

Abstract
Animal models of human diseases are critical for dissecting mechanisms of pathophysiology and developing therapies. In the context of cystic fibrosis (CF), mouse models have been the dominant species by which to study CF disease processes in vivo for the past two decades. Although much has been learned through these CF mouse models, limitations in the ability of this species to recapitulate spontaneous lung disease and several other organ abnormalities seen in CF humans have created a need for additional species on which to study CF. To this end, pig and ferret CF models have been generated by somatic cell nuclear transfer and are currently being characterized. These new larger animal models have phenotypes that appear to closely resemble human CF disease seen in newborns, and efforts to characterize their adult phenotypes are ongoing. This chapter will review current knowledge about comparative lung cell biology and cystic fibrosis transmembrane conductance regulator (CFTR) biology among mice, pigs, and ferrets that has implications for CF disease modeling in these species. We will focus on methods used to compare the biology and function of CFTR between these species and their relevance to phenotypes seen in the animal models. These cross-species comparisons and the development of both the pig and the ferret CF models may help elucidate pathophysiologic mechanisms of CF lung disease and lead to new therapeutic approaches.
Keywords: Lung biology, tracheal xenograft, CFTR processing, pig, ferret, mouse
1. Introduction
Animal models that reproduce the human cystic fibrosis (CF) disease phenotypes are required to effectively develop methods to treat the disease. These models also serve to increase our understanding of disease pathophysiology, cystic fibrosis transmembrane conductance regulator (CFTR) processing and channel function, testing of therapeutic molecules, and development of gene therapy approaches. It would be ideal if a single model was available that completely modeled the human disease (see Table 19.1); however, species differences are clearly apparent in the three CF models generated to date.
Table 19.1.
General species characteristics of CFTR
| Species | Avg. mass | CFTR AA No. | CFTR identitya (%) | Days of gestation | Avg. litter size | Sexual maturation | Avg. life expectancy (years) | SMG abundance |
|---|---|---|---|---|---|---|---|---|
| Human | ~80 kg | 1480 | 100 | 280 | 1 | 10–16 years | ~78 | Abundant |
| Mouse | ~25 g | 1476 | 78 | 21 | 6 | 6–8 weeks | 2 | Rare |
| Pig | ~90 kg | 1482 | 92 | 114 | 10 | 6–8 months | 10–15 | Abundant |
| Ferret | ~2–3 kg | 1484 | 91 | 42 | 8 | 4–6 months | 8–10 | Abundant |
AA, amino acid; SMG, submucosal gland
Protein sequence identity compared to the human CFTR sequence
As has been the case for CF mouse models, differences in the severity of various aspects of CF organ disease in the pig and ferret models will likely inform new biologic discoveries about CFTR functions in organ physiology and how dysfunction of these processes lead to disease in humans. The purpose of this chapter is to briefly review the general phenotypes of the CF mouse, pig, and ferret models, and to provide methods for comparative analysis of CFTR biology between these models. More detailed reviews of CF mouse models have been reported elsewhere (1–5), and the methods of construction of the CF pig and ferret models have also been previously reported (6, 7) and will not be a focus of the chapter. Comparative differences and similarities among these models will greatly enhance our understanding of the disease and accelerate the development of a cure for CF. It is important to emphasize that differences among species in their ability to model CF (see Table 19.2) will likely help to educate the field on what factors influence phenotypic variability seen in CF patients.
Table 19.2.
Cystic fibrosis phenotype across species
| Species | Spontaneous lung infections | Exocrine pancreas | Gastrointestinal | Liver | Micro-gallbladder | Fertility |
|---|---|---|---|---|---|---|
| Human | Birth: infrequent Adult: severe (49) |
Birth: ~72–90% ADD ~3% EPD Adult: ~83% EPD (41, 42, 50) |
Birth: ~10–15% MI ~80% MA Adult: MA, OB (51–55) |
Birth: ~50% ELFTs Adult: ~10–20% FBC (56, 57) |
Birth: ~23% (52, 58) | Male infertility: ~95% CBAVD (59, 60) |
| Mouse | None | Birth: none Adult: mild ADDa (4, 61) |
Birth: OB at weaninga Adult: MAa (4,61, 62) |
Birth: normal Adult: mild FBCa (4,61, 63) |
Birth: normal | Reduced female fertilitya (26) |
| Pig | Adult: severe (40) | Birth: 100% EPD (38, 64) | Birth: 100% MI (38, 64) | Birth: 20% FBC (38, 64) | Birth: 100% (38, 64) | CBAVD: variable at birth (40, 40a) |
| Ferret | Newborn: frequent (39) Adult: severe (unpublished) |
Birth: 100% ADD 0% EPD (39) | Birth: 75% MI 100% MA (39) | Birth: 100% ELFTs (39) | Birth: normal (39) | CBAVD: present at birth (39) |
MI, meconium ileus; TBD, to be determined; ADD, mild lesions associated with exocrine acinar duct dilatation; EPD, severe lesions associated with exocrine pancreas destruction; ELFTs, elevated liver function tests; FBC, focal biliary cirrhosis; MA, malabsorption; OB, intestinal obstruction; CBAVD, congenital bilateral absence of vas deferens
Observed only on certain background strains and/or CFTR genotypes
1.1. The Murine Models of CF
Murine models of CF have existed since the early 1990s, contributing invaluably to the current understanding of CF. To our knowledge, at least 14 mouse models of CF exist, including null and mutant forms of CFTR (8–21). The degree to which these models recapitulate various organ pathologies seen in human CF disease varies. Generally, the severity of the phenotypes in each of these models varies slightly based on the levels of CFTR mRNA, as a result of the gene targeting method used and the genetic background of the mouse (1, 4, 5). Briefly, most of the models display one or more of the following phenotypes including severe abnormalities in the gastrointestinal tract, failure to thrive, decreased rates of survival due to intestinal complications, and hyperinflammatory responses in the airway. Furthermore, most of these CF mouse models retain defects in cAMP-inducible chloride permeability in the nasal epithelium as seen in humans. Though not extensively studied in each model, reports have suggested decreased mucociliary clearance (22–24), reduced fertility (25, 26), mild pancreatic dysfunction (27–29), and liver abnormalities (28). However, these models thus far lack the development of significant spontaneous lung disease as observed in humans with CF. Furthermore, gut obstruction phenotypes seen in CF mice at weaning are clinically different from meconium ileus seen in newborn CF infants and suggest some level of biologic differences in the developmental control of chloride movement in the gut by CFTR between mice and humans.
Although the nasal bioelectric defects seen in the CF mouse models appear to closely resemble those in the human nasal epithelium (5), the bioelectric characteristics in the tracheal airways of mice and humans diverge significantly with murine models demonstrating cAMP-inducible changes in chloride permeability despite the absence of CFTR (30, 31). Potential explanations for this observation include differences in airway cell biology (i.e., different types of secretory cells are found in the proximal airway of humans [goblet cells] and mice [Clara cells]), differences in the distribution of submucosal glands (i.e., throughout the cartilaginous airways in humans and located only to the proximal regions of the trachea in mice), and the presence of alternative non-CFTR chloride channels in mice capable of activation in response to cAMP that are not found in humans (5, 30).
Much effort has been placed to develop methods capable of studying bacterial clearance defects in CF mouse lung (2, 4, 32, 33). Many groups with variable success have attempted inoculation of these CF mouse models with bacteria, mainly Pseudomonas aeruginosa, to reproduce the human lung disease phenotype. A variety of inoculation methods have been attempted from aerosolization of free bacteria to insertion of bacteria-laden agar beads. Some studies report decreased survival of the CF mice compared to their littermates, while others indicate no difference in clearance between genotypes (2, 4). However, some groups have reproducibly observed excessive inflammatory response and higher mortality to inoculation of the CF mouse lung with bacteria-laden agar beads, despite no differences in bacterial clearance (32, 33).
Another interesting model recently reported by Hodges and colleagues (21, 34) is the development of a conditional CFTR knockout mouse model. This model is being used to direct tissue-specific deletion of CFTR following crossing with transgenic mice that directs Cre recombinase expression under tissue-specific promoters. This system will allow for a systematic and directed evaluation of CFTR function at the level of individual organs.
1.2. New Methods for Generating Larger Animal Models of CF Disease
Given the observed phenotype in CF mouse models, it is clear that additional larger animal models of CF would be of utility to the field. Several parameters influence the choice of alternative species to model CF including (1) the types of cells in the airway in comparison to human (2), the distribution of submucosal glands which are thought to play an important role in CF airway disease (3), conservation of CFTR structure and function (4), the composite of alternative chloride channels in the airway, and (5) the reproductive parameters of the species which will make it feasible to rapidly perform research studies. Generation of larger CF animal models such as pig (35), ferret (36), and sheep (37) has been considered, but the technology to manipulate the genomes of these animals has lagged behind until recently. Recently, methods for generating both pig and ferret CF models have been developed using recombinant adeno-associated virus (rAAV)-mediated gene targeting of exon 10 (6, 7). The phenotypes of newborn CFTR knockout pigs and ferrets have also recently been described (38, 39) and will be discussed in more detail below.
1.2.1. The Porcine Models of CF
Generation of the CFTR−/− and ΔF508 alleles was accomplished by rAAV gene targeting of male porcine fetal fibroblasts (7). The targeted nuclei were subsequently used for somatic cell nuclear transfer, and CFTR+/− male piglets were born and bred to homozygosity. Due to the lack of reports on the heterozygous ΔF508 piglets, this section will focus mostly on the CFTR−/− piglets. Rogers et al. (38) recently reported a detailed description of the newborn CFTR−/− piglet phenotype. In summary, these pigs were born with near-Mendelian ratios of 1:2:1 with no differences in the newborn birth weight or in appearance between genotypes. CFTR-deficient piglets lacked CFTR mRNA and therefore expressed no protein. Nasal transepithelial potential difference (TEPD) displayed a lack of cAMP-inducible chloride permeability and an elevated baseline TEPD in the CFTR−/− piglets similar to that seen in CF humans and mice lacking functional CFTR. All of the CFTR-deficient piglets developed meconium ileus (MI) and atretic microcolon distal to the obstruction, resulting in failure to pass stool and gain weight after birth. If untreated by surgery, the MI was lethal in 100% of the CFTR−/− animals. To live beyond the first few days after birth, all animals required an ileostomy bypassing the obstruction. Rogers et al. also reported adipose infiltration of the pancreas and complete exocrine pancreatic insufficiency (or destruction) at birth in all CFTR−/− animals. These CFTR-deficient piglets also presented with focal biliary cirrhosis and developed a mucus- and bile-filled micro-gallbladder. No overt abnormalities were seen in the lungs, airways, submucosal glands, male reproductive tract, and other non-CF-related organs. Encouraging to the CF field was the recent report that aged (>2 months) CFTR−/− and CFTR−/ΔF508 piglets developed a CF-like lung phenotype (40). We anticipate future reports describing the lung phenotype of ΔF508/ΔF508 homozygous pigs.
1.2.2. The Ferret Model of CF
The generation of CFTR null ferrets was described in detail by Sun et al. (6). rAAV was used to introduce a stop codon and a neomycin cassette into exon 10 of the CFTR gene in primary ferret fibroblasts. Infected fibroblasts were cloned and selected by serial dilution into G418 followed by PCR screening for the targeting events. Due to early senescence of gene-targeted primary ferret fibroblasts (not an issue with the generation of the CF pig models), it was necessary to rejuvenate gene-targeted fibroblast clones by somatic cell nuclear transfer. Primary fibroblasts were then expanded from 21-day fetuses and used for a second round of somatic cell nuclear transfer. Using this process, eight CFTR+/− male ferret founders were obtained and expanded to generate CFTR+/− breeder pairs.
Similar to the CF piglets, there was no prenatal lethality associated with CFTR deficiency in ferret kits. Newborn CFTR-deficient newborn ferrets (kits) failed to thrive compared to their wild-type and heterozygous littermates. Approximately 75% of the CFTR-deficient kits failed to pass meconium due to MI and died within the first 36–48 h of life due to intestinal perforation and sepsis (39). Interestingly, this variable penetrance of MI seen in the CF kits is significantly different than the CF porcine model in which 100% of piglets presented with MI (see Table 19.2). Interestingly, the CFTR-deficient kits that passed stool (~25%) also failed to thrive and died within the first week of life, despite the fact that their intestinal tract was grossly and microscopically normal. The reason for death of these animals appeared to be due to malabsorption, demonstrating a histologic depletion of fat stores and progressive decline in blood cholesterol with age that was not corrected by pancreatic enzyme replacement. CFTR-deficient kits histologically demonstrated mild pancreatic disease at birth, presenting with exocrine acinar ducts that were swollen with inspissated secretions. Such findings are more similar to the human CF pancreatic phenotype at birth (41, 42) and contrasted with the CF pig model in which nearly complete exocrine pancreatic destruction was reported (38). CF kits also demonstrated early signs of functional liver disease (as evident by elevated liver function tests) despite liver histology not overtly different from controls. Unlike the CF pig model, CF newborn ferrets had a histologically normal gallbladder and the majority of kits that escaped MI presented with bronchopulmonary pneumonia at the time of death. It is currently unclear if the bronchopulmonary pneumonia seen in nutritionally compromised CF kits was secondary to their compromised health status and the inability to clear aspirated material from the lung. Based on the clinical blood chemistries seen in CFTR-deficient kits, drug and nutritional therapy to enhance fat absorption by the intestine was undertaken and significantly improved weight gain and survival (39). This treatment involved oral gavages with ursodeoxycholic acid (to treat apparent liver disease indicated by elevated liver enzymes in the blood), omeprazole (to raise gastrointestinal track pH), and elemental diet. This treatment regime improved liver function tests and raised serum cholesterol. Despite improved weight gain using these treatments, CF kits still often developed lung infections within the first four weeks of life that where characterized predominantly by steptococcus and staphylococcus infections. Rearing animals on antibiotics has also been used to enhance survival during the preweaning period. Although the adult CF ferret lung phenotype remains under investigation, a slow progressive and fatal lung disease has been shown to occur in this model (unpublished data). The finding that CFTR-deficient ferrets are susceptible to lung infections both early and late in life is encouraging.
1.3. Future Directions for Larger CF Animal Models
The creation of ferret and pig CF models will undoubtedly enhance CF research for decades to come. Comparative aspects of disease between CF mouse, ferret, and pig models should enlighten mechanisms of CFTR function and CF pathophysiology responsible for diverse disease phenotypes seen in CF patients. Despite the promise that comes with the new CF ferret and pig models, there remain significant barriers to their widespread use in research. Foremost among these barriers are the severe intestinal phenotypes at birth in both models. In the CF pig, the requirement for surgery to treat MI will significantly impair the use of this model for the average researcher. Similarly, since 75% of CF ferrets also present with a lethal MI phenotype at birth, the cost of implementing this model will be quite high until this problem can be solved. Two approaches are currently under investigation to reduce the severity of MI in these models. In the context of the ferret, the variable penetrance of MI suggests that there may be heritable influences on the severity of early intestinal disease in this model. To this end, the CF ferret model is being bred into different genetic lines in an effort to determine if a colony of CF ferrets with reduced penetrance of MI can be generated. Second, gut-corrected transgenic CFTR−/− ferrets that express ferret CFTR under the rat fatty acid-binding protein (FABP) promoter have been generated and shown to correct MI in newborn CF kits (39). This has proven feasibility that a transgenic complementation approach combined with somatic cell nuclear transfer can further improve both the CF ferret and pig models. This approach has been used successfully in CF mouse models to prevent intestinal obstruction at weaning (12). Future development of the gut-corrected CF ferret model will require regenerating the model on a heterozygous background for breeding expansion.
A second intriguing aspect of future ferret and pig models of CF pertains to the ability of a particular species to model certain mutations of CFTR. As discussed in more detail below, analysis of pig ΔF508-CFTR demonstrates that this protein is partially processed to the apical membrane of airway epithelia where it retains some level of function (43, 44). These findings suggest that the pig may not be the best model on which to study the ΔF508-CFTR mutation. It is currently unclear if ferret ΔF508-CFTR will retain similar processing defects as seen with the human mutant protein and a conclusive answer to this question in a ferret ΔF508-CFTR model is in progress (45).
Three methods useful in evaluating CF animal models will be discussed in this chapter. The first area will review methods for evaluating TEPD in tracheal xenografts. The second area will review antibodies that are suitable for studying alternative species of CFTR. The last area will review methods for studying species-specific processing of CFTR by metabolic pulse-chase labeling. We will use ferret as a model for most of the methods discussed, since other species have already been published elsewhere.
1.4. Cross-Species Analysis of Tracheal TEPD in a Tracheal Xenograft Model
Analysis of the bioelectric properties of cAMP-inducible chloride channels in the airway is critical to characterizing the ability of a particular species to model CF. TEPD measurements are an effective method to study chloride channel defects and have been extensively used in human and mouse models to study CF. Typically, this assay has been performed on the nasal epithelium since it is readily accessible in a live host. However, given the fact that the CF mouse retains nasal but not tracheal chloride transport defects seen in CF humans, methods to directly assess the bioelectric properties of the trachea in CF animal models are needed. Tracheal TEPD measurements can be obtained ex vivo in a tracheal xenograft model. Freshly excised tracheas cannulated with flexible plastic tubing and inserted subcutaneously in athymic Nu/Nu mice have allowed analysis of tracheal bioelectric properties in a vascularized airway free from infection. This system has been extremely useful in characterizing TEPD in these models due to the early intestinal complications in the newborn ferret and pig CF models. Perfusion of pharmacological ion channel agonists and antagonists allows for a systematic measurement of the bioelectric properties in these new models. This chapter will focus on the protocol for making tracheal TEPD measurements using this ex vivo system. Xenograft cassette design, implantation of xenografts, and maintenance of the xenografts will not be discussed in detail because these general methods have been detailed in an earlier edition of this book (46).
1.5. Cross-Species Analysis of CFTR Processing
As new CF models are developed with specific CFTR mutations, a clear understanding of comparative CFTR biology is paramount. Topics relevant to modeling CFTR mutations in a new species include how closely each of the following resembles that of the human mutant CFTR: (1) the efficiency of folding, (2) the efficiency of detection by endoplasmic reticulum associated protein degradation (ERAD), (3) the stability at the plasma membrane, and (4) the activity of the channel at plasma membrane. Reports have already shown that ΔF508-CFTR processing differences exist among mice, pigs, and humans (43, 44, 47). CFTR is composed of five structural domains, including two membrane-spanning domains, two nucleotide-binding domains, and a regulatory domain. With the exception of the regulatory domain, the other two domains are highly conserved between most model species and humans, more so in pigs and ferrets than in mice. Differences in the primary protein structure among species may dictate the ability of these species to correctly model the CF disease seen in humans. The mouse, pig, and ferret CFTRs are 77, 92, and 91 identical to the human CFTR, respectively (Table 19.1). The identification of species-specific differences in CFTR processing may also help to inform new approaches to enhance processing of human mutant CFTR, by identifying molecular targets responsible for variation among species (i.e., differences in chaperone interactions). This section will focus on CFTR antibody optimization across species and comparative metabolic pulse-chase experiments.
2. Materials
2.1. TEPD Measurements in a Tracheal Xenograft Model
Tracheal xenograft at least 4 weeks post-transplantation (see Note 1).
Ketamine (100 mg/kg) and xylazine (20 mg/kg) in PBS.
Multi-range, variable-rate infusion pump (Orion Research, Cat. no. 001967).
pH/mV meter (Fisher Scientific, Cat. no. 13-636-AB15P).
Calomel reference electrodes (Fisher Scientific, Cat. no. 13-620-51).
21-Gauge × 0.75-in. butterfly infusion set (Abbott Laboratories, Cat. no. 4492).
Computer with data acquisition software (CyberComm Pro 2.3; Fisher Scientific) for recording PD in millivolts.
10-mL Disposable syringes with 21-gauge × 1.5-in. needles.
Manifold pump tubing (PVC Solvent Flexible tubing; Fisher, Cat. no. 14-190-139).
Silicone tubing (Bio-Rad Laboratories, Cat. no. 7318211).
Agar Nobel (Difco Laboratories, Detroit, MI, Cat. no. 0142-01).
1 M KCl.
Hemostat.
HEPES phosphate-buffered Ringer’s (HPBR) solution: 10 mM HEPES (pH 7.4), 140 mM NaCl, 5 mM KCl, 1.2 mM MgSO4, 1.2 mM Ca gluconate, 2.4 mM K2HPO4, and 0.4 mM KH2PO4.
Chloride-free HPBR solution: 10 mM HEPES (pH 7.4), 140 mM Na gluconate, 5 mM K gluconate, 1.2 mM MgSO4, 1.2 mM Ca gluconate, 2.4 mM K2HPO4, and 0.4 mM KH2PO4.
Ham’s F12 medium.
-
PD buffer sequence:
HPBR solution, 100 μM 4,4′-diisothiocyanatostilbene-2,2′-disulfonic acid (DIDS).
HPBR solution, 100 μM DIDS, 100 μM amiloride.
Chloride-free HPBR solution, 100 μM DIDS, 100 μM amiloride.
Chloride-free HPBR solution, 100 μM DIDS, 100 μM amiloride, 200 μM 8-cpt-cAMP.
Chloride-free HPBR solution, 100 μM DIDS, 100 μM amiloride, 200 μM 8-cpt-cAMP, 50 μM CFTRINHGlyH-101.
2.2. Cross-Species Analysis of CFTR Processing Requires Antibodies that Efficiently Bind Across Species
Immunoprecipitation.
-
In vitro phosphorylation:
PKA phosphorylation buffer (per sample): 50 mM KH2PO4 (pH 6.8), 2 μg BSA, 2 μg protein kinase A (PKA; Calbiochem, La Jolla, CA) diluted to 20 μL using ddH2O.
ATP phosphorylation buffer (per sample): 50 mM KH2PO4 (pH 6.8), 4 μg BSA, 10 mM MgCl2, and 3.6 μCi (6000 Ci/mmol) [γ–32P]-ATP (Perkin Elmer, Waltham, MA) diluted to 40 μL using ddH2O.
Thermal-controlled shaker.
2.3. Cross-Species Analysis of the Processing Efficiency and Stability of CFTR by [35S]Methionine Pulse Chase
Starvation media, DMEM lacking methionine and cysteine (Invitrogen, Cat. no. 21013024).
[35S]Methionine and [35S]cysteine EasyTag Express 35S protein labeling mix (Perkin Elmer, Cat. no. NEG772007MC).
Activated charcoal-loaded syringe.
Chase media, DMEM containing 10% FBS, 1% penicillin and streptomycin, and 2 mM cold methionine and cysteine.
Ice-cold PBS.
RIPA buffer (150 mM NaCl, 20 mM Tris–HCl, 1% Triton X-100, 0.1% SDS, 0.5% deoxycholate, pH 8.0) containing protease inhibitors (Roche, Cat. no. 34342).
Refrigerated tabletop centrifuge.
1.5- and 2.0-mL Eppendorf tubes.
Anti-CFTR antibodies M3A7 and MM13-4 (Millipore, Cat. nos. 05-581 and 05-533, respectively) and anti-HA high-affinity antibody (Roche, Cat. no. 11867431001).
Protein G DynaBeads® and separation magnet (Invitrogen, Cat. nos. 100.04D and 123.21D).
7.5% SDS-PAGE gels.
Gel fixative solution (10% glacial acetic acid, 25% isopropanol, and 65% ddH2O).
Amplify fluorographic reagent (GE Healthcare, Cat. no. NAMP100).
Gel dryer.
Phosphoscreen, phosphoimager, and image analysis software (ex. ImageJ. or ImageQuant).
3. Methods
3.1. TEPD Measurements in a Tracheal Xenograft Model
Newborn tracheas from CF and non-CF pigs and ferrets are obtained at birth sterilely and connected to flexible tubing through a series of adapters and stents to keep the trachea extended to normal length. These cassettes are then implanted subcutaneously into the flanks of athymic Nu/Nu mice (a host that will not reject the tissue). A schematic view of this ex vivo tracheal xenograft model is shown in Fig. 19.1. Importantly, these grafts become vascularized by 2–3 weeks and have ports that allow for lumenal access for TEPD measurements. Details on the methods for generating these cassettes and for the surgical implantation are described elsewhere (46). TEPD measurements of the xenografted tracheal airways can be used to assess changes in the permeability to various ions in response to antagonists and agonists of epithelial ion channels such as the epithelial sodium channel (ENaC) and CFTR.
Fig. 19.1.
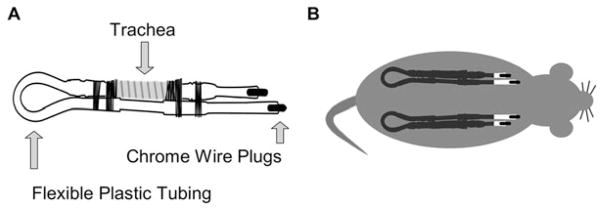
Tracheal xenograft design and transplantation. (a) The cassette is composed of flexible plastic tubing, freshly excised newborn pig or ferret trachea, and chrome wire plugs (see Note 1). The trachea is fastened to the tubing using silk sutures. (b) The xenograft cassettes are inserted subcutaneously into the flanks of Nu/Nu athymic mice. The xenografts vascularize within 2–3 weeks and continue to mature and develop until ready for bioelectric characterization by measuring PD (4–5 weeks). PD recordings are made weekly until 8–9 weeks post-transplantation. Usually a CFTR−/− xenograft is transplanted in parallel to either a CFTR+/− or a CFTR+/+ xenograft.
After 4–5 weeks post-transplantation, the xenografts are fully differentiated and ready for TEPD analysis using the equipment shown in Fig. 19.2. The xenograft-bearing mouse (typically with two xenografts) is anesthetized by intraperitoneal injection of ketamine (100 mg/kg) and xylazine (20 mg/kg) in PBS. Once anesthetized, the mouse is placed on a sterile drape and the chrome wire caps from the xenograft exit ports are removed using sterile forceps.
The xenograft is gently flushed with 1 mL of Ham’s F12 medium using a 1-mL syringe and a butterfly needle.
The Ham’s F12 medium is removed by flushing the xenograft with air. This is done by removing the syringe from the butterfly needle, refilling it with air, returning the syringe to the butterfly needle, and gently forcing the air through the xenograft.
The syringe is removed and the butterfly needle is left on the distal port as a drain for the perfused TEPD buffer solutions.
10-mL syringes are filled with TEPD buffers and fitted on the syringe pump. A length of manifold pump tubing, which is to be placed on the needle of the first syringe, is first fitted onto the medial port of the xenograft by means of a shortened pipette tip (20–200 μL).
Calomel electrodes, immersed in 1 M KCl, are connected to the pH/mV meter and to the butterfly electrodes.
Butterfly electrodes are prepared by filling 21-gauge butterfly tubing with 5% noble agar in 1 M KCl (see Note 2). The positive butterfly electrode is inserted into the perfusion tubing just external to the xenograft port. This is done by directly inserting the 21-gauge needle through the tubing. The negative electrode is inserted subcutaneously in the back of the mouse.
Millivolt recordings are obtained from the pH/mV meter and data linked directly to a computer, equipped with data acquisition software. Measurements are taken every second. Example TEPD recordings and histological sections for ferret CFTR−/− and CFTR+/+ xenografts are depicted in Fig. 19.3.
Typically, the xenograft is sequentially perfused with 2 mL of each of the TEPD buffers through the syringe pump at a flow rate of 200 μL/min (10 min, see Note 3 and 4).
After recording is complete, the xenograft is gently flushed with 1 mL of Ham’s F-12 medium using a butterfly needle and a 1-mL syringe. The syringe is then disconnected from the xenograft. The syringe is refilled with air by gently forcing air through the xenograft, restoring an air–liquid interface. The chrome wire inserts are replaced into the tubing ports.
Steps 1–10 are repeated (optional) on the xenograft on the other side of the animal if two xenografts are implanted.
Xenografts can be routinely measured up to two times per week and are typically irrigated with F12 media, followed by air, the day before each measurement to remove excess mucous.
Fig. 19. 2.
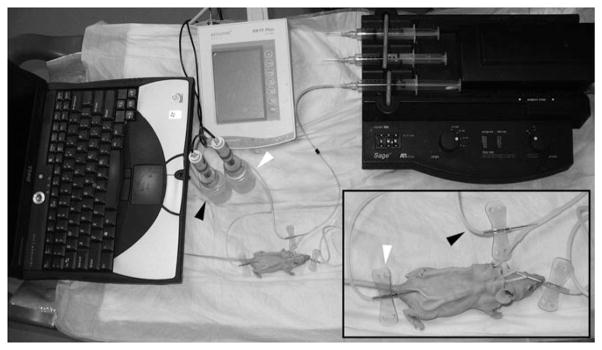
Potential difference instrumentation and setup. Measuring TEPD in this ex vivo model requires the following equipment: computer with data acquisition software, pH/mV meter, calomel electrodes, and syringe pump. The pH/mV is connected to the calomel electrodes that are connected to the anesthetized mouse by means of butterfly electrodes (see Note 2). The positive electrode is inserted into the perfusion tubing (black arrows) allowing access to the luminal surface of the trachea, while the negative electrode is inserted subcutaneously (white arrows).
Fig. 19.3.

TEPD analysis of ferret CF and non-CF tracheal xenografts. (a) Representative TEPD tracings of newborn ferret CFTR+/+ (dark line) and CFTR−/− (light line) tracheal xenografts. The buffer conditions change with ion channel agonists and antagonists are indicated above the tracing. (b) Reproducibility of sequential TEPD measurements taken in the same ferret CFTR+/+ and CFTR−/− xenografts at week intervals as indicated. Buffer conditions were the same as shown in (a) with the buffer number marked arrowheads. (c) Histological H&E sections of ferret CFTR+/+ (top) and CFTR−/− (bottom) xenografts. Note the intact pseudostratified ciliated epithelium (empty arrows) and the presence of submucosal glands (solid arrows) in both genotypes.
3.2. Cross-Species Analysis of CFTR Processing Requires Antibodies that Efficiently Bind to Conserved Epitopes Between Species
When comparing biologic properties among different species of CFTR, it is imperative that the antibodies used react similarly across the species. This can be achieved by screening available antibodies against each species of CFTR to be compared and/or adding a common epitope tag such as HA to the fourth extracellular loop (which has not been shown to affect CFTR function). Summarized in Table 19.3 are results of an antibody screen for comparative studies between human and ferret CFTRs using Western blotting and immunoprecipitation. This section will focus on our methods of immunoprecipitation of CFTR using DynaBeads® and the subsequent detection by phosphorylation of CFTR with protein kinase A (PKA) and [γ–32P]ATP as previously described with modifications (43, 48). A brief description of our Western blot protocol is contained in Note 5.
Table 19.3.
Human and ferret CFTR antibody optimization
| Name | Source | CFTR region | WB dilution | WB human | WB ferret | IP dilution | IP human | IP ferret |
|---|---|---|---|---|---|---|---|---|
| M3A7 | Millipore | N-Term | 1:1000 | +++ | +++ | 2 μg/mL | +++ | +++ |
| MM13-4 | Millipore | C-Term | 1:1000 | +++ | ++ | 2 μg/mL | +++ | +++ |
| 217 | CFFT | R Domain | 1:1000 | +++ | ++ | ND | ND | ND |
| 660 | CFFT | NBD1 | 1:500 | +++ | +++ | ND | ND | ND |
| 596 | CFFT | NBD2 | 1:1000 | +++ | +++ | ND | ND | ND |
| 570 | CFFT | R Domain | 1:1000 | +++ | +++ | ND | ND | ND |
| 3G11 | CFC | NBD1 | 1:1000 | ++ | ++ | 10 μg/mL | ++ | ++ |
| 10B6.2 | CFC | NBD1 | 1:500 | ++ | − | ND | ND | ND |
| L12B4 | Millipore | Pre-NBD1 | 1:1000 | ++ | ++ | 2 μg/mL | +++ | +++ |
| 13–1 | R&D Systems | R Domain | 1:1000 | + | + | ND | ND | ND |
| 24–1 | R&D Systems | C-Term | 1:1000 | + | + | ND | ND | ND |
| Mr. Pink | CFC | NBD1 | 1:500 | + | + | 7.5 μL | ND | ND |
| H-182 | Santa Cruz | N-Term | 1:200 | + | + | ND | ND | ND |
WB, Western blot; IP, immunoprecipitation; CFFT, cystic fibrosis foundation therapeutics; CFC, CFTR folding consortium; ND, not determined; −, no interaction; +, weak binding; ++, moderate binding; +++, strong binding. All WB and IPs came from a large pool of cell lysate derived from HT1080 cells expressing either human or ferret CFTR
-
Immunoprecipitation
Total protein (1 mg), from cell lysate derived from cells transiently transfected with human or ferret CFTR or EGFP expression plasmid (see Note 6), is aliquoted to a 2-mL Eppendorf tube and diluted to 1 mL using RIPA buffer containing freshly added protease inhibitors. Tween 20 is then added to a final concentration of 0.1%.
Primary antibody is incubated with the dilution specified in Table 19.3 for at least 2 h at 4°C with rotation. Washed protein G DynaBeads® (50 μL) is added to the solution and the mixture is incubated overnight at 4°C (see Note 7 and 8).
The beads are washed three times with PBS containing 0.1% Tween 20, placing the tube on a magnet between each wash to remove the supernatant.
-
In vitro phosphorylation of CFTR:
Prepare PKA and ATP phosphorylation buffers.
The beads from the finished IP protocol are washed once with PBS and the supernatant is removed by placing the tube on the magnet.
The beads are resuspended in 20 μL of the PKA phosphorylation buffer.
Add 40 μL of the ATP phosphorylation buffer and shake for 30 min at 30°C (see Note 9).
The beads are washed four times with PBS containing 0.1% Tween 20.
Beads, antibody, and CFTR are dissociated by adding 2× SDS-PAGE loading buffer, incubating at 37°C with continual shaking.
Place the tube in the magnet and load the supernatant on a 7.5% SDS-PAGE gel.
Resolve by electrophoresis (see Note 10).
Fix the gel by transferring the gel to a disposable plastic container filled with fixative solution (10% glacial acidic acid, 25% isopropanol, and 65% ddH2O) and gently agitate for 30 min.
Dry the gel by transferring to a piece of filter paper, cover with plastic wrap, and dry on a vacuum gel drier at 80°C for 45 min.
Expose to a pre-cleared phosphoscreen and scan several days later using a phosphoimager.
3.3. Cross-Species Analysis of the Processing Efficiency and Stability of CFTR by [35S]Methionine and Cysteine Pulse Chase
The glycosylation characteristics of CFTR serve as an excellent endpoint for assessing the processing CFTR. The immature form of CFTR resides in the endoplasmic reticulum (ER) and is called band B (~150 kDa), while the fully glycosylated mature protein resides at the plasma membrane and is called band C (~170–180 kDa). Note that the migratory apparent molecular weight of CFTR can vary slightly across species and may be due to slightly altered glycosylation. Metabolic pulse-chase experiments using radioactive amino acids, [35S]methionine and [35S]cysteine, serve as an excellent method to characterize the rate, stability, and efficiency of processing from band B to C of wild-type and mutant CFTRs across species. An example of a metabolic pulse-chase autoradiograph and quantification thereof for ferret wild-type CFTR is shown in Fig. 19.4.
Fig. 19.4.
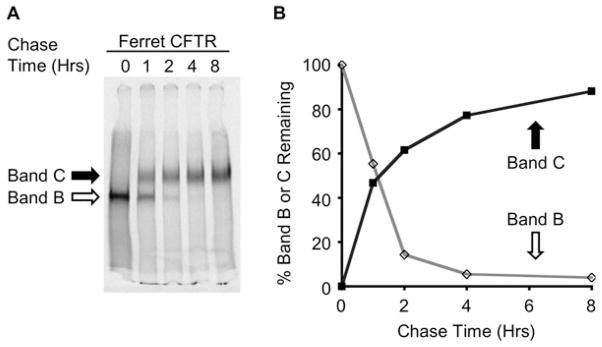
Metabolic [35S]methionine pulse chase of ferret CFTR processing. (a) HT1080 cells transiently expressing ferret CFTR were starved of methionine and cysteine (30 min), labeled with [35S]methionine and [35S]cysteine (15 min), and chased with media containing cold methionine and cysteine for the given time points as described under Section 3.3. (b) Densitometric quantification of (a) Empty points representing band BT/band B0 × 100. Solid data points representing band CT/band B0 × 100 (see Note 13).
Depletion of intracellular pools of methionine and cysteine: Gently wash the cells expressing CFTR two times with warm starvation media. Incubate the cells at 37°C for 30 min in starvation media (2 mL).
Metabolic labeling of CFTR: Gently aspirate the starvation media. Transfer cells to an area designated for the use of radioactive materials (see Note 9). Add 2 mL of starvation media containing 0.2 mCi/mL of [35S]methionine and cysteine. Incubate at 37°C for 15 min.
Metabolic chase of labeled CFTR: Aspirate the labeling media and place the cells on ice. Wash three times in cold PBS. Add warm chase media (4 mL) and incubate at 37°C for the desired amounts of time (see Note 11).
Harvesting the cells: Place cells on ice and remove the chase media by aspiration. Proceed to gently wash the cells three times with ice-cold PBS (1 mL). Lyse the cells on ice in 1 mL of RIPA buffer containing protease inhibitors for 5–30 min. Transfer the lysate to a 1.5-mL Eppendorf tubes and spin at 16,000×g for 10 min at 4°C (see Note 12). Freeze the samples until last chase time point has been harvested.
Immunoprecipitation of labeled CFTR: Thaw lysate and add Tween 20 to a final concentration of 0.1%. Then add 2 μg of anti-CFTR antibodies M3A7, L12B4, and MM13-4 and rotate for 3–5 h at 4°C. Wash 50 μL (30 μg/mL) of DynaBeads® with PBS containing 0.1% Tween 20, pelleting between each wash. Resuspend the beads in the original volume and add to the antibody/antigen containing lysate. Rotate at 4°C overnight. Wash the lysate with PBS (0.1% Tween 20) six times. Dissociate the protein and antibody by adding 2× SDS loading buffer (30 μL) and shake at 37°C for 30 min.
Electrophoresis and autoradiography: Load the lysate on 7.5% SDS-PAGE gels and electrophorese overnight (30 V). Fix the gel for 30 min by gently shaking at room temperature in the fixative solution. Replace the fixative solution with Amplify and incubate for 30 min at room temperature with gentle shaking. Dry the gel using a gel dryer and expose to a phosphoscreen for several days. Develop/scan the phospho-screen using a phosphoimager and imaging software.
Densitometric analysis: Quantify the total CFTR signal for bands B and C for each of the experimental time points (see Note 13).
Acknowledgments
This work was supported by grants from the NHLBI (RC1HL099516), NIDDK (P30DK054759, R37DK047967), NHLBI (HL091842) and the Cystic Fibrosis Foundation (ENGELH08XX0), as well as by the Roy J. Carver Chair in molecular medicine. We also gratefully acknowledge Drs Christine Blaumueller and Monali Sawai for editorial contributions.
Footnotes
Freshly isolated trachea from newborn ferret or pig is cannulated to the previously described xenograft cassette (46). In brief, the cassette consists of a combination of silastic (Dow Corning, Midland, MI) and Teflon (Thomas Scientific, Swedesboro, NJ) tubing attached to barb-to-barb connectors (Bio-Rad Laboratories, Hercules, CA). The trachea is ligated to the connectors and the tubing ports capped with chromel A steel wire (Hoskins MFG, Novi, MI). These cassettes are inserted subcutaneously into the flanks of male Nu/Nu athymic mice.
Butterfly electrodes are made by dissolving the agar noble (5%) in 1 M KCl by heating. The butterfly needle/tubing is filled with this hot solution by negative pressure generated by a 30-mL syringe. These electrodes are submerged in 1 M KCl in a 100-mm dish for up to 1 year at 4°C.
To accurately calculate changes in potential difference between buffers, it is important to wait for a stable millivolt reading before switching to the next buffer. This usually occurs within 10 min but depends upon the xenograft.
It is critical that the fluid-filled tubing and the xenograft are devoid of any air bubbles. The manifold tubing is therefore clamped with a hemostat when changing buffers. The hemostat is removed once the tubing is attached to the next buffer. Failure to keep air from the system will result in moments of infinite TEPD spikes as the air moving through the xenograft disrupts the electrical conductivity.
Equal amounts of protein from cellular lysate from cells expressing human or ferret CFTR (see Note 6) were resolved electrophoretically on 6% SDS-PAGE gels. The protein was transferred to nitrocellulose membranes and the membrane blocked in PBS containing 0.1% casein and 0.2% Tween 20. CFTR primary antibodies were then added at the dilution indicated in Table 19.3 and incubated for 1 h at room temperature. The blots were washed, probed with secondary antibodies conjugated to an IR dye, and imaged using an Odyssey IR scanner.
Transient expression of human and ferret CFTRs in HT1080 cells was achieved by electroporation using the BTX-830/630B system (Harvard Apparatus, Holliston, MA). The following electroporation conditions were used for HT1080 (4 pulses, 230 mV, 1 ms interval) and BHK21 (1 pulse, 260 mV, 1 ms interval) cell lines.
Incubation of the antigen/antibody and the DynaBeads® for 2 h at room temperature is also sufficient. It is also critical that the DynaBeads® be maintained in a 0.01–0.1% Tween 20 solution to avoid bead clumping.
Alternatively, one can prebind the CFTR antibodies and the DynaBeads® by incubating with rotation for 30 min at room temperature. The unbound antibody is then washed away and the antibody-bound DynaBeads® can be directly added to the cell lysate and rotated for 2 h at room temperature.
From this step forward, everything must be carried out in the radiation room according to manufacturer’s guidelines. Everything must be discarded appropriately and appropriate personal protective equipment used.
To achieve adequate separation between bands B and C on a 7.5% mini gel, we run the ladder off to 100 kDa. This is done by running the gel at 120 V for ~3 h or 30 V overnight.
It is recommended that several time points from 0 to 12 h be included to adequately study the rate of disappearance of band B and appearance and disappearance of band C. It may be helpful to also include time points up to 48 h to ascertain differences in protein stability.
To avoid non-specific pull down of other labeled proteins, keep the lysis time less than 30 min and spin the lysate at 4°C. This avoids protein degradation and lysis of nuclei in the sample.
Quantify the lane background using two regions for each lane (below band B and above band C). Subtract the lane background from the regions representing bands B and C. To analyze the disappearance of band B, measure the percentage of band B over time divided by the initial amount of band B (band BT/band B0). To analyze the appearance/disappearance of the higher molecular weight, fully processed band C, it is important to measure the intensity of band C over time divided by the initial band B labeling intensity (band CT/band B0). Due to background issues throughout the lane at time 0 of the chase (likely due to undegraded fragments of CFTR generated during the labeling process), band C0 is set to 0.
References
- 1.Davidson DJ, Dorin JR. The CF mouse: an important tool for studying cystic fibrosis. Expert Rev Mol Med. 2001;3:1–27. doi: 10.1017/S1462399401002551. [DOI] [PubMed] [Google Scholar]
- 2.Davidson DJ, Rolfe M. Mouse models of cystic fibrosis. Trends Genet. 2001;17:S29–S37. doi: 10.1016/s0168-9525(01)02452-0. [DOI] [PubMed] [Google Scholar]
- 3.Dickinson P, Dorin JR, Porteous DJ. Modelling cystic fibrosis in the mouse. Mol Med Today. 1995;1:140–148. doi: 10.1016/s1357-4310(95)80092-1. [DOI] [PubMed] [Google Scholar]
- 4.Egan ME. How useful are cystic fibrosis mouse models? Drug Discovery Today: Disease Models. 2009;6:35–41. doi: 10.1016/j.ddmod.2009.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Grubb BR, Boucher RC. Pathophysiology of gene-targeted mouse models for cystic fibrosis. Physiol Rev. 1999;79:S193–S214. doi: 10.1152/physrev.1999.79.1.S193. [DOI] [PubMed] [Google Scholar]
- 6.Sun X, Yan Z, Yi Y, Li Z, Lei D, Rogers CS, et al. Adeno-associated virus-targeted disruption of the CFTR gene in cloned ferrets. J Clin Invest. 2008;118:1578–1583. doi: 10.1172/JCI34599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rogers CS, Hao Y, Rokhlina T, Samuel M, Stoltz DA, Li Y, et al. Production of CFTR-null and CFTR-DeltaF508 heterozygous pigs by adeno-associated virus-mediated gene targeting and somatic cell nuclear transfer. J Clin Invest. 2008;118:1571–1577. doi: 10.1172/JCI34773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dorin JR, Dickinson P, Alton EW, Smith SN, Geddes DM, Stevenson BJ, et al. Cystic fibrosis in the mouse by targeted insertional mutagenesis. Nature. 1992;359:211–215. doi: 10.1038/359211a0. [DOI] [PubMed] [Google Scholar]
- 9.Snouwaert JN, Brigman KK, Latour AM, Malouf NN, Boucher RC, Smithies O, et al. An animal model for cystic fibrosis made by gene targeting. Science. 1992;257:1083–1088. doi: 10.1126/science.257.5073.1083. [DOI] [PubMed] [Google Scholar]
- 10.O’Neal WK, Hasty P, McCray PB, Jr, Casey B, Rivera-Perez J, Welsh MJ, et al. A severe phenotype in mice with a duplication of exon 3 in the cystic fibrosis locus. Hum Mol Genet. 1993;2:1561–1569. doi: 10.1093/hmg/2.10.1561. [DOI] [PubMed] [Google Scholar]
- 11.Ratcliff R, Evans MJ, Cuthbert AW, MacVinish LJ, Foster D, Anderson JR, et al. Production of a severe cystic fibrosis mutation in mice by gene targeting. Nat Genet. 1993;4:35–41. doi: 10.1038/ng0593-35. [DOI] [PubMed] [Google Scholar]
- 12.Zhou L, Dey CR, Wert SE, DuVall MD, Frizzell RA, Whitsett JA. Correction of lethal intestinal defect in a mouse model of cystic fibrosis by human CFTR. Science. 1994;266:1705–1708. doi: 10.1126/science.7527588. [DOI] [PubMed] [Google Scholar]
- 13.Colledge WH, Abella BS, Southern KW, Ratcliff R, Jiang C, Cheng SH, et al. Generation and characterization of a delta F508 cystic fibrosis mouse model. Nat Genet. 1995;10:445–452. doi: 10.1038/ng0895-445. [DOI] [PubMed] [Google Scholar]
- 14.Hasty P, O’Neal WK, Liu KQ, Morris AP, Bebok Z, Shumyatsky GB, et al. Severe phenotype in mice with termination mutation in exon 2 of cystic fibrosis gene. Somat Cell Mol Genet. 1995;21:177–187. doi: 10.1007/BF02254769. [DOI] [PubMed] [Google Scholar]
- 15.van Doorninck JH, French PJ, Verbeek E, Peters RH, Morreau H, Bijman J, et al. A mouse model for the cystic fibrosis delta F508 mutation. EMBO J. 1995;14:4403–4411. doi: 10.1002/j.1460-2075.1995.tb00119.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zeiher BG, Eichwald E, Zabner J, Smith JJ, Puga AP, McCray PB, Jr, et al. A mouse model for the delta F508 allele of cystic fibrosis. J Clin Invest. 1995;96:2051–2064. doi: 10.1172/JCI118253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Delaney SJ, Alton EW, Smith SN, Lunn DP, Farley R, Lovelock PK, et al. Cystic fibrosis mice carrying the missense mutation G551D replicate human genotype–phenotype correlations. EMBO J. 1996;15:955–963. [PMC free article] [PubMed] [Google Scholar]
- 18.Rozmahel R, Wilschanski M, Matin A, Plyte S, Oliver M, Auerbach W, et al. Modulation of disease severity in cystic fibrosis transmembrane conductance regulator deficient mice by a secondary genetic factor. Nat Genet. 1996;12:280–287. doi: 10.1038/ng0396-280. [DOI] [PubMed] [Google Scholar]
- 19.Dickinson P, et al. Generation of a CF mutant mouse possessing the G480C mutation. 22nd European CF Conference Berlin Book of Abstracts, PS7; 1998. p. 143. [Google Scholar]
- 20.Du M, Liu X, Welch EM, Hirawat S, Peltz SW, Bedwell DM. PTC124 is an orally bioavailable compound that promotes suppression of the human CFTR-G542X nonsense allele in a CF mouse model. Proc Natl Acad Sci USA. 2008;105:2064–2069. doi: 10.1073/pnas.0711795105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hodges CA, Cotton CU, Palmert MR, Drumm ML. Generation of a conditional null allele for Cftr in mice. Genesis. 2008;46:546–552. doi: 10.1002/dvg.20433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zahm JM, Gaillard D, Dupuit F, Hinnrasky J, Porteous D, Dorin JR, et al. Early alterations in airway mucociliary clearance and inflammation of the lamina propria in CF mice. Am J Physiol. 1997;272:C853–C859. doi: 10.1152/ajpcell.1997.272.3.C853. [DOI] [PubMed] [Google Scholar]
- 23.Kent G, Iles R, Bear CE, Huan LJ, Griesenbach U, McKerlie C, et al. Lung disease in mice with cystic fibrosis. J Clin Invest. 1997;100:3060–3069. doi: 10.1172/JCI119861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cowley EA, Wang CG, Gosselin D, Radzioch D, Eidelman DH. Mucociliary clearance in cystic fibrosis knockout mice infected with Pseudomonas aeruginosa. Eur Respir J. 1997;10:2312–2318. doi: 10.1183/09031936.97.10102312. [DOI] [PubMed] [Google Scholar]
- 25.Reynaert I, Van Der Schueren B, Degeest G, Manin M, Cuppens H, Scholte B, et al. Morphological changes in the vas deferens and expression of the cystic fibrosis transmembrane conductance regulator (CFTR) in control, deltaF508 and knockout CFTR mice during postnatal life. Mol Reprod Dev. 2000;55:125–135. doi: 10.1002/(SICI)1098-2795(200002)55:2<125::AID-MRD1>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 26.Hodges CA, Palmert MR, Drumm ML. Infertility in females with cystic fibrosis is multifactorial: evidence from mouse models. Endocrinology. 2008;149:2790–2797. doi: 10.1210/en.2007-1581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Beharry S, Ackerley C, Corey M, Kent G, Heng YM, Christensen H, et al. Long-term docosahexaenoic acid therapy in a congenic murine model of cystic fibrosis. Am J Physiol Gastrointest Liver Physiol. 2007;292:G839–G848. doi: 10.1152/ajpgi.00582.2005. [DOI] [PubMed] [Google Scholar]
- 28.Durie PR, Kent G, Phillips MJ, Ackerley CA. Characteristic multi-organ pathology of cystic fibrosis in a long-living cystic fibrosis transmembrane regulator knockout murine model. Am J Pathol. 2004;164:1481–1493. doi: 10.1016/S0002-9440(10)63234-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Freedman SD, Kern HF, Scheele GA. Pancreatic acinar cell dysfunction in CFTR(−/−) mice is associated with impairments in luminal pH and endocytosis. Gastroenterology. 2001;121:950–957. doi: 10.1053/gast.2001.27992. [DOI] [PubMed] [Google Scholar]
- 30.Grubb BR, Paradiso AM, Boucher RC. Anomalies in ion transport in CF mouse tracheal epithelium. Am J Physiol. 1994;267:C293–C300. doi: 10.1152/ajpcell.1994.267.1.C293. [DOI] [PubMed] [Google Scholar]
- 31.Liu X, Yan Z, Luo M, Engelhardt JF. Species-specific differences in mouse and human airway epithelial biology of recombinant adeno-associated virus transduction. Am J Respir Cell Mol Biol. 2006;34:56–64. doi: 10.1165/rcmb.2005-0189OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.van Heeckeren AM, Schluchter MD, Xue W, Davis PB. Response to acute lung infection with mucoid Pseudomonas aeruginosa in cystic fibrosis mice. Am J Respir Crit Care Med. 2006;173:288–296. doi: 10.1164/rccm.200506-917OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Heeckeren A, Walenga R, Konstan MW, Bonfield T, Davis PB, Ferkol T. Excessive inflammatory response of cystic fibrosis mice to bronchopulmonary infection with Pseudomonas aeruginosa. J Clin Invest. 1997;100:2810–2815. doi: 10.1172/JCI119828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hodges CA, Drumm ML. CF: tissue by tissue. Pediatr Pulmonol. 2009;44:188–189. [Google Scholar]
- 35.Rogers CS, Abraham WM, Brogden KA, Engelhardt JF, Fisher JT, McCray PB, Jr, et al. The porcine lung as a potential model for cystic fibrosis. Am J Physiol Lung Cell Mol Physiol. 2008;295:L240–L263. doi: 10.1152/ajplung.90203.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li Z, Engelhardt JF. Progress toward generating a ferret model of cystic fibrosis by somatic cell nuclear transfer. Reprod Biol Endocrinol. 2003;1:83. doi: 10.1186/1477-7827-1-83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Williams SH, Sahota V, Palmai-Pallag T, Tebbutt SJ, Walker J, Harris A. Evaluation of gene targeting by homologous recombination in ovine somatic cells. Mol Reprod Dev. 2003;66:115–125. doi: 10.1002/mrd.10340. [DOI] [PubMed] [Google Scholar]
- 38.Rogers CS, Stoltz DA, Meyerholz DK, Ostedgaard LS, Rokhlina T, Taft PJ, et al. Disruption of the CFTR gene produces a model of cystic fibrosis in newborn pigs. Science. 2008;321:1837–1841. doi: 10.1126/science.1163600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sun X, Sui H, Fisher JT, Yan Z, Liu X, Cho HJ, Joo NS, Zhang Y, Zhou W, Yi Y, Kinyon JM, Lei-Butters DC, Griffin MA, Naumann P, Luo M, Ascher J, Wang K, Frana T, Wine JJ, Meyerholz DK, Engelhardt JF. Disease phenotype of a ferret CFTR-knockout model of cystic fibrosis. J Clin Invest. 2010;120:3149–3160. doi: 10.1172/JCI43052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Stoltz DA, Meyerholz DK, Pezzulo AA, Ramachandran S, Rogan MP, Davis GJ, Hanfland RA, Wohlford-Lenane C, Dohrn CL, Bartlett JA, Nelson GAt, Chang EH, Taft PJ, Ludwig PS, Estin M, Hornick EE, Launspach JL, Samuel M, Rokhlina T, Karp PH, Ostedgaard LS, Uc A, Starner TD, Horswill AR, Brogden KA, Prather RS, Richter SS, Shilyansky J, McCray PB, Jr, Zabner J, Welsh MJ. Cystic fibrosis pigs develop lung disease and exhibit defective bacterial eradication at birth. Sci Transl Med. 2010;2:29ra31. doi: 10.1126/scitranslmed.3000928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40a.Pierucci-Alves F, Akoyev V, Stewart J, Wang L, Schultz BD. CFTR−/− pigs exhibit CBAVD phenotype at birth. Pediatr Pulmonol. 2010;45:291. [Google Scholar]
- 41.Oppenheimer EH, Esterly JR. Cystic fibrosis of the pancreas. Morphologic findings in infants with and without diagnostic pancreatic lesions. Arch Pathol. 1973;96:149–154. [PubMed] [Google Scholar]
- 42.Oppenheimer EH, Esterly JR. Pathology of cystic fibrosis review of the literature and comparison with 146 autopsied cases. Perspect Pediatr Pathol. 1975;2:241–278. [PubMed] [Google Scholar]
- 43.Ostedgaard LS, Rogers CS, Dong Q, Randak CO, Vermeer DW, Rokhlina T, et al. Processing and function of CFTR-DeltaF508 are species-dependent. Proc Natl Acad Sci USA. 2007;104:15370–15375. doi: 10.1073/pnas.0706974104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Liu Y, Wang Y, Jiang Y, Zhu N, Liang H, Xu L, et al. Mild processing defect of porcine DeltaF508-CFTR suggests that DeltaF508 pigs may not develop cystic fibrosis disease. Biochem Biophys Res Commun. 2008;373:113–118. doi: 10.1016/j.bbrc.2008.06.009. [DOI] [PubMed] [Google Scholar]
- 45.Yan Z, Lei-Butters D, Sun X, Yi Y, Fisher JT, Engelhardt JF. Progress toward generating a ΔF508-CFTR cystic fibrosis ferret model. Pediatr Pulmonol. 2009;44:284. [Google Scholar]
- 46.Filali M, Zhang Y, Ritchie TC, Engelhardt JF. Xenograft model of the CF airway. Methods Mol Med. 2002;70:537–550. doi: 10.1385/1-59259-187-6:537. [DOI] [PubMed] [Google Scholar]
- 47.French PJ, van Doorninck JH, Peters RH, Verbeek E, Ameen NA, Marino CR, et al. A delta F508 mutation in mouse cystic fibrosis transmembrane conductance regulator results in a temperature-sensitive processing defect in vivo. J Clin Invest. 1996;98:1304–1312. doi: 10.1172/JCI118917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ostedgaard LS, Zeiher B, Welsh MJ. Processing of CFTR bearing the P574H mutation differs from wild-type and deltaF508-CFTR. J Cell Sci. 1999;112:2091–2098. doi: 10.1242/jcs.112.13.2091. [DOI] [PubMed] [Google Scholar]
- 49.Vawter GF, Shwachman H. Cystic fibrosis in adults: an autopsy study. Pathol Annu. 1979;14:357–382. [PubMed] [Google Scholar]
- 50.Sturgess JM. Structural and developmental abnormalities of the exocrine pancreas in cystic fibrosis. J Pediatr Gastroenterol Nutr. 1984;3(Suppl 1):S55–S66. doi: 10.1097/00005176-198400031-00011. [DOI] [PubMed] [Google Scholar]
- 51.Gaillard D, Bouvier R, Scheiner C, Nessmann C, Delezoide AL, Dechelotte P, et al. Meconium ileus and intestinal atresia in fetuses and neonates. Pediatr Pathol Lab Med. 1996;16:25–40. [PubMed] [Google Scholar]
- 52.Oppenheimer EH, Esterly JR. Observations in cystic fibrosis of the pancreas. II. Neonatal intestinal obstruction. Bull Johns Hopkins Hosp. 1962;111:1–13. [PubMed] [Google Scholar]
- 53.Bronstein MN, Sokol RJ, Abman SH, Chatfield BA, Hammond KB, Hambidge KM, et al. Pancreatic insufficiency, growth, and nutrition in infants identified by newborn screening as having cystic fibrosis. J Pediatr. 1992;120:533–540. doi: 10.1016/s0022-3476(05)82478-3. [DOI] [PubMed] [Google Scholar]
- 54.Weber AM, Roy CC, Chartrand L, Lepage G, Dufour OL, Morin CL, et al. Relationship between bile acid malabsorption and pancreatic insufficiency in cystic fibrosis. Gut. 1976;17:295–299. doi: 10.1136/gut.17.4.295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.O’Brien S, Mulcahy H, Fenlon H, O’Broin A, Casey M, Burke A, et al. Intestinal bile acid malabsorption in cystic fibrosis. Gut. 1993;34:1137–1141. doi: 10.1136/gut.34.8.1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lindblad A, Glaumann H, Strandvik B. Natural history of liver disease in cystic fibrosis. Hepatology. 1999;30:1151–1158. doi: 10.1002/hep.510300527. [DOI] [PubMed] [Google Scholar]
- 57.Feranchak AP, Sokol RJ. Cholangiocyte biology and cystic fibrosis liver disease. Semin Liver Dis. 2001;21:471–488. doi: 10.1055/s-2001-19030. [DOI] [PubMed] [Google Scholar]
- 58.Chaudry G, Navarro OM, Levine DS, Oudjhane K. Abdominal manifestations of cystic fibrosis in children. Pediatr Radiol. 2006;36:233–240. doi: 10.1007/s00247-005-0049-2. [DOI] [PubMed] [Google Scholar]
- 59.Popli K, Stewart J. Infertility and its management in men with cystic fibrosis: review of literature and clinical practices in the UK. Hum Fertil. 2007;10:217–221. doi: 10.1080/14647270701510033. [DOI] [PubMed] [Google Scholar]
- 60.Oates RD, Amos JA. The genetic basis of congenital bilateral absence of the vas deferens and cystic fibrosis. J Androl. 1994;15:1–8. [PubMed] [Google Scholar]
- 61.Guilbault C, Saeed Z, Downey GP, Radzioch D. Cystic fibrosis mouse models. Am J Respir Cell Mol Biol. 2007;36:1–7. doi: 10.1165/rcmb.2006-0184TR. [DOI] [PubMed] [Google Scholar]
- 62.Hardcastle J, Harwood MD, Taylor CJ. Absorption of taurocholic acid by the ileum of normal and transgenic deltaf508 cystic fibrosis mice. J Pharm Pharmacol. 2004;56:445–452. doi: 10.1211/0022357022881. [DOI] [PubMed] [Google Scholar]
- 63.Freudenberg F, Broderick AL, Yu BB, Leonard MR, Glickman JN, Carey MC. Pathophysiological basis of liver disease in cystic fibrosis employing a deltaf508 mouse model. Am J Physiol Gastrointest Liver Physiol. 2008;294:G1411–G1420. doi: 10.1152/ajpgi.00181.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Meyerholz DK, Stoltz DA, Pezzulo AA, Welsh MJ. Pathology of gastrointestinal organs in a porcine model of cystic fibrosis. Am J Pathol. 2010;76:1377–1389. doi: 10.2353/ajpath.2010.090849. [DOI] [PMC free article] [PubMed] [Google Scholar]