

Abstract
Background
CaMKII activation is pro-arrhythmic in heart failure where myocardium is stretched. However, the arrhythmogenic role of CaMKII in stretched ventricle has not been well understood.
Objective
We tested abnormal impulse inducibility by stretch current in myocytes isolated from CaMKIIδ knockout (KO) mouse left ventricle (LV) where CaMKII activity is reduced by ≈ 62%.
Methods and Results
Action potentials (APs) were recorded by whole-cell patch clamp, and abnormal impulses were induced in LV myocytes by a simulation of stretch-activated-channel (SAC) current. SAC activation failed to induce abnormal impulses in wild type (WT) myocytes but steadily produced early afterdepolarizations (EADs) and automaticity in KO myocytes in which an increase in L-type calcium channel (LTCC) current (ICa) and a reduction of sarcoplasmic reticulum (SR) Ca2+ leak and action potential duration (APD) were observed. The abnormal impulses were not suppressed by CaMKII inhibitor AIP whereas a low concentration of nifedipine eliminated abnormal impulses without shortening APD, implicating ICa in promoting stretch-induced abnormal impulses. In addition, APD prolongation by LTCC opener S(−)Bay K 8644 or isoproterenol facilitated abnormal impulse induction in WT ventricular myocytes even in the presence of CaMKII inhibitor AIP, whereas APD prolongation by K+ channel blocker 4-aminopyridine promoted abnormal impulses in KO myocytes but not in WT myocytes.
Conclusion
ICa activation plays a central role in stretch-induced abnormal impulses and APD prolongation is arrhythmogenic only when ICa is highly activated. At increased ICa activation, CaMKII inhibition cannot suppress abnormal impulse induction.
Keywords: Stretch, Arrhythmias, calcium channel, myocyte, action potential, CaMKII
INTRODUCTION
The abnormal impulses observed in cardiomyocytes include early after-depolarization (EAD), delayed after-depolarization (DAD) and automaticity, which play important roles in triggering lethal ventricular arrhythmias1;2.
There is increasing recognition that CaMKII is pro-arrhythmic in cardiomyopathy where cardiac repolarization is significantly prolonged3. Up-regulation of CaMKII activity is a general feature of heart failure (HF) ventricular myocytes in patients4 and in animal models5;6. However, the macroscopic arrhythmic mechanisms for CaMKII-related arrhythmias in HF have not been well understood. In addition, a failing heart is characterized by persistent myocardium stretch, which is known to activate stretch-activated channel (SAC) current that depolarizes myocytes at diastole. Thus, activation of SAC current may be an important arrhythmogenic substrate in HF. Excessive CaMKII activation facilitates ICa, prolongs APD, and increases sarcoplasmic reticulum (SR) Ca2+ leak. However, it is unclear which alterations are more relevant to CaMKII-mediated arrhythmias in the stretched myocardium.
We have developed a SAC model in which an ionic current that represents SAC activation is injected into the isolated myocyte and induces myocyte depolarization. The injected current and the cell depolarization are regulated by the coupling conductance (Gsac) between the electrically coupled myocyte and the SAC model7. Here, we used this SAC model to study the effect of stretch current on abnormal impulse induction in ventricular myocytes isolated from CaMKIIδ knockout (KO) mouse left ventricle (LV), where CaMKII activity and SR Ca2+ leak are reduced but ICa is increased.
METHODS
Isolation of ventricular myocytes
Mouse LV myocytes were isolated enzymatically by a protocol described previously8 with collagenase (Worthington type II). Only calcium-tolerant, quiescent and rod-shaped cells showing clear cross striations were used for recording.
Action potential recording
APs were recorded in Tyrode’s solution at ≈37°C using whole-cell current clamp methods as we recently reported7. Series resistance was carefully compensated after recording of the membrane potential was established. APs were initiated by current pulses of 2 ms duration and amplitude of 10–15% suprathreshold.
Whole-cell outward K+ current recording
All electrophysiological recordings were performed at 36 ± 0.5 °C. Outward K+ currents were recorded using the whole-cell patch clamp technique in voltage clamp configuration as we reported recently9.
Intracellular [Ca2+]i transient and SR Ca2+ leak measurements
Myocytes were loaded with 2 μM fura-2/AM for 10 min and fluorescence measurements were recorded with a dual-excitation fluorescence photomultiplier system (IonOptix) at 37 °C. For SR Ca2+ leak measurement, Na/Ca-exchange was blocked in 0 Na+/0 Ca2+ Tyrode solution and the ryanodine receptor was blocked by tetracaine (1 mM) as we reported recently10.
SAC Model
This SAC model was described in detail in our recent work7. The stretch-activated current is calculated as Isac=Gsac*(Vm − Esac), where Gsac is the stretch-activated conductance, Vm is cell membrane potential and Esac is reversal potential of stretch-activated current.
Statistical methods
Statistical analysis was performed using Sigmastat for Windows (V3.5, Jandel Scientific). Paired and unpaired t-tests and one-way ANOVA were used for comparisons and p<0.05 was regarded as significant. Mann-Whitney Rank Sum tests were performed if tests for normality or equal variance failed.
Use of vertebrate animals
All procedures on mice were performed in accordance with protocols approved by the institution’s Animal Care and Use Committee.
RESULTS
Shortened APD in CaMKIIδ KO LV
We isolated myocytes from the subendocardial (SEN) and subepicardial (SEP) regions of LV and recorded APs in these myocytes. We found that APD was significantly longer in SEN than in SEP myocytes in both WT and KO LV (Table 1). Meanwhile, both APD30 and APD90 were significantly shortened in KO compared to WT LV. APD30 in the SEP and SEN myocytes from KO LV was shortened by 38.4% and 15.3%, while APD90 was shortened by 20.1% and 22.1%, respectively (Table 1). These results are consistent with those reported from another CaMKII inhibition mouse model in which the APD was shortened and the underlying ionic mechanism was predicted to be the up-regulation of repolarization potassium current11. Indeed, we have observed a significant increase in the outward K+ currents in KO myocytes (Figure 1A). The peak outward current was 54.1 ± 4.2 pA/pF for SEP (n=18) and 30.6 ± 2.5 pA/pF for SEN myocytes (n=16) in WT and 65.6 ± 3.3 pA/pF for SEP (n=23) and 40.4 ± 3.2 pA/pF for SEN (n=21) in KO, respectively (p<0.05 for both SEP and SEN myocytes). We then calculated the mean density for each component of outward K+ current (Ito, IK,slow and Iss) from the peak K+ current traces recorded at +60 mV in each individual myocyte and found that the differences in outward current were mainly caused by the increased Ito in the KO myocytes (26.4±2.8 pA/pF for SEP and 12.3±1.5 pA/pF for SEN in WT vs. 33.7±2.4 pA/pF for SEP and 18.7±2.1 pA/pF for SEN in KO, respectively).
Table 1.
AP Characteristics in WT and CaMKIIδ KO Mouse LV
| Parameters | WT (n=12) | CaMKIIδ KO (n=12) | ||
|---|---|---|---|---|
| SEN (n=14) | SEP (n=23) | SEN (n=12) | SEP (n=17) | |
| RMP (mV) | −76.7±0.8 | −75.5±0.6 | −76.4±0.9 | −76.9±0.5 |
| AP Peak (mV) | 48.1±0.9 | 47.7±1.8 | 49.8±0.7 | 45.0±1.5 |
| dV/dt (V/s) | 372.2±22.8 | 352.9±14.7 | 362.7±18.4 | 343.5±10.5 |
| APD30 (ms) | 2.36 ± 0.09 | 1.81±0.12* | 2.00 ± 0.11# | 1.18 ± 0.05*# |
| APD90 (ms) | 35.76 ± 1.96 | 29.61 ± 1.24* | 27.84±1.72# | 23.52±1.20*# |
Values = mean ± S.E.M.; RMP: resting membrane potential;
p < 0.05, compared to SEN;
p < 0.05, compared to WT.
Figure 1. Abnormal impulse inducibility in ventricular myocytes.
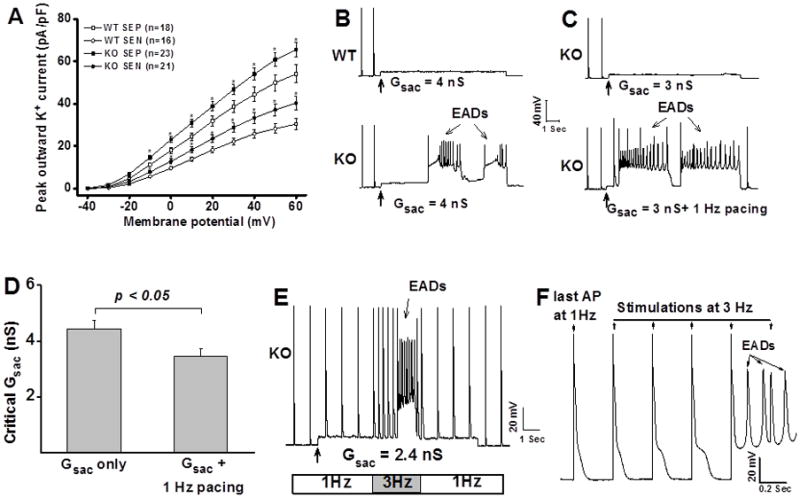
A: I–V relationship of outward K+ current recorded from WT and KO LV myocytes. SEP: subepicardial myocytes; SEN: subendocardial myocytes; *: p<0.05, compared with WT myocytes. B: Application of Gsac 4.0 nS induced EADs in CaMKIIδ KO myocytes but not in WT myocytes. C: Pacing facilitates EAD induction in KO myocytes. D: Pacing reduced the critical Gsac for EAD induction in KO myocytes. Error bars denote S.E.M. E: Fast-pacing promotes EAD induction in KO myocytes. F: Replotting of APs from panel E showing progressive APD prolongation and EAD induction at fast-pacing in KO myocytes.
Enhanced susceptibility to abnormal impulses in CaMKIIδ KO LV
Myocytes were paced at 1Hz. We found that myocytes isolated from WT and KO LV have similar AP thresholds (1.6 ± 0.1nA for WT vs. 1.7 ± 0.1nA for KO myocytes, n = 40 for each group) and that application of Gsac induced a similar depolarization level. For example, application of Gsac 2.0 nS produced depolarization of 4.67 ± 0.36 mV in WT (n = 29) and 4.21 ± 0.46 mV in KO myocytes (n = 23), respectively (p > 0.05), indicating similar input resistance for these myocytes. Therefore, we use the critical Gsac value to assess the susceptibility to abnormal impulse.
Our results showed that application of Gsac successfully induced EADs or automaticity in more than 90% of KO myocytes (30/32) but rarely induced abnormal impulses in WT myocytes (4/30). No difference in susceptibility to abnormal impulse was found between the SEP and SEN myocytes in both genotypes. Figure 1 B shows an example of recording traces in which application of Gsac 4.0 nS induced EADs in KO myocyte but failed to produce EAD in WT myocytes although Gsac induced similar levels of depolarization in these myocytes. These results suggest that KO myocytes are highly susceptible to cardiac stretch-induced arrhythmias.
We then applied Gsac to myocytes that are continuously paced at 1 Hz to mimic the physiological beating condition. We found that pacing did not promote abnormal impulse induction in WT myocytes (data not shown) but significantly facilitated abnormal impulse induction in KO myocytes. As shown in Figure 1C, application of Gsac 3.0 nS induced depolarization in a quiescent KO myocyte but no EAD was induced. However, for the same myocyte, EADs were produced by the same value of Gsac at 1 Hz pacing. In a total of 12 KO LV myocytes, pacing reduced the critical Gsac from 4.4 ± 0.3 nS to 3.4 ± 0.3 nS (p < 0.05), a 23% reduction (Figure 1D).
To mimic the premature excitation, we also tested the Gsac-induced abnormal impulses in response to a sudden switch of pacing rate from 1Hz to 3Hz. We found that fast pacing significantly facilitates abnormal impulse induction in KO myocytes. As shown in Figure 1E, at a relatively low value of Gsac (2.4 nS)in a KO myocyte, no EAD was induced at 1 Hz pacing, but EADs were successfully induced at 3 Hz. After the pacing rate was returned to 1Hz, EAD was no longer inducible. To understand the mechanism, we have reploted the Figure 1E by expanding the scale to include the last AP at 1 Hz, 3 APs at 3 Hz and part of the EADs. As shown in the expanded Figure (Figure 1F), increasing pacing rate from 1 Hz to 3 Hz progressively prolonged APDs until EADs were initiated. These results indicate that in stretched KO myocytes, the enhanced susceptibility to EADs at fast pacing is associated with frequency-dependent APD prolongation. Prolonged APD allows a larger Ca2+ entering in the myocytes that possess a potentiated ICa.
The enhanced susceptibility to abnormal impulses in CaMKIIδ KO LV is associated with ICa up-regulation but unrelated to intracellular Ca2+ handling or signaling
Surprisingly, our results demonstrated an increased susceptibility to SAC-induced abnormal impulses in KO myocytes where the total CaMKII activity was reduced by ≈ 62%12. This indicates that CaMKII activity may not be required for stretch-induced abnormal impulses in ventricular myocytes. To test this, CaMKII activity was totally inhibited in KO LV myocytes by internal diffusion of autoinhibitory peptide AIP (10μM)8. In a total of 15 KO myocytes, AIP failed to suppress abnormal impulse induction, which is manifested by unchanged induction rate (20/23 for control vs. 14/15 for AIP treatment) and critical Gsac (5.11 ± 0.61 nS for control vs. 5.05 ± 0.34 nS for AIP treatment, p > 0.05). These results suggest that the increase in abnormal impulses in KO myocytes is independent of CaMKII signaling.
We have recently reported that in CaMKIIδ KO LV, ICa density is significantly increased due to the up-regulation of the pore-forming α-subunits Cav1.212. To test whether the enhanced susceptibility to abnormal impulse in KO ventricular myocytes is associated with increased ICa, we employed a low dose LTCC blocker nifedipine. Indeed, perfusion with 2 μM of nifedipine totally abolished SAC-induced abnormal impulses in all tested KO myocytes (n=19). A representative recording was shown in Figure 2A. This concentration of nifedipine, however, did not induce significant change in APD. APD90 was 25.0 ± 1.0 ms in normal Tyrode’s solution and 24.7 ± 1.2 ms after exposure to nifedipine (2 μM) (n = 19, p > 0.05). This is because in murine ventricle, Ito current is the major repolarization current where ICa plays a minor role in determining APD. These results suggest that the potentiated ICa plays a crucial role in the enhanced susceptibility to abnormal impulse seen in KO ventricular myocytes. Although other ion currents, such as the later INa, may also be altered in KO LV, they may only play a minor role (if any) in promoting abnormal impulses because the abnormal impulse was totally suppressed by a low dose of ICa blocker.
Figure 2. Cellular substrates that affect abnormal impulse induction.
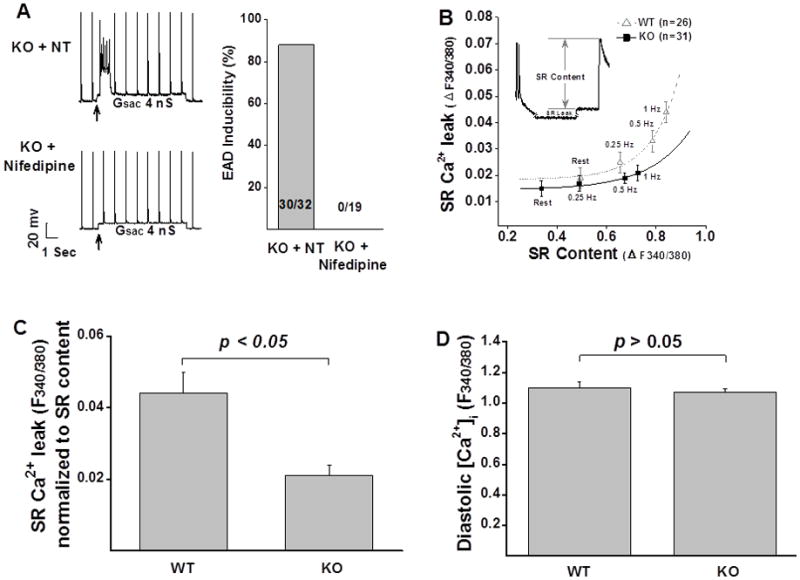
A: Low concentration of the LTCC blocker nifedipine eliminated abnormal impulses in KO myocytes. B: SR Ca2+ leak was reduced in KO myocytes at all tested frequencies, compared to that recorded from WT myocytes. C: Mean values of normalized SR Ca2+ leak measured in WT (n=10) and KO (n=15) myocytes. D: There is no difference in diastolic Ca2+ between WT (n=18) and KO myocytes (n=24). Error bars denote S.E.M.; NT: normal Tyrode’s solution.
Considering that up-regulation of ICa may induce changes in SR Ca2+ handling and intracellular Ca2+ signaling, one may raise an important question whether the increased susceptibility to abnormal impulses in KO myocytes is linked to ICa-induced changes in [Ca2+]i handling. But this is unlikely the case because the Ca2+ transient in KO myocytes was similar to that recorded in the WT myocytes at baseline and even lower at higher frequencies12. Furthermore, we found that the diastolic SR Ca2+ leak was significantly reduced in KO LV myocytes at all tested stimulation frequencies (Figure 2B). SR Ca2+ leak normalized to SR contents was 0.021 ± 0.003 F340/380 (n = 15) in KO vs. 0.044 ± 0.006 F340/380 in WT LV myocytes (n = 10, p < 0.05, Figure 2 C). There was no difference in the levels of diastolic [Ca2+]i between WT and KO LV myocytes (1.122 ± 0.014 F340/380, n = 24 for KO vs. 1.130 ± 0.015 F340/380, n = 18 for WT) (p > 0.05, Figure 2D). These data are consistent with previous studies showing that overexpression of Ca2+ channel β2a increases EAD independent of SR Ca2+ load13.
APD prolongation and abnormal impulses
APD prolongation is a general feature of ventricular myocytes in structural heart diseases, and it has been proposed to be a cause of EAD. In KO myocytes, the susceptibility to EAD was increased while APD was actually shortened compared to WT myocytes, indicating that APD prolongation is not required for stretch-induced EAD. To test the role of APD prolongation in facilitating EADs, we used a K+ channel blocker 4-aminopyridine (4-AP) to inhibit outward K+ currents and prolong APD. We found that 2 mM 4-AP prolonged APD90 by 172.6% in WT myocytes (from 30.6 ± 3 ms to 83.4 ± 5.9 ms, n=10) and 161% in KO myocytes (from 24.0 ± 4.3 ms to 62.6 ± 6.1 ms, n=10) (p > 0.05, WT vs. KO). APD prolongation by 4-AP greatly promoted EAD induction in KO myocytes (the critical Gsac was reduced from 1.95 ± 0.13 nS to 1.45 ± 0.14 nS, n=15, p < 0.05) but not in WT myocytes (no abnormal impulse was induced, as shown in Figure 3A). These results indicate that APD prolongation promotes EADs in stretched myocytes only when ICa is potentiated. In line with this, in WT myocytes, the LTCC opener S(−)Bay K8644 (external perfusion of 1μM S(−)Bay K8644 for 10 minutes) prolonged APD (by 247 ± 25.2%, n=8) and induced EADs in all tested myocytes (Figure 3B). These results suggest that whether APD prolongation can promote abnormal impulse in LV myocytes depends largely on the activation level of ICa.
Figure 3. APD prolongation and abnormal impulse induction.
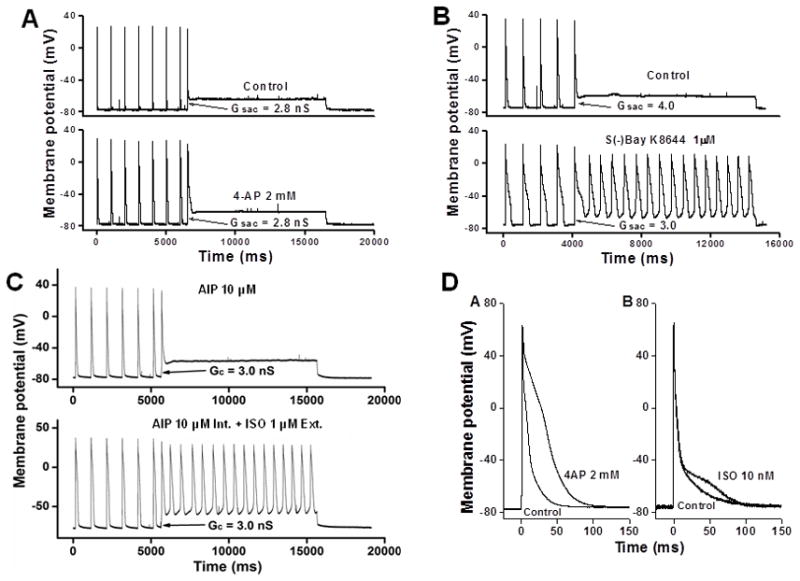
A: Application of the K+ channel blocker 4-AP significantly prolonged APD but failed to promote abnormal impulses in WT ventricular myocyte. B: In the presence of the LTCC opener S(−)Bay K8644, automaticity was induced in WT ventricular myocytes at a lowered Gsac level. C: SAC-induced automaticity in the presence of AIP and ISO. D: APD prolongation by 4-AP and ISO, respectively.
β-adrenergic stimulation promotes abnormal impulse induction, independent of CaMKII activation
In normal hearts, β-adrenergic receptor (β-AR)/PKA pathway is the primary ICa regulator. CaMKII, as a down-stream target of β-AR/PKApathway, plays a major role in ICa regulation in the setting of HF8. Because PKA and CaMKII regulate Ca2+ channels by phosphorylating channel subunits at different sites, CaMKII inhibition may not totally suppress β-adrenergic stimulation-induced abnormal impulses. To test this, we recorded APs in WT ventricular myocytes in which CaMKII activity has been inhibited by internal diffusion of AIP (10 μM). After the maximum effect of CaMKII inhibition was reached (≈10 minutes of internal equilibration with AIP8), Gsac was applied to these myocytes in normal Tyrode’s solution and then in the presence of isoproterenol (1 μM of ISO for 10 minutes). Gsac application failed to induce abnormal impulses in these myocytes in normal Tyrode’s solution. However, in all myocytes with external perfusion of ISO, Gsac activation successfully induced either EADs or automaticity (n=12) (Figure 3C). These results suggest that β-adrenergic stimulation facilitates abnormal impulse induction independent of CaMKII activation. Although ISO significantly prolonged APD in ventricular myocytes, these results are unlikely caused by APD prolongation because 1 μM of ISO prolonged APD90 by only 57.4% (from 40.1 ± 5.1 ms to 63.1 ± 6.7 ms, n=12), while 2 mM of 4-AP prolonged APD90 by 172.6% (from 30.6 ± 3 ms to 83.4 ± 5.9 ms, n=10) (Figure 3D) but failed to promote abnormal impulses. In other words, ISO likely promotes abnormal impulses in ventricular myocytes through ICa activation but not APD prolongation.
Role of ICa in promoting abnormal impulses in stretched human ventricular myocyte model
To understand whether the role of ICa activation in promoting abnormal impulse induction in stretched ventricle applies to human heart, we used a human ventricular myocyte simulation (Beukelmann human ventricular myocyte model14) in stretch studies. As shown in Figure 4, at Gsac of 4.0, ICa factor 1.9 did not produce abnormal impulse (Figure 4, upper panel). However, at the same Gsac level, increasing ICa factor to 2.0 caused automaticity in this human ventricular myocyte model after a long delay of depolarization (Figure 4, mid-panel). The depolarization time for the onset of automaticity is 958 ms and the cycle length of automaticity is 1397 ms. Further increasing ICa factor to 2.4 produced a faster automaticity with the cycle length of 1317 ms and a faster depolarization with the depolarization time of 559 ms (Figure 4, lower panel). These data from human ventricular myocyte model further implicate ICa activation in promoting abnormal impulse induction (a doubled ICa magnitude may be required) and on the other hand indicate that insights gleaned from animal cells may be relevant to human.
Figure 4. Role of ICa in promoting abnormal impulses in stretched human ventricular myocyte model.
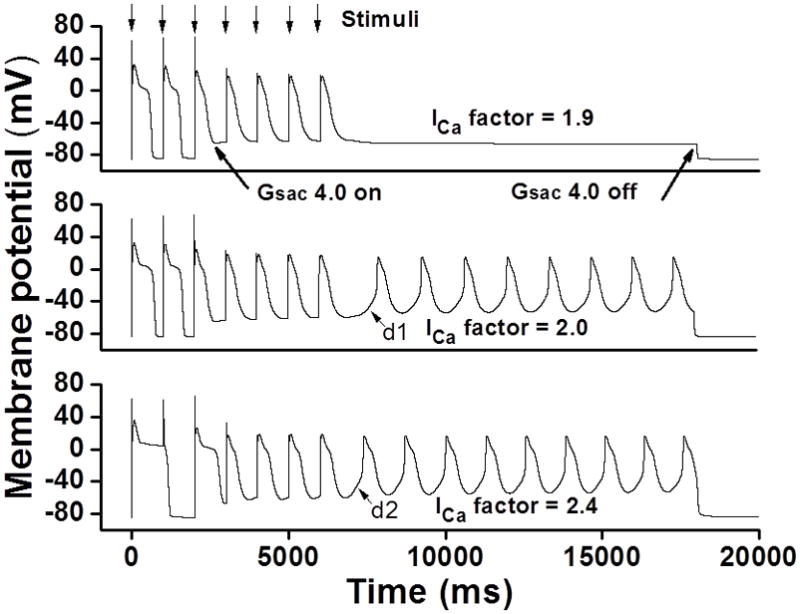
Upper panel: ICa factor of 1.9 did not induce abnormal impulse. Mid-panel: Increasing ICa factor to 2.0 caused automaticity after a long depolarization delay (d1). Lower panel: Further increasing ICa factor to 2.4 produced a faster automaticity with a faster depolarization for the onset of automaticity (d2).
DISCUSSION
In general, the arrhythmic mechanisms in structural heart disease involve both cellular and tissue level remodeling. Studies at the cellular level consistently demonstrate APD prolongation and increased abnormal impulses, which predispose to the initiation of ventricular arrhythmias in patients with cardiomyopathy15;16. The retrograde activation of ventricular myocardium (reentry) is thought to be an important mechanism for the ventricular arrhythmias that are associated with APD prolongation, such as the long-QT syndrome and drug-induced QT prolongation17. However, Pogwizd et al16 demonstrated in large sample clinical mapping studies that most ventricular arrhythmias in nonischemic cardiomyopathy are initiated by nonreentrant mechanisms. Furthermore, up to 50% of ventricular arrhythmias in ischemic cardiomyopathy were initiated by after-depolarizations, although the proportions remain controversial18. These results implicate the increased susceptibility to abnormal impulses in the induction of ventricular arrhythmias in cardiomyopathy. However, our understanding of the molecular substrates for the increased abnormal impulses is very limited. Our results at cellular level indicate that ICa activation plays a central role in promoting abnormal impulses in stretched ventricular myocytes.
APD prolongation and abnormal impulses
AP repolarization is governed by a delicate balance between inward and outward membrane currents of which the inward currents favor APD prolongation, while the outward currents shift APD toward shortening. Studies in HF animal models and HF patients consistently reveal APD prolongation due to reduction of outward K+ currents and changes in ICa density and inactivation19. APD prolongation lengthens Ca2+ channel opening window and thus preserves contractile force but also increases the risk of Ca2+ overload, which contributes to the triggered abnormal impulses. However, whether APD prolongation directly contributes to abnormal impulse induction remains debatable. For example, Wu et al. reported that inhibition of CaMKII by KN93 abolished EADs and suppressed ventricular arrhythmias in CaMKIV transgenic mice without shortening the prolonged APD. In the current study, we observed significant increase in propensity of abnormal impulses in CaMKIIδ KO mouse ventricular myocytes that actually have shortened APD (due to increased outward K+ currents). Our data are consistent with the results reported from another CaMKII inhibition mouse model in which the APD was shortened and the underlying ionic mechanism was predicted to be the up-regulation of repolarization potassium current11. Furthermore, inhibition of outward K+ currents in WT ventricular myocytes caused significant prolongation of APD but failed to promote abnormal impulses, whereas ISO significantly facilitated EAD induction with much less APD prolongation. These data demonstrate that whether APD prolongation can induce abnormal impulse depends largely on the ICa activation status in that cell.
CaMKII activity and arrhythmias
Excessive CaMKII activation is pro-arrhythmic in HF20. However, the arrhythmogenic mechanisms remain elusive. CaMKII is the key contributor to the remodeling of Ca2+ handling proteins in HF, the most important among them are ICa remodeling and SR Ca2+ leak. In addition, CaMKII also contributes to the HF-related alteration of INa21. All these alterations are reported to be arrhythmogenic20. Our results in CaMKIIδ KO mice LV showed an increased abnormal impulses even when CaMKII is totally inhibited by AIP, suggesting that CaMKII activity may not be directly associated with abnormal impulse induction. Instead, an increased ICa activation plays an important role herein because abnormal impulses were totally suppressed by ICa blocker. Furthermore, Ca2+ transient was unchanged and SR Ca2+ leak was reduced in KO LV (although ICa was increased). It is unlikely that the increase in abnormal impulse is related to the changes of Ca2+ transient and SR Ca2+ leak by the increased ICa. Taken together, our results suggest that CaMKII mediated ICa facilitation is likely a key substrate for the increased abnormal impulses seen in HF ventricular myocytes. Considering that CaMKII activation is the main contributor to the ICa potentiation in failing LV8, it is understandable that CaMKII inhibition is efficient in suppression of ventricular arrhythmias in HF. However, since β-adrenergic receptor (β-AR) stimulation plays an independent role in promoting abnormal impulse induction, CaMKII inhibition itself may not be sufficient in suppression of abnormal impulses under conditions of increased β-adrenergic activation (e.g., increased workload in the setting of HF) and β-adrenergic inhibition (β-receptor blocker) may be valuable herein. This is consistent with the results from clinical trials that β-blocking agents are helpful in preventing sudden cardiac death and ventricular arrhythmias in HF patients22;23.
Myocardium stretch and abnormal impulses
It has been shown that mechanical stretch induces depolarization in isolated ventricular myocytes from mice, rats, guinea-pigs and human24–26. In most cases, the magnitude of depolarization depends on the degree of stretch27. Our results demonstrated that stretch current plays an important role in promoting abnormal impulses in ventricular myocytes when ICa is potentiated. In the setting of HF, LTCC is highly phosphorylated by excessive activation of CaMKII8, implicating myocardium stretch in the enhanced susceptibility to abnormal impulses in HF. Indeed, reducing myocardial wall tension by left ventricular offloading using intra-aortic balloon counterpulsation significantly reduced ventricular arrhythmias in patients with medically refractory ventricular arrhythmias28.
Here, we used a SAC model to mimic myocardium stretch-activated channel current, in the absence of actual physical stretch. Previous studies have demonstrated that physical stretch can alter kinase activity29, increase intracellular calcium30 and change troponin C affinity31. One of the advantages of this SAC model is that we can exclusively assess the effect of stretch-induced membrane potential changes on abnormal impulse induction (in the absence of other intracellular changes) which is impossible when using actual physical stretch.
Kamkin et al.25 showed that stretching an isolated rat ventricular myocyte for 8 μm induced a SAC current of 269 pA at holding potential of −45 mV. For those few WT myocytes in which abnormal impulses were induced, the critical Gsac was 7.55 ± 0.9 nS. For the average resting membrane potential (RMP) of −80 mV and Esac of −10 mV, this conductance generates a current of 528 pA, which is much higher than the feasible range of physical stretch (nearly doubled)7;25. In KO myocytes, the critical Gsac for abnormal impulse induction was 3.4 ± 0.3 nS, which generates a stretch current of 238 pA. This current level can be readily produced by physical stretch13.
In most studies, β-adrenergic agonist has been used as a major tool for abnormal impulse induction in resting myocytes. Owing to the technical difficulty, abnormal impulse has rarely studied in stretched myocytes, although myocytes in the heart are in fact stretched during heart beats. In the setting of HF, myocyte stretch presents in both systole and diastole due to the ventricular dilation and increase in the pre- and after-load. To our knowledge, this is the first report showing the critical role of enhanced ICa activation, independent of action potential prolongation, CaMKII activation and intracellular Ca2+ handling, in abnormal impulse induction in stretched ventricular myocytes, and this has been confirmed in human ventricular myocyte model.
Conclusion
In this study, we have dissected the role of APD prolongation, CaMKII activation, ICa potentiation and β-adrenergic stimulation in triggering abnormal impulses in stretched ventricular myocytes using SAC activation. We demonstrated that CaMKII activation may not be required for abnormal impulse induction in stretched ventricular myocytes. The pro-arrhythmic role of CaMKII is likely mediated by ICa activation. Moreover, APD prolongation itself is incapable of triggering abnormal impulses unless ICa is highly activated, and the same is myocardial stretch. In addition, we have shown that β-AR stimulation and CaMKII activation promote abnormal impulse independently in stretched ventricular myocytes, suggesting that inhibition of both β-AR and CaMKII may be required for effective suppression of ventricular arrhythmias in heart failure.
Acknowledgments
This work was supported by grants from NIH (HL-088168, HL-083271), American Health Assistant Foundation (H2007-019) and Emory University (pilot grant 280249).
We thank Dr. Eric N. Olson (University of Texas Southwestern Medical Center at Dallas) for sharing CaMKIIδ KO mice with us.
Footnotes
No disclosures.
Reference List
- 1.January CT, Fozzard HA. Delayed afterdepolarizations in heart muscle: mechanisms and relevance. Pharmacol Rev. 1988;40:219–227. [PubMed] [Google Scholar]
- 2.Fozzard HA. Afterdepolarizations and triggered activity. Basic Res Cardiol. 1992;87(Suppl 2):105–13. doi: 10.1007/978-3-642-72477-0_10. [Review] [DOI] [PubMed] [Google Scholar]
- 3.Wu Y, Temple J, Zhang R, Dzhura I, Zhang W, Trimble R, Roden DM, Passier R, Olson EN, Colbran RJ, Anderson ME. Calmodulin kinase II and arrhythmias in a mouse model of cardiac hypertrophy. Circulation. 2002;106:1288–1293. doi: 10.1161/01.cir.0000027583.73268.e7. [DOI] [PubMed] [Google Scholar]
- 4.Hoch B, Meyer R, Hetzer R, Krause EG, Karczewski P. Identification and expression of delta-isoforms of the multifunctional Ca2+/calmodulin-dependent protein kinase in failing and nonfailing human myocardium. Circ Res. 1999;84:713–721. doi: 10.1161/01.res.84.6.713. [DOI] [PubMed] [Google Scholar]
- 5.Hagemann D, Bohlender J, Hoch B, Krause EG, Karczewski P. Expression of Ca2+/calmodulin-dependent protein kinase II delta-subunit isoforms in rats with hypertensive cardiac hypertrophy. Mol Cell Biochem. 2001;220:69–76. doi: 10.1023/a:1010899724222. [DOI] [PubMed] [Google Scholar]
- 6.Currie S, Smith GL. Calcium/calmodulin-dependent protein kinase II activity is increased in sarcoplasmic reticulum from coronary artery ligated rabbit hearts. FEBS Lett. 1999;459:244–248. doi: 10.1016/s0014-5793(99)01254-5. [DOI] [PubMed] [Google Scholar]
- 7.Wang Y, Joyner RW, Wagner MB, Cheng J, Lai D, Crawford BH. Stretch-activated channel activation promotes early afterdepolarizations in rat ventricular myocytes under oxidative stress. Am J Physiol Heart Circ Physiol. 2009;296:H1227–H1235. doi: 10.1152/ajpheart.00808.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang Y, Tandan S, Cheng J, Yang C, Nguyen L, Sugianto J, Johnstone JL, Sun Y, Hill JA. Ca2+/calmodulin-dependent protein kinase II-dependent remodeling of Ca2+ current in pressure overload heart failure. J Biol Chem. 2008;283:25524–25532. doi: 10.1074/jbc.M803043200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang Y, Cheng J, Chen G, Rob F, Naseem RH, Nguyen L, Johnstone JL, Hill JA. Remodeling of outward K+ currents in pressure-overload heart failure. J Cardiovasc Electrophysiol. 2007;18:869–875. doi: 10.1111/j.1540-8167.2007.00864.x. [DOI] [PubMed] [Google Scholar]
- 10.Cheng J, Xu L, Lai D, Guilbert A, Lim HJ, Keskanokwong T, Wang Y. CaMKII inhibition in heart failure, beneficial, harmful, or both. Am J Physiol Heart Circ Physiol. 2012;302:H1454–H1465. doi: 10.1152/ajpheart.00812.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang R, Khoo MS, Wu Y, Yang Y, Grueter CE, Ni G, Price EE, Jr, Thiel W, Guatimosim S, Song LS, Madu EC, Shah AN, Vishnivetskaya TA, Atkinson JB, Gurevich VV, Salama G, Lederer WJ, Colbran RJ, Anderson ME. Calmodulin kinase II inhibition protects against structural heart disease. Nat Med. 2005;11:409–417. doi: 10.1038/nm1215. [DOI] [PubMed] [Google Scholar]
- 12.Xu L, Lai D, Cheng J, Lim HJ, Keskanokwong T, Backs J, Olson EN, Wang Y. Alterations of L-type calcium current and cardiac function in CaMKII{delta} knockout mice. Circ Res. 2010;107:398–407. doi: 10.1161/CIRCRESAHA.110.222562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Koval OM, Guan X, Wu Y, Joiner ML, Gao Z, Chen B, Grumbach IM, Luczak ED, Colbran RJ, Song LS, Hund TJ, Mohler PJ, Anderson ME. CaV1.2 beta-subunit coordinates CaMKII-triggered cardiomyocyte death and afterdepolarizations. Proc Natl Acad Sci U S A. 2010;107:4996–5000. doi: 10.1073/pnas.0913760107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Priebe L, Beuckelmann DJ. Simulation study of cellular electric properties in heart failure. Circ Res. 1998;82:1206–1223. doi: 10.1161/01.res.82.11.1206. [DOI] [PubMed] [Google Scholar]
- 15.Zeng J, Rudy Y. Early afterdepolarizations in cardiac myocytes: mechanism and rate dependence. Biophys J. 1995;68:949–964. doi: 10.1016/S0006-3495(95)80271-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pogwizd SM, McKenzie JP, Cain ME. Mechanisms underlying spontaneous and induced ventricular arrhythmias in patients with idiopathic dilated cardiomyopathy. Circulation. 1998;98:2404–2414. doi: 10.1161/01.cir.98.22.2404. [DOI] [PubMed] [Google Scholar]
- 17.Antzelevitch C, Yan GX, Shimizu W. Transmural dispersion of repolarization and arrhythmogenicity: the Brugada syndrome versus the long QT syndrome. J Electrocardiol. 1999;32 (Suppl):158–165. doi: 10.1016/s0022-0736(99)90074-2. [DOI] [PubMed] [Google Scholar]
- 18.Rubart M, Zipes DP. Mechanisms of sudden cardiac death. J Clin Invest. 2005;115:2305–2315. doi: 10.1172/JCI26381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tomaselli GF, Zipes DP. What causes sudden death in heart failure? Circ Res. 2004;95:754–763. doi: 10.1161/01.RES.0000145047.14691.db. [DOI] [PubMed] [Google Scholar]
- 20.Erickson JR, Anderson ME. CaMKII and its role in cardiac arrhythmia. J Cardiovasc Electrophysiol. 2008;19:1332–1336. doi: 10.1111/j.1540-8167.2008.01295.x. [DOI] [PubMed] [Google Scholar]
- 21.Maltsev VA, Reznikov V, Undrovinas NA, Sabbah HN, Undrovinas A. Modulation of late sodium current by Ca2+, calmodulin, and CaMKII in normal and failing dog cardiomyocytes: similarities and differences. Am J Physiol Heart Circ Physiol. 2008;294:H1597–H1608. doi: 10.1152/ajpheart.00484.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Adamson PB, Gilbert EM. Reducing the risk of sudden death in heart failure with beta-blockers. J Card Fail. 2006;12:734–746. doi: 10.1016/j.cardfail.2006.08.213. [DOI] [PubMed] [Google Scholar]
- 23.Pitt B. The role of beta-adrenergic blocking agents in preventing sudden cardiac death. Circulation. 1992;85:I107–I111. [PubMed] [Google Scholar]
- 24.Kamkin A, Kiseleva I, Isenberg G. Ion selectivity of stretch-activated cation currents in mouse ventricular myocytes. Pflugers Arch. 2003;446:220–231. doi: 10.1007/s00424-003-1018-y. [DOI] [PubMed] [Google Scholar]
- 25.Kamkin A, Kiseleva I, Isenberg G. Stretch-activated currents in ventricular myocytes: amplitude and arrhythmogenic effects increase with hypertrophy. Cardiovasc Res. 2000;48:409–420. doi: 10.1016/s0008-6363(00)00208-x. [DOI] [PubMed] [Google Scholar]
- 26.Zeng T, Bett GC, Sachs F. Stretch-activated whole cell currents in adult rat cardiac myocytes. Am J Physiol. 2000;278:H548–H557. doi: 10.1152/ajpheart.2000.278.2.H548. [DOI] [PubMed] [Google Scholar]
- 27.Youm JB, Han J, Kim N, Zhang YH, Kim E, Joo H, Hun LC, Joon KS, Cha KA, Earm YE. Role of stretch-activated channels on the stretch-induced changes of rat atrial myocytes. Prog Biophys Mol Biol. 2006;90:186–206. doi: 10.1016/j.pbiomolbio.2005.06.003. [DOI] [PubMed] [Google Scholar]
- 28.Fotopoulos GD, Mason MJ, Walker S, Jepson NS, Patel DJ, Mitchell AG, Ilsley CD, Paul VE. Stabilisation of medically refractory ventricular arrhythmia by intra-aortic balloon counterpulsation. Heart. 1999;82:96–100. doi: 10.1136/hrt.82.1.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sadoshima J, Izumo S. The cellular and molecular response of cardiac myocytes to mechanical stress. Annu Rev Physiol. 1997;59:551–571. doi: 10.1146/annurev.physiol.59.1.551. [DOI] [PubMed] [Google Scholar]
- 30.Calaghan SC, White E. The role of calcium in the response of cardiac muscle to stretch. Prog Biophys Mol Biol. 1999;71:59–90. doi: 10.1016/s0079-6107(98)00037-6. [DOI] [PubMed] [Google Scholar]
- 31.Tavi P, Han C, Weckstrom M. Mechanisms of stretch-induced changes in [Ca2+]i in rat atrial myocytes: role of increased troponin C affinity and stretch-activated ion channels. Circ Res. 1998;83:1165–1177. doi: 10.1161/01.res.83.11.1165. [DOI] [PubMed] [Google Scholar]