

Abstract
Roux-en-Y gastric bypass surgery (RYGB) remains to be the most effective long-term treatment for obesity and its associated comorbidities, but the specific mechanisms involved remain elusive. Because RYGB patients appear to no longer be preoccupied with thoughts about food and are satisfied with much smaller meals and calorically dilute foods, brain reward mechanisms could be involved. Just as obesity can produce maladaptive alterations in reward functions, reversal of obesity by RYGB could normalize these changes or even further reset the food reward system through changes in gut hormone secretion, aversive conditioning and/or secondary effects of weight loss. Future studies with longitudinal assessments of reward behaviors and their underlying neural circuits before and after surgery will be necessary to uncover the specific mechanisms involved. Such new insights could be the base for future `knifeless' pharmacological and behavioral approaches to obesity.
Keywords: brain, gut hormones, food addiction, weight loss
Introduction
Factors important for the control energy balance can be divided into those that are compensated and those that are not compensated. Among the compensated ones are internal signals of energy repletion and depletion such as metabolic substrates, hormones and neural signals that function in a negative-feedback manner to achieve energy homeostasis. Among the non-compensated ones are mainly external factors such as availability, cost/benefit ratio, conditioned cues, palatability and social constraints. Using this general intake model, it has been shown by mathematical modeling that body weight/adiposity settles at a higher level when exposed to such uncompensated factors.1 This outcome is well known from the animal literature, showing diet-induced obesity, and in humans, exposed to the modern food environment of plenty. Importantly, the new level of body weight appears to be defended by engaging the same strong homeostatic mechanisms preventing weight loss at normal levels of body weight. Therefore, for most over-weight and obese subjects, dieting has not been successful as a weight-reducing strategy. All the weight lost is typically regained sooner or later, and the cycle of loss and regain is often repeated many times.
Clearly, the correct treatment of this type of obesity would be to remove the uncompensated factor; however, in the face of the enormous corporate, political and social pressures, it is unrealistic to expect that this will be achieved in the near future. In the meantime, we will have to rely to a large degree on development of drugs and other symptomatic treatments of obesity. Bariatric surgery is one of these treatments that has recently gained strong momentum, not only judging by the number of surgeries performed but also by its promise to reveal potential secrets of the still incompletely understood physiological mechanisms contributing to the controls of body weight/adiposity. Uncovering these secrets might lead to `knifeless' obesity surgery in the future.
Remarkably and in stark contrast to weight loss induced by restrained eating, weight loss induced by bariatric surgeries, particularly Roux-en-Y gastric bypass (RYGB), is sustained for up to 15 years or longer, suggesting that the strong biological mechanisms defending weight loss are either neutralized or overshadowed by the surgical intervention. Ideally, gastric bypass surgery would somehow reset the level of defended body weight, so that the patient, happy with the reduced amount of food, is not `eternally' hungry.
Preliminary data and anecdotal reports suggest that bariatric surgery patients `lose the desire' to eat and are no longer preoccupied with thoughts about food and eating. They seem satisfied with much smaller meals and calorically dilute foods.2–5 It is thus possible that RYGB leads to changes in the way the palatability of foods is perceived and how the brain computes food reward. Limited investigations in obese subjects show that obesity is associated with alterations in neural and behavioral mechanisms of food reward,6,7 and parallels have been drawn between drug and food addiction.8–11 Thus, bariatric surgery might be successful because it reverses, or at least neutralizes, obesity-induced changes in reward functions.
The aim of this article is, therefore, to review the existing literature on obesity-associated reward dysfunctions and bariatric surgery-induced changes in taste and reward functions, including our own recent observations in a rat model of RYGB, and to explore potential mechanisms involved in these changes (Figure 1).
Figure 1.
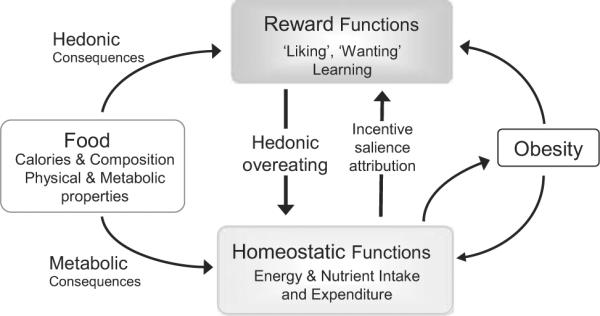
Schematic diagram depicting the relationship between obesity and food reward.
Altered reward mechanisms in obesity
Do genetic and other preexisting differences in reward functions cause obesity?
Although environmental pressure undoubtedly pushes the general population to higher food intake and body weight, this simple explanation does not account for the fact that not all subjects exposed to the same environment gain weight. This suggests that preexisting differences make some individuals more vulnerable to the increased availability of palatable food and food cues. Preexisting differences could be determined by genetic and epigenetic alterations, and by early life experience through developmental programming. Among the 20 or so major genes (clear evidence from at least two independent studies) linked to the development of obesity,12 none are directly implicated in known mechanisms of reward functions. However, because the combined effect of these genes only accounts for less than about 5% of human obesity, it is very likely that many important genes have not yet been discovered, some of which could operate within the reward system.
A comparison of lean and obese subjects carrying different alleles of either the dopamine D2-receptor or mu-opioid receptor genes reveals differences in behavioral and neural responses to palatable food.13–16 In selectively bred lines of obesity-prone and obesity-resistant rats, several differences in mesolimbic dopamine signaling have been reported.17 Mesolimbic dopamine signaling is also severely suppressed in leptin-deficient ob/ob mice and rescued by systemic leptin replacement.18 However, in genetically leptin-deficient humans, viewing pictures of palatable foods elicited exaggerated neural activity in the absence of leptin that was abolished after leptin administration.19 Furthermore, positron emission tomography neuroimaging showed reduced dopamine D2 receptor availability, mostly in the dorsal and lateral, but not in ventral, striatum.10 On the basis of this last observation, the reward-deficiency hypothesis was coined, suggesting that increased food intake is an attempt to generate more reward in compensation for reduced mesolimbic dopamine signaling.10,20
Our own studies in rats indicate that obesity changes food-reward behaviors in a nutrient-concentration-specific manner, with reduced `liking' of dilute sucrose and corn oil, but increased `liking' of the highest concentrations. In addition, diet-induced obese rats compared with chow-fed, lean outbred Sprague–Dawley rats, as well as young, preobese genetically select lines of obesity-prone compared with obesity-resistant rats, exhibit reduced `wanting' as measured by slower completion speed in the incentive runway and lower break points in the progressive ratio lever-press paradigm.
In summary, differences in mesolimbic dopamine signaling are most strongly implicated in altered food anticipatory and consummatory behaviors and obesity. However, it is still not clear to what extent preexisting differences and/or secondary effects determine these behavioral alterations and cause obesity. Only longitudinal studies in genetically defined populations will provide more conclusive answers.
Is the obese state secondarily changing reward mechanisms and accelerating the process?
Obesity is associated with dysregulated signaling systems, such as leptin and insulin resistance, as well as increased signaling through proinflammatory cytokines and pathways activated by oxidative and endoplasmic reticulum stress.21 Clearly, the obesity-induced toxic internal environment does not spare the brain.22–27 Obesity-induced brain insulin resistance is believed to have a direct effect on development of Alzheimer's disease, now also called Type 3 diabetes,28 as well as other neurodegenerative diseases.29
A number of recent studies directed attention to the hypothalamus, where high-fat diets disturb the delicate relationship between glial cells and neurons through increased endoplasmatic reticulum and oxidative stress, leading to stress–response pathways with generally cytotoxic effects.25 The end effects of these changes are central insulin and leptin resistance and impaired hypothalamic regulation of energy balance, further favoring the development of obesity and, in turn, neurodegeneration. However, these toxic effects do not stop at the level of the hypothalamus, but can also affect brain areas involved in reward processing. The obese, leptin-deficient mouse is much more sensitive to chemically induced neurodegeneration such as metamphetamine-induced dopamine nerve terminal degeneration as indicated by reduced striatal dopamine levels.30 Obesity and hypertriglyceridemia produce cognitive impairment in mice, including reduced lever pressing for food reward,23 and epidemiological studies show an association of body mass index and risk of Parkinson's disease and cognitive decline.31 Obesity-prone rats allowed to become obese on regular chow, or fed amounts of high-fat diet so as not to gain extra body weight, exhibited significantly reduced operant responding (progressive ratio break point) for sucrose, amphetamine-induced-conditioned place preference and dopamine turnover in the nucleus accumbens.17 These results suggest that both obesity per se and high-fat diet can cause alterations in mesolimbic dopamine signaling and reward behaviors.
In summary, it seems clear that the obesity-induced internal `toxic' environment does not stop at the level of the brain, and within the brain does not stop at the reward circuitry. Similar to brain areas involved in homeostatic energy balance regulation, such as the hypothalamus, and in cognitive control, such as the hippocampus and neocortex, reward circuitry in corticolimbic and other areas is likely to be affected by obesity-induced changes in peripheral signals to the brain and local brain signaling through inflammatory, oxidative and mitochondrial stress pathways.
Altered reward mechanisms after bariatric surgeries
Few studies have addressed this question, but there is a sense of rapidly increasing interest in the field. In one study, RYGB patients showed heightened acuity for sweet taste, with some patients complaining that the food was too sweet,32 but in another study there was increased acuity for bitter and sour tastes and a trend toward reduction in salt and sweet detection.33 In other human studies, preference for high-carbohydrate foods34 and high-fat foods3,34 was decreased after RYGB. In addition, RYGB patients were reported to lose the desire or motivation to eat.34
In an attempt to further investigate the underlying behavioral and neural processes responsible for such changes, we have used a rat model of RYGB.35,36 The model closely replicates the major effects of RYGB seen in obese human patients on food intake, body weight loss and secretion of gut hormones. We find a shift from high to low concentrations of sweet and oily food stimuli in their capacity to generate pleasure and stimulate intake, compared with sham-operated obese animals.36 Under non-food-deprived conditions, diet-induced obese rats largely ignore low concentrations of sweet and oily stimuli and are less motivated to work for a food reward compared with chow-fed lean rats. Rather, obese rats seem to respond only to easily accessible very sweet and oily stimuli. All obesity-induced changes were completely reversed after RYGB.36 In addition, preference for high-fat diet steadily decreased over a 5-month postsurgical period, and preference for normal (low-fat) chow increased.35 Very similar findings regarding sucrose sensitivity and preference were reported in a simultaneously published paper by Hajnal et al.37 after RYGB in Otsuka Long Evans Tokushima Fatty (OLETF) rats lacking cholecystokinin-1 receptors. Together, these observations strongly suggest that at least one mechanism by which RYGB surgery reduces total energy intake and body weight is by changing taste perception and hedonic neural processing; however, the specific mechanisms involved are not known at this point.
Potential mechanisms for RYGB-induced changes in taste sensitivity and food hedonics
Some changes in reward functions secondary to the obese state may have their origin in altered hormone levels and sensitivities such as leptin and insulin. In the obese state, circulating levels of leptin and insulin are high, together with resistance to receptor-mediated signaling capacity. Sensitivity of both systems is reestablished after weight loss induced by RYGB and other interventions. As leptin signaling does not appear to be necessary for at least the initial RYGB-induced weight loss as demonstrated in fatty Zucker rats,38 it is unlikely to have a major role in changes of reward functions during the initial weight loss period after RYGB. However, given the recent evidence for direct leptin actions on reward processing areas of the brain,18,19,39,40 its involvement during later stages of reduced weight maintenance cannot be ruled out, and effects of obesity-associated changes in insulin sensitivity on reward processing41 have not been investigated. Furthermore, besides leptin, adipose tissue secretes a number of other hormones and cytokines that change with obesity status and could thus potentially mediate effects of body weight changes on brain reward functions. As discussed above, inflammatory cytokines are known to affect cognitive brain functions in obesity and are also likely to affect brain reward processing.
Gut hormones have obtained the most attention as possible mediators for the beneficial effects of RYGB on glucose homeostasis and body weight. The lower gut hormones glucagon-like peptide-1 and peptide YY, which are both substantially elevated after RYGB, are the leading candidates for the rapid amelioration of glucose homeostasis and reduced appetite. Additional factors secreted by the small intestine such as oleylethanolamide, N-acetyl-phospahatidylethanolamine, apolipoprotein A-IV and ghrelin, secreted mainly from the bypassed stomach, are other potential candidates. These hormones and factors could affect reward processing by functioning directly on taste and olfactory pathways42–44 and reward circuits,45–47 or by first functioning on primary visceral afferents relaying the information to these circuits.
Finally, it is also very likely that at least part of the hypophagia is the result of aversive conditioning during the early postsurgical period, when the rearranged gut is suddenly faced with unusual challenges. Rapidly ingested liquid food can lead to dumping and solid food to obstruction, both associated with very unpleasant feelings. Over time, such negative feelings associated with particular foods can lead to the conditioning of food aversions.
Conclusions
Although it appears plausible that RYGB-induced changes in gut hormone secretion, aversive conditioning and/or secondary effects of weight loss reset the brain reward system, the specific mechanisms and pathways involved are completely unknown. Future studies with longitudinal assessments of reward behaviors and their underlying neural mechanisms, before and at several time points after surgery, and in pair-fed controls, will be essential.
Footnotes
Conflict of interest H-R Berthoud received consulting fees from Mars Petfoods Inc. and AstraZeneca. AC Shin declares no conflict of interest.
References
- 1.de Castro JM. The control of food intake of free-living humans: putting the pieces back together. Physiol Behav. 2010;100:446–453. doi: 10.1016/j.physbeh.2010.04.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kenler HA, Brolin RE, Cody RP. Changes in eating behavior after horizontal gastroplasty and Roux-en-Y gastric bypass. Am J Clin Nutr. 1990;52:87–92. doi: 10.1093/ajcn/52.1.87. [DOI] [PubMed] [Google Scholar]
- 3.Olbers T, Bjorkman S, Lindroos A, Maleckas A, Lonn L, Sjostrom L, et al. Body composition, dietary intake, and energy expenditure after laparoscopic Roux-en-Y gastric bypass and laparoscopic vertical banded gastroplasty: a randomized clinical trial. Ann Surg. 2006;244:715–722. doi: 10.1097/01.sla.0000218085.25902.f8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ernst B, Thurnheer M, Wilms B, Schultes B. Differential changes in dietary habits after gastric bypass versus gastric banding operations. Obes Surg. 2009;19:274–280. doi: 10.1007/s11695-008-9769-3. [DOI] [PubMed] [Google Scholar]
- 5.Thomas JR, Marcus E. High and low fat food selection with reported frequency intolerance following Roux-en-Y gastric bypass. Obes Surg. 2008;18:282–287. doi: 10.1007/s11695-007-9336-3. [DOI] [PubMed] [Google Scholar]
- 6.Stice E, Spoor S, Ng J, Zald DH. Relation of obesity to consummatory and anticipatory food reward. Physiol Behav. 2009;97:551–560. doi: 10.1016/j.physbeh.2009.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Geiger BM, Haburcak M, Avena NM, Moyer MC, Hoebel BG, Pothos EN. Deficits of mesolimbic dopamine neurotransmission in rat dietary obesity. Neuroscience. 2009;159:1193–1199. doi: 10.1016/j.neuroscience.2009.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Davis C, Carter JC. Compulsive overeating as an addiction disorder. A review of theory and evidence. Appetite. 2009;53:1–8. doi: 10.1016/j.appet.2009.05.018. [DOI] [PubMed] [Google Scholar]
- 9.Avena NM, Rada P, Hoebel BG. Evidence for sugar addiction: behavioral and neurochemical effects of intermittent, excessive sugar intake. Neurosci Biobehav Rev. 2008;32:20–39. doi: 10.1016/j.neubiorev.2007.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Volkow ND, Wang GJ, Fowler JS, Telang F. Overlapping neuronal circuits in addiction and obesity: evidence of systems pathology. Philos Trans R Soc Lond B Biol Sci. 2008;363:3191–3200. doi: 10.1098/rstb.2008.0107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kelley AE, Berridge KC. The neuroscience of natural rewards: relevance to addictive drugs. J Neurosci. 2002;22:3306–3311. doi: 10.1523/JNEUROSCI.22-09-03306.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rankinen T, Zuberi A, Chagnon YC, Weisnagel SJ, Argyropoulos G, Walts B, et al. The human obesity gene map: the 2005 update. Obesity (Silver Spring) 2006;14:529–644. doi: 10.1038/oby.2006.71. [DOI] [PubMed] [Google Scholar]
- 13.Davis CA, Levitan RD, Reid C, Carter JC, Kaplan AS, Patte KA, et al. Dopamine for `wanting' and opioids for `liking': a comparison of obese adults with and without binge eating. Obesity (Silver Spring) 2009;17:1220–1225. doi: 10.1038/oby.2009.52. [DOI] [PubMed] [Google Scholar]
- 14.Davis C, Levitan RD, Kaplan AS, Carter J, Reid C, Curtis C, et al. Reward sensitivity and the D2 dopamine receptor gene: a case-control study of binge eating disorder. Prog Neuropsychopharmacol Biol Psychiatry. 2008;32:620–628. doi: 10.1016/j.pnpbp.2007.09.024. [DOI] [PubMed] [Google Scholar]
- 15.Stice E, Spoor S, Bohon C, Small DM. Relation between obesity and blunted striatal response to food is moderated by TaqIA A1 allele. Science. 2008;322:449–452. doi: 10.1126/science.1161550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Felsted JA, Ren X, Chouinard-Decorte F, Small DM. Genetically determined differences in brain response to a primary food reward. J Neurosci. 2010;30:2428–2432. doi: 10.1523/JNEUROSCI.5483-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Davis JF, Tracy AL, Schurdak JD, Tschop MH, Lipton JW, Clegg DJ, et al. Exposure to elevated levels of dietary fat attenuates psychostimulant reward and mesolimbic dopamine turnover in the rat. Behav Neurosci. 2008;122:1257–1263. doi: 10.1037/a0013111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fulton S, Pissios P, Manchon RP, Stiles L, Frank L, Pothos EN, et al. Leptin regulation of the mesoaccumbens dopamine pathway. Neuron. 2006;51:811–822. doi: 10.1016/j.neuron.2006.09.006. [DOI] [PubMed] [Google Scholar]
- 19.Farooqi IS, Bullmore E, Keogh J, Gillard J, O'Rahilly S, Fletcher PC. Leptin regulates striatal regions and human eating behavior. Science. 2007;317:1355. doi: 10.1126/science.1144599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Blum K, Braverman ER, Holder JM, Lubar JF, Monastra VJ, Miller D, et al. Reward deficiency syndrome: a biogenetic model for the diagnosis and treatment of impulsive, addictive, and compulsive behaviors. J Psychoactive Drugs. 2000;32(Suppl):i–iv. 1–112. doi: 10.1080/02791072.2000.10736099. [DOI] [PubMed] [Google Scholar]
- 21.Ahima RS, Qi Y, Singhal NS, Jackson MB, Scherer PE. Brain adipocytokine action and metabolic regulation. Diabetes. 2006;55(Suppl 2):S145–S154. doi: 10.2337/db06-s018. [DOI] [PubMed] [Google Scholar]
- 22.Posey KA, Clegg DJ, Printz RL, Byun J, Morton GJ, Vivekanandan-Giri A, et al. Hypothalamic proinflammatory lipid accumulation, inflammation, and insulin resistance in rats fed a high-fat diet. Am J Physiol Endocrinol Metab. 2009;296:E1003–E1012. doi: 10.1152/ajpendo.90377.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Farr SA, Yamada KA, Butterfield DA, Abdul HM, Xu L, Miller NE, et al. Obesity and hypertriglyceridemia produce cognitive impairment. Endocrinology. 2008;149:2628–2636. doi: 10.1210/en.2007-1722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kleinridders A, Schenten D, Konner AC, Belgardt BF, Mauer J, Okamura T, et al. MyD88 signaling in the CNS is required for development of fatty acid-induced leptin resistance and diet-induced obesity. Cell Metab. 2009;10:249–259. doi: 10.1016/j.cmet.2009.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.De Souza CT, Araujo EP, Bordin S, Ashimine R, Zollner RL, Boschero AC, et al. Consumption of a fat-rich diet activates a proinflammatory response and induces insulin resistance in the hypothalamus. Endocrinology. 2005;146:4192–4199. doi: 10.1210/en.2004-1520. [DOI] [PubMed] [Google Scholar]
- 26.de la Monte SM. Insulin resistance and Alzheimer's disease. BMB Rep. 2009;42:475–481. doi: 10.5483/bmbrep.2009.42.8.475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dugan LL, Ali SS, Shekhtman G, Roberts AJ, Lucero J, Quick KL, et al. IL-6 mediated degeneration of forebrain GABAergic interneurons and cognitive impairment in aged mice through activation of neuronal NADPH oxidase. PLoS One. 2009;4:e5518. doi: 10.1371/journal.pone.0005518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.de la Monte SM, Wands JR. Alzheimer's disease is type 3 diabetes-evidence reviewed. J Diabetes Sci Technol. 2008;2:1101–1113. doi: 10.1177/193229680800200619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Skaper SD. The brain as a target for inflammatory processes and neuroprotective strategies. Ann N Y Acad Sci. 2007;1122:23–34. doi: 10.1196/annals.1403.002. [DOI] [PubMed] [Google Scholar]
- 30.Sriram K, Benkovic SA, Miller DB, O'Callaghan JP. Obesity exacerbates chemically induced neurodegeneration. Neuroscience. 2002;115:1335–1346. doi: 10.1016/s0306-4522(02)00306-8. [DOI] [PubMed] [Google Scholar]
- 31.Hu G, Jousilahti P, Nissinen A, Antikainen R, Kivipelto M, Tuomilehto J. Body mass index and the risk of Parkinson disease. Neurology. 2006;67:1955–1959. doi: 10.1212/01.wnl.0000247052.18422.e5. [DOI] [PubMed] [Google Scholar]
- 32.Burge JC, Schaumburg JZ, Choban PS, DiSilvestro RA, Flancbaum L. Changes in patients' taste acuity after Roux-en-Y gastric bypass for clinically severe obesity. J Am Diet Assoc. 1995;95:666–670. doi: 10.1016/S0002-8223(95)00182-4. [DOI] [PubMed] [Google Scholar]
- 33.Scruggs DM, Buffington C, Cowan GS., Jr Taste acuity of the morbidly obese before and after gastric bypass surgery. Obes Surg. 1994;4:24–28. doi: 10.1381/096089294765558854. [DOI] [PubMed] [Google Scholar]
- 34.Naslund E, Melin I, Gryback P, Hagg A, Hellstrom PM, Jacobsson H, et al. Reduced food intake after jejunoileal bypass: a possible association with prolonged gastric emptying and altered gut hormone patterns. Am J Clin Nutr. 1997;66:26–32. doi: 10.1093/ajcn/66.1.26. [DOI] [PubMed] [Google Scholar]
- 35.Zheng H, Shin AC, Lenard NR, Townsend RL, Patterson LM, Sigalet DL, et al. Meal patterns, satiety, and food choice in a rat model of Roux-en-Y gastric bypass surgery. Am J Physiol Regul Integr Comp Physiol. 2009;297:R1273–R1282. doi: 10.1152/ajpregu.00343.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shin AC, Zheng H, Townsend RL, Sigalet DL, Berthoud HR. Meal-induced hormone responses in a rat model of Roux-en-Y gastric bypass surgery. Endocrinology. 2010;151:1588–1597. doi: 10.1210/en.2009-1332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hajnal A, Kovacs P, Ahmed TA, Meirelles K, Lynch CJ, Cooney RN. Gastric bypass surgery alters behavioral and neural taste functions for sweet taste in obese rats. Am J Physiol Gastrointest Liver Physiol. 2010;299:G967–G979. doi: 10.1152/ajpgi.00070.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Xu Y, Ohinata K, Meguid MM, Marx W, Tada T, Chen C, et al. Gastric bypass model in the obese rat to study metabolic mechanisms of weight loss. J Surg Res. 2002;107:56–63. doi: 10.1006/jsre.2002.6508. [DOI] [PubMed] [Google Scholar]
- 39.Rosenbaum M, Sy M, Pavlovich K, Leibel RL, Hirsch J. Leptin reverses weight loss-induced changes in regional neural activity responses to visual food stimuli. J Clin Invest. 2008;118:2583–2591. doi: 10.1172/JCI35055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hommel JD, Trinko R, Sears RM, Georgescu D, Liu ZW, Gao XB, et al. Leptin receptor signaling in midbrain dopamine neurons regulates feeding. Neuron. 2006;51:801–810. doi: 10.1016/j.neuron.2006.08.023. [DOI] [PubMed] [Google Scholar]
- 41.Figlewicz DP. Adiposity signals and food reward: expanding the CNS roles of insulin and leptin. Am J Physiol Regul Integr Comp Physiol. 2003;284:R882–R892. doi: 10.1152/ajpregu.00602.2002. [DOI] [PubMed] [Google Scholar]
- 42.Shigemura N, Ohta R, Kusakabe Y, Miura H, Hino A, Koyano K, et al. Leptin modulates behavioral responses to sweet substances by influencing peripheral taste structures. Endocrinology. 2004;145:839–847. doi: 10.1210/en.2003-0602. [DOI] [PubMed] [Google Scholar]
- 43.Julliard AK, Chaput MA, Apelbaum A, Aime P, Mahfouz M, Duchamp-Viret P. Changes in rat olfactory detection performance induced by orexin and leptin mimicking fasting and satiation. Behav Brain Res. 2007;183:123–129. doi: 10.1016/j.bbr.2007.05.033. [DOI] [PubMed] [Google Scholar]
- 44.Getchell TV, Kwong K, Saunders CP, Stromberg AJ, Getchell ML. Leptin regulates olfactory-mediated behavior in ob/ob mice. Physiol Behav. 2006;87:848–856. doi: 10.1016/j.physbeh.2005.11.016. [DOI] [PubMed] [Google Scholar]
- 45.Diano S, Farr SA, Benoit SC, McNay EC, da Silva I, Horvath B, et al. Ghrelin controls hippocampal spine synapse density and memory performance. Nat Neurosci. 2006;9:381–388. doi: 10.1038/nn1656. [DOI] [PubMed] [Google Scholar]
- 46.Abizaid A, Liu ZW, Andrews ZB, Shanabrough M, Borok E, Elsworth JD, et al. Ghrelin modulates the activity and synaptic input organization of midbrain dopamine neurons while promoting appetite. J Clin Invest. 2006;116:3229–3239. doi: 10.1172/JCI29867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jerlhag E, Egecioglu E, Dickson SL, Douhan A, Svensson L, Engel JA. Ghrelin administration into tegmental areas stimulates locomotor activity and increases extracellular concentration of dopamine in the nucleus accumbens. Addict Biol. 2007;12:6–16. doi: 10.1111/j.1369-1600.2006.00041.x. [DOI] [PubMed] [Google Scholar]