

Abstract
We describe Vivaldi (VIsualization and VALidation DIsplay; http://pdbe.org/vivaldi), a web-based service for the analysis, visualization, and validation of NMR structures in the Protein Data Bank (PDB). Vivaldi provides access to model coordinates and several types of experimental NMR data using interactive visualization tools, augmented with structural annotations and model-validation information. The service presents information about the modeled NMR ensemble, validation of experimental chemical shifts, residual dipolar couplings, distance and dihedral angle constraints, as well as validation scores based on empirical knowledge and databases. Vivaldi was designed for both expert NMR spectroscopists and casual non-expert users who wish to obtain a better grasp of the information content and quality of NMR structures in the public archive. © Proteins 2013. © 2012 Wiley Periodicals, Inc.
Keywords: Protein Data Bank, nuclear magnetic resonance, chemical shifts, distance constraints, angular constraints, residual dipolar couplings
INTRODUCTION
The Protein Data Bank (PDB)1,2 archive is a rich repository of data and information on the structure and function of biologically relevant macromolecules and their complexes. The archive currently contains over 84,500 entries (referencing over 28,000 unique UniProt3 accession codes), of which almost 10,000 NMR-derived structures (almost 5000 unique UniProt codes, Table I). The PDB archive is managed by the Worldwide Protein Data Bank organization (wwPDB),4 which consists of the Research Collaboratory for Structural Bioinformatics (RCSB)5 and the BioMagResBank (BMRB)6 in the USA, the Protein Data Bank Japan (PDBj),7 and the Protein Data Bank in Europe (PDBe; http://pdbe.org/).8 Atomic models deposited to the PDB are curated by RCSB, PDBe, and PDBj, while experimental NMR data are processed by BMRB. As one of the founding wwPDB partners, PDBe provides annotated data on three-dimensional (3D) structures of biomacromolecules to the scientific community as well as advanced services based on these structures.
Table I.
Data Coverage
| Source | PDB archive | NMR entries | |
|---|---|---|---|
| Total number of entries | PDB1,2 | 84,508 | 9616 |
| Unique UniProt3 accession codes referenced | SIFTS49 | 28,104 | 4822 |
| Pfam50 sequence families referenced | SIFTS | 6143 | 2455 |
| CATH51 domain architectures present | SIFTS | 2549 | 660 |
| SCOP52 domain architectures present | SIFTS | 4191 | 1150 |
| Cluster analysis | OLDERADO14 | − | 7289 |
| Chemical shift analysis | VASCO28 | − | 3361 |
| Distance constraints | BMRB-NRG36 | − | 5978 |
| Dihedral constraints | BMRB-NRG | − | 3850 |
| RDCs | BMRB-NRG | − | 602 |
| Validation scores | NRG-CING47 | − | 9491 |
Coverage of the protein universe in the PDB, and data available in Vivaldi for NMR entries (as of September 12, 2012). Up to date data coverage statistics can be found at http://pdbe.org/nmrstats/.
As the PDB archive continues to grow, the provision of adequate validation tools to its users becomes increasingly important.9 In many cases, the archive contains multiple structures of the same or similar molecules, for example, solved by competing groups or by different experimental techniques, with different ligands or containing mutations. Choosing the most appropriate structure in such cases is not an easy task for experts, let alone for non-specialist users. Even if only a single structure is known of a molecule of interest, it is still essential to assess whether the structure is suitable for the intended use.
A number of validation software packages are available,10–12 but a single robust and widely-accepted validation score or standard set of validation criteria is yet to be defined for NMR-derived biomacromolecular structures. Therefore, a wwPDB NMR validation task force (VTF) has been convened to define standard validation criteria that will be applied by all wwPDB partners to all depositions of NMR structures (http://www.wwpdb.org/workshop/2010/nmr_validation.html), in analogy with the wwPDB X-ray VTF.13 In an effort to make information about the quality of NMR structures in the PDB accessible to the wider scientific community, PDBe has developed an interactive web-based tool called Vivaldi (Visualization and Validation Display; http://pdbe.org/vivaldi; Fig.1).
Figure 1.
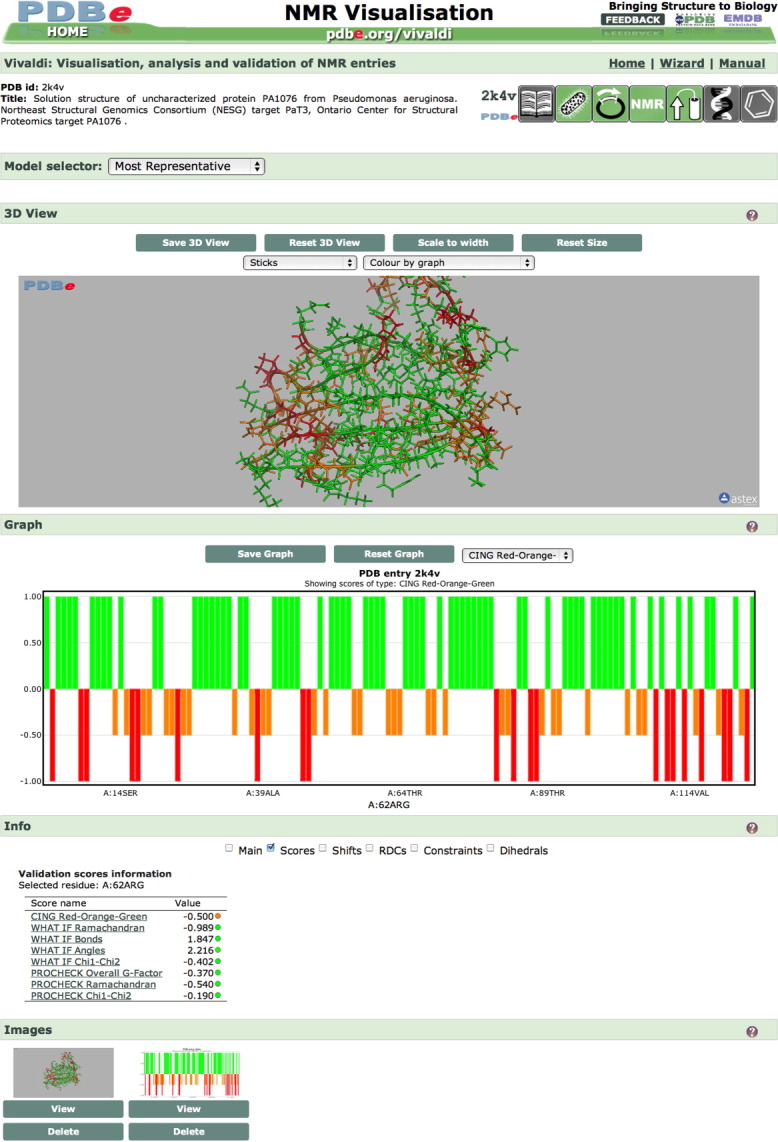
Layout of a Vivaldi page showing the default view for protein PA1076 from Pseudomonas aeruginosa (PDB entry 2k4v).15 The page contains a header including PDBprints,9 an interactive 3D viewer (OpenAstexViewer),48 a graph and a textual information section. The most representative model according to OLDERADO14 cluster analysis is displayed in the 3D viewer and the graph shows the NRG-CING47 red-orange-green (ROG) scores for this protein.
MATERIALS AND METHODS
Several types of information are accessible through the Vivaldi service, including information about the homogeneity of the modeled structure ensemble, validation of the available experimental NMR data, and knowledge-based validation of the modeled conformations. The decisions on which sources of data and which validation criteria to include in Vivaldi were based on three factors: (a) accessibility, (b) the sources being well-established and published, and (c) data coverage of the PDB archive. In the future, Vivaldi will be modified to accommodate the recommendations of the wwPDB NMR VTF.
Typically, an NMR entry in the PDB archive contains 20 models of a macromolecule or a complex. For all intents and purposes, these are deposited in an arbitrary order and therefore no single model can be assumed to be more important than the others without analyzing the structure ensemble in more detail. One such type of analysis is performed by OLDERADO,14 which is used by PDBe to determine core domains within proteins and to cluster together individual models of the ensemble. OLDERADO identifies the most representative model of each cluster and the most representative model of the entire ensemble. With the Vivaldi web service, users can interactively explore the results of this cluster and domain analysis, overlay different models and visualize similarities and differences within and between clusters. Furthermore, the most representative model as identified by OLDERADO is used throughout Vivaldi as the one displayed by default (rather than, for instance, the first model of the ensemble). Supporting Information Figure S1(a) shows representative cluster models as identified by OLDERADO for PDB entry 2k4v,15 while Supporting Information Figure S1(b) highlights the domains on the most representative model of the ensemble.
To help users assess the local variability in the deposited ensemble of structures, Vivaldi calculates a simple dihedral order parameter,16 S2, for each residue in protein and nucleic acid molecules [Eq.(1)] and presents this information as a graph of S2 vs. residue number [Supporting Information Fig. S1(c)]. Selecting a residue in the graph displays a table with the dihedral angle values, pie-charts for the relevant dihedral angles and, if appropriate, a Ramachandran plot [Supporting Information Fig. S1(d)].
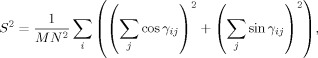 |
(1) |
where γ are backbone dihedral angles (φ and ψ for proteins and α–ζ for nucleic acids), index i runs over the M different dihedral angles and index j runs over the N conformers of the ensemble. Values of S2 close to 1 indicate that the dihedral angles show little variation across the ensemble.
Chemical shifts are arguably the most studied NMR parameters with respect to the covalent structure, local conformations, and immediate spatial surroundings. The complex dependence of the chemical shift on an atom's chemical environment has resulted in a wide variety of shift prediction and validation tools based on different approaches (e.g., quantum-chemical calculations and neural networks) and taking into account different aspects of the chemical environment (e.g., solvent effects, aromatic ring currents, and hydrogen bonds). Some of the more recent software packages include AVS,17 CamShift,18 LACS,19,20 TALOS+,21 ShiftX2,22,23 SPARTA+,24,25 CheShift,26 DANGLE,27 and VASCO.28 As deposition of chemical shift information has been mandatory for NMR structures since December 2010, chemical shifts are becoming an increasingly important source of data for validating PDB structures.
For Vivaldi, VASCO28 validates experimental chemical shifts of proteins in a two-step process. First, the optimal referencing offset for each nucleus type (1H, 15N, 13Caliphatic, 13Caromatic, and 13C′) is calculated by comparing the reported chemical shifts to a set of protein chemical shifts that were correctly referenced by hand. Second, for each atom a database of chemical shifts is queried to identify atoms in a similar environment, defined as a combination of atom and residue type, secondary structure, and solvent accessibility. If a sufficient number of similar atoms are found, the Z-score of the reported chemical shift is calculated and if the absolute Z-score exceeds a value of 3 (99.7% confidence interval assuming a true normal distribution) the shift is labeled as an outlier.
Vivaldi shows these outliers as solid spheres in its 3D viewer and as bars in the interactive graph [Fig.2(a,b)] with default coloring for both the spheres and the bars varying smoothly from green (Z-score < 2) via yellow (Z-score = 3.5) to red (Z-score > 5) depending on the degree of the deviation from the expected range. Aromatic ring currents, which can have a profound influence on chemical shifts, are not explicitly taken into account by VASCO. Therefore, aromatic rings in the structure are highlighted in the 3D viewer as stick models. Furthermore, the user can select a residue by clicking on a bar in the graph or on an atom in the 3D viewer and thereby obtain a table of the original chemical shift values and the VASCO scores for the nuclei in that residue.
Figure 2.

Vivaldi visualization of experimental data for protein PA1076 from Pseudomonas aeruginosa (PDB entry 2k4v).15 (a,b) Presentation of VASCO28 analysis of chemical shifts. (a) Nuclei with unusual chemical shifts are shown as spheres in the 3D viewer; aromatic residues are shown as sticks, to remind the user that the effect of aromatic ring currents is not explicitly accounted for in the VASCO analysis. (b) The same information is displayed in an interactive graph vs. residue number. (c,d) Analysis of distance constraints. (c) The most representative model of the ensemble is shown, colored by rigid-body domains as determined by OLDERADO.14 All long-range distance constraints (five or more residues apart in sequence) are shown as green sticks in the 3D viewer. (d) Graph of the number of long-range distance constraints per residue. The shaded area of the graph corresponds to helix 91–105 (UniProt numbering). (e) The chemical groups, for example N–HN, for which RDCs are available are shown as balls and sticks. Principal axes of the alignment tensor are also displayed. (f) Correlation plot of calculated vs. experimental RDCs. For each datapoint, the middle bar shows the calculated RDC averaged over the ensemble, and the box represents the standard deviation, while the whiskers are the minimum and maximum values calculated from the ensemble.
Distance and dihedral angle constraints, together with residual dipolar couplings (RDCs) constitute the bulk of the experimental data used during structure calculation for the majority of NMR-derived macromolecular structures in the PDB. Although these types of experimental data are used directly for structure calculation (hence, the models are expected to fit them rather well), validation can still pinpoint problems. The Queen29 and PSVS10 validation suites provide analyses of distance constraints, whereas PALES,30 REDCAT,31 and MODULE232 can be used to analyze RDC data.
Vivaldi retrieves pre-processed CCPN projects,33 which include all the constraints, from the Database Of Converted Restraints (DOCR),34 which is part of the NMR Restraints Grid (NRG)35,36 at BMRB. Distance constraints are further processed to classify them into groups, that is, intra-residual, short (1 or 2 residues apart in sequence), medium (3 or 4 residues apart), and long-range (5 or more residues apart), as well as to identify constraint violations for each group. Vivaldi can show violated and satisfied constraints, graphically in charts and in the 3D viewer as well as in tables [Fig.2(c,d)]. Any individual constraint can be selected to show the atoms involved, the actual inter-atomic distance(s) from the displayed model(s) and the constraint's lower and upper limit. Weighted summations (d−6) were used for constraints involving ambiguous assignments or magnetically equivalent groups (e.g., methyl groups).37
Dihedral-angle constraints are commonly obtained from an analysis of backbone chemical shifts by programs such as TALOS+21 and DANGLE27 or, less frequently today, from experimental measurements of scalar coupling constants. As with distance constraints, Vivaldi presents these constraints and their violations interactively in the 3D viewer and as graphs. Selecting a residue with reported dihedral angle constraints will display a table as well as a Ramachandran plot and/or pie-charts for each dihedral angle as applicable, highlighting the constraint limits and angles calculated from the displayed models in a fashion similar to that shown in Supporting Information Figure S1(d).
RDC38,39 constraints are obtained from the same resource at BMRB as the distance and torsion angle constraints. Alignment tensors, which are generally not deposited together with the RDC constraints, are calculated locally by fitting one tensor per model in the NMR ensemble per alignment medium. If multiple RDC types have been deposited for a single alignment medium (e.g., N–HN, Cα–Hα, N–Cα), Vivaldi checks whether the RDC constraints were deposited as their original experimental values or as scaled values to match N–HN RDC values. The optimal alignment tensor is obtained by minimizing the RMSD between the experimental and recalculated RDC values for the alignment tensor.
The Vivaldi 3D viewer can show the principal axes of the fitted alignment tensor as three orthogonal bars tied to the molecule's orientation in the viewer and the RDC constraints are visualized by showing the atoms involved as spheres [Fig.2(e)]. Further, fitted and experimental RDC values are presented as bar and scatter plots [Fig. 2(f)]. Details about the fitted tensor, that is tensor magnitude (Da) and rhombicity (R), as well as three values relating to the goodness-of-fit, that is RMS deviation (Hz), Pearson R-value, and Cornilescu Q-value,40 are shown in a separate information section. When a residue is selected from a bar chart or in the 3D viewer, experimental and fitted values of all corresponding RDC constraints are listed.
Empirical knowledge about the structure of biological macromolecules can be used to validate local conformations and the overall fold. A number of programs, such as WHATIF,41 PROCHECK,42 WHATCHECK,43 and Verify3D44 can be used to assess the quality of macromolecular structures. More recent software packages including PSVS,10 ProSA-web,45 MolProbity,46 and CING12,47 combine and extend them to gain further insight into the quality of an atomistic structure model.
Residue-based quality scores are extracted from the external NRG-CING database, including a general ROG score that labels residues as red, orange, or green depending on a series of quality measures. Several PROCHECK42 and WHATIF41 scores are also shown by Vivaldi. As for any type of information, residue-based scores can be visualized as a graph, in a table or in the 3D viewer. Figure1 shows the CING ROG score for PDB entry 2k4v.15
For any NMR entry in the PDB, Vivaldi can be launched from the corresponding entry page at PDBe or from a direct shortcut URL (http://pdbe.org/vivaldi/NNNN where NNNN is the four-character PDB code of the entry). Alternatively, the homepage of the NMR resource at PDBe (http://pdbe.org/nmr) provides a quick-access form into which the PDB code of interest can be entered. Finally, Vivaldi can be accessed from its own webpage (http://pdbe.org/vivaldi) and the information that is displayed can be customized by means of a user-friendly wizard. On the Vivaldi page of a PDB entry, the available information can be accessed, analyzed and visualized in a multitude of ways. High-quality images from the 3D viewer and the interactive graphs can be saved for use in publications, presentations, or teaching resources. The amount and type of information that is available for a particular entry depends on what type of experimental data (if any) was deposited, whether an ensemble or a single model was deposited, and so forth. Table I lists all the types of information presented in Vivaldi, its sources, and the number of PDB entries for which it is available. All available data for each entry is assembled and presented in a consistent and intuitive way that makes it useful to both NMR experts and non-expert users.
Generating the information used by Vivaldi involves collection and format conversion of relevant data on the one hand and data presentation on the other. The first aspect is dealt with by automated Python scripts that carry out weekly checks for new, updated or removed NMR entries in the PDB and for updated validation reports. These scripts import, check, and match data in different formats and ensure their internal consistency. This is necessary as curation and annotation of coordinates and constraints is performed separately in current wwPDB curation practice. Finally, the gathered data are output as objects in JSON files, which are used for presentation to the end user. A modified version of the OpenAstexViewer 3.0 Java applet48 accommodating some of the specific features of Vivaldi is used as 3D display engine, while the Flotr JavaScript Plotting Library (http://solutoire.com/flotr) is used to generate charts and graphs.
RESULTS AND DISCUSSION
Table I summarizes the types of data that can be encountered for NMR structures and the number of PDB entries for which each type of data is available in Vivaldi at the time of manuscript submission (weekly updated statistics are available from the PDBe website http://pdbe.org/nmrstats). While all NMR entries can be viewed in Vivaldi and almost all have at least a geometric validation report in the NRG-CING database, only about 60% have associated experimental constraints data deposited and processed for further analysis or visualization and only about a third have VASCO reports on chemical shift outliers at present. These numbers are set to improve, since deposition of both the experimental chemical shifts and constraints data is now mandatory (http://wwpdb.org/policy.html).
The major motivation for developing Vivaldi is to provide the users of the PDB archive (expert NMR spectroscopists and non-experts alike) with an easily accessible way to view and analyze NMR structures, their associated experimental data, and validation-related information. To demonstrate its capabilities, we present two use cases that demonstrate how Vivaldi can help evaluate the suitability of a particular entry for a user's needs.
Example 1. Analysis of RDCs in the context of other data for uncharacterized protein PA1076 from Pseudomonas aeruginosa, PDB entry 2k4v.15 The VASCO analysis of the chemical shifts [Fig.2(a,b)] indicates that the 23 atoms with unusual chemical shift values are spread evenly throughout the structure and many of them are in the vicinity of aromatic rings. This information confirms that the structure and chemical shift values are compatible. Furthermore, OLDERADO analysis suggests that the majority of the protein residues form one rigid body domain [Fig. 2(c), orange], including residues 91–105 (UniProt3 sequence numbering), which form an α-helix. However, review of the long-range distance constraints indicates that there is little support for the exact positioning of this helix [Fig. 2(d)]. Indeed, solving the structure of PA1076 with only distance and dihedral angle constraints did not allow for a reliable placement of the helix (data not shown). Introducing RDC data [Fig. 2(e,f)] allowed the positioning of helix 91–105 with more confidence.
Example 2. Selecting a structure from a set of identical or similar proteins: comparing two structures for protein HP_0495 from Helicobacter pylori (PDB entries 2h9z53 and 2joq15), solved by different structural genomics projects. While in this case the two entries only differ by their purification tags, a similar investigation could be performed for homologous proteins. To compare two structures, one needs to launch two instances of Vivaldi in separate browser windows. In this case, the two structures are similar, but the mostly red and orange ROG scores from the NRG-CING database for entry 2h9z suggest that there may be problems with this structure [Fig.3(c)], whereas the ROG scores for entry 2joq [Fig. 3(a)] are mostly green. Comparison of several knowledge-based scores available in Vivaldi confirms that both the Ramachandran statistics (not shown) and unusually short distance analysis [Fig. 3(d,e)] are slightly less favorable for 2h9z. However, analysis of the number of long-range distance constraints available for each residue reveals that there are significant local differences between the two structures in the residue range 24–40 (UniProt3 sequence numbering), which spans between Strands 1 and 2 of the central β-sheet and includes helix I (residues 27–34). Only one residue from this range (Leu 34) has a significant number of long-range distance constraints (highlighted in the graph by a single green bar) to other parts of the molecule in 2h9z, while 2joq contains 10 residues that each have 10 or more long-range distance constraints (green bars), which would presumably help position helix I and surrounding loops relative to the rest of the protein with more confidence [Fig. 3(f,g)]. While a detailed analysis of the differences between the two structures is beyond the scope of this paper, use of Vivaldi reveals these differences and may direct further investigation. Indeed, overlaying the representative models from 2h9z and 2joq [Fig. 3(b)] shows that the positioning of helix I and surrounding loops differs significantly and the PDBeFold55 service excludes this region (residues 23–42 and 45–51) from the structural alignment of the two models. While it is not trivial to pinpoint the exact reasons for these differences, it is interesting to note that BMRB entry 15,101 associated with 2h9z has only 485 assigned chemical shift values, of which 159 belong to 1H nuclei, whereas BMRB entry 15,190 associated with 2joq has 1157 assigned chemical shifts, of which 653 belong to 1H nuclei. Completeness of assignments and consequently the number and level of ambiguity of distance constraints strongly depend on the number, types, and resolutions of collected spectra, as well as on the usually labor-intensive step of spectral analysis. At this point one can only speculate if the observed differences are due to genuine differences in experimental conditions (spectra for 2h9z were collected at 35°C, and for 2joq at 25°C), to different types and resolutions of collected data, to variations in data analysis protocols or to some combination of these factors.
Figure 3.

Comparison of two NMR solution structures for protein HP_0495 from Helicobacter pylori (PDB codes 2joq, left, and 2h9z, right). (a,c) The 3D viewers show the most representative model of each ensemble, colored by the NRG-CING red-orange-green scores, which are calculated for the entire ensemble. (b) Superposition of the most representative models from the two structures, highlighting the differences in the conformation of helix I and adjacent loops. Image created in VMD54 (d,e). Graphs of per-residue WHATIF scores reporting unusual short distances in the entire ensemble. (f,g) Graphs of the number of long-range distance constraints for each residue. Long-range constraints are defined as connecting two atoms that are five or more residues apart in the amino-acid sequence.
CONCLUSION
A powerful interactive web-based tool has been developed to facilitate the analysis, visualization and validation of NMR structure ensembles from the PDB. It combines structural, experimental, and validation data from a variety of sources and presents them in a consistent fashion. A user-friendly interface, including a 3D viewer, tightly coupled with charts and tables, enables both experts and non-experts in NMR structure determination to analyze NMR ensembles and to assess their quality.
Acknowledgments
The authors wish to thank Dr W.F. Vranken for discussions on the concept and technical details of the Vivaldi service, Drs J.F. Doreleijers, Ch. Schulte and E. Ulrich for technical assistance in obtaining data from the BMRB NMR Restraints Grid, and Prof. M. Sattler for valuable comments on an early version of Vivaldi. Mrs P. Haslam's help in preparation of the manuscript is greatly appreciated. We would also like to acknowledge our colleagues in the PDBe team for testing and constructive criticism, as well as technical assistance in implementing and maintaining the service. Finally, we wish to thank the structural biology community for depositing their macromolecular structures and experimental data in the PDB and BMRB archives.
Supplementary material
Additional Supporting Information may be found in the online version of this article.
REFERENCES
- 1.Berman HM. The Protein Data Bank: a historical perspective. Acta Crystallogr A. 2008;64:88–95. doi: 10.1107/S0108767307035623. [DOI] [PubMed] [Google Scholar]
- 2.Bernstein FC, Koetzle TF, Williams GJB, Meyer EF, Brice MD, Rodgers JR, Kennard O, Shimanouchi T, Tasumi M. Protein Data Bank—computer-based archival file for macromolecular structures. J Mol Biol. 1977;112:535–542. doi: 10.1016/s0022-2836(77)80200-3. [DOI] [PubMed] [Google Scholar]
- 3.UniProt Consortium. Reorganizing the protein space at the Universal Protein Resource (UniProt) Nucleic Acids Res. 2012;40:D71–D75. doi: 10.1093/nar/gkr981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Berman H, Henrick K, Nakamura H, Markley JL. The worldwide Protein Data Bank (wwPDB): ensuring a single, uniform archive of PDB data. Nucleic Acids Res. 2007;35:D301–D303. doi: 10.1093/nar/gkl971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. The Protein Data Bank. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ulrich EL, Akutsu H, Doreleijers JF, Harano Y, Ioannidis YE, Lin J, Livny M, Mading S, Maziuk D, Miller Z, Nakatani E, Schulte CF, Tolmie DE, Wenger RK, Yao HY, Markley JL. BioMagResBank. Nucleic Acids Res. 2008;36:D402–D408. doi: 10.1093/nar/gkm957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kinjo AR, Suzuki H, Yamashita R, Ikegawa Y, Kudou T, Igarashi R, Kengaku Y, Cho H, Standley DM, Nakagawa A, Nakamura H. Protein Data Bank Japan (PDBj): maintaining a structural data archive and resource description framework format. Nucleic Acids Res. 2012;40:D453–D460. doi: 10.1093/nar/gkr811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Velankar S, Alhroub Y, Best C, Caboche S, Conroy MJ, Dana JM, Fernandez Montecelo MA, van Ginkel G, Golovin A, Gore SP, Gutmanas A, Haslam P, Hendrickx PMS, Heuson E, Hirshberg M, John M, Lagerstedt I, Mir S, Newman LE, Oldfield TJ, Patwardhan A, Rinaldi L, Sahni G, Sanz-Garcia E, Sen S, Slowley R, Suarez-Uruena A, Swaminathan GJ, Symmons MF, Vranken WF, Wainwright M, Kleywegt GJ. PDBe: Protein Data Bank in Europe. Nucleic Acids Res. 2012;40:D445–D452. doi: 10.1093/nar/gkr998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Velankar S, Kleywegt GJ. The Protein Data Bank in Europe (PDBe): bringing structure to biology. Acta Crystallogr D. 2011;67:324–330. doi: 10.1107/S090744491004117X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bhattacharya A, Tejero R, Montelione GT. Evaluating protein structures determined by structural genomics consortia. Proteins. 2007;66:778–795. doi: 10.1002/prot.21165. [DOI] [PubMed] [Google Scholar]
- 11.Berjanskii M, Liang YJ, Zhou JJ, Tang P, Stothard P, Zhou Y, Cruz J, MacDonell C, Lin GH, Lu P, Wishart DS. PROSESS: a protein structure evaluation suite and server. Nucleic Acids Res. 2010;38:W633–W640. doi: 10.1093/nar/gkq375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Doreleijers JF, Sousa da Silva AW, Krieger E, Nabuurs SB, Spronk CAEM, Stevens TJ, Vranken WF, Vriend G, Vuister GW. CING: an integrated residue-based structure validation program suite. J Biomol NMR, NMR. 2012;54:267–283. doi: 10.1007/s10858-012-9669-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Read RJ, Adams PD, Arendall WB, Brunger AT, Emsley P, Joosten RP, Kleywegt GJ, Krissinel EB, Lutteke T, Otwinowski Z, Perrakis A, Richardson JS, Sheffler WH, Smith JL, Tickle IJ, Vriend G, Zwart PH. A new generation of crystallographic validation tools for the Protein Data Bank. Structure. 2011;19:1395–1412. doi: 10.1016/j.str.2011.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kelley LA, Sutcliffe MJ. OLDERADO: on-line database of ensemble representatives and domains. Protein Sci. 1997;6:2628–2630. doi: 10.1002/pro.5560061215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lemak A, Gutmanas A, Chitayat S, Karra M, Fares C, Sunnerhagen M, Arrowsmith CH. A novel strategy for NMR resonance assignment and protein structure determination. J Biomol NMR. 2011;49:27–38. doi: 10.1007/s10858-010-9458-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hyberts SG, Goldberg MS, Havel TF, Wagner G. The solution structure of Eglin-C based on measurements of many NOEs and coupling-constants and its comparison with X-ray structures. Protein Sci. 1992;1:736–751. doi: 10.1002/pro.5560010606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Moseley HNB, Sahota G, Montelione GT. Assignment validation software suite for the evaluation and presentation of protein resonance assignment data. J Biomol NMR. 2004;28:341–355. doi: 10.1023/B:JNMR.0000015420.44364.06. [DOI] [PubMed] [Google Scholar]
- 18.Kohlhoff KJ, Robustelli P, Cavalli A, Salvatella X, Vendruscolo M. Fast and accurate predictions of protein NMR chemical shifts from interatomic distances. J Am Chem Soc. 2009;131:13894–13895. doi: 10.1021/ja903772t. [DOI] [PubMed] [Google Scholar]
- 19.Wang LY, Eghbalnia HR, Bahrami A, Markley JL. Linear analysis of carbon-13 chemical shift differences and its application to the detection and correction of errors in referencing and spin system identifications. J Biomol NMR. 2005;32:13–22. doi: 10.1007/s10858-005-1717-0. [DOI] [PubMed] [Google Scholar]
- 20.Wang LY, Markley JL. Empirical correlation between protein backbone N-15 and C-13 secondary chemical shifts and its application to nitrogen chemical shift re-referencing. J Biomol NMR. 2009;44:95–99. doi: 10.1007/s10858-009-9324-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shen Y, Delaglio F, Cornilescu G, Bax A. TALOS+ : a hybrid method for predicting protein backbone torsion angles from NMR chemical shifts. J Biomol NMR. 2009;44:213–223. doi: 10.1007/s10858-009-9333-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Han B, Liu YF, Ginzinger SW, Wishart DS. SHIFTX2: significantly improved protein chemical shift prediction. J Biomol NMR. 2011;50:43–57. doi: 10.1007/s10858-011-9478-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Neal S, Nip AM, Zhang HY, Wishart DS. Rapid and accurate calculation of protein H-1, C-13 and N-15 chemical shifts. J Biomol NMR. 2003;26:215–240. doi: 10.1023/a:1023812930288. [DOI] [PubMed] [Google Scholar]
- 24.Shen Y, Bax A. Protein backbone chemical shifts predicted from searching a database for torsion angle and sequence homology. J Biomol NMR. 2007;38:289–302. doi: 10.1007/s10858-007-9166-6. [DOI] [PubMed] [Google Scholar]
- 25.Shen Y, Bax A. SPARTA+: a modest improvement in empirical NMR chemical shift prediction by means of an artificial neural network. J Biomol NMR. 2010;48:13–22. doi: 10.1007/s10858-010-9433-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vila JA, Arnautova YA, Martin OA, Scheraga HA. Quantum-mechanics-derived C-13(alpha) chemical shift server (CheShift) for protein structure validation. Proc Natl Acad Sci USA. 2009;106:16972–16977. doi: 10.1073/pnas.0908833106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cheung MS, Maguire ML, Stevens TJ, Broadhurst RW. DANGLE: a Bayesian inferential method for predicting protein backbone dihedral angles and secondary structure. J Magn Reson. 2010;202:223–233. doi: 10.1016/j.jmr.2009.11.008. [DOI] [PubMed] [Google Scholar]
- 28.Rieping W, Vranken WF. Validation of archived chemical shifts through atomic coordinates. Proteins. 2010;78:2482–2489. doi: 10.1002/prot.22756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nabuurs SB, Spronk CAEM, Krieger E, Maassen H, Vriend G, Vuister GW. Quantitative evaluation of experimental NMR restraints. J Am Chem Soc. 2003;125:12026–12034. doi: 10.1021/ja035440f. [DOI] [PubMed] [Google Scholar]
- 30.Zweckstetter M, Bax A. Prediction of sterically induced alignment in a dilute liquid crystalline phase: aid to protein structure determination by NMR. J Am Chem Soc. 2000;122:3791–3792. [Google Scholar]
- 31.Valafar H, Prestegard JH. REDCAT: a residual dipolar coupling analysis tool. J Magn Reson. 2004;167:228–241. doi: 10.1016/j.jmr.2003.12.012. [DOI] [PubMed] [Google Scholar]
- 32.Dosset P, Hus JC, Marion D, Blackledge M. A novel interactive tool for rigid-body modeling of multi-domain macromolecules using residual dipolar couplings. J Biomol NMR. 2001;20:223–231. doi: 10.1023/a:1011206132740. [DOI] [PubMed] [Google Scholar]
- 33.Vranken WF, Boucher W, Stevens TJ, Fogh RH, Pajon A, Llinas P, Ulrich EL, Markley JL, Ionides J, Laue ED. The CCPN data model for NMR spectroscopy: development of a software pipeline. Proteins. 2005;59:687–696. doi: 10.1002/prot.20449. [DOI] [PubMed] [Google Scholar]
- 34.Doreleijers JF, Nederveen AJ, Vranken W, Lin JD, Bonvin AMJJ, Kaptein R, Markley JL, Ulrich EL. BioMagResBank databases DOCR and FRED containing converted and filtered sets of experimental NMR restraints and coordinates from over 500 protein PDB structures. J Biomol NMR. 2005;32:1–12. doi: 10.1007/s10858-005-2195-0. [DOI] [PubMed] [Google Scholar]
- 35.Doreleijers JF, Mading S, Maziuk D, Sojourner K, Yin L, Zhu J, Markley JL, Ulrich EL. BioMagResBank database with sets of experimental NMR constraints corresponding to the structures of over 1400 biomolecules deposited in the Protein Data Bank. J Biomol NMR. 2003;26:139–146. doi: 10.1023/a:1023514106644. [DOI] [PubMed] [Google Scholar]
- 36.Doreleijers JF, Vranken WF, Schulte C, Lin JD, Wedell JR, Penkett CJ, Vuister GW, Vriend G, Markley JL, Ulrich EL. The NMR restraints grid at BMRB for 5,266 protein and nucleic acid PDB entries. J Biomol NMR. 2009;45:389–396. doi: 10.1007/s10858-009-9378-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Neuhaus D, Williamson MP. The nuclear Overhauser effect in structural and conformational analysis. NY: Wiley-VCH; 2000. [Google Scholar]
- 38.Bax A, Kontaxis G, Tjandra N. Dipolar couplings in macromolecular structure determination. Method Enzymol. 2001;339:127–174. doi: 10.1016/s0076-6879(01)39313-8. [DOI] [PubMed] [Google Scholar]
- 39.Losonczi JA, Andrec M, Fischer MWF, Prestegard JH. Order matrix analysis of residual dipolar couplings using singular value decomposition. J Magn Reson. 1999;138:334–342. doi: 10.1006/jmre.1999.1754. [DOI] [PubMed] [Google Scholar]
- 40.Cornilescu G, Marquardt JL, Ottiger M, Bax A. Validation of protein structure from anisotropic carbonyl chemical shifts in a dilute liquid crystalline phase. J Am Chem Soc. 1998;120:6836–6837. [Google Scholar]
- 41.Vriend G. What If—a molecular modeling and drug design program. J Mol Graphics. 1990;8:52–56. doi: 10.1016/0263-7855(90)80070-v. [DOI] [PubMed] [Google Scholar]
- 42.Laskowski RA, Rullmann JAC, MacArthur MW, Kaptein R, Thornton JM. AQUA and PROCHECK-NMR: programs for checking the quality of protein structures solved by NMR. J Biomol NMR. 1996;8:477–486. doi: 10.1007/BF00228148. [DOI] [PubMed] [Google Scholar]
- 43.Hooft RWW, Vriend G, Sander C, Abola EE. Errors in protein structures. Nature. 1996;381:272–272. doi: 10.1038/381272a0. [DOI] [PubMed] [Google Scholar]
- 44.Eisenberg D, Luthy R, Bowie JU. VERIFY3D: assessment of protein models with three-dimensional profiles. Macromol Crystallogr B. 1997;277:396–404. doi: 10.1016/s0076-6879(97)77022-8. [DOI] [PubMed] [Google Scholar]
- 45.Wiederstein M, Sippl MJ. ProSA-web: interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Res. 2007;35:W407–W410. doi: 10.1093/nar/gkm290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chen VB, Arendall WB, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, Richardson DC. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr D. 2010;66:12–21. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Doreleijers JF, Vranken WF, Schulte C, Markley JL, Ulrich EL, Vriend G, Vuister GW. NRG-CING: integrated validation reports of remediated experimental biomolecular NMR data and coordinates in wwPDB. Nucleic Acids Res. 2012;40:D519–D524. doi: 10.1093/nar/gkr1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hartshorn MJ. AstexViewer (TM): a visualisation aid for structure-based drug design. J Comput Aid Mol Des. 2002;16:871–881. doi: 10.1023/a:1023813504011. [DOI] [PubMed] [Google Scholar]
- 49.Velankar S, McNeil P, Mittard-Runte V, Suarez A, Barrell D, Apweiler R, Henrick K. E-MSD: an integrated data resource for bioinformatics. Nucleic Acids Res. 2005;33:D262–D265. doi: 10.1093/nar/gki058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Finn RD, Mistry J, Tate J, Coggill P, Heger A, Pollington JE, Gavin OL, Gunasekaran P, Ceric G, Forslund K, Holm L, Sonnhammer ELL, Eddy SR, Bateman A. The Pfam protein families database. Nucleic Acids Res. 2010;38:D211–D222. doi: 10.1093/nar/gkp985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cuff AL, Sillitoe I, Lewis T, Clegg AB, Rentzsch R, Furnham N, Pellegrini-Calace M, Jones D, Thornton J, Orengo CA. Extending CATH: increasing coverage of the protein structure universe and linking structure with function. Nucleic Acids Res. 2011;39:D420–D426. doi: 10.1093/nar/gkq1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Andreeva A, Howorth D, Chandonia JM, Brenner SE, Hubbard TJP, Chothia C, Murzin AG. Data growth and its impact on the SCOP database: new developments. Nucleic Acids Res. 2008;36:D419–D425. doi: 10.1093/nar/gkm993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Seo MD, Park SJ, Kim HJ, Lee BJ. Solution structure of hypothetical protein, HP0495 (Y495_HELPY) from Helicobacter pylori. Proteins. 2007;67:1189–1192. doi: 10.1002/prot.21346. [DOI] [PubMed] [Google Scholar]
- 54.Humphrey W, Dalke A, Schulten K. VMD: Visual molecular dynamics. J Mol Graph Model. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 55.Krissinel E, Henrick K. Secondary-structure matching (SSM), a new tool for fast protein structure alignment in three dimensions. Acta Crystallogr D. 2004;60:2256–2268. doi: 10.1107/S0907444904026460. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.