

Abstract
This study reports an amelioration of abnormal motor behaviors in tetrahydrobiopterin (BH4)-deficient Spr −/− mice by the dietary supplementation of tyrosine. Since BH4 is an essential cofactor for the conversion of phenylalanine into tyrosine as well as the synthesis of dopamine neurotransmitter within the central nervous system, the levels of tyrosine and dopamine were severely reduced in brains of BH4-deficient Spr −/− mice. We found that Spr −/− mice display variable ‘open-field’ behaviors, impaired motor functions on the ‘rotating rod’, and dystonic ‘hind-limb clasping’. In this study, we report that these aberrant motor deficits displayed by Spr −/− mice were ameliorated by the therapeutic tyrosine diet for 10 days. This study also suggests that dopamine deficiency in brains of Spr −/− mice may not be the biological feature of aberrant motor behaviors associated with BH4 deficiency. Brain levels of dopamine (DA) and its metabolites in Spr −/− mice were not substantially increased by the dietary tyrosine therapy. However, we found that mTORC1 activity severely suppressed in brains of Spr −/− mice fed a normal diet was restored 10 days after feeding the mice the tyrosine diet. The present study proposes that brain mTORC1 signaling pathway is one of the potential targets in understanding abnormal motor behaviors associated with BH4-deficiency.
Introduction
Tetrahydrobiopterin (BH4) has been established as an obligatory cofactor for aromatic amino acid hydroxylases including phenylalanine hydroxylases (PAH), tyrosine hydroxylase (TH), tryptophan hydroxylase (TPH), and all isoforms of nitric oxide synthase (NOS) [1]. Since TH and TPH metabolize tyrosine and tryptophan to produce dopamine and serotonin neurotransmitters within the central nervous system, BH4 deficiency has often been implicated to be associated with many neurological disorders such as Parkinson’s disease, autism, depression, Alzheimer’s disease, and phenylketonurea [1]–[3].
Studies have suggested that there is interplay between the dysfunction of dopaminergic neurons and behavioral abnormalities in BH4-deficient mice. Yang et al. [4] reported severely impaired locomotive activities along with dramatically reduced levels of dopamine in brains of BH4-deficient Spr −/− mice. More recently, a correlation among the suppression of dopaminergic development, subsequent deficiency in brain dopamine levels, and motor dysfunction was evidenced in BH4-deficient Spr −/− mice [5]. Many behavioral disorders are associated with abnormal neurotransmitter activities. A typical example of these disorders is Parkinson’s disease, a progressive neurodegenerative disease, which is characterized by a deficit in the neurotransmitter dopamine (DA) [6]. The pathological hallmarks of Parkinson’s disease include the loss of dopaminergic neurons in the substantia nigra pars compacta (SNpc) and corresponding decrease in the amount of dopamine production in the striatum (STR), which plays a pivotal role in normal motor function [7]. Major symptoms of Parkinson’s disease thus include tremors in hands, arms, feet, and face, slowness of movement (bradykinesia or akinesia), and difficulties in balance and coordination (rigidity) [8], [9].
Previously, we have demonstrated that BH4 deficiency results in the inactivation of mTORC1 pathway in brains of BH4-deficient Spr −/− mice due to paucity of a specific amino acid, tyrosine [10]. An evolutionarily conserved protein kinase complex, mTORC1, regulates cell proliferation, cell size, and cell cycle [11], [12]. Activation of mTORC1 pathway stimulates protein synthesis as well as ribosomal biosynthesis, but inactivates catabolic processes such as autophagy [13]–[15]. Recent advances in the study of mTORC1 signaling delineate a potential link between neurological disorders and impaired mTORC1 signaling in the brain. For example, mTORC1 signaling is known to be modified in some pathological states of the nervous system including tuberous sclerosis, cortical dysplasia and neurodegenerative disorders [16]–[18].
Herein, we first report the effects of dietary supplementation of a specific amino acid on the improvement of abnormal motor functions in BH4-deficient mice generated by the knockout of the gene encoding sepiapterin reductase (SR), which catalyzes BH4 synthesis.
Materials and Methods
Experimental Mice and the Dietary Tyrosine Supplementation
Spr −/− mice on a mixed C57BL6/sv129 hybrid background were generated as described elsewhere [4]. Both Spr +/+ and Spr −/− mice from the same mother were weaned at 20 days after birth and fed a normal diet ad libitum for 5 days to acclimate mice to normal mouse food. For the dietary tyrosine therapy, Spr −/− mice were pretreated with ‘high dose of BH4’ (122 µg/g body weight/day), L-DOPA (13.5 µg/g body weight/day), 5-hydroxytryptophan (9.5 µg/g body weight/day), carbidopa (2.5 µg/g body weight/day), ascorbic acid (100 µg/g body weight/day), and N-acetyl-L-cysteine (50 µg/g body weight/day) by oral administration as previously adopted by Elzaouk et al. [19]. After the acclimatization period, Spr +/+ or Spr −/− mice 25 days of age were fed either a normal or the therapeutic tyrosine diet in which 5.6% tyrosine (w/w) was added to the normal diet for 10 days. This dietary trial was based on the study by Matalon et al. [20] who has found that a dietary formula enriched in tyrosine is effective in reducing blood phenylalanine concentration in mice. The effects of the dietary tyrosine therapy on motor behaviors, brain tyrosine levels, and dopamine in 35-d-old experimental mice were then examined. All animal experiments were approved by Institutional Animal Care and Use Committee (IACUC) at Korea Advanced Institute of Science and Technology (KA2010-01). For leucine supplementation, 25-d-old mice pretreated with ‘high-dose of BH4’ solution were fed the leucine-enriched diet in which 3.6% leucine (w/w) was added to a normal diet for 10 days under ad libitum conditions. To explore for the effects of dopamine on motor behaviors, TH protein expression and brain mTORC1 activity, 25-d-old mice were treated with dopamine precursor, L-DOPA (13.5 µg/g body weight/day) was coadministered with carbidopa (2.5 µg/g body weight/day) for 10 days to inhibit peripheral aromatic-L-amino acid decarboxylase (AADC), an important enzyme that metabolizes L-DOPA into dopamine [21].
Determination of Phenylalanine and Tyrosine Levels in Brains of Experimental Mice
Concentrations of free phenylalanine and tyrosine in the cerebra separated from brains of Spr +/+ or Spr −/− mice fed either a normal or the tyrosine diet were measured. Levels of each amino acids were determined by reversed phase high-performance liquid chromatograpy (RP-HPLC) using the Pico-tag method. Phenylisothiocyanate-derived free amino acids were analyzed on a Pico-tag amino acid analysis column with monitoring of the elutes at 254 nm.
Levels of Brain Dopamine and its Metabolites
Samples of caudate putamen separated from brains of 35-d-old Spr +/+ or Spr −/− mice fed either a normal or the tyrosine diet, were homogenized in 0.1 M perchloric acid and the acid soluble fraction was collected. Quantification of striatal dopamine (DA), 3,4-dihydroxyphenylacetic acid (DOPAC), and homovanillic acid (HVA) were measured by HPLC using C18 column (Novapack Coorporation) and detected electrochemically [22].
Behavioral Analyses
Open-field test
Mice were gently placed at the center of the open-field test arena (40×40×40 cm) in a dark room and allowed to explore for a period of 30 min or 1 h. Motor activities were monitored at 5 min intervals in a 1 h time period by using digital video recording. Etho Vision (Noldus) was employed to analyze the video data [23].
Rotating rod test
Coordination of motor behaviors were monitored by the rotating rod performance [24]. Mice were placed onto a rotating rod (4 cm in diameter: manufactured by Ugo Basile Biological Research Apparatus, model 47600) that started at 4 rpm and incrementally accelerated at each 20 sec to a final 40 rpm and the latency to fall was recorded. Each experimental mouse performed nine trials over a span of three days (3 trials/day with an interval of 1 h).
Hind-limb clasping test
Experimental mice were picked up and suspended by the tail for 25 sec while being videotaped. The duration of hind-limb clasping near the body was measured for each mouse [5].
Western Blotting
Cerebral or midbrain proteins obtained from 35-d-old experimental mice were separated by 10% SDS-PAGE, blotted onto a nitrocellulose membrane, blocked with 5% BSA, incubated with anti-phospho S6K antibody (Thr389, #9234, 1∶500, Cell Signaling Technology), anti-S6K antibody (SC-8414, 1∶1,000, Santa Cruz Biotechnology), anti-phospho S6 antibody (Ser235/236, #2211, 1∶1,000, Cell Signaling Technology), anti-S6 antibody (#2317, 1∶500, Cell Signaling Technology), anti-TH antibody (1∶3,000, Millipore) or anti-actin antibody (SC1616, 1∶1,000, Santa Cruz Biotechnology), followed by an additional incubation with horseradish peroxide-conjugated anti-rabbit-IgG or anti-mouse-IgG antibodies (Jackson Laboratory). Immunoreactivity was visualized using horseradish peroxide substrate (Dae Myung Science).
Results
Recovery of Tyrosine Concentrations in Brains of Spr −/− Mice by the Tyrosine Diet
Since BH4 function is required for phenylalanine hydroxylase, which converts phenylalanine into tyrosine, it was anticipated that the availabilities of phenylalanine and tyrosine in brains of Spr −/− mice would be deranged compared to the wild type control Spr +/+ mice. Phenylalanine levels in brains of Spr −/− mice fed a normal diet were remarkably higher than levels in brains of control Spr +/+ mice, while tyrosine levels in brains of Spr −/− mice were much lower than those in the wild type Spr +/+ counterparts. Brain tyrosine levels were increased in both Spr +/+ and Spr −/− mice after the therapy relative to mice that did not receive tyrosine therapy. Contrarily, concentrations of brain phenylalanine were reduced in brains of Spr −/− mice given the dietary tyrosine therapy compared to Spr −/− mice fed a normal diet (Figure 1).
Figure 1. Restoration of brain tyrosine levels in BH4-deficient Spr −/− mice after the dietary tyrosine supplementation.
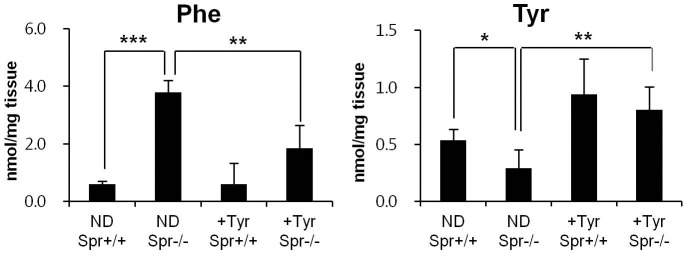
Concentrations of phenylalanine and tyrosine in the cerebra separated from brains of Spr +/+ or Spr −/− mice either fed a normal diet (ND) or the tyrosine diet (+Tyr) for 10 days are shown (n ≥5 for each experimental group). Data represent mean ± SD. Statistical significance was determined by Student’s t test, * P<0.05, ** P<0.01, *** P<0.001.
Amelioration of Abnormal Motor Behaviors by the Tyrosine Therapy in BH4-deficient Spr −/− Mice
We examined whether the motor impediments displayed by BH4-deficient mice whose brain tyrosine levels are insufficient can be improved by the dietary tyrosine supplementation. Spr −/− mice fed a normal diet displayed abnormal motor activities in the open-field during a 30 min or 1 h time period. Some Spr −/− mice fed a normal diet tended to avoid the center of the open-field, which is represented by their stereotypic circling behaviors (Figure 2A, b and d). The distance of spontaneous locomotive movements by experimental mice across the 5 min observation period in the open-field environment during the 1 h time period is shown in Figure 2B. The open-field test revealed that the dietary tyrosine supplementation did not affect the motor behaviors displayed by wild-type Spr +/+ mice. However, locomotive movements by Spr −/− mice fed a normal diet varied among the mice. Some Spr −/− mice fed a normal diet displayed only basal levels of motor activities when compared to control Spr +/+ mice fed a normal diet. Other Spr −/− mice fed a normal diet developed hyperactive movements in the open-field test (Figure 2B). Surprisingly, these controversial motor behaviors displayed by Spr −/− mice fed a normal diet were markedly normalized by the dietary supplementation of tyrosine for 10 days. Variance (S2) of total distance of locomotive movements tracked over a time period of 30 min or 1 h by Spr −/− mice fed a normal diet (S2 = 2535.4, S2 = 23625.6) became much smaller after the therapeutic tyrosine diet for 10 days (S2 = 586.9, S2 = 2831.5) (Table S1). In like manner, Spr −/− mice fed the therapeutic tyrosine diet performed comparably well on the rotating rod task after the dietary tyrosine therapy, compared with the performance by Spr −/− mice fed a normal diet (Figure 3). The rescue of motor dysfunction by the dietary tyrosine supplementation in Spr −/− mice was also shown by the hind-limb clasping test. Dystonic postures of hind-limbs with self-clasping were observed in most Spr −/− mice, whereas almost none of control Spr +/+ mice exhibited hind-limb clasping phenotype. We observed that the hind-limb clasping phenotype in Spr −/− was substantially ameliorated after the dietary supplementation of tyrosine for 10 days (Figure 4).
Figure 2. Abnormal open-field motor behaviors in Spr −/− mice were ameliorated by the dietary supplementation of tyrosine.

Each experimental mouse was placed at the center of a square box (40×40×50 cm) made of white acryl in the dark. Each experimental group of mice was prepared as described in Materials and Methods. Newborn Spr −/− mice pretreated with ‘high dose of BH4’ for 25 days [19]. Spr +/+ or Spr −/− mice 25 days of age were fed either a normal diet or the tyrosine diet for additional 10 days. A group of 35-d-old experimental mice fed either a normal diet (ND) or the tyrosine diet (+Tyr) was employed for the open-field test. (A) The representative locomotive patterns of wild-type control Spr +/+ or mutant Spr −/− mice fed either a normal diet or the tyrosine diet are shown. The open-field test revealed a development of stereotypic circling behaviors in Spr −/− group of mice fed a normal diet. (B) Spontaneous movements during a time period of 30 min or 1 h were monitored by digital video recording. The distances traveled by experimental mice across the 5 min of observation period are shown.
Figure 3. Improved rotating rod performance by dietary tyrosine therapy in Spr −/− mice.
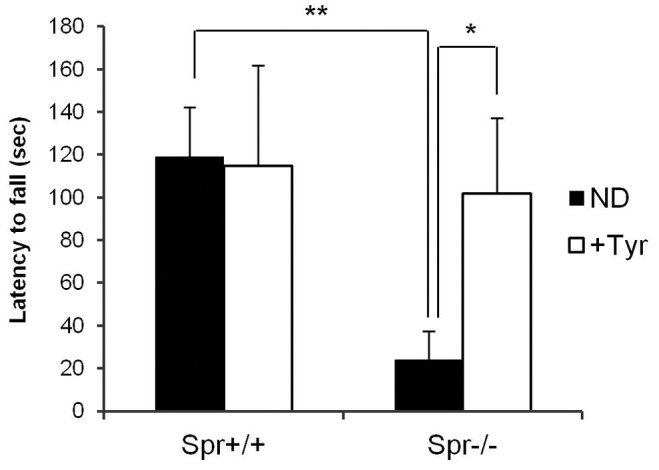
Mice were placed onto a rotating rod and the latency to fall was recorded. Each mouse performed 9 trials over a span of 3 days. Rotating rod performance by Spr +/+ mice (n = 5) or Spr −/− mice (n = 5) fed a normal diet (ND) was compared with that by wild-type Spr +/+ mice (n = 5) or Spr −/− mice (n = 6) that received the dietary tyrosine therapy (+Tyr). Data represent average time of latency to fall ± SD, * P<0.05, ** P<0.01.
Figure 4. Hind-limb clasping test.
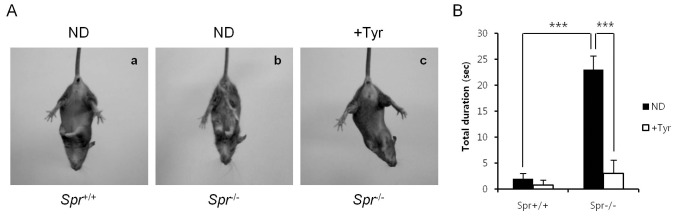
Each experimental group of mice was suspended by the tail for 25 sec, and their abilities of hind-limb clasping were monitored by video recording. (A) Representative hind-limb clasping observed in Spr −/− mice fed a normal diet (ND) (b) but not in Spr +/+ mice fed a normal diet (ND) (a) and Spr −/− mice fed the tyrosine diet (+Tyr) (c) is shown. (B) Total duration of hind-limb clasping during a 25 sec in Spr +/+ mice fed a normal diet, Spr +/+ mice fed the tyrosine diet and Spr −/− mice fed either a normal or the tyrosine diet (n = 5 each) is shown. Values represent average time of duration ± SD, *** P<0.001.
Brain Dopamine Levels were not Changed by the Dietary Tyrosine Therapy in Spr −/− Mice
Dopamine (DA) is a classical neurotransmitter of the central nervous system and is essential for the body movement. We examined the hypothesis that the improved behavioral phenotypes in Spr −/− mice after the tyrosine diet are associated with the increase of brain dopamine levels. We measured levels of DA and its metabolites in the caudate putamen separated from brains of Spr +/+ or Spr −/− mice fed either a normal or the therapeutic tyrosine diet. In accordance with the previous reports [4], [5], the levels of DA and its metabolites, 3,4-dihydroxyphenylacetic acid (DOPAC) and homovanillic acid (HVA) in the caudate putamen from brains of Spr −/− mice fed a normal diet were severely lower than the levels measured from the brains of control Spr +/+ mice. In contrast to what we have expected, the levels of DA and its metabolites in brains of Spr −/− mice fed the therapeutic tyrosine diet were only slightly increased or above the detection limit. Despite the fact that the brain tyrosine levels in Spr −/− mice were restored after the dietary tyrosine therapy (Figure 1), the levels of brain DA and its metabolites were not substantially recovered in Spr −/− mice fed the tyrosine diet (Figure 5). In addition, the protein expression of tyrosine hydroxylase (TH), a rate-limiting enzyme for the production of DA [25], was investigated in the caudate putamen from experimental mice (Figure 5D). The amounts of TH protein were severely reduced in midbrains of Spr −/− mice fed either a normal or the tyrosine diet. The brain levels of TH in Spr −/− mice failed to recover after the tyrosine therapy. Neither the leucine diet nor the administration of dopamine precursor L-DOPA restored the diminished levels of TH protein in brains of Spr −/− mice.
Figure 5. Levels of brain DA and its metabolites are irrelevant to the dietary tyrosine supplementation in Spr −/− mice.

(A), (B), and (C) Spr +/+ or Spr −/− mice were fed a normal (ND) or the tyrosine diet (+Tyr). Mice were analyzed for brain levels of DA (A), DOPAC (B), and HVA (C) in their caudate putamen (n ≥3). Values are means ± SD, ** P<0.01, *** P<0.001, compared with respective values in Spr +/+ mice. (D) The brain levels of TH proteins in experimental mice were monitored by Western blot analysis. Spr +/+ or Spr −/− mice were fed a normal diet or treated with the dietary tyrosine, L-DOPA, or leucine therapy for 10 days as described in Materials and Methods.
Enhanced Brain mTORC1 Activities in Spr −/− Mice by the Dietary Tyrosine Therapy
We explored whether the improved behavioral phenotypes in Spr −/− mice fed the therapeutic tyrosine diet correlate with brain mTORC1 activity. We compared mTORC1 activities in the cerebra from brains of Spr +/+ or Spr −/− mice either fed a normal diet or the tyrosine diet by monitoring the phosphorylation of S6 and S6K. Phosphorylation of S6 at Ser235/236 and S6K at Thr389, which represent the activation of mTORC1 activity, was almost completely suppressed in brains of Spr −/− mice fed a normal diet. Interestingly, mTORC1 activities became activated in brains of Spr −/− mice after the tyrosine therapy for 10 days. Data also reveal that brain mTORC1 activities were not affected by the oral administration of L-DOPA in Spr −/− mice (Figure 6).
Figure 6. The dietary tyrosine supplementation enhances mTORC1 activity in brains of Spr −/− mice.

(A) Immunoblotting was performed on the cerebral homogenates separated from brains of 35-d-old Spr +/+ or Spr −/− mice. Each group of experimental mice was fed a normal diet (ND) or treated with the dietary tyrosine, or L-DOPA therapy for 10 days. Phosphorylation of S6 and S6K in the brain homogenates was examined to evaluate mTORC1 activity. The representative Western blot demonstrating mTORC1 recovery in Spr −/− mice by dietary tyrosine supplementation is shown. (B) The ratios of band intensities of pS6K/S6K and pS6/S6 were quantified by using image J software. Values are means ± SD (n ≥4 for each group of experimental mice). * P<0.05, ** P<0.01.
Discussion
The present study indicates that the dietary supplementation of tyrosine ameliorates the abnormal motor behaviors exhibited by BH4-deficient mouse model. The cerebral concentrations of phenylalanine in Spr −/− mice were significantly higher than those in the wild-type Spr +/+ control mice. Phenylalanine shares a common transporter to the brain with other LNAAs (large neutral amino acids) including tyrosine and also competes for the transport with other LNAAs across the blood brain barrier [26]. Amongst LNAAs, phenylalanine has the lowest Michaelis constant (Km) and is preferentially transported by the LNAA carrier protein [27]. Dietary supplementation of high-dose of tyrosine into Spr −/− mice not only elevated the cerebral levels of tyrosine but also lowered the cerebral phenylalanine levels (Figure 1).
Through a series of behavioral tests, we found that BH4-deficient Spr −/− mice display motor deficits such as variable and instable open-field behaviors (Figure 2), impaired motor coordination on rotating rod (Figure 3) and dystonic hind-limb clasping (Figure 4). The spontaneous locomotor activities in Spr −/− mice fed a normal diet in the open-field environment have provided conflicting results. These mice exhibited both hypo- and hyper-activity in the open-field test (Figure 2). Variance of total distance traveled in the open-field by Spr −/− mice was much greater than that by Spr +/+ mice (Table S1). These complicated results seem to represent low baseline motor activity early during the initiation period of observation in Spr −/− mice as well as the impairment in the habituation to the test environment following the movement execution (Figure 2). The dietary effects on the amelioration of these motor deficits in BH4-deficient Spr −/− mice seem to be tyrosine-specific. We also examined the effects of the dietary leucine supplementation on the motor behaviors displayed by Spr −/− mice. The variable motor movements in the open-field and impaired motor coordination on the rotating rod displayed by Spr −/− mice were not improved by the dietary supplementation of leucine (Figure S1A–C).
Many behavioral disorders often are associated with abnormal neurotransmitter activity. For example, Parkinson’s disease is characterized by the degeneration of dopamine (DA) neurons in the brain [6]–[9]. Regulation of motor behaviors can be linked to a disruption of the nigrostriatal DA system and accounts for many motor impairments caused by the disruption of dopaminergic transmission [28], [29]. Bradykinesia or akinesia, the main neuropathological feature of Parkinson’s disease [30], and hyperactivity or hyperkinesia, a core feature of attention deficit hyperactivity disorder (ADHD) [31], are extremely oppositional phenotypes of abnormal motor behaviors. Interestingly, the neuropathological features of these two neurological disorders commonly include decreased DA activity in the brain [7], [30], [32]. We examined whether the improved motor activities by the dietary tyrosine supplementation in Spr −/− mice is linked with the increased brain DA levels in the experimental mice. The DA levels in the caudate putamen from brains of Spr −/− mice fed a normal diet were much lower than the levels in control Spr +/+ mice. However, our data in Figure 5 do not support the hypothesis that the amelioration of abnormal motor behaviors in Spr −/− mice after the dietary tyrosine therapy stems from the recovery of brain DA concentrations since there was only modest increase in the DA levels in the caudate putamen from brains of Spr −/− mice after the dietary tyrosine supplementation. Our notion that amelioration of abnormal motor behavior is irrelevant to the brain DA levels in Spr −/− mice fed the tyrosine diet is strongly supported by independent experiments showing the dispensable roles of dietary supplementation of L-DOPA in open-field behaviors and rotating rod performances by Spr −/− mice (Figure S2). Since the brain TH protein and DA contents are known to be affected by BH4 deficiency, we performed Western blot analysis to examine whether the TH protein levels in the midbrains of Spr −/− mice were restored after the tyrosine therapy. Our data in Figure 5D indicate that brain TH protein levels in BH4-deficient Spr −/− mice are not affected by the dietary tyrosine treatment. The result further implies that the reduced levels of tyrosine conversion into L-DOPA and subsequent DA in Spr −/− mice were not influenced by tyrosine treatment (Figure 5A).
How the increased brain tyrosine levels in Spr −/− mice can biochemically correlate the improvement of abnormal motor behaviors in BH4-deficient mouse model is the least known aspect of this study. We have noticed that brain mTORC1 activities were pronouncedly increased in BH4-deficient Spr −/− mice by the dietary tyrosine treatment (Figure 6). Brain mTORC1 activity was restored neither by the leucine treatment (Figure S1D) nor by L-DOPA treatment (Figure 6) in these mutant mice suggesting tyrosine-dependent activation of brain mTORC1 activity in Spr −/− mice. The involvement of mTORC1 function in motor behaviors has been evidenced in some other mouse models. Both the deteriorative and beneficial effects of mTORC1 function on motor behaviors have been reported. Research by Santini et al. [33] provided evidence for the negative effects of mTORC1 signaling on motor function. They showed that the administration of rapamycin, an mTORC1 inhibitor, prevents the development of dyskinesia in a mouse model of Parkinsonism. The involvement of activated mTORC1 signaling in the behavioral abnormalities was further supported by the work showing the amelioration of behavioral abnormalities by rapamycin in Pten-knockout mice where the signals for cell growth, proliferation, and survival through phosphatidylinositol 3-kinase (PI3K) pathway were stimulated [34]. On the other hand, mTORC1 signaling has been implicated to be necessary for standardized motor behavior in mice. For example, S6K1-knockout mice generated by the knockout of the gene encoding mTORC1 substrate protein S6K1 have neurological and behavioral abnormalities [24]. Some human neurological disorders are also known to be associated with the dysregulation of mTORC1 signaling. Autistic disorders characterized by cognitive impairment and autism have been proposed to be the results of direct or indirect dysregulation of mTORC1 signaling [35]. Aberrant mTORC1 signaling observed in the postmorterm samples from brains of patients with Alzheimer’s disease, a neurodegenerative disorder characterized by gradual and severe loss of neurological function including memory and reasoning, implicates dysregulation of mTORC1 signaling as a biochemical feature of Alzheimer’s disease [36], [37]. Other studies have also postulated that human patients with neurological disorders display behavioral impairments and have dysregulated mTORC1 signaling [24], [34], [38], [39].
Amino acid availability appears to be necessary for the activation of mTORC1 and Rag GTPase, an amino acid-specific regulator of mTORC1 pathway [40], [41]. Since mTORC1 signaling is regarded as the most important regulator of protein synthesis and its degradation, dysregulation of mTORC1 signaling may result in significant synaptic plasticity, autophagy and ubiquitin-mediated proteolysis in the brain [42]. Thus, it is a reasonable expectation that the availability of brain amino acids, which signal mTORC1 pathway in the brain may play an important role in the behavioral deficits associated with many neurodegenerative diseases.
Supporting Information
Dietary effect of leucine on motor behaviors displayed by Spr −/− mice. For dietary leucine supplementation, Spr +/+ or Spr −/− mice 25 days of age were fed the leucine supplemented diet in which 3.6% leucine (w/w) was added to normal diet for 10 days under ad libitum conditions. (A, B) Abnormal open-field behaviors in Spr −/− mice were not improved by the dietary supplementation of leucine. The experimental details were the same as in Figure 2. (C) Dietary supplementation of leucine fails to improve rotating rod performance displayed by Spr −/− mice. The experimental details were the same as in Figure 3. ***P<0.001. (D) Dietary leucine supplementation has no effect on brain mTORC1 activity in Spr −/− mice. Brain mTORC1 activities were measured using the phosphorylation of S6 as a readout. Other experimental procedures were the same as in Figure 6.
(TIF)
Dietary effect of L-DOPA on motor behaviors by Spr −/− mice. Both strains of Spr +/+ and Spr −/− mice 25 days of age were orally administrated L-DOPA (13.5 µg/g body weight/day) for 10 days. Open-field behaviors (A) or rotating rod performance (B) displayed by the experimental mice (n = 2 for each experimental group) are shown. Experimental conditions were the same as in Figure 2 and Figure 3. ***P<0.001.
(TIF)
Hypervariable distances moved by Spr −/− mice in the open-field test become normalized after the tyrosine therapy.
(DOCX)
Acknowledgments
We thank S.P. Oh for the Spr −/− mouse strain. Authors are grateful to J.S. Choi (Korea Basic Science Institute) for amino acid analysis.
Funding Statement
This project was supported by grants from National Research Foundation of Korea (2012R1A1A2007843) and KAIST Future Systems Healthcare Project. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Thony B, Auerbach G, Blau N (2000) Tetrahydrobiopterin biosynthesis, regeneration and functions. Biochem J 347: 1–16. [PMC free article] [PubMed] [Google Scholar]
- 2. Foxton RH, Land JM, Heales SJR (2007) Tetrahydrobiopterin availability in Parkinson's and Alzheimer's disease; potential pathogenic mechanisms. Neurochem Res 32: 751–756. [DOI] [PubMed] [Google Scholar]
- 3. Ichinose H, Nomura T, Sumi-Ichinose C (2008) Metabolism of tetrahydrobiopterin: its relevance in monoaminergic neurons and neurological disorders. Chem Rec 8: 378–385. [DOI] [PubMed] [Google Scholar]
- 4. Yang SK, Lee YJ, Kim JM, Park S, Peris J, et al. (2006) A murine model for human sepiapterin-reductase deficiency. Am J Hum Genet 78: 575–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Homma D, Sumi-Ichinose C, Tokuoka H, Ikemoto K, Nomura T, et al. (2010) Partial biopterin deficiency disturbs postnatal development of the dopaminergic system in the brain. J Bio Chem 286: 1445–1452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Dauer W, Przedborski S (2003) Parkinson's disease: mechanisms and models. Neuron 39: 889–909. [DOI] [PubMed] [Google Scholar]
- 7. Savitt JM, Dawson VL, Dawson TM (2006) Diagnosis and treatment of Parkinson's disease: molecules to medicine. J Clin Invest 116: 1744–1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Xia R, Mao ZH (2012) Progression of motor symptoms in Parkinson's disease. Neurosci Bull 28: 39–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Poewe W, Mahlknecht P (2009) The clinical progression of Parkinson's disease. Parkinsonism Relat Disord 15S: S28–32. [DOI] [PubMed] [Google Scholar]
- 10. Kwak SS, Suk JK, Choi JH, Yang SK, Kim JW, et al. (2011) Autophagy induction by tetrahydrobiopterin deficiency. Autophagy 7: 1323–1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wullschleger S, Loewith R, Hall MN (2006) TOR signaling in growth and metabolism. Cell 124: 471–484. [DOI] [PubMed] [Google Scholar]
- 12. Sarbassov DD, Ali SM, Sabatini DM (2005) Growing roles for the mTOR pathway. Curr Opin Cell Biol 17: 596–603. [DOI] [PubMed] [Google Scholar]
- 13. Guertin DA, Sabatini DM (2007) Defining the Role of mTOR in Cancer. Cancer Cell 12: 9–22. [DOI] [PubMed] [Google Scholar]
- 14. Jung CH, Jun CB, Ro SH, Kim YM, Otto NM, et al. (2009) ULK-Atg13-FIP200 complexes mediate mTOR signaling to the autophagy machinery. Mol Biol Cell 20: 1992–2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Levine B, Kroemer G (2008) Autophagy in the pathogenesis of disease. Cell 132: 27–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bekinschtein P, Katche C, slipczuk LN, Iqaz LM, Cammarota M, et al. (2007) mTOR signaling in the hippocampus is necessary for memory formation. Neurobiol Learn Mem 87: 303–307. [DOI] [PubMed] [Google Scholar]
- 17. Dash PK, Orsi SA, Moore AN (2006) Spatial memory formation and memory-enhancing effect of glucose involves activation of the tuberous sclerosis complex-Mammalian target of rapamycin pathway. J Neurosci 26: 8048–8056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Swiech L, Perycz M, Malik A, Jaworski J (2008) Role of mTOR in physiology and pathology of the nervous system. Biochim Biophys Acta 1784: 116–132. [DOI] [PubMed] [Google Scholar]
- 19. Elzaouk L, Leimbacher W, Turri M, Ledermann B, Burki K, et al. (2003) Dwarfism and low insulin-like growth factor-1 due to dopamine depletion in Pts−/− mice by feeding neurotransmitter precursors and H4-biopterin. J Bio Chem 278: 28303–28311. [DOI] [PubMed] [Google Scholar]
- 20. Matalon R, Michals-Matalon K, Bhatia G, Grechanina E, Novikov P, et al. (2006) Large neurtal amino acids in the treatment of phenylketonuria (PKU). J Inherit Metab Dis 29: 732–738. [DOI] [PubMed] [Google Scholar]
- 21. Gilbert JA, Frederick LM, Ames MM (2000) The aromatic-L-amino acid decarboxylase inhibitor carbidopa is selectively cytotoxic to human pulmonary carcinoid and small cell lung carcinoma cells. Clin Cancer Res 6: 4365–4372. [PubMed] [Google Scholar]
- 22. Kim ST, Son HJ, Choi JH, Ji IJ, Hwang O (2009) Vertical grid test and modified horizontal grid test are sensitive methods for evaluating motor dysfunctions in the MPTP mouse model for Parkinson's disease. Brain Res 1306: 176–183. [DOI] [PubMed] [Google Scholar]
- 23. Kim J, Woo J, Park YG, Chae S, Jo S, et al. (2011) Thalamic T-type Ca2+ channels mediate frontal lobe dysfunctions caused by a hypoxia-like damage in the prefrontal cortex. J Neurosci 31: 4063–4073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Antion MD, Merhav M, Hoeffer CA, Reis G, Kozma SC, et al. (2008) Removal of S6K1 and S6K2 leads to divergent alterations in learning, memory, and synaptic plasticity. Learn Mem 15: 29–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Fitzpatrick PF (1999) Tetrahydropterin-dependent amino acid hydroxylases. Annu Rev Biochem 68: 355–381. [DOI] [PubMed] [Google Scholar]
- 26. Oldendorf WH, Szabo J (1976) Amino acid assignment to one of three blood-brain barrier amino acid carriers. Am J Physiol 230: 94–98. [DOI] [PubMed] [Google Scholar]
- 27. Matalon R, Surendran S, Matalon KM, Tyring S, Quast M, et al. (2003) Future role of large neutral amino acids in transport of phenylalanine into the brain. Pediatrics 112: 1570–1574. [PubMed] [Google Scholar]
- 28. Cousins MS, Salamone JD (1996) Involvement of ventrolateral striatal dopamine in movement initiation and execution: a microdialysis and behavioral investigation. Neuroscience 70: 849–859. [DOI] [PubMed] [Google Scholar]
- 29. W H (1996) Impairments of movement initiation and execution induced by a blockade of dopamine D1 or D2 receptors are reversed by a blockade of N-methyl-D-aspartate receptors. Neuroscience 73: 121–130. [DOI] [PubMed] [Google Scholar]
- 30. Shen LH, Liao MH, Tseng YC (2012) Recent advances in imaging of dopaminergic neurons for evaluation of neuropsychiatric disorders. J Biomed Biotechnol 2012: 259349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Krain AL, Castellanos FX (2006) Brain development and ADHD. Clin Psychol Rev 26: 433–444. [DOI] [PubMed] [Google Scholar]
- 32. Avale ME, Falzone TL, Gelman DM, Low MJ, Grandy DK, et al. (2004) The dopamine D4 receptor is essential for hyperactivity and impaired behavioral inhibition in a mouse model of attention deficit/hyperactivity disorder. Mol Psychiatry 9: 718–726. [DOI] [PubMed] [Google Scholar]
- 33. Santini E, Heiman M, Greengard P, Valjent E, Fisone G (2009) Inhibition of mTOR signaling in Parkinson's disease prevents L-DOPA-induced dyskinesia. Sci Signal 2: ra36. [DOI] [PubMed] [Google Scholar]
- 34. Zhou J, Blundell J, Ogawa S, Kwon CH, Zhang W, et al. (2009) Pharmacological inhibition of mTORC1 suppresses anatomical, cellular, and behavioral abnormalities in neural-specific Pten knock-out mice. J Neurosci 29: 1773–1783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kelleher lll RJ, Bear MF (2008) The autistic neuron: troubled translation? Cell 135: 401–406. [DOI] [PubMed] [Google Scholar]
- 36. Li X, Alafuzoff I, Soninen H, Winblad B, Pei JJ (2005) Levels of mTOR and its downstream targets 4E-BP1, eEF2, and eEF2 kinase in relationships with tau in Alzheimer's disease brain. FEBS J 272: 4211–4220. [DOI] [PubMed] [Google Scholar]
- 37. Chano T, Okabe H, Hulette CM (2007) RB1CC1 insufficiency causes neuronal atrophy through mTOR signaling alteration and involved in the pathology of Alzheimer's diseases. Brain Res 1168: 97–105. [DOI] [PubMed] [Google Scholar]
- 38. Hoeffer CA, Klann E (2010) mTOR signaling: at the crossroads of plasticity, memory and disease. Trends Neurosci 33: 67–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Talos DM, Sun H, Zhou X, Fitzgerald EC, Jackson MC, et al. (2012) The interaction between early life epilepsy and autistic-like behavioral consequences: a role for the mammalian target of rapamycin (mTOR) pathway. PLoS One 7: e35885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kim E, Goraksha-Hicks P, Li L, Neufeld TP, Guan KL (2008) Regulation of TORC1 by Rag GTPase in nutrient response. Nat Cell Biol 10: 935–945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Sancak Y, Peterson TR, Shaul YD, Lindquist RA, Thoreen CC, et al. (2008) The Rag GTPase bind raptor and mediate amino acid signaling to mTORC1. Science 320: 1496–1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Manning BD, Cantley LC (2007) AKT/PKB signaling: navigating downstream. Cell 129: 1261–1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Dietary effect of leucine on motor behaviors displayed by Spr −/− mice. For dietary leucine supplementation, Spr +/+ or Spr −/− mice 25 days of age were fed the leucine supplemented diet in which 3.6% leucine (w/w) was added to normal diet for 10 days under ad libitum conditions. (A, B) Abnormal open-field behaviors in Spr −/− mice were not improved by the dietary supplementation of leucine. The experimental details were the same as in Figure 2. (C) Dietary supplementation of leucine fails to improve rotating rod performance displayed by Spr −/− mice. The experimental details were the same as in Figure 3. ***P<0.001. (D) Dietary leucine supplementation has no effect on brain mTORC1 activity in Spr −/− mice. Brain mTORC1 activities were measured using the phosphorylation of S6 as a readout. Other experimental procedures were the same as in Figure 6.
(TIF)
Dietary effect of L-DOPA on motor behaviors by Spr −/− mice. Both strains of Spr +/+ and Spr −/− mice 25 days of age were orally administrated L-DOPA (13.5 µg/g body weight/day) for 10 days. Open-field behaviors (A) or rotating rod performance (B) displayed by the experimental mice (n = 2 for each experimental group) are shown. Experimental conditions were the same as in Figure 2 and Figure 3. ***P<0.001.
(TIF)
Hypervariable distances moved by Spr −/− mice in the open-field test become normalized after the tyrosine therapy.
(DOCX)