, and 1D4 persisted in the presence of VWF (Figure 7; supplemental Figure 5). Because VWF accelerates fVIII light-chain cleavage by thrombin, group E mAbs potentially could act indirectly by inhibiting the binding of fVIII to VWF. However, the binding of fVIII to VWF was not affected by group E mAbs B161, B66, or 1D4 (supplemental Figure 6).
Figure 7.

Effect of group D and E anti-A2 mAbs on thrombin-catalyzed proteolytic cleavage of the full-length fVIII/VWF complex. fVIII (100 nM) was incubated for 10 minutes at 37°C with VWF (1000 nM monomer equivalents) and then incubated for an additional 10 minutes in the presence of increasing mAb concentrations. Thrombin (0.5 nM) was added for 10 minutes and the reaction products were analyzed by SDS-PAGE as described in “Effect of anti-A2 mAbs on thrombin-catalyzed proteolytic cleavage of fVIII.” Ctrl, fVIII control not exposed to thrombin or anti-A2 mAbs; Stds, Molecular weight standards are 250, 150, 100, 75, 50, 37, and 25 kDa.
Discussion
In this study, we characterized a panel of 26 anti-fVIII A2 domain mAbs that were produced by hemophilia A mice in response to adjuvant-free clinically relevant intravenous doses of human fVIII and 3 previously described anti-A2 mAbs: 413, GMA-012, and CLB-CAg 9. We used competition ELISA to interrogate all pairwise interactions of the mAb set. Competition ELISA, which was described soon after the advent of mAb technology,23 is a powerful method to map overlapping mAb epitopes. The use of Venn diagrams to represent overlapping epitopes (Figure 1B) was described as early as 1994.24 More recently, competition ELISA has been used to map mAb epitopes resulting from the polyclonal response to influenza hemagglutinin,25,26 Plasmodium falciparum,27 norovirus,28 and West Nile virus antigens.29
Our analysis identified a basis set of 5 anti-A2 mAbs—4A4, 2-93, G48, 2-54, and 1D4 (Figure 1A-B)—defined as mAbs that do not compete with each other, but as a group compete with all other Abs. The 5 basis set groups—A, B, C, D, and E—accounted for 26 of the 29 mAbs that were studied. The 3 remaining mAbs fit into >1 group and are designated group AB, BCD, and DE mAbs. The Venn diagram produced by the overlapping epitopes yielded a continuous spectrum of epitopes. Homolog-scanning mutagenesis identified antigenic A2 sequences (Figure 2), which allowed physical placement of the Venn map onto the A2 surface. Our results indicate that the overlapping epitopes cover most of the A2 surface and contrast with our earlier study of the immune response to the human fVIII C2 domain in the murine hemophilia A model.30 In that study, continuously overlapping epitopes were identified that corresponded to only 1 face of the C2 β-sandwich.
Eight of the 9 group A mAbs mapped to the extensively characterized Arg484-Ile508 epitope (Table 1).10,11 413 in this group is a noncompetitive inhibitor of the intrinsic fXase complex.6 Group A mAbs are characterized by specific inhibitory activities that reach ∼40 000 BU/mg of IgG. This corresponds to near stoichiometric inhibition in which every mAb in the system binds and inhibits a fVIIIa molecule.
Groups AB, B, BCD, and C contained 11 of the 29 mAbs. All of these mAbs possessed either weak or undetectable inhibitory activity. Noninhibitory anti-fVIII Abs conceivably could contribute to pathogenicity by increasing the clearance rate of fVIII or contributing to Fc receptor–mediated inflammation. Most patients with anti-fVIII Abs measured by ELISA have positive inhibitor titers as measured by the Bethesda assay,31 indicating that these noninhibitory Abs occur as part of an immune response that also contains inhibitory Abs. Nonetheless, the results of this study suggest that noninhibitory Abs make up a significant portion of the immune response. Thus, immune complexes must be considered in the overall pathogenicity of anti-A2 Abs.
Homolog-scanning mutagenesis revealed that group D and group E mAbs recognize major epitope determinants in the C-terminal cupredoxin-like A2 subdomain. Our results are consistent with western blotting studies of GMA-012 and deletion mapping studies of CLB-CAg, which located the epitopes recognized by these mAbs to the C-terminal A2 subdomain.32,33 Most of these mAbs interfere with proteolytic activation cleavages of fVIII catalyzed by thrombin. Our results must be interpreted in light of the complex pathway that leads to fVIII activation. Following rapid removal of the B domain following cleavage at Arg740, 2 pathways potentially lead to the formation of the A1/A2/A3-C1-C2 fVIIIa heterotrimer, depending on whether cleavage at Arg1689 occurs followed by cleavage at Arg372 or vice versa22 (right and left paths, respectively, in Figure 5). In the presence of VWF, which is the physiologically relevant situation, Arg1689 cleavage is accelerated severalfold, suggesting that the right-sided pathway dominates fVIII activation in vivo.22 This VWF cofactor effect is due to the ability of fVIII to bind more tightly to thrombin in the presence of VWF.22 Arg1689 cleavage results in the dissociation of VWF from fVIII. This event appears to be necessary for normal hemostasis because VWF competes with phospholipid membranes for binding to fVIII,34,35 preventing assembly of the intrinsic fXase complex in the absence of dissociation of fVIII.
Arg372 cleavage generates the A2 subunit, which is critical for fVIIIa function.36,37 Group D mAb 2-54, which has a specific activity of 34 000 BU/mg (Table 1), inhibits A2 subunit formation (Figures 6 and 7; supplemental Figure 3). 2-54 also inhibits fXa formation in a purified intrinsic fXase system (Figure 4B), which correlates well with its degree of inhibition of A2 formation (supplemental Figure 3). 2-54 also inhibits fVIII light-chain cleavage (Figure 6), which, however, is alleviated in the presence of VWF (Figure 7). This indicates that the binding of fVIII to VWF produces a conformational change that allows thrombin to outcompete 2-54 for binding to fVIII. Overall, these results suggest that in the physiologically relevant setting in which VWF is present, inhibitory group D Abs function as anticoagulants by blocking cleavage at Arg372. Group D Abs bind the C-terminal end of the A2 domain, which evidently is remote from Arg372. This suggests that they allosterically regulate thrombin recognition at Arg372.
CLB-CAg 9 and GMA-012 displayed little or no anticoagulant activity consistent with previous reports (Table 1).33,38 Both mAbs also inhibited production of the A2 but not the A1 subunit, indicating that they interfere with cleavage at Arg740, but not Arg372. Site-directed mutagenesis of Arg740 produces an activatable fVIII molecule,37 indicating that cleavage at this site is not necessary for fVIII activation. Donath et al39 concluded that the inhibition of light-chain cleavage by CLB-CAg 9 indicates that the C-terminal A2 subdomain contributes to fVIII activation. However, inhibition of light-chain cleavage by CLB-CAg 9 and GMA-012 was alleviated by VWF (Figure 7), which is consistent with the lack of observed anticoagulant activity.
Group E mAbs B66 and 1D4 displayed relatively high specific activities of 4000 and 7000 BU/mg, respectively. Both mAbs inhibited light-chain cleavage, but not heavy-chain cleavage (Figure 6). In contrast to group D mAbs, this inhibition persisted in the presence of VWF (Figure 7). Thus, inhibition of light-chain cleavage, which prevents dissociation of fVIIIa from VWF, appears to contribute to the anticoagulant properties of B66 and 1D4. B66 and 1D4 also may inhibit fVIIIa directly. 1D4 completely inhibited fXa formation in a VWF-free intrinsic fXase assay (Figure 4B), which correlated with complete inhibition of fVIII light-chain cleavage (supplemental Figure 3). A1/A2/LC heterotrimers that lack Arg1689 cleavage have substantial fIXa cofactor activity.40,41Furthermore, although mutations at Arg1689 cause hemophilia A, dissociation of Arg1689 mutant fVIII/VWF complexes with disulfide-bond reducing agents can produce fVIII procoagulant activity in vitro.42,43 Thus, cleavage at Arg1689, while necessary for dissociation of fVIII from VWF, is not necessary for significant fVIIIa activation per se. These results suggest that B66 and 1D4 possess both VWF-dependent and VWF-independent inhibitory properties.
The Arg484-Ile508 immunodominant epitope that is recognized by 413 and other group A murine mAbs is also recognized by human inhibitors.6 Murine anti-A2 mAbs potentially can be used as probes to determine whether novel epitopes characterized in the present study are also targeted by the human immune system. For example, we have identified a novel class of human C2 inhibitors by ELISA using biotinylated murine anti-human C2 mAbs as competitive ligands testing human inhibitor plasmas.44 Recently, murine anti-PF4/heparin mAbs were used to identify epitopes involved in the pathogenesis of heparin-induced thrombocytopenia using a similar approach.45
In summary, our results confirm that the Arg484-Ile508 region is an immunodominant fVIII A2 epitope and identify 2 additional mechanisms by which fVIII inhibitors act, namely inhibition of cleavage at Arg372 and activation of the fVIII cofactor function and inhibition of cleavage at Arg1689 and dissociation of fVIII from VWF. Additionally, our epitope-mapping results reveal that the anti-A2 immune response in the murine hemophilia A model is structurally complex.
Supplementary Material
Acknowledgments
The authors thank Dr Jan Voorberg for his generous gift of CLB-CAg 9.
This work was supported by National Institutes of Health grants U54 HL112309 and K08 HL102262 and Hemophilia of Georgia, Inc (S.L.M.), and National Institutes of Health grants U54 HL112309, R01 HL082609, and R01 HL040921 and Hemophilia of Georgia, Inc (P.L.).
Footnotes
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: R.C.M., J.F.H., and E.T.P. designed and performed research, analyzed data, and co-wrote the manuscript; S.L.M. designed research, analyzed data, and co-wrote the manuscript; and P.L. designed research, analyzed data, and co-wrote the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Pete Lollar, Emory Children’s Center, Room 426D, 2015 Uppergate Dr, Atlanta, GA 30322; e-mail: jlollar@emory.edu.
References
- 1.Lusher JM, Arkin S, Abildgaard CF, et al. Kogenate Previously Untreated Patient Study Group. Recombinant factor VIII for the treatment of previously untreated patients with hemophilia A. Safety, efficacy, and development of inhibitors. N Engl J Med. 1993;328(7):453–459. doi: 10.1056/NEJM199302183280701. [DOI] [PubMed] [Google Scholar]
- 2.Bray GL, Gomperts ED, Courter S, et al. The Recombinate Study Group. A multicenter study of recombinant factor VIII (recombinate): safety, efficacy, and inhibitor risk in previously untreated patients with hemophilia A. Blood. 1994;83(9):2428–2435. [PubMed] [Google Scholar]
- 3.Lusher JM, Lee CA, Kessler CM, et al. ReFacto Phase 3 Study Group. The safety and efficacy of B-domain deleted recombinant factor VIII concentrate in patients with severe haemophilia A. Haemophilia. 2003;9(1):38–49. doi: 10.1046/j.1365-2516.2003.00708.x. [DOI] [PubMed] [Google Scholar]
- 4.Zaitseva I, Zaitsev V, Card G, et al. The X-ray structure of human serum ceruloplasmin at 3.1 Angstroms: nature of the copper centres. J Biol Inorg Chem. 1996;1:15–23. [Google Scholar]
- 5.Lollar P, Hill-Eubanks DC, Parker CG. Association of the factor VIII light chain with von Willebrand factor. J Biol Chem. 1988;263(21):10451–10455. [PubMed] [Google Scholar]
- 6.Lollar P, Parker ET, Curtis JE, et al. Inhibition of human factor VIIIa by anti-A2 subunit antibodies. J Clin Invest. 1994;93(6):2497–2504. doi: 10.1172/JCI117259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fay PJ, Beattie T, Huggins CF, et al. Factor VIIIa A2 subunit residues 558-565 represent a factor IXa interactive site. J Biol Chem. 1994;269(32):20522–20527. [PubMed] [Google Scholar]
- 8.Lollar P, Parker CG. pH-dependent denaturation of thrombin-activated porcine factor VIII. J Biol Chem. 1990;265(3):1688–1692. [PubMed] [Google Scholar]
- 9.Prescott R, Nakai H, Saenko EL, et al. Recombinate and Kogenate Study Groups. The inhibitor antibody response is more complex in hemophilia A patients than in most nonhemophiliacs with factor VIII autoantibodies. Blood. 1997;89(10):3663–3671. [PubMed] [Google Scholar]
- 10.Scandella D, Timmons L, Mattingly M, et al. A soluble recombinant factor VIII fragment containing the A2 domain binds to some human anti-factor VIII antibodies that are not detected by immunoblotting. Thromb Haemost. 1992;67(6):665–671. [PubMed] [Google Scholar]
- 11.Healey JF, Lubin IM, Nakai H, et al. Residues 484-508 contain a major determinant of the inhibitory epitope in the A2 domain of human factor VIII. J Biol Chem. 1995;270(24):14505–14509. doi: 10.1074/jbc.270.24.14505. [DOI] [PubMed] [Google Scholar]
- 12.Healey JF, Parker ET, Barrow RT, et al. The humoral response to human factor VIII in hemophilia A mice. J Thromb Haemost. 2007;5(3):512–519. doi: 10.1111/j.1538-7836.2007.02373.x. [DOI] [PubMed] [Google Scholar]
- 13.Fay PJ, Haidaris PJ, Smudzin TM. Human factor VIIIa subunit structure. Reconstruction of factor VIIIa from the isolated A1/A3-C1-C2 dimer and A2 subunit. J Biol Chem. 1991;266(14):8957–8962. [PubMed] [Google Scholar]
- 14.Parker ET, Doering CB, Lollar P. A1 subunit-mediated regulation of thrombin-activated factor VIII A2 subunit dissociation. J Biol Chem. 2006;281(20):13922–13930. doi: 10.1074/jbc.M513124200. [DOI] [PubMed] [Google Scholar]
- 15.Summers RJ, Meeks SL, Healey JF, et al. Factor VIII A3 domain substitution N1922S results in hemophilia A due to domain-specific misfolding and hyposecretion of functional protein. Blood. 2011;117(11):3190–3198. doi: 10.1182/blood-2010-09-307074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kasper CK, Aledort LM, Counts RB, et al. Letter: a more uniform measurement of factor VIII inhibitors. Thromb Diath Haemorrh. 1975;34(3):869–872. [PubMed] [Google Scholar]
- 17.Barrow RT, Lollar P. Neutralization of antifactor VIII inhibitors by recombinant porcine factor VIII. J Thromb Haemost. 2006;4(10):2223–2229. doi: 10.1111/j.1538-7836.2006.02135.x. [DOI] [PubMed] [Google Scholar]
- 18.Lubin IM, Healey JF, Scandella D, et al. Elimination of a major inhibitor epitope in factor VIII. J Biol Chem. 1994;269(12):8639–8641. [PubMed] [Google Scholar]
- 19.Shen BW, Spiegel PC, Chang C-H, et al. The tertiary structure and domain organization of coagulation factor VIII. Blood. 2008;111(3):1240–1247. doi: 10.1182/blood-2007-08-109918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ngo JC, Huang M, Roth DA, et al. Crystal structure of human factor VIII: implications for the formation of the factor IXa-factor VIIIa complex. Structure. 2008;16(4):597–606. doi: 10.1016/j.str.2008.03.001. [DOI] [PubMed] [Google Scholar]
- 21.Hoyer LW, Gawryl MS, de la Fuente B. Immunochemical characterization of factor VIII inhibitors. In: Hoyer LW, editor. Factor VIII Inhibitors. New York, NY: Alan R. Liss; 1984. pp. 73–85. [PubMed] [Google Scholar]
- 22.Hill-Eubanks DC, Lollar P. von Willebrand factor is a cofactor for thrombin-catalyzed cleavage of the factor VIII light chain. J Biol Chem. 1990;265(29):17854–17858. [PubMed] [Google Scholar]
- 23.Pierres M, Devaux C, Dosseto M, et al. Clonal analysis of B- and T-cell responses to Ia antigens. I. Topology of epitope regions on I-Ak and I-Ek molecules analyzed with 35 monoclonal alloantibodies. Immunogenetics. 1981;14(6):481–495. doi: 10.1007/BF00350120. [DOI] [PubMed] [Google Scholar]
- 24.Morris CA, Underwood PA, Bean PA, et al. Relative topography of biologically active domains of human vitronectin. Evidence from monoclonal antibody epitope and denaturation studies. J Biol Chem. 1994;269(38):23845–23852. [PubMed] [Google Scholar]
- 25.Corti D, Suguitan AL, Jr, Pinna D, et al. Heterosubtypic neutralizing antibodies are produced by individuals immunized with a seasonal influenza vaccine. J Clin Invest. 2010;120(5):1663–1673. doi: 10.1172/JCI41902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Corti D, Voss J, Gamblin SJ, et al. A neutralizing antibody selected from plasma cells that binds to group 1 and group 2 influenza A hemagglutinins. Science. 2011;333(6044):850–856. doi: 10.1126/science.1205669. [DOI] [PubMed] [Google Scholar]
- 27.de Silva HD, Saleh S, Kovacevic S, et al. The antibody response to Plasmodium falciparum merozoite surface protein 4: comparative assessment of specificity and growth inhibitory antibody activity to infection-acquired and immunization-induced epitopes. Malar J. 2011;10:266. doi: 10.1186/1475-2875-10-266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Parra GI, Abente EJ, Sandoval-Jaime C, et al. Multiple antigenic sites are involved in blocking the interaction of GII.4 norovirus capsid with ABH histo-blood group antigens. J Virol. 2012;86(13):7414–7426. doi: 10.1128/JVI.06729-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lelli D, Moreno A, Brocchi E, et al. West Nile virus: characterization and diagnostic applications of monoclonal antibodies. Virol J. 2012;9:81. doi: 10.1186/1743-422X-9-81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Meeks SL, Healey JF, Parker ET, et al. Antihuman factor VIII C2 domain antibodies in hemophilia A mice recognize a functionally complex continuous spectrum of epitopes dominated by inhibitors of factor VIII activation. Blood. 2007;110(13):4234–4242. doi: 10.1182/blood-2007-06-096842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ling M, Duncan EM, Rodgers SE, et al. Low detection rate of antibodies to non-functional epitopes on factor VIII in patients with hemophilia A and negative for inhibitors by Bethesda assay. J Thromb Haemost. 2003;1(12):2548–2553. doi: 10.1046/j.1538-7836.2003.00477.x. [DOI] [PubMed] [Google Scholar]
- 32.Fay PJ, Smudzin TM, Walker FJ. Activated protein C-catalyzed inactivation of human factor VIII and factor VIIIa. Identification of cleavage sites and correlation of proteolysis with cofactor activity. J Biol Chem. 1991;266(30):20139–20145. [PubMed] [Google Scholar]
- 33.Leyte A, Mertens K, Distel B, et al. Inhibition of human coagulation factor VIII by monoclonal antibodies. Mapping of functional epitopes with the use of recombinant factor VIII fragments. Biochem J. 1989;263(1):187–194. doi: 10.1042/bj2630187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Andersson LO, Brown JE. Interaction of factor VIII-von Willebrand factor with phospholipid vesicles. Biochem J. 1981;200(1):161–167. doi: 10.1042/bj2000161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lajmanovich A, Hudry-Clergeon G, Freyssinet JM, et al. Human factor VIII procoagulant activity and phospholipid interaction. Biochim Biophys Acta. 1981;678(1):132–136. doi: 10.1016/0304-4165(81)90056-8. [DOI] [PubMed] [Google Scholar]
- 36.Arai M, Inaba H, Higuchi M, et al. Direct characterization of factor VIII in plasma: detection of a mutation altering a thrombin cleavage site (arginine-372----histidine). Proc Natl Acad Sci U S A. 1989;86(11):4277–4281. doi: 10.1073/pnas.86.11.4277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pittman DD, Kaufman RJ. Proteolytic requirements for thrombin activation of anti-hemophilic factor (factor VIII). Proc Natl Acad Sci U S A. 1988;85(8):2429–2433. doi: 10.1073/pnas.85.8.2429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fay PJ, Scandella D. Human inhibitor antibodies specific for the factor VIII A2 domain disrupt the interaction between the subunit and factor IXa. J Biol Chem. 1999;274(42):29826–29830. doi: 10.1074/jbc.274.42.29826. [DOI] [PubMed] [Google Scholar]
- 39.Donath MJ, Lenting PJ, Van Mourik JA, et al. Kinetics of factor VIII light-chain cleavage by thrombin and factor Xa. A regulatory role of the factor VIII heavy-chain region Lys713-Arg740. Eur J Biochem. 1996;240(2):365–372. doi: 10.1111/j.1432-1033.1996.0365h.x. [DOI] [PubMed] [Google Scholar]
- 40.Hill-Eubanks DC, Parker CG, Lollar P. Differential proteolytic activation of factor VIII-von Willebrand factor complex by thrombin. Proc Natl Acad Sci U S A. 1989;86(17):6508–6512. doi: 10.1073/pnas.86.17.6508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Regan LM, Fay PJ. Cleavage of factor VIII light chain is required for maximal generation of factor VIIIa activity. J Biol Chem. 1995;270(15):8546–8552. doi: 10.1074/jbc.270.15.8546. [DOI] [PubMed] [Google Scholar]
- 42.Aly AM, Arai M, Hoyer LW. Cysteamine enhances the procoagulant activity of factor VIII-East Hartford, a dysfunctional protein due to a light chain thrombin cleavage site mutation (arginine-1689 to cysteine). J Clin Invest. 1992;89(5):1375–1381. doi: 10.1172/JCI115725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Arai M, Higuchi M, Antonarakis SE, et al. Characterization of a thrombin cleavage site mutation (Arg 1689 to Cys) in the factor VIII gene of two unrelated patients with cross-reacting material-positive hemophilia A. Blood. 1990;75(2):384–389. [PubMed] [Google Scholar]
- 44.Meeks SL, Healey JF, Parker ET, et al. Nonclassical anti-C2 domain antibodies are present in patients with factor VIII inhibitors. Blood. 2008;112(4):1151–1153. doi: 10.1182/blood-2008-01-132639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sachais BS, Litvinov RI, Yarovoi SV, et al. Dynamic antibody-binding properties in the pathogenesis of HIT. Blood. 2012;120(5):1137–1142. doi: 10.1182/blood-2012-01-407262. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.