

Abstract
The exact therapeutic mechanism of action of antipsychotic drugs remains unclear. Recent evidence has shown that second-generation antipsychotic drugs (SGAs) are differentially associated with metabolic side effects compared to first-generation antipsychotic drugs (FGAs). Their proclivity to cause metabolic disturbances correlates, to some degree, with their comparative efficacy. This is particularly the case for clozapine and olanzapine. In addition, the insulin signaling pathway is vital for normal brain development and function. Abnormalities of this pathway have been found in persons with schizophrenia and antipsychotic drugs may ameliorate some of these alterations. This prompted us to hypothesize that the therapeutic antipsychotic and adverse metabolic effects of antipsychotic drugs might be related to a common pharmacologic mechanism. This article reviews insulin metabolism in the brain and related abnormalities associated with schizophrenia with the goals of gaining insight into antipsychotic drug effects and possibly also into the pathophysiology of schizophrenia. Finally, we speculate about one potential mechanism of action (that is, functional selectivity) that would be consistent with the data reviewed herein and make suggestions for the future investigation that is required before a therapeutic agent based on these data can be realized.
Keywords: antipsychotic, functional selectivity, insulin, mechanism of action, metabolic side effects, schizophrenia
Introduction
Schizophrenia is a chronic, debilitating psychiatric illness characterized by positive, negative and cognitive symptoms.1 Antipsychotic drugs have been the primary treatment for approximately half of a century, beginning with the discovery and introduction of chlorpromazine.2 However, despite the extensive use and study of these agents, questions remain regarding their exact mechanism of action.3 Much of the study to date has focused on the receptor profiles of these medications.3 Interest in dopamine-2 (D2) receptor blockade stemmed, in part, from evidence that there existed a close relationship between the potency of conventional agents at the dopamine receptor and clinical potency.4,5 Serotonergic and glutamatergic mechanisms have also been implicated, in part, because of the perceived advantages of the second-generation antipsychotic drugs (SGAs; for example, risperidone and olanzapine) compared to the first-generation antipsychotic drugs (FGAs; for example, haloperidol and chlorpromazine) and because of the schizophrenia-like symptoms that occur when substances of abuse targeting these receptors are used.3,6,7 Nevertheless, although many theories have been proposed, no antipsychotic effect has been observed in an agent without D2 receptor affinity,8 with the exception of the very recent data suggesting anti-psychotic properties of an mGlu2/3 receptor agonist.9
The uncertainty regarding the precise mechanisms of action of antipsychotic drugs, and in particular the role of serotonin antagonism, has been compounded by the recent data that suggest that SGAs, with the possible exception of clozapine and to a lesser extent olanzapine, have not been wholly proven to be more effective than FGAs.10–14 Further complicating the efforts by clinicians and researchers to understand the neurochemical and cellular modifications necessary for an antipsychotic effect is the lack of a complete understanding of the pathogenesis, pathophysiology and genetics of schizophrenia.7
The objective of this article is to review the evidence and propose a plausible basis for a unitary mechanism of action for the differential metabolic and therapeutic effects of antipsychotic drugs. This will be fundamentally based upon clinical observations of the positive relationship between the effectiveness of antipsychotic drugs and their proclivity for metabolic side effects (that is, primarily for SGAs). We hypothesize that the therapeutic and adverse metabolic effects of anti-psychotic drugs (as typified by clozapine and olanzapine) are related. We also suggest that there are abnormalities of the insulin signaling pathway in schizophrenia that are affected by antipsychotic drugs. Several lines of evidence support this hypothesis. We first describe the relationship between the comparative effectiveness and metabolic disturbances of antipsychotic drugs, which forms the basis for the hypothesis of this article. Thereafter, this article focuses on the role of the insulin signaling pathway in neurodevelopment and brain function, new data on interactions between the dopamine and N-methyl-D-aspartate (NMDA) systems and the insulin signaling pathway, abnormalities of the insulin signaling pathway in the brains of individuals with schizophrenia and the data indicating that antipsychotic drugs can ameliorate some of these deficits. Finally, we speculate how one mechanism of action (that is, functional selectivity, a phenomenon by which ligands that bind to the same receptor can differentially recruit intra-cellular signaling pathways15) could account for the positive relationship between the effectiveness of antipsychotic drugs and their proclivity for metabolic side effects and discuss implications for future research and clinical practice.
The relationship between the comparative effectiveness and metabolic disturbances of antipsychotic drugs
With the introduction of clozapine and demonstration of its superior efficacy when compared to FGAs16 came the hope that subsequent SGAs would also exhibit superior efficacy. Although this was largely thought to be the case, recent data suggest that there is less clinical difference in effectiveness between FGAs and SGAs than previously believed, with the possible exceptions of clozapine and, to a lesser extent, olanzapine (Table 1).10–13,17
Table 1.
Comparisons between second-generation anti-psychotics on effectiveness, degree of metabolic disturbances and magnitude of effects on glycogen synthase kinase
| Clinical and biochemical measures | Comparisons between SGAs |
|---|---|
| Effectiveness | Clz > Olz ≥ Risp ≃ Quet ≃ Zip ≃ Ari |
| Degree of metabolic disturbances | Clz ≥ Olz > Risp ≃ Quet ≥ Zip ≃ Ari |
| Magnitude of effects on GSK | Clz ≃ Olz > Risp ≃ Quet ≥ Zip |
Abbreviations: Ari, aripiprazole; Clz, clozapine; GSK, glycogen synthase kinase; Olz, olanzapine; Quet, quetiapine; Risp, risperidone; SGAs, second-generation anti-psychotic drugs; Zip, ziprasidone.
However, there are several distinguishing characteristics between FGAs and SGAs. High-affinity and sustained D2 receptor blockade is overtly responsible for some of the major side effects (that is, extra-pyramidal side effects, EPS) of FGAs and, to some degree, antipsychotic activity.4,8,18 By contrast, clozapine, the prototype SGA, has relatively low affinity for the D2 receptor, induces few to no EPS and is considered to possibly be the most effective anti-psychotic, but has a high liability for precipitating metabolic disturbances.11,19,20 In addition, it has become apparent that several other SGAs, in addition to clozapine, have a marked propensity to induce metabolic disturbances, including hyperglycemia, hyperlipidemia, obesity, insulin resistance and sometimes frank diabetes mellitus.20–23
Following the introduction of chlorpromazine and the first neuroleptics, EPS were thought to be a prerequisite for antipsychotic efficacy.2,24 Clozapine and other SGAs violated this principle by having antipsychotic efficacy with little or no EPS,3 hence the term ‘atypical’ antipsychotic. Subsequently, SGAs were observed to have an increased risk of metabolic side effects. Therefore, if one considers the relationship between potency (that is, of dopamine receptor antagonism) and clinical efficacy of FGAs in relation to their ability to produce EPS, one might similarly consider a relationship between potency (that is, of a nontraditional mechanism of action) and efficacy of SGAs in relation to their ability to produce metabolic side effects. Although this apparent association between antipsychotic efficacy and metabolic side effects liability could be coincidental, it is also possible that they are due to a common mechanism. What mechanism could be responsible for this relationship? As all antipsychotic drugs are dopamine receptor blockers but only some of them produce metabolic side effects and to significantly varying degrees, this would implicate either selective interactions with the dopaminergic system missed by current methods of visualizing receptor occupancy and drug–receptor interactions or an extra-dopaminergic mechanism. However, to date there is no explanation of alternative therapeutic mechanisms of action beyond D2 receptor blockade that involve any other neuroreceptor, with the exception of the very recent data suggesting antipsychotic properties of an mGlu2/3 receptor agonist,9 even though theories involving the 5-HT2A, 5-HT1A and NMDA receptors, among others, have been proposed.3 Hence, there is great interest in examining novel or nontraditional mechanisms of action that could explain the superior efficacy of clozapine and olanzapine and their side effect liabilities.
Such speculation is reinforced by the striking realization that the most effective of the SGAs (that is, clozapine and olanzapine) have the greatest metabolic side effect liabilities, followed variably by quetiapine and risperidone, although ziprasidone and aripiprazole are least implicated in these effects (Table 1).20,25,26 However, this is not meant to oversimplify the relationship between schizophrenia and disturbances in glucose metabolism. In fact, this relationship is quite complex and not well understood.27 For example, although evidence suggests that the prevalence of diabetes mellitus is high in individuals with schizophrenia, there is debate27 as to whether abnormalities in glucose metabolism are actually present at the first episode28 or develop in the course of the illness, which complicates understanding the precise contribution of antipsychotic drugs to alterations in glucose metabolism in individuals with schizophrenia. Nevertheless, it is clear that the increased use of SGAs is associated with increasing rates of diabetes mellitus.21 Therefore, the recognition of this intriguing relationship between the therapeutic efficacy and magnitude of metabolic disturbances of SGAs warrants examination and is the basis for the hypothesis of this paper that the therapeutic and metabolic side effects of antipsychotic drugs are related.
Possible mechanisms of antipsychotic-induced disordered glucose metabolism
Before discussing the possible mechanisms of anti-psychotic-induced disturbances in glucose metabolism, we briefly review the metabolic side effects of antipsychotic medications. SGAs have been associated with clinically significant weight gain, sometimes greater than 20 lb with higher doses of olanzapine.29,30 Some have also been associated with increased levels of blood glucose, glycosylated hemoglobin and insulin, along with insulin resistance, and sometimes frank type 2 diabetes mellitus.29,30 Finally, some SGAs have been associated with unfavorable effects on blood lipid profiles, such as increased total cholesterol, low-density lipoprotein, and triglyceride levels and decreased levels of high-density lipoprotein levels.29,30 In general, clozapine and olanzapine have been most associated with these preceding adverse effects, followed variably by risperidone and quetiapine, whereas ziprasidone and aripiprazole have been associated with the least or no such adverse metabolic risks.29,30
Although hyperglycemia, insulin resistance, hyperlipidemia and obesity are all aspects of the metabolic profiles of SGAs, the focus of this hypothesis is on the phenomena of hyperglycemia and insulin resistance given the fundamental role that glucose utilization and the insulin signaling pathway play in normal brain development and function along with the pathophysiology of schizophrenia (as discussed in the following sections). This is done with the understanding that these various metabolic disturbances are likely related, rather than isolated, occurrences.31
As with the association between schizophrenia and diabetes mellitus, the mechanism of disordered glucose metabolism associated with SGAs is not clearly understood32 and is likely multifactorial. One suggested mechanism of these disturbances is via inhibition of glucose transporters.32,33 Impaired glucose regulation centrally and peripherally, along with associated alterations in insulin production in the pancreas, may also contribute.32–35 Alteration of the phosphoinositide-3-kinase (PI3K)/Akt insulin signaling pathway is another potential mechanism.33 Interactions between antipsychotic drugs and serotonergic, histaminergic, muscarinic and α-adrenergic receptors along with leptin might also contribute to these phenomena, either primarily or as secondary effects on weight and/or adiposity.32,34,36 Some of these mechanisms, such as blockade of histamine-1 receptors37 and inhibition of glucose transport,38–40 are correlated with antipsychotic-associated metabolic disturbances, such as their potential to induce orexigenic effects and hyperglycemia. Of these potential mechanisms, the insulin signaling cascade has been implicated in the pathophysiology of schizophrenia and is targeted by antipsychotic drugs and will therefore be the focus of this paper.
Insulin, insulin-like growth factor and the insulin signaling pathway
The insulin and insulin-like growth factor (IGF)-1 receptors share common evolutionary origins and have similar structures and signaling pathways.41 IGF-1 receptor expression in the brain begins as early as neural tube development, and IGF-1 and insulin receptor expression become widespread, particularly in neurons.41 Abnormalities of IGF have been suggested to make individuals more susceptible to schizophrenia,42 possibly by making those who already have a predisposition to schizophrenia more vulnerable to environmental insults during neurodevelopment.43
The effects of insulin/IGF are mediated by a signal transduction cascade, beginning with a tyrosine kinase receptor (Figure 1).44 After insulin binds the extracellular aspect of its receptor, a conformational change takes place, which results in autophosphorylation of the receptor.44 Multiple different insulin receptor substrates transmit the signal to different effectors in the cell, such as PI3K.44 Several of the lipid products of PI3K activate another group of kinases, such as Akt and protein kinase C (PKC), which are highly implicated in glucose metabolism and transport and the effects of insulin.44 Akt, also known as protein kinase B, requires phosphorylation for activation.44
Figure 1.

A simplified version of the insulin signaling pathway. Insulin or insulin-like growth factor (IGF) binds to the insulin/IGF receptor causing autophosphorylation and activation. This leads to activation of phosphoinositide-3-kinase (PI3kinase), and the end result is to phosphorylate and activate Akt and phosphorylate and deactivate glycogen synthase kinase-3 (GSK-3). Active GSK-3 deactivates glycogen synthase and eukaryotic initiation factor 2B (eIF2B) and activates insulin receptor substrate-1 (IRS-1; an inhibitor of the insulin receptor). PIP3, phosphoinositide 3,4,5-tris-phosphate; PKC, protein kinase C (adapted and modified with permission from Gould and Manji49).
Akt has a variety of effects in the cell including regulation of the cell cycle, regulation of cell death as an antiapoptotic factor and DNA synthesis.45 The PI3K/Akt pathway has also been implicated as a mediator of the effects of neuregulin, a gene that has been associated with schizophrenia,46 on neuronal development and survival.47 Interestingly, Akt1-deficient mice show abnormal dendritic architecture of layer V of their prefrontal cortex.48 These mice also exhibit impaired working memory under D2, but not dopamine-1 (D1), challenge.48 The significance of these findings is underscored by the abnormalities in dendritic architecture and working memory, along with the potential role of the D2 receptor, in schizophrenia.1 Finally, Akt appears to be associated with the trafficking of glucose transporters to the neuronal membrane.41
Another substrate of Akt is glycogen synthase kinase (GSK). GSK has been associated with mood stabilization and implicated as a possible target of lithium.49 More recently, GSK has been associated with schizophrenia as well.50,51 GSK is expressed widely throughout the human brain and is observed as early as embryonic day 10 in rats.52 Two isoforms (GSK-3α and β) have been identified52 that are constitutively active.49 In the PI3K/Akt pathway, Akt phosphorylates GSK-3α at Ser21 and GSK-3β at Ser9, resulting in their inhibition.53 Cholinergic, serotonergic and glutamatergic neurotransmission, also implicated in the pathophysiology of schizophrenia, may also regulate the phosphorylation state of GSK (reviewed in Jope and Roh50).
GSK has multiple effects on both neurodevelopment and glucose metabolism.52 Through the PI3K/Akt pathway GSK is involved in neurotrophic effects49 and the regulation of apoptosis.52 GSK also influences other crucial neuronal processes including neural plasticity, microtubule dynamics, neurite outgrowth, neurogenesis and transcription factors/gene expression (for example, via transcription factors such as cyclic AMP response element-binding protein).50 GSK is one of the main mediators of the Wnt signaling cascade, which is important in synaptic arrangement and neural tube development/patterning.52 Interestingly, preliminary findings suggest that components of the Wnt pathway may be abnormal in schizophrenia (reviewed in Jope and Roh50). Furthermore, the protein products of approximately 10 genes that have been associated with schizophrenia (for example, Akt1, disrupted in schizophrenia-1 (DISC1), NRG1) appear to be involved in GSK signaling.51 Finally, GSK has effects on glucose and insulin metabolism as inhibition (that is, phosphorylation) of GSK activates glycogen synthase, which leads to the synthesis of glycogen.49,52
In summary, in addition to its well-established role in maintaining euglycemia, the insulin signaling pathway and its components may be significant in neuronal growth, synaptic development and neuronal survival/apoptosis during brain development. Therefore, alterations of this pathway may have profound implications for the pathogenesis, pathophysiology and treatment of neurodevelopmental disorders of the brain.
The role of the insulin signaling pathway in schizophrenia: insights from the dopamine and NMDA receptor hypofunction hypotheses
Traditionally, dopamine receptors have been associated with G-protein regulation and cyclic AMP/protein kinase A signaling.54 Recently, a cyclic AMP-independent mechanism of dopaminergic behaviors and signaling at the D2 receptor was identified, which involves the Akt/GSK pathway.54 The model used was dopamine transporter (DAT) knockout mice, which have increased availability of dopamine and exhibit hyperactive behaviors and stereotypic movements.55 DAT knockout mice had decreased levels of both phospho-Thr308-Akt, phospho-Ser21-GSK-3α and phospho-Ser9-GSK-3β in their striatum, abnormalities which were ameliorated by dopamine depletion and by D2 (but not D1) receptor antagonism.55 Remarkably, inhibition of GSK-3 in DAT knockout mice also inhibited the dopamine-dependent behaviors.55 Finally, dopamine agonism decreased the phosphorylation of both Akt and GSK-3 in normal mouse striatum, and GSK-3β mutant mice had reduced behavioral response to dopamine agonism.55
Later research elucidated the mechanism of D2 receptor-mediated Akt regulation (Figure 2).56 Namely, the binding of dopamine to the D2 receptor leads to the formation of a complex consisting of β-arrestin 2, Akt and protein phosphatase 2A.56 β-Arrestin 2 serves as a scaffold for both Akt and protein phosphatase 2A, thereby facilitating dephosphorylation of Akt.56
Figure 2.
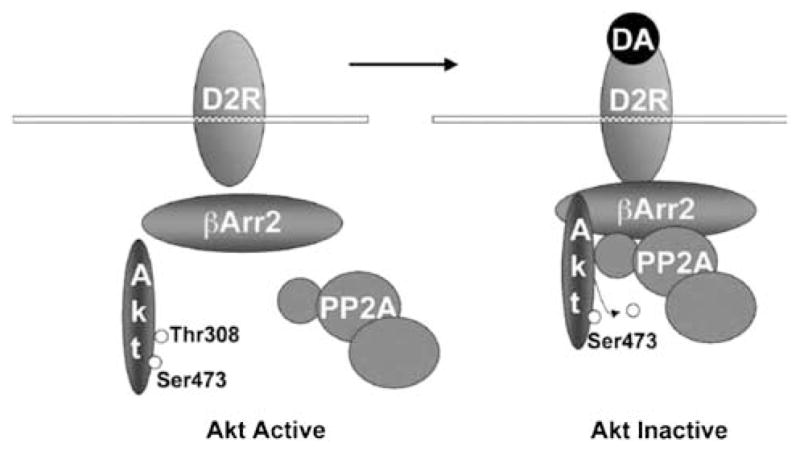
Working model of the Akt/β-arrestin 2 (βArr2)/protein phosphatase 2A (PP2A) signaling complex formed in response to activation of dopamine (DA) receptors (D2R) leading to Akt inactivation in the mouse striatum (adapted and modified with permission from Beaulieu et al.56).
Data from studies on the effects of NMDA receptor antagonists on the insulin signaling pathway also indicate abnormalities of insulin signaling in schizophrenia.57 Phencyclidine (PCP) or ketamine use can lead to schizophrenia-like symptoms in humans.58 In neuronal culture, PCP can be toxic.57 In rats, PCP administration led to decreased phospho-Ser473-Akt but stable total Akt in frontal cortex, hippocampus and striatum.57 Similar results were observed in rat neuronal culture,57 indicating that PCP leads to decreased activity of Akt. Activating L-type calcium channels (which favors cell survival) or NMDA receptors blocked both PCP-induced neurotoxicity and reduction in phospho-Ser473-Akt.57 In both cases, adding a specific inhibitor of Akt restored the neurotoxicity of PCP, indicating that the PI3K/Akt pathway was primarily responsible for protecting against the PCP-induced neurotoxicity.57 As expected, PCP administration also led to decreased phospho-Ser9-GSK-3β in rat frontal cortex, hippocampus and striatum, along with in rat neuronal culture and no change in total GSK-3β, indicating increased activity of GSK-3β.57 Selective inhibition of GSK-3β eliminated the neurotoxic effects of PCP57 again implicating a role for this pathway in the neurotoxic effects of PCP.
The next sections of this article review the reported abnormalities of the insulin signaling pathway in schizophrenia and how antipsychotic drugs may ameliorate these alterations and possibly lead to therapeutic efficacy in schizophrenia. However, based on the data presented to this point, several predictions can be made. First, an overactive dopaminergic system in schizophrenia,1 or hypofunction of the NMDA system,58 would predictably lead to decreased phosphorylation and activity of Akt and decreased phosphorylation and increased activity of GSK. For antipsychotic drugs (that is, D2 receptor antagonists) to be effective, they would predictably have to reverse these effects (that is, lead to increased phosphorylation and activity of Akt and increased phosphorylation and decreased activity of GSK).
Abnormalities of the insulin signaling pathway in schizophrenia and how antipsychotic drugs might affect these alterations
The data on the insulin signaling pathway in schizophrenia and the effects of antipsychotic drugs are complicated for several reasons. First, human studies often involve subjects who have already been exposed to a variety of FGAs and/or SGAs. In addition, the presence or absence of the disease state (that is, schizophrenia) potentially complicates the comparison and analysis of studies involving human neuropathologic or cell culture data. Further, comparisons are required between human and nonhuman data and between studies involving different cell types. Finally, issues regarding specimen processing (as discussed below)50,59 add another level of complexity to the interpretation of this data. In spite of these limitations, a review of the available studies is informative. Inconsistencies and confounded effects will be identified and discussed as they are presented.
The insulin receptor, phosphoinositides and protein kinase C
Genetic alterations of various components of the PI3K pathway have been reported in individuals with schizophrenia,60 including in Chinese61 and African American62 populations. Relationships between other components of phosphoinositide signaling, such as phosphatidylinositol-4-phosphate 5-kinase II-α (PIP5-K2A), with schizophrenia have also variably been observed,63,64 though one study of IGF-1 in Caucasian populations did not find any association with schizophrenia.65
There is little neuropathologic data suggesting abnormalities of the more proximal components of the insulin signaling pathway. In a sample of individuals with schizophrenia, all of whom had been treated with FGAs, except for one who had been on clozapine, a neuropathologic examination showed decreased density of PKC in the parahippocampus when compared to controls, with no correlation between PKC density and antipsychotic dose.66 In a similar analysis, no differences were found in the striatum,67 or in the platelets of another group of individuals with schizophrenia when PKC activity was compared to that in control subjects.68 In addition, while haloperidol exposure in rats increased the activity of PKC in the frontal cortex in one investigation,69 another demonstrated that rats treated with 4 weeks of haloperidol or clozapine had no changes in PKC levels in frontal cortex, basal ganglia or olfactory tubercle.70 These data indicate little correlation between antipsychotic drugs and the proximal components of the insulin signaling pathway and therefore corroborate observations that the interaction between the D2 receptor/antipsychotic drugs and the insulin signaling pathway may occur at the level of Akt.56 Although another study suggests that PKC is involved in the augmenting effects of clozapine on NMDA signaling in pyramidal cells of the medial prefrontal cortex,71 this study did not specifically address the interaction of this phenomenon with the insulin signaling pathway.
Other data are consistent with a state of insulin resistance and hyperinsulinemia after treatment with antipsychotic drugs. Olanzapine has been shown to inhibit the insulin-stimulated activation of PI3K in a rat skeletal muscle cell line72 and upregulates insulin-2 in rat frontal cortex.73 In addition, Zhao et al.74 examined the insulin receptor in dorsolateral prefrontal cortex from individuals with schizophrenia, most of whom had been receiving FGAs and several of whom had been receiving SGAs. Both the total and activated amounts of the insulin receptor were found to be reduced in these individuals.74
Akt
Akt and schizophrenia
The potential relevance of genetic abnormalities of Akt1 in individuals with schizophrenia was strongly supported by the finding that an Akt1 haplotype associated with lower levels of Akt1 was related to schizophrenia in a Northern European population.75 Positive relationships between Akt1 and schizophrenia were also reported in Caucasian,76 Iranian,77 Japanese78 and Chinese79 populations, but not in two other Japanese populations59,80 or a Taiwanese population,81 and variably in a Caucasian population.82 In addition, undertransmission of some haplotypes of Akt1 was found in an Irish sample.83
There is limited data on Akt in schizophrenia. In addition, the phosphorylation level of Akt is closely related to the perimortem condition of the tissue (for example, temperature, pH),59 which could serve as a potential confound of these data. However, the available data suggest decreased levels of total and phosphorylated Akt in schizophrenia, which is consistent with theories of schizophrenia that postulate overactive dopaminergic systems in mesolimbic areas of the brain1 or NMDA receptor hypofunction.58 One study examined dorsolateral prefrontal cortex from individuals with schizophrenia, most of whom had been receiving FGAs and several of whom had been receiving SGAs, and demonstrated decreased levels of both total and phospho-Ser473-Akt.74 Similar decreases in Akt1 expression were found in dorsolateral prefrontal cortex in another sample,83 whereas other studies reported no alterations in frontal cortex Akt159,84 or phosphorylated Akt in individuals with schizophrenia.59 Meanwhile, Emamian et al.75 observed decreased levels of total Akt1 in lymphocyte-derived cell lines, frontal cortex and hippocampus from individuals with schizophrenia. Finally, in primary neuronal culture, inhibition of DISC1, a gene implicated in schizophrenia, led to decreased phosphorylation of Akt but stable levels of total Akt.85
Akt and antipsychotic drugs
In contrast to the effects of DISC1 inhibition, clozapine has been reported to increase phosphorylation of Akt in neuroblastoma cells.86 A similar analysis in mouse neuroblastoma cells demonstrated that 5 days of both clozapine and chlorpromazine increased phospho-Ser473-Akt without increasing total Akt levels.87 Olanzapine has been similarly shown to increase levels of phospho-Ser473-Akt in PC12 cells although total Akt remained the same.88 This effect was dependent on PI3K, suggests an additional mechanism of antipsychotic effects on Akt and may be related to differences in cell line.88 Chronic treatment of mice with haloperidol did not alter the total levels of Akt1 in the frontal cortex but did increase phosphorylated Akt1 at both Thr308 (acute and chronic) and Ser473 (chronic only).75 This was consistent with a study of mice treated with clozapine for 68 days in which although insulin receptor activity and Akt levels were decreased, the amount, and thus activity, of phospho-Ser473-Akt was unchanged.74
Curiously, chlorpromazine, clozapine and fluphenazine did not lead to increased phosphorylation of Akt in PC12 cells.88 In another study, fluphenazine, chlorpromazine and haloperidol decreased the nerve growth factor-induced phosphorylation of Akt at Ser473 in PC12 cells,89 which is consistent with another study that showed that acute haloperidol treatment of rat neurons decreased levels of phospho-Ser473-Akt.90 These data begin to underscore and elucidate differences in efficacy between antipsychotic drugs (that is, more effects on Akt of olanzapine and clozapine than other antipsychotic drugs). However, these data are limited by the nearly cytotoxic doses of antipsychotic drugs used in some of these experiments,34,91 which may contribute to their discrepant effects on Akt.
Although most of the preceding data suggest that SGAs generally increase levels of phosphorylated Akt, opposite effects were noted in glioblastoma cells in which clozapine inhibited Akt phosphorylation,92 possibly due to differences in cell line. In a rat skeletal muscle cell line, olanzapine inhibited the insulin-stimulated phosphorylation of Akt,72 which is consistent with a state of peripheral insulin resistance; a known side effect of olanzapine.29
In summary, at least in a number of studies, anti-psychotic drugs appear to increase phosphorylation of Akt. These effects are antagonistic to what occurs in schizophrenia (that is, decreased Akt phosphorylation) and predictable based on both the dopamine and NMDA receptor hypofunction hypotheses.
GSK
GSK and schizophrenia
Although genetic relationships have been reported between GSK-3β and the paranoid subtype of schizophrenia in an Italian population93 and marginally so in a Chinese population,94 significant relationships were not found for schizophrenia in general in Italian,93 Finnish,95 Japanese,96 Chinese94 and Korean97 populations.
Multiple investigations have examined the levels and activities of GSK-3 isoforms in individuals with schizophrenia and in animal models. The data suggest decreased levels and increased activity of GSK-3 in schizophrenia. These results are consistent with the data reviewed above showing decreased activity of Akt and are consistent with predictions based on the dopamine and NMDA receptor hypofunction hypotheses of schizophrenia.1,58 However, the results of studies of GSK in schizophrenia are mixed. This is not unexpected for the reasons discussed previously, as well as because the study of GSK in post-mortem tissues is highly problematic given the marked changes in phosphorylation state that occur post mortem and in response to anesthesia (that is, in nonhuman brain tissue).50,59 Nevertheless, the available data are informative and are reviewed below.
In one neuropathologic study of individuals with schizophrenia, bipolar disorder and unipolar depression, only the individuals with schizophrenia had decreased levels of GSK-3β and decreased activity of GSK-3 in frontal cortex, whereas no alteration in the level of GSK-3β in occipital cortex was observed.98,99 In addition, no correlation was noted between lifetime antipsychotic exposure and GSK-3β levels.98 These results are consistent with a later study showing that GSK-3β mRNA, but not GSK-3α mRNA, levels were decreased in the dorsolateral prefrontal cortex from individuals with schizophrenia compared with a comparison group of normal controls, individuals with bipolar disorder and individuals with unipolar depression; no correlations were noted with lifetime antipsychotic consumption, and no differences were noted between those individuals treated and not treated with lithium.100 When the phosphorylation levels of GSK-3β at Ser9 (that is, the site phosphorylated by Akt1) were measured in frontal cortex and lymphocytes from individuals with schizophrenia, decreased levels were found, although levels of the GSK-3β form phosphorylated at Tyr216 were not abnormal in either the lymphocytes or frontal cortex.75 Other neuropathologic studies of individuals with schizophrenia reported that GSK-3β levels were decreased in the prefrontal cortex from these individuals101 and that phospho-Ser9-GSK-3β levels were decreased in the frontal cortex of these individuals.84 Decreased levels of GSK-3β were also observed in the cerebrospinal fluid of individuals with schizophrenia, with no correlation with antipsychotic dose.102 Zhao et al.74 examined dorsolateral prefrontal cortex from individuals with schizophrenia, most of whom had been receiving FGAs and several of whom had been receiving SGAs, and found increased levels of total GSK-3 (α and β) while the phosphorylated forms of both enzymes remained unaltered, indicating increased activity of these enzymes.74
Data from animal studies have supported and extended these abnormalities of GSK in schizophrenia. In rats exposed to hippocampal damage, reduced frontal GSK-3β levels were observed prior to but not after puberty, whereas no differences at either age were observed in total GSK-3 (α and β) activity.103 This is of particular relevance to schizophrenia because of the robust evidence suggesting abnormalities of the hippocampus in schizophrenia.104 A study of GSK-3β levels in rat frontal cortex showed no response to acute or chronic stress, suggesting that the altered GSK-3β levels observed in schizophrenia may be disease related and not a nonspecific stress response.105 Similarly, rats exposed to 21 days of haloperidol, chlorpromazine or clozapine had no alterations in GSK-3β levels or total GSK-3 (α and β) activity in their frontal cortex,106 again suggesting that the changes observed in patients were not simply a response to antipsychotic treatment.
However, other neuropathologic and biochemical studies have found inconsistent results. Two such investigations found no differences in GSK-3β in prefrontal cortex of individuals with schizophrenia.107,108 A third study reported no abnormalities of GSK-3α or -β mRNA levels, GSK-3β protein levels or total GSK-3 (α and β) activity in frontal cortex from individuals with schizophrenia, but did find reductions in these measures in hippocampus (that is, GSK-3β protein levels only after omitting outliers).109 Similarly, no alterations in frontal cortex GSK-3β or phosphorylated GSK-3β were found in a recent study of individuals with schizophrenia.59 Finally, no differences in GSK-3α and -β mRNA, GSK-3β protein or GSK-3 (α and β) activity were observed in the lymphocytes of individuals with schizophrenia.75,110
GSK and antipsychotic drugs
Recent investigations in animal models have indicated that antipsychotic drugs have important effects on GSK. Subchronic treatment of rats with haloperidol and risperidone, but not clozapine, increased GSK-3 protein levels in striatum, whereas all three of these drugs increased GSK-3 protein levels in the prefrontal cortex and ventral midbrain.111,112 In addition, subchronic haloperidol and risperidone increased phospho-Ser9-GSK-3β levels in the prefrontal cortex, whereas chronic, but not acute, haloperidol and risperidone also increased GSK-3 (α and β) levels in prefrontal cortex and ventral midbrain, indicating that repeated treatment may be necessary for the observed changes.111,112 Further, as with Akt1, chronic treatment with haloperidol increased the levels of phospho-Ser9-GSK-3β in mice.75 In another study, rats treated with haloperidol had decreased phospho-Ser9-GSK-3β in frontal cortex whereas those treated with clozapine had increased levels.113 As noted previously, the discrepancy between these two studies may be related to the different animal models used (rats versus mice), the different doses of haloperidol used (10 versus 1 mg kg−1), the different lengths of exposure in each study (21 versus 12 days)75,113 or confounds related to the use of anesthetic agents and post-mortem processing of tissues.50
Li et al.114 showed that one dose of risperidone, but not haloperidol, increased phospho-Ser9-GSK-3β and phospho-Ser21-GSK-3α levels in mouse cortex, striatum (not significant for GSK-3α), hippocampus and cerebellum, although total GSK-3β remained the same.114 Some of the effects of risperidone were augmented by co-treatment with imipramine or fluoxetine.114 Further, no effects of risperidone were noted on either phospho-Ser473-Akt or phospho-Thr308-Akt, suggesting that the Akt pathway was not involved,114 a finding inconsistent with previous data. In the same study, a similar analysis was performed comparing the effects of clozapine, olanzapine, quetiapine and ziprasidone on phospho-Ser9-GSK-3β levels in the brains of mice and found that clozapine and olanzapine had the greatest effect, followed by quetiapine, whereas ziprasidone had the least effect.114 Interestingly, these results are consistent with data that suggest that clozapine and olanzapine are the most likely of the SGAs to cause metabolic disturbances and are possibly also the most effective (Table 1).12–14,20,25,26
Antipsychotic drugs have been found to have mixed effects on GSK in different cell and tissue culture environments. Clozapine has been found to increase levels of phospho-Ser9-GSK-3β in neuroblastoma cells.86 Similar results were observed in another study of mouse neuroblastoma cells exposed to 5 days of antipsychotic; active, nonphosphorylated GSK-3β decreased after exposure to both chlorpromazine and clozapine.87 In mice that had been treated with clozapine for 68 days it was observed that although insulin receptor activity decreased and phospho-Ser21-GSK-3α and phospho-Ser9-GSK-3β remained the same, total GSK-3α and -β levels, and thus activity, were decreased.74 Opposite effects were noted in glioblastoma cells in which clozapine had the effect of dephosphorylating GSK-3β at Ser9,92 possibly related to differences in cell line. Olanzapine inhibited the insulin-stimulated phosphorylation (that is, inactivation) of GSK-3 (α and β) in a rat skeletal muscle cell line.72 Though apparently discrepant with previous findings in neural cells, this phenomenon would be consistent with a state of peripheral insulin resistance; a known side effect of olanzapine.29
In summary, the effect of antipsychotic drugs on GSK is relatively consistent and is to increase phosphorylation (and inactivation). This is consistent with predictions based on the dopamine and NMDA receptor hypofunction hypotheses of schizophrenia.
Summary of the abnormalities of the insulin signaling pathway in schizophrenia and the effects of antipsychotic drugs
In summary, there are limited and mixed data on the insulin signaling pathway in schizophrenia and how antipsychotic drugs interact with it. Further, as stated above, the synthesis and analysis of this data is complicated by a number of important factors. Nevertheless, several general observations and speculations can be made. First, limited genetic analyses have implicated alterations in Akt1, certain phosphoinositide-related genes and GSK-3β in some populations of individuals with schizophrenia. Second, the data on inherent abnormalities of the insulin signaling pathway in schizophrenia are inconclusive for multiple reasons, not the least of which is prior anti-psychotic exposure, though they suggest decreased levels of Akt, phosphorylated Akt and GSK, along with increased activity of GSK, in schizophrenia. Antipsychotic drugs appear in most cases to increase phosphorylation levels of Akt and GSK, thereby decreasing the activity of GSK. These observations are strikingly consistent with the known operation of the insulin signaling pathway (Figure 1). They are also consistent with both the overactive dopaminergic stimulation and NMDA receptor hypofunction hypothetical models of schizophrenia. In addition, anti-psychotic drugs appear to reverse these abnormalities. Finally, although both FGAs and SGAs have been implicated in these effects, preliminary evidence suggests that these effects might occur more or to a greater degree with SGAs and in particular for those SGAs that are considered to be both the most effective and to be the most associated with metabolic disturbances (that is, clozapine, and, to a lesser extent, olanzapine; Table 1). Altogether, these data support our hypothesis that the unique therapeutic and adverse effects of antipsychotic drugs (and in particular clozapine and olanzapine) are related and suggest that the insulin signaling pathway may be one focal point for such a phenomenon. However, the relevance of the insulin signaling pathway for the effects of antipsychotic drugs remains speculative.
Functional selectivity as an example of a potential mechanism for coupling antipsychotic drug therapeutic and metabolic side effects
Much investigation is needed before a compelling unitary mechanism of antipsychotic drug action for therapeutic efficacy and metabolic side effects can be put forward. However, the fact that antipsychotic drugs with the greatest efficacy have the greatest metabolic effects is curious and prompts speculation. In addition, the recent findings that the D2 receptor and antipsychotic drug effects are intertwined with the insulin signaling pathway may be relevant and make speculation about a potential unitary mechanism of action possible.
One possibility is that clozapine and olanzapine have different targets than other antipsychotic drugs, and therefore D2 receptor blockade is irrelevant to their antipsychotic effect. This seems unlikely given that no antipsychotic effect has been observed in any agent without D2 receptor affinity,8 with the exception of the very recent data suggesting antipsychotic properties of an mGlu2/3 receptor agonist.9 Alternatively, although D2 receptor blockade may still be required for their therapeutic effects, these drugs may gain their superior response profile by acting at a non-D2 receptor target, such as at histamine-1,37 serotonergic or muscarinic receptors.36 Another possibility is that different antipsychotic drugs selectively interact with the D2 receptor in ways that lead to differences in signaling but are missed by our current methods of visualizing receptor occupancy and drug–receptor interactions. This model suggests that it is the ability of an antipsychotic drug to enhance signaling via the insulin signaling pathway (that is, via Akt/GSK) rather than traditional D2 receptor mechanisms (that is, via the cyclic AMP pathway) that leads to greater efficacy. Similarly, this model suggests that these unique, qualitative interactions between antipsychotic drugs and the D2 receptor, by contrast to their quantitative interactions (for example, potency or strength of stimulus which would suggest ‘all-or-nothing’ effects on signaling pathways),15 may more clearly explain their differential therapeutic effects. This would also contribute to our understanding of why the medications that are more prone to affect this pathway (for example, clozapine, olanzapine) are also more associated with metabolic side effects, with the caveat that this might be one of multiple factors affecting proclivity for metabolic side effects, as reviewed above.
The notion that the binding of different psychotropic agents to the same G-protein-coupled receptor can cause different behavioral and physiologic effects is intriguing but not novel. Indeed, evidence has begun to emerge suggesting that ligands may be able to induce different and selective conformational changes in their receptors, which in turn leads to the recruitment of different signaling pathways.15,115,116 Structurally distinct ligands may also be able to differentially activate signaling pathways based on the existence of certain key signal transducing proteins, such as arrestins,117 proteins also involved in D2 receptor signaling.56 This phenomenon has been given numerous names, including ‘functional selectivity,’ ‘agonist-directed trafficking of receptor stimulus,’ ‘biased agonism’ and ‘stimulus trafficking,’15,116 and investigative techniques, such as transcriptome analysis,115 are already being used to address these effects. In particular, functional selectivity has been observed at the D2 receptor.15,118 However, to our knowledge, no studies to date have explored the functional selectivity of D2 receptor signaling as the basis for the effects of antipsychotic drugs on the Akt/GSK signaling pathway. Therefore, although this phenomenon is intriguing, its pertinence remains unclear.
On the basis of the information presented herein, several directions of future inquiry can be suggested. First and foremost is the need to develop a better characterization of the insulin signaling pathway in schizophrenia. One way to do this, in addition to neuropathologic examination, would be to use cerebrospinal fluid and/or the peripheral tissues of neuroleptic-naive patients.51 The use of peripheral tissues would be feasible given the relative consistencies that have been reported between measures in peripheral tissues and cerebrospinal fluid with neuropathologic data.75,102 Obtaining such measures both before and after treatment with antipsychotic drugs and other pharmacologic compounds known to affect the insulin signaling pathway (for example, lithium) would also shed light on the effects of anti-psychotic drugs on this pathway. Our understanding of the degree to which antipsychotic drugs participate in functional selectivity could also be assessed in cell culture by exposing D2 receptor-expressing cells to antipsychotic drugs at clinically therapeutic concentrations and measuring the amounts and activities of various components of the different dopamine signaling pathways (for example, cyclic AMP/protein kinase A pathway, Akt/GSK pathway). Finally, the roles of the dopamine and glutamate systems, along with other neurotransmitter systems (for example, serotonergic, cholinergic) implicated in schizophrenia, on the insulin signaling pathway need to be further clarified to understand the likely complex role of these pathways in schizophrenia and before drug development targeted at this pathway can be most efficiently accomplished and understood.
Conclusions and therapeutic implications
In summary, there is a variety of evidence that supports the hypothesis that the therapeutic and adverse effects of antipsychotic drugs (and in particular clozapine and olanzapine) are related. These data also suggest that there exist abnormalities of the insulin signaling pathway in schizophrenia and that antipsychotic drug effects on this pathway are therapeutic in schizophrenia. Importantly, this hypothesis was developed based on clinical observations of the differential therapeutic effectiveness and proclivity for metabolic side effects of SGAs and appears to be consistent with current hypotheses about the role of the dopamine and NMDA receptor systems in the pathophysiology and treatment of schizophrenia. However, the data that associate antipsychotic action with the insulin signaling pathway remain preliminary and sometimes contradictory.
The development of this hypothesis based on a theoretical unitary mechanism of therapeutic and metabolic side effects is not meant to, nor could it, fully explain two likely complicated and multi-factorial phenomena. For example, our hypothesis does not fully explain the development of peripheral insulin resistance. It remains unclear whether it is the presence of the disease state (that is, schizophrenia), the presence or absence of D2 receptors, or some other mechanism that explains why antipsychotic drugs have therapeutic effects centrally and detrimental effects peripherally. Rather, this hypothesis and article are meant to assemble relevant basic science, translational and clinical data with the hope of providing a theoretical framework for one potential avenue of future drug development. Given the range of effects that components of the insulin signaling pathway may have on neurodevelopment and brain function, it is possible that any one of these may have an important role in antipsychotic effects. Further investigation is crucial to foster the much-needed development of novel pharmacologic agents based on these observations.
Acknowledgments
This work was supported in part by grants from the NIH and from the Lieber Center for Schizophrenia Research and Treatment. We thank Dr Karen Duff for her helpful comments on this paper.
References
- 1.Lewis DA, Lieberman JA. Catching up on schizophrenia: natural history and neurobiology. Neuron. 2000;28:325–334. doi: 10.1016/s0896-6273(00)00111-2. [DOI] [PubMed] [Google Scholar]
- 2.Lehmann HE, Ban TA. The history of the psychopharmacology of schizophrenia. Can J Psychiatry. 1997;42:152–162. doi: 10.1177/070674379704200205. [DOI] [PubMed] [Google Scholar]
- 3.Miyamoto S, Duncan GE, Marx CE, Lieberman JA. Treatments for schizophrenia: a critical review of pharmacology and mechanisms of action of antipsychotic drugs. Mol Psychiatry. 2005;10:79–104. doi: 10.1038/sj.mp.4001556. [DOI] [PubMed] [Google Scholar]
- 4.Creese I, Burt DR, Snyder SH. Dopamine receptor binding predicts clinical and pharmacological potencies of antischizophrenic drugs. Science. 1976;192:481–483. doi: 10.1126/science.3854. [DOI] [PubMed] [Google Scholar]
- 5.Seeman P, Chau-Wong M, Tedesco J, Wong K. Brain receptors for antipsychotic drugs and dopamine: direct binding assays. Proc Natl Acad Sci USA. 1975;72:4376–4380. doi: 10.1073/pnas.72.11.4376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Aghajanian GK, Marek GJ. Serotonin model of schizophrenia: emerging role of glutamate mechanisms. Brain Res Brain Res Rev. 2000;31:302–312. doi: 10.1016/s0165-0173(99)00046-6. [DOI] [PubMed] [Google Scholar]
- 7.Tamminga CA, Holcomb HH. Phenotype of schizophrenia: a review and formulation. Mol Psychiatry. 2005;10:27–39. doi: 10.1038/sj.mp.4001563. [DOI] [PubMed] [Google Scholar]
- 8.Remington G. Understanding antipsychotic ‘atypicality’: a clinical and pharmacological moving target. J Psychiatry Neurosci. 2003;28:275–284. [PMC free article] [PubMed] [Google Scholar]
- 9.Patil ST, Zhang L, Martenyi F, Lowe SL, Jackson KA, Andreev BV, et al. Activation of mGlu2/3 receptors as a new approach to treat schizophrenia: a randomized Phase 2 clinical trial. Nat Med. 2007;13:1102–1107. doi: 10.1038/nm1632. [DOI] [PubMed] [Google Scholar]
- 10.Jones PB, Barnes TR, Davies L, Dunn G, Lloyd H, Hayhurst KP, et al. Randomized controlled trial of the effect on quality of life of second- vs first-generation antipsychotic drugs in schizophrenia: Cost Utility of the Latest Antipsychotic Drugs in Schizophrenia Study (CUtLASS 1) Arch Gen Psychiatry. 2006;63:1079–1087. doi: 10.1001/archpsyc.63.10.1079. [DOI] [PubMed] [Google Scholar]
- 11.Lieberman JA. Comparative effectiveness of antipsychotic drugs. A commentary on: Cost Utility of the latest Antipsychotic Drugs in Schizophrenia Study (CUtLASS 1) and Clinical Antipsychotic Trials of Intervention Effectiveness (CATIE) Arch Gen Psychiatry. 2006;63:1069–1072. doi: 10.1001/archpsyc.63.10.1069. [DOI] [PubMed] [Google Scholar]
- 12.Lieberman JA, Stroup TS, McEvoy JP, Swartz MS, Rosenheck RA, Perkins DO, et al. Clinical Antipsychotic Trials of Intervention Effectiveness (CATIE) investigators. Effectiveness of antipsychotic drugs in patients with chronic schizophrenia. N Engl J Med. 2005;353:1209–1223. doi: 10.1056/NEJMoa051688. [DOI] [PubMed] [Google Scholar]
- 13.Tandon R, Fleischhacker WW. Comparative efficacy of anti-psychotics in the treatment of schizophrenia: a critical assessment. Schizophr Res. 2005;79:145–155. doi: 10.1016/j.schres.2005.07.025. [DOI] [PubMed] [Google Scholar]
- 14.Volavka J, Czobor P, Sheitman B, Lindenmayer JP, Citrome L, McEvoy JP, et al. Clozapine, olanzapine, risperidone, and haloperidol in the treatment of patients with chronic schizophrenia and schizoaffective disorder. Am J Psychiatry. 2002;159:255–262. doi: 10.1176/appi.ajp.159.2.255. [DOI] [PubMed] [Google Scholar]
- 15.Urban JD, Clarke WP, von Zastrow M, Nichols DE, Kobilka B, Weinstein H, et al. Functional selectivity and classical concepts of quantitative pharmacology. J Pharmacol Exp Ther. 2007;320:1–13. doi: 10.1124/jpet.106.104463. [DOI] [PubMed] [Google Scholar]
- 16.Kane J, Honigfeld G, Singer J, Meltzer H. Clozapine for the treatment-resistant schizophrenic. A double-blind comparison with chlorpromazine. Arch Gen Psychiatry. 1988;45:789–796. doi: 10.1001/archpsyc.1988.01800330013001. [DOI] [PubMed] [Google Scholar]
- 17.Geddes J, Freemantle N, Harrison P, Bebbington P. Atypical anti-psychotics in the treatment of schizophrenia: systematic overview and meta-regression analysis. BMJ. 2000;321:1371–1376. doi: 10.1136/bmj.321.7273.1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Glazer WM. Extrapyramidal side effects, tardive dyskinesia, and the concept of atypicality. J Clin Psychiatry. 2000;61(Suppl 3):16–21. [PubMed] [Google Scholar]
- 19.Caroff SN, Mann SC, Campbell EC, Sullivan KA. Movement disorders associated with atypical antipsychotic drugs. J Clin Psychiatry. 2002;63(Suppl 4):12–19. [PubMed] [Google Scholar]
- 20.Newcomer JW. Abnormalities of glucose metabolism associated with atypical antipsychotic drugs. J Clin Psychiatry. 2004;65 (Suppl 18):36–46. [PubMed] [Google Scholar]
- 21.Basu A, Meltzer HY. Differential trends in prevalence of diabetes and unrelated general medical illness for schizophrenia patients before and after the atypical antipsychotic era. Schizophr Res. 2006;86:99–109. doi: 10.1016/j.schres.2006.04.014. [DOI] [PubMed] [Google Scholar]
- 22.Casey DE. Dyslipidemia and atypical antipsychotic drugs. J Clin Psychiatry. 2004;65(Suppl 18):27–35. [PubMed] [Google Scholar]
- 23.Wirshing DA. Schizophrenia and obesity: impact of antipsychotic medications. J Clin Psychiatry. 2004;65(Suppl 18):13–26. [PubMed] [Google Scholar]
- 24.Patel JK, Pinals DA, Breier A. Schizophrenia and other psychoses. In: Tasman A, Kay J, Lieberman JA, editors. Psychiatry. 2. Wiley; Chichester: 2003. pp. 1131–1206. [Google Scholar]
- 25.Baptista T, Kin NM, Beaulieu S, de Baptista EA. Obesity and related metabolic abnormalities during antipsychotic drug administration: mechanisms, management and research perspectives. Pharmacopsychiatry. 2002;35:205–219. doi: 10.1055/s-2002-36391. [DOI] [PubMed] [Google Scholar]
- 26.Gardner DM, Baldessarini RJ, Waraich P. Modern antipsychotic drugs: a critical overview. CMAJ. 2005;172:1703–1711. doi: 10.1503/cmaj.1041064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Holt RI, Bushe C, Citrome L. Diabetes and schizophrenia 2005: are we any closer to understanding the link? J Psychopharmacol. 2005;19(6 Suppl):56–65. doi: 10.1177/0269881105058379. [DOI] [PubMed] [Google Scholar]
- 28.Spelman LM, Walsh PI, Sharifi N, Collins P, Thakore JH. Impaired glucose tolerance in first-episode drug-naïve patients with schizophrenia. Diabet Med. 2007;24:481–485. doi: 10.1111/j.1464-5491.2007.02092.x. [DOI] [PubMed] [Google Scholar]
- 29.Newcomer JW. Metabolic considerations in the use of anti-psychotic medications: a review of recent evidence. J Clin Psychiatry. 2007;68(Suppl 1):20–27. [PubMed] [Google Scholar]
- 30.Newcomer JW, Haupt DW. The metabolic effects of antipsychotic medications. Can J Psychiatry. 2006;51:480–491. doi: 10.1177/070674370605100803. [DOI] [PubMed] [Google Scholar]
- 31.Reaven GM. The metabolic syndrome: is this diagnosis necessary? Am J Clin Nutr. 2006;83:1237–1247. doi: 10.1093/ajcn/83.6.1237. [DOI] [PubMed] [Google Scholar]
- 32.Ferraioli A, Shirley KL, David P. The role of atypical antipsychotics in glucose/insulin dysregulation and the evolving role of the psychiatrist in a new era of drug treatment options. CNS Spectr. 2004;9:849–861. doi: 10.1017/s1092852900002261. [DOI] [PubMed] [Google Scholar]
- 33.Dwyer DS, Donohoe D, Lu XH, Aamodt EJ. Mechanistic connections between glucose/lipid disturbances and weight gain induced by antipsychotic drugs. Int Rev Neurobiol. 2005;65:211–247. doi: 10.1016/S0074-7742(04)65008-2. [DOI] [PubMed] [Google Scholar]
- 34.Houseknecht KL, Robertson AS, Zavadoski W, Gibbs EM, Johnson DE, Rollema H. Acute effects of atypical antipsychotics on whole-body insulin resistance in rats: implications for adverse metabolic effects. Neuropsychopharmacology. 2007;32:289–297. doi: 10.1038/sj.npp.1301209. [DOI] [PubMed] [Google Scholar]
- 35.Sasaki N, Iwase M, Uchizono Y, Nakamura U, Imoto H, Abe S, et al. The atypical antipsychotic clozapine impairs insulin secretion by inhibiting glucose metabolism and distal steps in rat pancreatic islets. Diabetologia. 2006;49:2930–2938. doi: 10.1007/s00125-006-0446-6. [DOI] [PubMed] [Google Scholar]
- 36.Haupt DW, Newcomer JW. Abnormalities in glucose regulation associated with mental illness and treatment. J Psychosom Res. 2002;53:925–933. doi: 10.1016/s0022-3999(02)00471-3. [DOI] [PubMed] [Google Scholar]
- 37.Kim SF, Huang AS, Snowman AM, Teuscher C, Snyder SH. From the cover: antipsychotic drug-induced weight gain mediated by histamine H1 receptor-linked activation of hypothalamic AMP-kinase. Proc Natl Acad Sci USA. 2007;104:3456–3459. doi: 10.1073/pnas.0611417104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dwyer DS, Bradley RJ, Kablinger AS, Freeman AM., III Glucose metabolism in relation to schizophrenia and antipsychotic drug treatment. Ann Clin Psychiatry. 2001;13:103–113. doi: 10.1023/a:1016671725396. [DOI] [PubMed] [Google Scholar]
- 39.Dwyer DS, Ardizzone TD, Bradley RJ. Psychoactive drugs affect glucose transport and the regulation of glucose metabolism. Int Rev Neurobiol. 2002;51:503–530. doi: 10.1016/s0074-7742(02)51015-1. [DOI] [PubMed] [Google Scholar]
- 40.Dwyer DS, Donohoe D. Induction of hyperglycemia in mice with atypical antipsychotic drugs that inhibit glucose uptake. Pharmacol Biochem Behav. 2003;75:255–260. doi: 10.1016/s0091-3057(03)00079-0. [DOI] [PubMed] [Google Scholar]
- 41.Bondy CA, Cheng CM. Insulin-like growth factor-1 promotes neuronal glucose utilization during brain development and repair processes. Int Rev Neurobiol. 2002;51:189–217. doi: 10.1016/s0074-7742(02)51006-0. [DOI] [PubMed] [Google Scholar]
- 42.Gunnell D, Holly JM. Do insulin-like growth factors underlie associations of birth complications, fetal and pre-adult growth with schizophrenia? Schizophr Res. 2004;67:309–311. doi: 10.1016/S0920-9964(03)00180-4. [DOI] [PubMed] [Google Scholar]
- 43.Kalkman HO. The role of the phosphatidylinositide 3-kinase-protein kinase B pathway in schizophrenia. Pharmacol Ther. 2006;110:117–134. doi: 10.1016/j.pharmthera.2005.10.014. [DOI] [PubMed] [Google Scholar]
- 44.Bjornholm M, Zierath JR. Insulin signal transduction in human skeletal muscle: identifying the defects in type II diabetes. Biochem Soc Trans. 2005;33:354–357. doi: 10.1042/BST0330354. [DOI] [PubMed] [Google Scholar]
- 45.Chang F, Lee JT, Navolanic PM, Steelman LS, Shelton JG, Blalock WL, et al. Involvement of PI3K/Akt pathway in cell cycle progression, apoptosis, and neoplastic transformation: a target for cancer chemotherapy. Leukemia. 2003;17:590–603. doi: 10.1038/sj.leu.2402824. [DOI] [PubMed] [Google Scholar]
- 46.Munafo MR, Thiselton DL, Clark TG, Flint J. Association of the NRG1 gene and schizophrenia: a meta-analysis. Mol Psychiatry. 2006;11:539–546. doi: 10.1038/sj.mp.4001817. [DOI] [PubMed] [Google Scholar]
- 47.Li BS, Ma W, Jaffe H, Zheng Y, Takahashi S, Zhang L, et al. Cyclin-dependent kinase-5 is involved in neuregulin-dependent activation of phosphatidylinositol 3-kinase and Akt activity mediating neuronal survival. J Biol Chem. 2003;278:35702–35709. doi: 10.1074/jbc.M302004200. [DOI] [PubMed] [Google Scholar]
- 48.Lai WS, Xu B, Westphal KG, Paterlini M, Olivier B, Pavlidis P, et al. Akt1 deficiency affects neuronal morphology and predisposes to abnormalities in prefrontal cortex functioning. Proc Natl Acad Sci USA. 2006;103:16906–16911. doi: 10.1073/pnas.0604994103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gould TD, Manji HK. Glycogen synthase kinase-3: a putative molecular target for lithium mimetic drugs. Neuropsychopharmacology. 2005;30:1223–1237. doi: 10.1038/sj.npp.1300731. [DOI] [PubMed] [Google Scholar]
- 50.Jope RS, Roh MS. Glycogen synthase kinase-3 (GSK3) in psychiatric diseases and therapeutic interventions. Curr Drug Targets. 2006;7:1421–1434. doi: 10.2174/1389450110607011421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lovestone S, Killick R, Di Forti M, Murray R. Schizophrenia as a GSK-3 dysregulation disorder. Trends Neurosci. 2007;30:142–149. doi: 10.1016/j.tins.2007.02.002. [DOI] [PubMed] [Google Scholar]
- 52.Kozlovsky N, Belmaker RH, Agam G. GSK-3 and the neurodevelopmental hypothesis of schizophrenia. Eur Neuropsychopharmacol. 2002;12:13–25. doi: 10.1016/s0924-977x(01)00131-6. [DOI] [PubMed] [Google Scholar]
- 53.Doble BW, Woodgett JR. GSK-3: tricks of the trade for a multitasking kinase. J Cell Sci. 2003;116:1175–1186. doi: 10.1242/jcs.00384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Beaulieu JM, Gainetdinov RR, Caron MG. The Akt-GSK-3 signaling cascade in the actions of dopamine. Trends Pharmacol Sci. 2007;28:166–172. doi: 10.1016/j.tips.2007.02.006. [DOI] [PubMed] [Google Scholar]
- 55.Beaulieu JM, Sotnikova TD, Yao WD, Kockeritz L, Woodgett JR, Gainetdinov RR, et al. Lithium antagonizes dopamine-dependent behaviors mediated by an AKT/glycogen synthase kinase 3 signaling cascade. Proc Natl Acad Sci USA. 2004;101:5099–5104. doi: 10.1073/pnas.0307921101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Beaulieu JM, Sotnikova TD, Marion S, Lefkowitz RJ, Gainetdinov RR, Caron MG. An Akt/beta-arrestin 2/PP2A signaling complex mediates dopaminergic neurotransmission and behavior. Cell. 2005;122:261–273. doi: 10.1016/j.cell.2005.05.012. [DOI] [PubMed] [Google Scholar]
- 57.Lei G, Xia Y, Johnson KM. The role of Akt-GSK-3beta signaling and synaptic strength in phencyclidine-induced neurodegeneration. Neuropsychopharmacology. 2007 doi: 10.1038/sj.npp.1301511. [e-pub ahead of print] [DOI] [PubMed] [Google Scholar]
- 58.Coyle JT. Glutamate and schizophrenia: beyond the dopamine hypothesis. Cell Mol Neurobiol. 2006;26:365–384. doi: 10.1007/s10571-006-9062-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ide M, Ohnishi T, Murayama M, Matsumoto I, Yamada K, Iwayama Y, et al. Failure to support a genetic contribution of AKT1 polymorphisms and altered AKT signaling in schizophrenia. J Neurochem. 2006;99:277–287. doi: 10.1111/j.1471-4159.2006.04033.x. [DOI] [PubMed] [Google Scholar]
- 60.Stopkova P, Saito T, Papolos DF, Vevera J, Paclt I, Zukov I, et al. Identification of PIK3C3 promoter variant associated with bipolar disorder and schizophrenia. Biol Psychiatry. 2004;55:981–988. doi: 10.1016/j.biopsych.2004.01.014. [DOI] [PubMed] [Google Scholar]
- 61.Duan S, Gao R, Xing Q, Du J, Liu Z, Chen Q, et al. A family-based association study of schizophrenia with polymorphisms at three candidate genes. Neurosci Lett. 2005;379:32–36. doi: 10.1016/j.neulet.2004.12.040. [DOI] [PubMed] [Google Scholar]
- 62.Saito T, Aghalar MR, Lachman HM. Analysis of PIK3C3 promoter variant in African-Americans with schizophrenia. Schizophr Res. 2005;76:361–362. doi: 10.1016/j.schres.2005.01.002. [DOI] [PubMed] [Google Scholar]
- 63.Jamra RA, Klein K, Villela AW, Becker T, Schulze TG, Schmael C, et al. Association study between genetic variants at the PIP5K2A gene locus and schizophrenia and bipolar affective disorder. Am J Med Genet B Neuropsychiatr Genet. 2006;141:663–665. doi: 10.1002/ajmg.b.30358. [DOI] [PubMed] [Google Scholar]
- 64.Schwab SG, Knapp M, Sklar P, Eckstein GN, Sewekow C, Borrmann-Hassenbach M, et al. Evidence for association of DNA sequence variants in the phosphatidylinositol-4-phosphate 5-kinase IIalpha gene (PIP5K2A) with schizophrenia. Mol Psychiatry. 2006;11:837–846. doi: 10.1038/sj.mp.4001864. [DOI] [PubMed] [Google Scholar]
- 65.Gunnell D, Lewis S, Wilkinson J, Georgieva L, Davey GS, Day IN, et al. IGF1, growth pathway polymorphisms and schizophrenia: a pooling study. Am J Med Genet B Neuropsychiatr Genet. 2007;144:117–120. doi: 10.1002/ajmg.b.30396. [DOI] [PubMed] [Google Scholar]
- 66.Dean B, Opeskin K, Pavey G, Hill C, Keks N. Changes in protein kinase C and adenylate cyclase in the temporal lobe from subjects with schizophrenia. J Neural Transm. 1997;104:1371–1381. doi: 10.1007/BF01294738. [DOI] [PubMed] [Google Scholar]
- 67.Opeskin K, Dean B, Pavey G, Hill C, Keks N, Copolov D. Neither protein kinase C nor adenylate cyclase are altered in the striatum from subjects with schizophrenia. Schizophr Res. 1996;22:159–164. doi: 10.1016/s0920-9964(96)00065-5. [DOI] [PubMed] [Google Scholar]
- 68.Hahn CG, Umapathy, Wang HY, Koneru R, Levinson DF, Friedman E. Lithium and valproic acid treatments reduce PKC activation and receptor-G protein coupling in platelets of bipolar manic patients. J Psychiatr Res. 2005;39:355–363. doi: 10.1016/j.jpsychires.2004.10.007. [DOI] [PubMed] [Google Scholar]
- 69.Borda T, Genaro AM, Cremaschi G. Haloperidol effect on intra-cellular signals system coupled to alpha1-adrenergic receptor in rat cerebral frontal cortex. Cell Signal. 1999;11:293–300. doi: 10.1016/s0898-6568(98)00065-5. [DOI] [PubMed] [Google Scholar]
- 70.Wan DC, Dean B, Pavey G, Copolov DL. Treatment with haloperidol or clozapine causes changes in dopamine receptors but not adenylate cyclase or protein kinase C in the rat forebrain. Life Sci. 1996;59:2001–2008. doi: 10.1016/s0024-3205(96)00551-6. [DOI] [PubMed] [Google Scholar]
- 71.Jardemark KE, Ninan I, Liang X, Wang RY. Protein kinase C is involved in clozapine’s facilitation of N-methyl-D-aspartate- and electrically evoked responses in pyramidal cells of the medial prefrontal cortex. Neuroscience. 2003;118:501–512. doi: 10.1016/s0306-4522(02)00976-4. [DOI] [PubMed] [Google Scholar]
- 72.Engl J, Laimer M, Niederwanger A, Kranebitter M, Starzinger M, Pedrini MT, et al. Olanzapine impairs glycogen synthesis and insulin signaling in L6 skeletal muscle cells. Mol Psychiatry. 2005;10:1089–1096. doi: 10.1038/sj.mp.4001729. [DOI] [PubMed] [Google Scholar]
- 73.Fatemi SH, Reutiman TJ, Folsom TD, Bell C, Nos L, Fried P, et al. Chronic olanzapine treatment causes differential expression of genes in frontal cortex of rats as revealed by DNA microarray technique. Neuropsychopharmacology. 2006;31:1888–1899. doi: 10.1038/sj.npp.1301002. [DOI] [PubMed] [Google Scholar]
- 74.Zhao Z, Ksiezak-Reding H, Riggio S, Haroutunian V, Pasinetti GM. Insulin receptor deficits in schizophrenia and in cellular and animal models of insulin receptor dysfunction. Schizophr Res. 2006;84:1–14. doi: 10.1016/j.schres.2006.02.009. [DOI] [PubMed] [Google Scholar]
- 75.Emamian ES, Hall D, Birnbaum MJ, Karayiorgou M, Gogos JA. Convergent evidence for impaired AKT1-GSK3beta signaling in schizophrenia. Nat Genet. 2004;36:131–137. doi: 10.1038/ng1296. [DOI] [PubMed] [Google Scholar]
- 76.Schwab SG, Hoefgen B, Hanses C, Hassenbach MB, Albus M, Lerer B, et al. Further evidence for association of variants in the AKT1 gene with schizophrenia in a sample of European sib-pair families. Biol Psychiatry. 2005;58:446–450. doi: 10.1016/j.biopsych.2005.05.005. [DOI] [PubMed] [Google Scholar]
- 77.Bajestan SN, Sabouri AH, Nakamura M, Takashima H, Keikhaee MR, Behdani F, et al. Association of AKT1 haplotype with the risk of schizophrenia in Iranian population. Am J Med Genet B Neuropsychiatr Genet. 2006;141:383–386. doi: 10.1002/ajmg.b.30291. [DOI] [PubMed] [Google Scholar]
- 78.Ikeda M, Iwata N, Suzuki T, Kitajima T, Yamanouchi Y, Kinoshita Y, et al. Association of AKT1 with schizophrenia confirmed in a Japanese population. Biol Psychiatry. 2004;56:698–700. doi: 10.1016/j.biopsych.2004.07.023. [DOI] [PubMed] [Google Scholar]
- 79.Xu MQ, Xing QH, Zheng YL, Li S, Gao JJ, He G, et al. Association of AKT1 gene polymorphisms with risk of schizophrenia and with response to antipsychotics in the Chinese population. J Clin Psychiatry. 2007;68:1358–1367. doi: 10.4088/jcp.v68n0906. [DOI] [PubMed] [Google Scholar]
- 80.Ohtsuki T, Inada T, Arinami T. Failure to confirm association between AKT1 haplotype and schizophrenia in a Japanese case–control population. Mol Psychiatry. 2004;9:981–983. doi: 10.1038/sj.mp.4001559. [DOI] [PubMed] [Google Scholar]
- 81.Liu YL, Fann CS, Liu CM, Wu JY, Hung SI, Chan HY, et al. Absence of significant associations between four AKT1 SNP markers and schizophrenia in the Taiwanese population. Psychiatr Genet. 2006;16:39–41. doi: 10.1097/01.ypg.0000180681.80546.f3. [DOI] [PubMed] [Google Scholar]
- 82.Norton N, Williams HJ, Dwyer S, Carroll L, Peirce T, Moskvina V, et al. Association analysis of AKT1 and schizophrenia in a UK case control sample. Schizophr Res. 2007;93:58–65. doi: 10.1016/j.schres.2007.02.006. [DOI] [PubMed] [Google Scholar]
- 83.Thiselton DL, Vladimirov VI, Kuo PH, McClay J, Wormley B, Fanous A, et al. AKT1 is associated with schizophrenia across multiple symptom dimensions in the Irish study of high density schizophrenia families. Biol Psychiatry. 2007;63:449–457. doi: 10.1016/j.biopsych.2007.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Amar S, Shaltiel G, Mann L, Shamir A, Dean B, Scarr E, et al. Possible involvement of post-dopamine D2 receptor signalling components in the pathophysiology of schizophrenia. Int J Neuropsychopharmacol. 2007;11:197–205. doi: 10.1017/S1461145707007948. [DOI] [PubMed] [Google Scholar]
- 85.Hashimoto R, Numakawa T, Ohnishi T, Kumamaru E, Yagasaki Y, Ishimoto T, et al. Impact of the DISC1 Ser704Cys polymorphism on risk for major depression, brain morphology and ERK signaling. Hum Mol Genet. 2006;15:3024–3033. doi: 10.1093/hmg/ddl244. [DOI] [PubMed] [Google Scholar]
- 86.Kang UG, Seo MS, Roh MS, Kim Y, Yoon SC, Kim YS. The effects of clozapine on the GSK-3-mediated signaling pathway. FEBS Lett. 2004;560:115–119. doi: 10.1016/S0014-5793(04)00082-1. [DOI] [PubMed] [Google Scholar]
- 87.Basta-Kaim A, Budziszewska B, Jaworska-Feil L, Tetich M, Kubera M, Leskiewicz M, et al. Antipsychotic drugs inhibit the human corticotropin-releasing-hormone gene promoter activity in neuro-2A cells—an involvement of protein kinases. Neuropsychopharmacology. 2006;31:853–865. doi: 10.1038/sj.npp.1300911. [DOI] [PubMed] [Google Scholar]
- 88.Lu XH, Bradley RJ, Dwyer DS. Olanzapine produces trophic effects in vitro and stimulates phosphorylation of Akt/PKB, ERK1/2, and the mitogen-activated protein kinase p38. Brain Res. 2004;1011:58–68. doi: 10.1016/j.brainres.2004.03.018. [DOI] [PubMed] [Google Scholar]
- 89.Lu XH, Dwyer DS. Second-generation antipsychotic drugs, olanzapine, quetiapine, and clozapine enhance neurite outgrowth in PC12 cells via PI3K/AKT, ERK, and pertussis toxin-sensitive pathways. J Mol Neurosci. 2005;27:43–64. doi: 10.1385/jmn:27:1:043. [DOI] [PubMed] [Google Scholar]
- 90.Ukai W, Ozawa H, Tateno M, Hashimoto E, Saito T. Neurotoxic potential of haloperidol in comparison with risperidone: implication of Akt-mediated signal changes by haloperidol. J Neural Transm. 2004;111:667–681. doi: 10.1007/s00702-004-0109-z. [DOI] [PubMed] [Google Scholar]
- 91.Dwyer DS, Lu XH, Bradley RJ. Cytotoxicity of conventional and atypical antipsychotic drugs in relation to glucose metabolism. Brain Res. 2003;971:31–39. doi: 10.1016/s0006-8993(03)02351-5. [DOI] [PubMed] [Google Scholar]
- 92.Shin SY, Choi BH, Ko J, Kim SH, Kim YS, Lee YH. Clozapine, a neuroleptic agent, inhibits Akt by counteracting Ca(2+)/calmodulin in PTEN-negative U-87MG human glioblastoma cells. Cell Signal. 2006;18:1876–1886. doi: 10.1016/j.cellsig.2006.02.004. [DOI] [PubMed] [Google Scholar]
- 93.Scassellati C, Bonvicini C, Perez J, Bocchio-Chiavetto L, Tura GB, Rossi G, et al. Association study of −1727 A/T, −50 C/T and (CAA)n repeat GSK-3beta gene polymorphisms with schizophrenia. Neuropsychobiology. 2004;50:16–20. doi: 10.1159/000077936. [DOI] [PubMed] [Google Scholar]
- 94.Meng J, Shi Y, Zhao X, Zhou J, Zheng Y, Tang R, et al. No significant association between the genetic polymorphisms in the GSK-3beta gene and schizophrenia in the Chinese population. J Psychiatr Res. 2007;42:365–370. doi: 10.1016/j.jpsychires.2007.01.005. [DOI] [PubMed] [Google Scholar]
- 95.Turunen JA, Peltonen JO, Pietiläinen OP, Hennah W, Loukola A, Paunio T, et al. The role of DTNBP1, NRG1, and AKT1 in the genetics of schizophrenia in Finland. Schizophr Res. 2007;91:27–36. doi: 10.1016/j.schres.2006.11.028. [DOI] [PubMed] [Google Scholar]
- 96.Ikeda M, Iwata N, Suzuki T, Kitajima T, Yamanouchi Y, Kinoshita Y, et al. No association of GSK3beta gene (GSK3B) with Japanese schizophrenia. Am J Med Genet B Neuropsychiatr Genet. 2005;134:90–92. doi: 10.1002/ajmg.b.30155. [DOI] [PubMed] [Google Scholar]
- 97.Lee KY, Ahn YM, Joo EJ, Jeong SH, Chang JS, Kim SC, et al. No association of two common SNPs at position −1727 A/T, −50 C/T of GSK-3 beta polymorphisms with schizophrenia and bipolar disorder of Korean population. Neurosci Lett. 2006;395:175–178. doi: 10.1016/j.neulet.2005.10.059. [DOI] [PubMed] [Google Scholar]
- 98.Kozlovsky N, Belmaker RH, Agam G. Low GSK-3beta immunoreactivity in postmortem frontal cortex of schizophrenic patients. Am J Psychiatry. 2000;157:831–833. doi: 10.1176/appi.ajp.157.5.831. [DOI] [PubMed] [Google Scholar]
- 99.Kozlovsky N, Belmaker RH, Agam G. Low GSK-3 activity in frontal cortex of schizophrenic patients. Schizophr Res. 2001;52:101–105. doi: 10.1016/s0920-9964(00)00174-2. [DOI] [PubMed] [Google Scholar]
- 100.Kozlovsky N, Shanon-Weickert C, Tomaskovic-Crook E, Kleinman JE, Belmaker RH, Agam G. Reduced GSK-3beta mRNA levels in postmortem dorsolateral prefrontal cortex of schizophrenic patients. J Neural Transm. 2004;111:1583–1592. doi: 10.1007/s00702-004-0166-3. [DOI] [PubMed] [Google Scholar]
- 101.Beasley C, Cotter D, Khan N, Pollard C, Sheppard P, Varndell I, et al. Glycogen synthase kinase-3beta immunoreactivity is reduced in the prefrontal cortex in schizophrenia. Neurosci Lett. 2001;302:117–120. doi: 10.1016/s0304-3940(01)01688-3. [DOI] [PubMed] [Google Scholar]
- 102.Kozlovsky N, Regenold WT, Levine J, Rapoport A, Belmaker RH, Agam G. GSK-3beta in cerebrospinal fluid of schizophrenia patients. J Neural Transm. 2004;111:1093–1098. doi: 10.1007/s00702-003-0127-0. [DOI] [PubMed] [Google Scholar]
- 103.Nadri C, Lipska BK, Kozlovsky N, Weinberger DR, Belmaker RH, Agam G. Glycogen synthase kinase (GSK)-3beta levels and activity in a neurodevelopmental rat model of schizophrenia. Brain Res Dev Brain Res. 2003;141:33–37. doi: 10.1016/s0165-3806(02)00639-9. [DOI] [PubMed] [Google Scholar]
- 104.Harrison PJ. The hippocampus in schizophrenia: a review of the neuropathological evidence and its pathophysiological implications. Psychopharmacology (Berl) 2004;174:151–162. doi: 10.1007/s00213-003-1761-y. [DOI] [PubMed] [Google Scholar]
- 105.Kozlovsky N, Belmaker RH, Agam G. Lack of effect of acute, subchronic, or chronic stress on glycogen synthase kinase-3beta protein levels in rat frontal cortex. Prog Neuropsychopharmacol Biol Psychiatry. 2002;26:1309–1312. doi: 10.1016/s0278-5846(02)00294-4. [DOI] [PubMed] [Google Scholar]
- 106.Kozlovsky N, Nadri C, Belmaker RH, Agam G. Lack of effect of mood stabilizers or neuroleptics on GSK-3 protein levels and GSK-3 activity. Int J Neuropsychopharmacol. 2003;6:117–120. doi: 10.1017/S1461145703003353. [DOI] [PubMed] [Google Scholar]
- 107.Beasley C, Cotter D, Everall I. An investigation of the Wnt-signalling pathway in the prefrontal cortex in schizophrenia, bipolar disorder and major depressive disorder. Schizophr Res. 2002;58:63–67. doi: 10.1016/s0920-9964(01)00376-0. [DOI] [PubMed] [Google Scholar]
- 108.Swatton JE, Sellers LA, Faull RL, Holland A, Iritani S, Bahn S. Increased MAP kinase activity in Alzheimer’s and Down syndrome but not in schizophrenia human brain. Eur J Neurosci. 2004;19:2711–2719. doi: 10.1111/j.0953-816X.2004.03365.x. [DOI] [PubMed] [Google Scholar]
- 109.Nadri C, Dean B, Scarr E, Agam G. GSK-3 parameters in postmortem frontal cortex and hippocampus of schizophrenic patients. Schizophr Res. 2004;71:377–382. doi: 10.1016/j.schres.2004.02.020. [DOI] [PubMed] [Google Scholar]
- 110.Nadri C, Kozlovsky N, Agam G, Bersudsky Y. GSK-3 parameters in lymphocytes of schizophrenic patients. Psychiatry Res. 2002;112:51–57. doi: 10.1016/s0165-1781(02)00191-9. [DOI] [PubMed] [Google Scholar]
- 111.Alimohamad H, Rajakumar N, Seah YH, Rushlow W. Anti-psychotics alter the protein expression levels of beta-catenin and GSK-3 in the rat medial prefrontal cortex and striatum. Biol Psychiatry. 2005;57:533–542. doi: 10.1016/j.biopsych.2004.11.036. [DOI] [PubMed] [Google Scholar]
- 112.Alimohamad H, Sutton L, Mouyal J, Rajakumar N, Rushlow WJ. The effects of antipsychotics on beta-catenin, glycogen synthase kinase-3 and dishevelled in the ventral midbrain of rats. J Neurochem. 2005;95:513–525. doi: 10.1111/j.1471-4159.2005.03388.x. [DOI] [PubMed] [Google Scholar]
- 113.Kozlovsky N, Amar S, Belmaker RH, Agam G. Psychotropic drugs affect Ser9-phosphorylated GSK-3 beta protein levels in rodent frontal cortex. Int J Neuropsychopharmacol. 2006;9:337–342. doi: 10.1017/S1461145705006097. [DOI] [PubMed] [Google Scholar]
- 114.Li X, Rosborough KM, Friedman AB, Zhu W, Roth KA. Regulation of mouse brain glycogen synthase kinase-3 by atypical antipsychotics. Int J Neuropsychopharmacol. 2007;10:7–19. doi: 10.1017/S1461145706006547. [DOI] [PubMed] [Google Scholar]
- 115.González-Maeso J, Weisstaub NV, Zhou M, Chan P, Ivic L, Ang R, et al. Hallucinogens recruit specific cortical 5-HT(2A) receptor-mediated signaling pathways to affect behavior. Neuron. 2007;53:439–452. doi: 10.1016/j.neuron.2007.01.008. [DOI] [PubMed] [Google Scholar]
- 116.Kenakin T. Ligand-selective receptor conformations revisited: the promise and the problem. Trends Pharmacol Sci. 2003;24:346–354. doi: 10.1016/S0165-6147(03)00167-6. [DOI] [PubMed] [Google Scholar]
- 117.Schmid CL, Raehal KM, Bohn LM. Agonist-directed signaling of the serotonin 2A receptor depends on beta-arrestin-2 interactions in vivo. Proc Natl Acad Sci USA. 2008;105:1079–1084. doi: 10.1073/pnas.0708862105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Gay EA, Urban JD, Nichols DE, Oxford GS, Mailman RB. Functional selectivity of D2 receptor ligands in a Chinese hamster ovary hD2L cell line: evidence for induction of ligand-specific receptor states. Mol Pharmacol. 2004;66:97–105. doi: 10.1124/mol.66.1.97. [DOI] [PubMed] [Google Scholar]