

Abstract
Purpose
Inhibitor of apoptosis proteins (IAPs) promote cancer cell survival and confer resistance to therapy. We report on the ability of second mitochondria-derived activator of caspases (SMAC) mimetic, birinapant, which acts as antagonist to cIAP1 and cIAP2, to restore the sensitivity to apoptotic stimuli such as tumor necrosis factor (TNF)-α in melanomas
Experimental Design
Seventeen melanoma cell lines, representing five major genetic subgroups of cutaneous melanoma, were treated with birinapant as a single agent or in combination with TNF-α. Effects on cell viability, target inhibition, and initiation of apoptosis were assessed and findings were validated in in 2D, 3D spheroid and in vivo xenograft models.
Results
When birinapant was combined with TNF-α, strong combination activity, i.e. neither compound was effective individually but the combination was highly effective, was observed in twelve out of eighteen cell lines. This response was conserved in spheroid models, whereas in vivo birinapant inhibited tumor growth without adding TNF-α in in vitro resistant cell lines. Birinapant combined with TNF-α inhibited the growth of a melanoma cell line with acquired resistance to BRAF inhibition to the same extent as in the parental cell line.
Conclusions
Birinapant in combination with TNF-α exhibits a strong anti-melanoma effect in vitro. Birinapant as a single agent shows in vivo anti-tumor activity, even if cells are resistant to single agent therapy in vitro. Birinapant in combination with TNF-α is effective in a melanoma cell line with acquired resistance to BRAF inhibitors.
Keywords: birinapant, IAP inhibitor, SMAC mimetic, TNF-α, melanoma
Introduction
Treatment options for metastasized melanoma, a disease with a low 5-year survival rate, have improved remarkably in the last two years. After a decades-long period in which the gold standard remained dacarbazine chemotherapy with modest response rates(1), two new therapies have been FDA-approved recently (2–4) and several are currently in late stage clinical development (5). Among small molecule inhibitors, the majority targets two major pathways: the mitogen-activated protein kinase (MAPK) and phosphatidylinositol 3-kinase (PI3K) pathways. Therapy through modulating T cell responses (4) is also highly promising, but unfortunately for all new therapies most patients eventually progress.
Anti- apoptotic mechanisms are also frequently at work in melanoma (6) and an attractive target for therapeutic intervention. For example, targeting the B-cell lymphoma 2 (Bcl-2) family of anti-apoptotic proteins using Bcl-2 antisense (7) and Bcl-2 homology domain 3 (BH3) mimetics (8, 9) has been promising pre-clinically, but will require additional studies to fully realize their potential in clinical trials.
One of the factors responsible for the difficulties in engaging the apoptotic cascade efficiently in melanoma is the up-regulation of conserved inhibitor of apoptosis proteins (IAPs) (10, 11). IAPs are a family of proteins defined by the presence of baculoviral IAP repeats (BIR). The best described members are XIAP, cIAP1, cIAP2, ML-IAP and survivin. These proteins are associated with chemoresistance and poor outcome in many cancer types (reviewed in (12)). IAPs themselves are controlled by the second mitochondria-derived activator of caspases (SMAC). SMAC is released from mitochondria upon onset of apoptosis (13) and binds directly to IAPs leading to their degradation (14, 15). A number of IAP inhibitors, designed to function as SMAC mimetics, have shown preclinical activity in a variety of cancer types (16–19). In contrast to XIAP, cIAP1 and cIAP2 can bind to caspases but lack binding domains capable of inhibiting caspases (20). Previous studies have shown that synthetic IAP antagonists are able to induce apoptosis in tumor cells primarily through inhibition of cIAP1 and or cIAP2, which results in changes in tumor necrosis factor receptor (TNFR) complex signaling. These effects were dependent on TNF-α signaling and caspase 8 activation (21). Moreover, SMAC mimetics can perturb nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) signaling downstream of TNFR (22).
Birinapant is a novel dimeric SMAC mimetic designed to specifically target cIAP1 and cIAP2 for degradation, resulting in a switch in TNFR signaling; activation of TNFR upon binding of TNF-α in the presence of cIAP1 and cIAP2 leads to NF-κB activation and increased cell proliferation. In contrast, following cIAP1 and cIAP2 inhibition TNF-α signaling leads to activation of caspase-8 and induction of apoptosis (12).
Immune cell infiltrates are often found in melanoma lesions, leading to chronic inflammation and (among other factors) elevated levels of TNF-α (23, 24). TNF-α can then be utilized by melanoma cells to promote cell growth, invasion, and metastasis (25). Binding of TNF-α to its receptor leads to activation of the TNFR complex-I, a membrane localized activator of the canonical NF-κB pathway comprised of TNFRSF1A associated via death domain (TRADD), the Ubiquitin ligases TRAF2, TRAF5, cIAP1 and cIAP2, and the protein kinase RIPK1 (26). This leads to increased cell proliferation via an ubiquitination cascade and downstream activation of NF-κB. After SMAC mimetic-induced degradation of cIAP1 and cIAP2, binding of TNF-α to its receptor leads to formation of a death complex and activation of caspases. This results in a reversal of the role of TNF-α from promoting proliferation to inducing cell death (12).
In the present study, birinapant in combination with TNF-α led to a reduction of viability in the majority of seventeen melanoma cell lines tested. This effect was dependent on TNF-α.
Our laboratory has previously published on melanoma cell lines with acquired resistance to BRAF inhibitors (27). Notably, birinapant in combination with TNF-α elicited the same anti-melanoma effect in a parental and BRAF inhibitor resistant cell line. This suggests a potential use of birinapant for the treatment of BRAF inhibitor refractory melanoma patients.
Materials & Methods
Cell lines and reagents
Human melanoma cell lines were cultured in Dulbecco’s modified Eagle’s medium supplemented with 5% fetal bovine serum and grown at 37°C in 5% CO2. All cell lines were periodically authenticated by DNA finger printing using Coriell’s microsatellite kit and tested for mycoplasma by PCR.
Genomic DNA from samples were analyzed for mutations in BRAF, CDK4, CTNNB1, FBX04, KIT, MEK1, MEK2, MET, and NRAS by the nucleotide extension assay using the iPlex platform (Sequenom, Inc, San Diego, CA) (28).
Birinapant was provided by TetraLogic (Malvern, PA). Birinapant is a novel bivalent, selective small molecule peptidomimetic of the Smac tetrapeptide that binds with high affinity to multiple members of the IAP family including XIAP and cIAP1. The dissociation constant (Kd) for XIAP and cIAP-1 was determined to be 45 nM and <1nM respectively (29). Birinapant was developed by TetraLogic Pharmaceuticals and designed based on the four N-terminal amino acids of Smac/DIABLO (Ala- Val-Pro-Ile) and caspase-9 (Ala-Thr-Pro-Phe) (30).
Human recombinant TNF-α was obtained from Invitrogen (Carlsbad, CA). TNF-α mAb MAB610 was obtained from R&D Systems (Minneapolis, MN).
cis-Diamineplatinum(II) dichloride (cisplatin) was obtained from Sigma- Aldrich (St. Louis, MO).
Immunoblot analysis
For immunoblot analyses, adherent cells were washed with cold PBS containing 100μM Na3VO4, scraped, collected by centrifugation, and quick-frozen in dry ice before lysis. Xenograft tumors were snap frozen in liquid nitrogen immediately after harvesting. Tumor chunks were ground on liquid N2 using a MM2 mixer mill (Retsch, Newtown, PA). Cells and powder were lysed and equal amounts of protein (10–40μg) were subjected to SDS-PAGE and proteins transferred onto PVDF membranes (Immobilon). Antibodies used were: cIAP1, AF8181; cIAP2, AF8171 (both R & D systems, Minneapolis, MN); PARP, 7150; GAPDH, 32233 (both Santa Cruz biotechnology, Santa Cruz, CA); Caspase 8, 9746; NF-κBp65, 3031 (both Cell Signaling, Danvers, MA); XIAP, ADI-AAM-050 (ENZO, Farmingdale, NY); NFKB2 p100- p52, 4882 (Cell Signaling, Danvers, MA); RIP1, 51-6559GR, RAC1, 610651 (both BD Biosciences, Franklin Lakes, NJ); ERK, sc-271270, pERK, sc 7976, (both Santa Cruz biotechnology, Santa Cruz, CA). Membranes were probed with primary antibodies overnight at 4°C, then further incubated with Alexa Fluor-labeled secondary antibodies (IRDye 680LT goat-anti mouse, IRDye 800CW goat-anti rabbit, donkey-anti mouse, donkey-anti rabbit, or donkey-anti goat IRDye 800CW or 680LT (LI-COR, Lincoln, NE) for 1h and scanned using the Odyssey system (LI-COR, Lincoln, NE).
In vitro drug sensitivity assays
For monolayer cell culture assays, cells were allowed to attach for 24h and subsequently incubated with birinapant and/or TNF-α for 24 or 72h. CellTiter 96® AQueous Non-Radioactive Cell Proliferation Assay (MTS) assay (Promega, Madison, WI) was performed according to the manufacturer’s description. For cell cycle analysis, melanoma cells were fixed in 70% ethyl alcohol and stained with propidium iodide. Samples were subsequently analyzed with an EPICS XL (Beckman-Coulter) apparatus. Annexin V staining was performed with an annexin V allophycocyanin conjugate (Invitrogen, Carlsbad, CA) according to the manufacturer’s description. Briefly, cells were treated with DMSO control, birinapant and/or TNF-α for 24h. Resuspended cells were washed and incubated with the conjugate for 15min and annexin-binding buffer was added. Samples were subsequently analyzed with an EPICS XL (Beckman-Coulter) apparatus.
451Lu and WM1366 melanoma cells were treated with birinapant 1uM in combination with TNF-α 1ng/ml. Cells were then incubated for 72h in the presence or absence of Z-VAD-FMK (R & D systems, Minneapolis, MN) a pan caspase inhibitor. Proliferation was assessed using the MTS assay.
451Lu and WM1366 melanoma cells were treated with birinapant 1uM in combination with TNF-α 1ng/ml. Cells were then incubated for 72h in the presence or absence of Necrostatin-1 (N9037, Sigma) a RIP1 kinase inhibitor. Proliferation was assessed using the MTS assay.
RIPK1 gene expression was evaluated using commercially available gene expression assays from Life Technologies. Briefly, RNA was isolated using Qiagen RNeasy kit, RNA was quantified and normalized for addition to cDNA reaction. cDNA was created using cDNA kit from Life Technologies (4368814) and real-time PCR was carried out for RIPK1 (assay ID Hs00169407_m1) and GUSB (assay ID Hs99999908_m1) as a housekeeping gene. For analysis, the relative quantity of RIPK1 to GUSB was calculated.
For three dimensional melanoma spheroid assays, cells were prepared as described previously (31, 32). Collagen-embedded spheroids were incubated with reagents for 72h. Spheroids were imaged using a Leica TCS SP2 confocal microscope after addition of a live/ dead viability assay kit using the fluorescent dyes calcein AM and ethidium homodimer-1 (Invitrogen, Carlsbad, CA). To assess viability, spheroids were grown in a 96 well format, and allowed to grow in the presence of birinapant and/or TNF-α for 72h, then AlamarBlue (Invitrogen, Carlsbad, CA) was added. Fluorescence was measured using an Envision Xcite plate reader (Perkin Elmer). All experiments were performed in triplicate.
To measure TNF-α levels in cell culture supernatants, cells were grown for three days to 70% confluence. Supernatants were collected and 200uL were tested in triplicate following the manufacturer’s instructions using the BD OptEIA Human TNFa ELISA Kit II (BD, Franklin Lakes, NJ).
Xenograft studies
All animal experiments were performed in accordance with Wistar IACUC protocol 111954 in NUDE mice. Ten animals each were inoculated s.c. with 1×106 451Lu or 1205Lu human melanoma cells in a suspension of matrigel (BD Matrigel™ Basement Membrane Matrix, Growth Factor Reduced) / complete media at a ratio of 1:1. After formation of palpable tumors, mice from both tumor models were randomized into two groups. Both groups were treated intra-peritoneal three times/ week with either vehicle control or birinapant 30mg/kg for 21 days. Birinapant was dissolved in 12.5% Captisol (Ligand Pharmaceuticals, La Jolla, CA) in distilled water. Tumor size was assessed twice weekly by caliper measurement. Subsequently, satellite groups of ten and fifteen mice were inoculated in the same fashion with 451Lu and 1205Lu tumor cells respectively. After tumors had reached a mean volume of 200mm3 animals were dosed with either birinapant or vehicle control as described above. After 48 hours and two doses, animals were sacrificed and tumors were harvested at four time points after the last treatment. Tumor samples were snap frozen in liquid nitrogen for subsequent protein analysis and preserved as FFPE blocks for immune-histochemistry.
Paraffin embedded 5umsections were stained with activated caspase-3 mAb (5A1E, Cell Signaling, Danvers, MA). Briefly, after de-paraffinization and endogen peroxidase blocking, antigen retrieval was performed via heat mediation with citrate buffer. The antibody was diluted 1:100 in Superblock and incubated over night at 4°C. After washing with D-PBS-T slides were incubated for 30min in ABComplex/HRP conj. Kit (DAKO, Copenhagen, Denmark). Following a further 3 washing steps the DAB-chromogen solution Kit (DAKO) was incubated on slides until visible color reaction was observed. Slides were washed and counterstained in diluted (1:4 in aqua dest.) Harris’ hematoxlin solution and mounted with Aquatex (Merck).
Statistics
IC50 values were calculated with GraphPad Prism 5 (GraphPad Software Inc.). Statistical signi cance was determined using two-sided Student’s t-test. P<0.05 was considered to be signi cant.
Results
The majority of cell lines exhibited strong combination activity of birinapant and TNF-α
Reflecting the heterogeneity of melanoma, we used a panel of seventeen human melanoma cell lines comprising major genetic subgroups of cutaneous melanoma, approximately reflecting the distribution seen in patients (33) (Supplementary Table S1).
In order to test birinapant as a single agent and calculate the half maximal inhibitory concentration (IC50) values in this cell line panel, cells were treated with birinapant at 1, 10, 100 and 1000nM for 72h and cell viability was assessed through the MTS assay. Only one cell line (WM9, BRAFV600E) out of seventeen showed a response to birinapant (Fig. 1, grey bars, TL32711). However, when birinapant was combined with TNF-α (1ng/ml), the majority of cell lines showed birinapant IC50 values in the low nanomolar range (Fig. 1, black bars). This response was independent of cell line genetic background. All cell lines were resistant to TNF-α alone (Supplementary Figure S1).
Figure 1. Melanoma cell lines can be sensitized to birinapant by co-treatment with TNF-α.

Seventeen human melanoma cell lines representing major mutational subgroups of cutaneous melanoma were treated with birinapant at concentrations up to 1uM, alone (grey bars) or in combination with TNF-α 1ng/ml (black bars). Values on the y-axis represent IC50 values as assessed by the MTS proliferation assay at 72h. Data labels indicate IC50 values from mean value of biological triplicates.
Three distinct response patterns following birinapant addition were observed. For subsequent experiments, four cell lines were therefore selected based on their response to birinapant in combination with TNF-α: 1205Lu, resistant to the combination therapy; WM9, sensitive to birinapant as a single agent; and two cell lines sensitive to birinapant only in combination with TNF-α: 451Lu (BRAFV600E) and WM1366 (NRASQ61L), (Fig. 2A).
Figure 2. Effect of birinapant alone or in combination with TNF-α.

Four cell lines showing varying responses to birinapant as seen in the whole panel of cell lines assessed: combination resistant (1205Lu), single agent sensitive (WM9), and combination sensitive (451Lu and WM1366). A, dose response curves after 72h incubation with birinapant (TL32711) at indicated doses alone or in combination with TNF-α 1ng/ml using the MTS assay. For the cell lines 451Lu and WM1366: p<0.05 for doses ≥10nM. Data are represented as mean of three or more experiments ± standard error of mean (SEM). B, immunoblots showing the same four cell lines after 24h incubation with birinapant 1uM, TNF-α 1ng/ml, or the combination of both, compared to untreated controls. Levels of target IAP proteins, cleaved PARP, NF-κB p65, NF-κB2 p100- p52, and RIP1 were assessed. GAPDH was included to ensure equal loading. C, relative number of cells in the sub G1 fraction as assessed by propidium iodide cell cycle analysis after 24h incubation with birinapant 1uM, TNF-α 1ng/ml or combination of both. Data are represented as mean ± SEM (n=3 biological replicates). D, relative number of annexin V positive cells as assessed by flow cytometry after 24h incubation with birinapant 1uM, TNF-α 1ng/ml or combination of both. Data are represented as mean ± SEM (n=3 biological replicates).
cIAP1 and cIAP2 downregulation was observed in all response types, but apoptosis occurred only in sensitive cell lines
Immunoblot analysis showed inhibition of the birinapant target protein cIAP1 in all four cell lines. Whereas cIAP2 was up-regulated in the 1205Lu and WM1366 cell lines upon treatment with TNF-α, and was again degraded in the combination with birinapant. No change in the levels of XIAP could be observed (Fig. 2B). After treatment with birinapant alone, apoptosis as assessed by cleavage of PARP could be observed in the single agent sensitive cell line only. When birinapant was combined with TNF-α, PARP cleavage was now detectable in the combination-sensitive cell lines 451Lu and WM1366 as well, corresponding to the growth inhibition observed in the MTS assay. No cleavage of PARP could be observed in the resistant cell line 1205Lu, even after combination treatment. Notably, a decrease in NF-κB p65 protein levels could only be observed in the single agent sensitive cell line, WM9, after treatment with birinapant alone or in combination with TNF-α. An increase in NF-κB2 p100 levels could be observed with TNF-α stimulation, and this could be brought back to baseline by the combination of birinapant and TNF-α. The p52 band of NF-κB2 was not detectable. No change in the protein levels of RIP1 kinase, which was found at similar levels in all four cell lines, could be observed upon treatment with birinapant or TNF-α alone, but RIP1 was depleted in three out of four cell lines when both agents where combined (Fig. 2B).
We then confirmed the induction of apoptosis seen at the protein level through cell cycle analysis by propidium iodide staining. A significant increase in sub G1 fractions, indicative of apoptosis, was seen in WM9 cells when treated with birinapant alone; in 451Lu and WM1366 cells only when birinapant was combined with TNF-α; but not in 1205Lu cells, even after combination treatment (Fig. 2C). To confirm the indication of apoptosis seen in the cell cycle analysis annexin V staining was performed. The relative increase in annexin V positive cells seen with birinapant alone only in WM9 and with birinapant in combination with TNF-α in WM9, 451Lu, and WM1366, but not in 1205Lu cells could confirm the pattern of apoptosis seen by cell cycle analysis (Fig. 2D).
To investigate a possible role of MAPK and RAC1 signaling in resistance to birinapant, phosphorylation status of ERK1/2 and protein levels of RAC1 upon treatment with birinapant at different time points in birinapant sensitive and resistant cell lines were assessed. We could observe a marked decrease in phosphorylation of ERK1/2 only at 24h and this decrease was seen in a birinapant sensitive as well as a birinapant resistant cohort of cell lines. RAC1 protein was found at similar levels in both sensitive and resistant cohorts and there was no significant change upon birinapant treatment (Supplementary Fig. S2).
An immunoblot performed in the 451Lu cell line demonstrated that birinapant could cause sustained cIAP1 protein degradation within one hour at a dose of 100nM, whereas XIAP levels were not affected (Fig. 3A).
Figure 3. Effect of birinapant is dependent on kinases and RIP1.

A, immunoblot showing the effect of birinapant on cIAP1 and XIAP protein levels at three different time points and concentrations in the melanoma cell line 451Lu. GAPDH was included to ensure even protein loading. B, the cell lines 451Lu and WM1366 were treated with birinapant at a dose of 1uM in combination with TNF-α 1ng/ml in the absence or presence of the pan caspase inhibitor Z-VAD-FMK. Relative cell proliferation after 72h was assessed using the MTS assay. Data are represented as mean ± SEM (n=3 biological replicates), P<0.05. C, the cell lines 451Lu and WM1366 were treated with birinapant at a dose of 1uM in combination with TNF-α 1ng/ml in the absence or presence of the RIP1 kinase inhibitor necrostatin-1 (Nec-1). Relative cell proliferation after 72h was assessed using the MTS assay. Data are represented as mean ± SEM (n=3 biological replicates), P<0.05. D, RIPK1 RNA expression levels in the panel of cell lines shown in Supplementary Table S1. Cell lines are stratified into birinapant sensitive (with or without addition of TNF-α) and birinapant in combination with TNF-α resistant. The left panel shows untreated cells and the right panel cells stimulated with TNF-α 1ng/ml for 24h. Notice the difference in scale of the y-axes.
To confirm that birinapant resistance was not caused by failure of target degradation, we performed immunoblots for cIAP1 and XIAP in a cohort of birinapant-TNF-α combination therapy resistant cell lines and found cIAP1 protein levels to be depressed to similar levels as seen in the birinapant sensitive cell line 451Lu (Supplementary Fig. S3).
Next, we investigated whether the observed cell death with birinapant at a dose of complete cIAP1 inhibition in combination with TNF-α was caspase dependent. Two combination sensitive cell lines (451Lu and WM1366) were treated with the combination of birinapant and TNF-α with or without addition of Z-VAD-FMK, a pan caspase inhibitor. In both cell lines, the inhibition of caspases led to a complete restoration of proliferation (Fig. 3B). In a similar experiment, we then further explored the role of RIP1 kinase, an essential part of the caspase initiating complex activated by TNF-α. By treating the two cell lines as above, now in the absence or presence of necrostatin-1, a RIP1 kinase inhibitor, we could again reverse the effect of birinapant and TNF-α and restore cell proliferation (Fig. 3C).
Since loss of RIP1 expression could therefore be a possible mechanism of resistance to birinapant, we assessed RIP1 expression in the whole panel of cell lines. However, no loss or decrease in RIP1 expression in the six birinapant resistant cell lines compared to the sensitive group could be observed. This was also true after stimulation with TNF-α 1ng/ml (Fig. 3D).
Three-dimensional spheroid models confirmed responses seen in adherent cell cultures
Cells grown in three-dimensional spheroid cultures were previously shown to have altered drug response profiles compared to adherent cell cultures (34), and are expected to more accurately predict in vivo efficacy due to similar architecture and micro-environmental signals (35, 36). The four previously selected cell lines were grown as three-dimensional spheroids in a collagen matrix and treated with birinapant alone or in combination with TNF-α. A live/ dead fluorescent cell stain was used to visually assess treatment effects using confocal microscopy (Fig. 4A): Spheroids of the birinapant single agent sensitive cell line, WM9, did indeed show an extensive reduction in live cells after addition of birinapant, but not after addition of TNF-α alone. The combination-sensitive cell lines, 451Lu and WM1366, retained the same response patterns in three-dimensional cultures: both showed a marked decrease in live cells and increase in dead cells only after treatment with birinapant in combination with TNF-α. In addition, the cell line that was completely resistant to the combination treatment in adherent cell culture, 1205Lu, showed only slight growth retardation when grown as spheroids in the presence of birinapant in combination with TNF-α.
Figure 4. Effect of birinapant on melanoma cells grown as three dimensional spheroids.
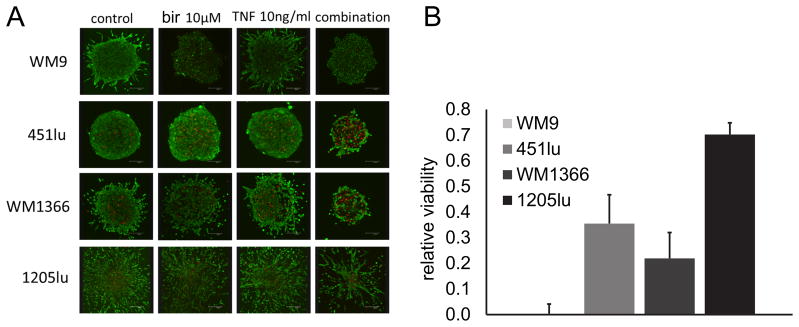
A, cell lines were grown as spheroids in a collagen mix for 72h with or without addition of birinapant 1uM or TNF-α 1ng/ml or the combination of both. Spheroids were stained with a Live (green)/ Dead (red) cell viability kit and imaged using confocal microscopy. Experiments were repeated three times, each time one spheroid per condition was imaged, representative images shown. B, spheroids were grown in 96 well plates with one spheroid/ well (n=6 technical replicates). Spheroids were then treated for 72h with birinapant 1uM, TNF-α 1ng/ml, or the combination of both. Viability was assessed with the Alamar Blue assay. Data is shown relative to untreated control. Mean of biological triplicates, error bars represent SEM.
To objectively quantify viability in this model, we assessed metabolic activity of spheroids after treatment with birinapant in combination with TNF-α using Alamar Blue. The viability results mirrored the responses seen in the Live/ Dead assay: a near total loss of viability in WM9, a dramatic decrease in viability in the combination sensitive cell lines (451Lu, WM1366) and only a slight reduction of viability in the 1205Lu cell line (Fig 4B).
Birinapant inhibits tumor growth in melanoma xenotransplantation models as a single agent
To investigate whether birinapant could inhibit melanoma tumor growth in an in vivo setting as a single agent, two cell lines were selected for xenotransplantation experiments: both were in vitro birinapant single agent resistant, but 451Lu did respond in vitro to the combination of birinapant with TNF-α, whereas 1205Lu did not respond to the combination treatment in vitro. Both cell lines were inoculated s.c. in NUDE mice and allowed to form palpable tumors before being randomized into vehicle control and birinapant treatment groups. During three weeks of dosing, birinapant showed an antitumor effect in both models, although the effect in the in vitro combination sensitive cell line was more sustained with abrogation of tumor growth in the birinapant treated animals. In contrast, 1205Lu tumors showed a marked slowing of tumor growth, but not abrogation of tumors (Fig 5A).
Figure 5. Effect of birinapant in vivo.

A, NUDE mice bearing 451Lu (left panel) or 1205Lu (right panel) xenografts were dosed three times per week intra-peritoneal with either birinapant at 30mg/kg or vehicle control. Dosing was started after formation of palpable tumors. The graphs show duration of treatment in days on the x-axes and fold change in tumor volume compared to first day of dosing on the y-axes. In the 451Lu xenografts, two animals of the birinapant group had un-measureable tumors at the end of study. Data represents mean ±SEM (n=5/ group), p<0.05 at end of treatment. B, NUDE mice bearing 451Lu or 1205Lu xenotransplants with a mean tumor volume of approximately 200mm3 were dosed twice at a 48h interval with either birinapant at 30mg/kg or vehicle control. At four time points (3h, 6h, 12h, and 24h) after last dosing tumors were harvested. Immunoblots of tumor protein lysates harvested at indicated time points are shown. Blots were probed for levels of cIAP1, XIAP, and cIAP2 (not measurable). GAPDH was included to insure equal loading. Quantifications of cIAP1 bands are included as bar graphs. Data represents mean +SEM. C, tumor tissues of control and birinapant treated animals were stained for activated caspase-3.
In a subsequent in vivo experiment, we then went on to confirm birinapant target inhibition in both models by immunoblot of tumor lysates. Animals were again inoculated with both xenograft models and tumors allowed to from. Animals were then pre-treated twice in an interval of 48h and tumors were harvested 3, 6, 12, and 24 hours after the second dosing. Compared to vehicle control, cIAP1 protein was reduced to low levels at 3h post and this effect was sustained for 24 hours in both models (Fig 5B). Staining for activated caspase-3 in biopsies of the same tumors showed a modest increase in apoptotic cells in the birinapant treated animals compared to vehicle control, 24h post treatment (Fig 5C).
To further investigate the combination activity between birinapant and TNF-α in vitro, the four cell lines previously selected based on their response profiles were again utilized. All four cell lines had comparable levels of TNF-α in cell culture supernatants (Supplementary Fig. S4) and adding high levels of TNF-α alone to all four cell lines had no effect on cell proliferation (Supplementary Fig. S5). The one cell line sensitive to birinapant as a single agent, WM9, provided an opportunity to further investigate the role of TNF-α in this setting. We therefore added a TNF-α blocking monoclonal antibody to WM9 cell cultures before treatment with birinapant. By gradually binding endogenous TNF-α in the supernatant with increasing doses of the antibody, it was possible to completely abrogate the effect of birinapant in a dose dependent manner (Fig. 6A). This indicates that, while no addition of exogenous TNF-α was necessary to observe a birinapant effect in this cell line, the observed anti-tumor effect was still dependent on endogenous TNF-α.
Figure 6. The role of TNF-α in the effectiveness of birinapant in vitro.

A, TNF-α is required for in vitro growth inhibition: The melanoma cell line WM9 was treated with birinapant at doses indicated on the lower x-axis for 72h. The same cell line was treated with birinapant at a fixed dose of 1uM in combination with a monoclonal blocking antibody binding to TNF-α (TNF-α mAb) at increasing doses as indicated on the upper x-axis. Viability was assessed after 72h via the MTS assay; absolute values are shown on the Y-axis. Data represents mean of three experiments ±SEM. B, combination activity between birinapant and TNF-α is schedule dependent: 451Lu cells were incubated either with birinapant at indicated doses for 36h and subsequently TNF-α 1ng/ml for 36h (birinapant -> TNF-α), with TNF-α 1ng/ml for 36h and subsequently birinapant at indicated doses (TNF-α-> birinapant), or the combination of both for the full 72h. Cell viability was assessed relative to untreated controls. Data represents mean of three experiments ±SEM. C, Effect of the combination therapy on a BRAF inhibitor resistant cell line. A cell line with acquired resistance to BRAF inhibition (451Lu-BR) shows the identical response to birinapant as its parental cell line (451Lu). Cells were treated with birinapant at indicated doses either alone (451Lu, 451Lu-BR) or in combination with TNF-α 1ng/ml (451Lu + TNF-α, 451Lu-BR + TNF-α) for 72h. Cell viability is shown relative to untreated controls. Data represents mean of three experiments ±SEM. D, Effect of cisplatin on melanoma viability in the absence or presence of birinapant. The birinapant resistant cell lines 451Lu and WM1366 were treated with cisplatin in increasing doses up to 20uM as a single agent or in combination with birinapant at a fixed dose of 100nM. Cell viability is shown relative to untreated controls. Data represents mean of both cell lines ±SEM.
Additional evidence on the causative role of TNF-α on the effect of birinapant was provided by varying the schedule of combining birinapant with TNF-α: a combination sensitive cell line treated with birinapant for 36h and subsequently incubated with TNF-α for 36h, showed significant growth inhibition. Conversely, when treated first with TNF-α for 36h and then with birinapant for 36h, cells were significantly less sensitive (Fig. 6B). Therefore, the combination activity between birinapant and TNF-α is schedule dependent. This corroborates the hypothesis that birinapant mediated IAP inhibition has to occur prior to activation of the TNFR in order to induce an anti-tumor effect.
Previously, our group has generated human melanoma cell lines with acquired resistance to BRAF inhibitors (27). The mechanism of resistance in these cell lines was RAF isoform switching and increased IGF-1R/PI3K signaling. We compared the cell lines 451Lu (BRAFV600E, BRAF inhibitor sensitive) and 451Lu-BR (BRAF inhibitor resistant) in regard to birinapant response. Cell viability was not affected after treatment with birinapant alone in neither the parental nor the resistant cell line. When birinapant was combined with TNF-α however, both cell lines showed a strong response that was identical between the sensitive and the resistant cell line (Fig 6C).
Next, we investigated whether birinapant could sensitize melanoma cells to chemotherapy. For this experiment, two birinapant single agent resistant cell lines were treated with cisplatin, a cytotoxic drug, with or without addition of birinapant. Cisplatin as a single agent reduced viability in both melanoma cell lines (mean of both cell lines) at increasing doses up to 20uM. Birinapant mediated cIAP1 degradation in combination with cisplatin did significantly (P<0.05) enhance sensitivity to cisplatin in both cell lines (Fig. 6D).
Discussion
A delicate balance between inducers and inhibitors of apoptosis exists in any given cell (37). In cancer cells, including melanoma, this equilibrium is often skewed in favor of survival, with IAPs being predominant (38, 39). Committing melanoma cells to apoptosis, which would be an ideal outcome for most therapies, therefore requires additional stimuli. Inhibition of IAPs for one can restore the balance between survival and death signals, thereby facilitating programmed cell death in melanoma.
Chronic inflammation in the tumor microenvironment of melanoma lesions often leads to elevated levels of TNF-α, at least in part provided by tumor-infiltrating immune cells such as macrophages (23–25). Levels of tumor-infiltrating macrophages have furthermore been shown to be associated with aggressive disease (40). All melanoma cell lines tested were resistant to treatment with exogenous TNF-α as a single agent. This was in line with previous clinical experiences, where minimal anti-tumor effect and significant toxicity was observed (41, 42). Birinapant mediated downregulation of cIAP1 by itself did not induce any anti-tumor effect in vitro. Since neither compound was effective alone, but both were highly effective in combination this satisfies the definition of “coalism” according to the so-called Saariselka agreement (43, 44). This was confirmed on a subset of cell lines to be apoptosis-mediated and dependent on caspases and RIPK1 activity.
Independent of mutational background (we assessed melanoma cell lines from major genetic subgroups), two patterns of responses emerged: cell lines that were either exquisitely sensitive or remarkably resistant to birinapant in combination with TNF-α at low or high doses of both compounds respectively. We confirmed downregulation of cIAP1 target protein in resistant and sensitive cell lines by western blot, but only the sensitive cell lines showed PARP cleavage, indicative of apoptosis. This observation could also be demonstrated by increased sub G1 cell fractions and increased annexin V positive cells, indicating an increase in apoptotic cells, in the combination-sensitive cell lines.
Since a subgroup of melanoma cell lines of all genetic backgrounds tested was found to be resistant to birinapant in combination with TNF-α, we investigated whether levels of phosphorylated ERK 1/2 or RAC1 were increased after birinapant treatment in these cell lines. These were recently described to play a role in resistance to SMAC mimetics (45, 46). Although we observed a decrease in ERK1/2 activation after 24h of birinapant treatment in most cell lines tested, we could not find any differential regulation of these two proteins in the resistant cell lines as compared to the sensitive subset. Another possible mechanism of resistance to SMAC mimetics is loss of RIP1 expression. We observed the effect of birinapant to be dependent on RIP1 kinase in concordance with previously published reports on compounds from the same class (47). Since there was no difference in RIP1 expression between birinapant resistant and sensitive cell lines in the panel used in this study, RIP1 expression is likely not a suitable biomarker to predict melanoma response to birinapant. Therefore, further investigation to define a biomarker for this subgroup is warranted since this would be highly useful in the clinical application of birinapant.
Our observations remained consistent across increasingly complex models from monolayer culture, to three dimensional matrix cultures, to human melanoma xenograft models. While the 3D spheroid models closely mirrored the effects seen in adherent monolayer culture, the in vivo xenotransplantation experiment results were reflecting the complexity of the in vivo setting. While in vitro 451Lu cells responded only to the combination of birinapant and TNF-α, birinapant was highly active as a single agent in the in vivo model abrogating tumor growth. In addition, a cell line resistant to birinapant in vitro even in combination with TNF-α still showed slower tumor growth when treated with birinapant in vivo compared to vehicle treated controls. This observation indicates the high complexity of melanoma growth in a tissue microenvironment providing a multitude of additional stimuli. Together, these results indicate a potential effectiveness of birinapant as a single agent in patients with melanoma.
Birinapant was effective as a single agent in vitro only in one of the seventeen cell lines tested. We therefore investigated the role of TNF-α signaling on the birinapant effect in this cell line. Blocking endogenous TNF-α in the supernatant completely abrogated the effect of birinapant in a dose dependent fashion, thereby demonstrating the dependency of birinapant on concurrent TNF-α stimulation in vitro.
This dependency was furthermore schedule-specific, as we could show in a cell line sensitive to birinapant only in combination with TNF-α. The effect of the combination therapy was preserved when sequentially added in the following order: first birinapant, with downregulation of cIAP1 and cIAP2, and subsequent TNF-α, with activation of the TNFR complex 2 and the apoptotic cascade. Adding these two compounds in the reverse order diminished their effectiveness significantly.
With the prospect of increased numbers of melanoma patients acquiring resistance to BRAF inhibitors, the question whether birinapant might be a feasible second line therapy for such cases was explored. Indeed, a cell line with acquired resistance to BRAF inhibition did not alter its sensitivity profile to birinapant when compared to the parental cell line. This suggests birinapant as a possible second line therapy in patients with acquired resistance to BRAF inhibitors.
Although targeted therapy with small molecules is showing impressive results in melanoma therapy, cytotoxic agents are still playing a major role in this disease when patients are either not eligible for kinase inhibitors and immune-therapy, or have relapsed on these drugs. We have therefore investigated the role of birinapant in sensitizing melanoma to cisplatin, a DNA damaging agent. Although the results seen in this study are encouraging, more combinations and schedules will have to be explored and these studies are currently ongoing.
Supplementary Material
Translational Relevance.
Although major advances have recently been made in the treatment of malignant melanoma, this disease remains largely incurable once metastasized. The main reason is the development of resistance, even to new therapies recently introduced into the clinic or currently in late stage testing. Additionally, the genetic heterogeneity of melanoma often limits the use of effective targeted therapies to specific subsets of patients. Novel small molecule inhibitors that could be applicable for all melanoma patients, regardless of genetic subgroup, would be highly beneficial to the current standard of care. We describe here a novel SMAC mimetic small molecule, birinapant, which shows anti-tumor activity in major subgroups of cutaneous melanoma. Moreover, birinapant could be useful in overcoming acquired resistance to BRAF inhibitors, a patient population that is in urgent need of new treatment strategies.
Acknowledgments
Grant Support: C.K. is a MAX KADE Foundation postdoctoral research exchange grant recipient. The studies were funded by NIH grants CA25874, CA047159, CA 114046, CA10815, and the Adelson Medical Research Foundation.
C.K. is on sabbatical from the Department of Dermatology, Medical University Vienna, Vienna, Austria and is thankful for this opportunity.
We thank the Microscopy Facility managed by James E. Hayden, the Flow Cytometry Facility managed by Jeffrey S. Faust, and the Animal Facility managed by Denise DiFrancesco, all at the Wistar Institute.
Footnotes
Disclosure of Potential Conflicts of Interest: Srinivas K. Chunduru, Yasuhiro Mitsuuchi, Eric M. Neiman, Christopher Benetatos and Mark McKinlay are employees of TetraLogic.
References
- 1.Serrone L, Zeuli M, Sega FM, Cognetti F. Dacarbazine-based chemotherapy for metastatic melanoma: thirty-year experience overview. JExpClinCancer Res. 2000;19:21–34. [PubMed] [Google Scholar]
- 2.Bollag G, Hirth P, Tsai J, Zhang J, Ibrahim PN, Cho H, et al. Clinical efficacy of a RAF inhibitor needs broad target blockade in BRAF-mutant melanoma. Nature. 2010;467:596–9. doi: 10.1038/nature09454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Flaherty KT, Puzanov I, Kim KB, Ribas A, McArthur GA, Sosman JA, et al. Inhibition of mutated, activated BRAF in metastatic melanoma. N Engl J Med. 2010;363:809–19. doi: 10.1056/NEJMoa1002011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hodi FS, O’Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363:711–23. doi: 10.1056/NEJMoa1003466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nikolaou VA, Stratigos AJ, Flaherty KT, Tsao H. Melanoma: New Insights and New Therapies. J Invest Dermatol. 2012 doi: 10.1038/jid.2011.421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hersey P, Zhang X. How Melanoma Cells Evade TRAIL-Induced Apoptosis. Nature Reviews Cancer. 2001:1. doi: 10.1038/35101078. [DOI] [PubMed] [Google Scholar]
- 7.Bedikian AY, Millward M, Pehamberger H, Conry R, Gore M, Trefzer U, et al. Bcl-2 Antisense (oblimersen sodium) Plus Dacarbazine in Patients With Advanced Melanoma: The Oblimersen Melanoma Study Group. Journal of Clinical Oncology. 2006;24:4738–45. doi: 10.1200/JCO.2006.06.0483. [DOI] [PubMed] [Google Scholar]
- 8.Dai Y, Grant S. Targeting Multiple Arms of the Apoptotic Regulatory Machinery. Cancer Research. 2007;67:2908–11. doi: 10.1158/0008-5472.CAN-07-0082. [DOI] [PubMed] [Google Scholar]
- 9.Nguyen M, Marcellus RC, Roulston A, Watson M, Serfass L, Murthy Madiraju SR, et al. Small molecule obatoclax (GX15-070) antagonizes MCL-1 and overcomes MCL-1-mediated resistance to apoptosis. Proceedings of the National Academy of Sciences. 2007;104:19512–7. doi: 10.1073/pnas.0709443104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hunter AM, LaCasse EC, Korneluk RG. The inhibitors of apoptosis (IAPs) as cancer targets. Apoptosis. 2007;12:1543–68. doi: 10.1007/s10495-007-0087-3. [DOI] [PubMed] [Google Scholar]
- 11.Tamm I, Kornblau SM, Segall H, Krajewski S, Welsh K, Kitada S, et al. Expression and prognostic significance of IAP-family genes in human cancers and myeloid leukemias. ClinCancer Res. 2000;6:1796–803. [PubMed] [Google Scholar]
- 12.Gyrd-Hansen M, Meier P. IAPs: from caspase inhibitors to modulators of NF-kappaB, inflammation and cancer. Nat Rev Cancer. 2010;10:561–74. doi: 10.1038/nrc2889. [DOI] [PubMed] [Google Scholar]
- 13.Du C, Fang M, Li Y, Li L, Wang X. Smac, a Mitochondrial Protein that Promotes Cytochrome c–Dependent Caspase Activation by Eliminating IAP Inhibition. Cell. 2000;102:33–42. doi: 10.1016/s0092-8674(00)00008-8. [DOI] [PubMed] [Google Scholar]
- 14.Wu G, Chai J, Suber TL, Wu J-W, Du C, Wang X, et al. Structural basis of IAP recognition by Smac/DIABLO. Nature. 2000;408:1008–12. doi: 10.1038/35050012. [DOI] [PubMed] [Google Scholar]
- 15.Liu Z, Sun C, Olejniczak ET, Meadows RP, Betz SF, Oost T, et al. Structural basis for binding of Smac/DIABLO to the XIAP BIR3 domain. Nature. 2000;408:1004–8. doi: 10.1038/35050006. [DOI] [PubMed] [Google Scholar]
- 16.Bockbrader KM, Tan M, Sun Y. A small molecule Smac-mimic compound induces apoptosis and sensitizes TRAIL- and etoposide-induced apoptosis in breast cancer cells. Oncogene. 2005;24:7381–8. doi: 10.1038/sj.onc.1208888. [DOI] [PubMed] [Google Scholar]
- 17.Li L, Thomas RM, Suzuki H, De Brabander JK, Wang X, Harran PG. A small molecule Smac mimic potentiates TRAIL- and TNFalpha-mediated cell death. Science. 2004;305:1471–4. doi: 10.1126/science.1098231. [DOI] [PubMed] [Google Scholar]
- 18.Lecis D, Drago C, Manzoni L, Seneci P, Scolastico C, Mastrangelo E, et al. Novel SMAC-mimetics synergistically stimulate melanoma cell death in combination with TRAIL and Bortezomib. British Journal of Cancer. 2010;102:1707–16. doi: 10.1038/sj.bjc.6605687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dineen SP, Roland CL, Greer R, Carbon JG, Toombs JE, Gupta P, et al. Smac Mimetic Increases Chemotherapy Response and Improves Survival in Mice with Pancreatic Cancer. Cancer Research. 2010;70:2852–61. doi: 10.1158/0008-5472.CAN-09-3892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Eckelman B, Salvesen G. The human anti-apoptotic proteins cIAP1 and cIAP2 bind but do not inhibit caspases. The Journal of biological chemistry. 2006;281:3254–60. doi: 10.1074/jbc.M510863200. [DOI] [PubMed] [Google Scholar]
- 21.Vince JE, Wong WW-L, Khan N, Feltham R, Chau D, Ahmed AU, et al. IAP Antagonists Target cIAP1 to Induce TNFα-Dependent Apoptosis. Cell. 2007;131:682–93. doi: 10.1016/j.cell.2007.10.037. [DOI] [PubMed] [Google Scholar]
- 22.Varfolomeev E, Blankenship JW, Wayson SM, Fedorova AV, Kayagaki N, Garg P, et al. IAP Antagonists Induce Autoubiquitination of c-IAPs, NF-κB Activation, and TNFα-Dependent Apoptosis. Cell. 2007;131:669–81. doi: 10.1016/j.cell.2007.10.030. [DOI] [PubMed] [Google Scholar]
- 23.Coussens LM, Werb Z. Inflammation and cancer. Nature. 2002;420:860–7. doi: 10.1038/nature01322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Balkwill F. Tumor necrosis factor or tumor promoting factor? Cytokine Growth Factor Rev. 2002;13:135–41. doi: 10.1016/s1359-6101(01)00020-x. [DOI] [PubMed] [Google Scholar]
- 25.Katerinaki E, Evans GS, Lorigan PC, MacNeil S. TNF-[alpha] increases human melanoma cell invasion and migration in vitro: the role of proteolytic enzymes. Br J Cancer. 2003;89:1123–9. doi: 10.1038/sj.bjc.6601257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Micheau O, Tschopp J. Induction of TNF Receptor I-Mediated Apoptosis via Two Sequential Signaling Complexes. Cell. 2003;114:181–90. doi: 10.1016/s0092-8674(03)00521-x. [DOI] [PubMed] [Google Scholar]
- 27.Villanueva J, Vultur A, Lee JT, Somasundaram R, Fukunaga-Kalabis M, Cipolla AK, et al. Acquired resistance to BRAF inhibitors mediated by a RAF kinase switch in melanoma can be overcome by cotargeting MEK and IGF-1R/PI3K. Cancer Cell. 2010;18:683–95. doi: 10.1016/j.ccr.2010.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Thomas RK, Baker AC, DeBiasi RM, Winckler W, LaFramboise T, Lin WM, et al. High-throughput oncogene mutation profiling in human cancer. Nat Genet. 2007;39:347–51. doi: 10.1038/ng1975. [DOI] [PubMed] [Google Scholar]
- 29.Allensworth JL, Sauer SJ, Lyerly HK, Morse MM, Devi GR. Smac Mimetic Birinapant Induces Apoptosis and Enhances TRAIL Potency in Inflammatory Breast Cancer Cells in an IAP-Dependent and TNF-α-Independent Mechanism. Breast Cancer Research and Treatment. doi: 10.1007/s10549-012-2352-6. submitted. [DOI] [PubMed] [Google Scholar]
- 30.Condon SM. Chapter 13 - The Discovery and Development of Smac Mimetics Small-Molecule Antagonists of the Inhibitor of Apoptosis Proteins. In: John EM, editor. Annual Reports in Medicinal Chemistry. Academic Press; 2011. pp. 211–26. [Google Scholar]
- 31.Meier F, Nesbit M, Hsu M, Martin B, Van Belle P, Elder D, et al. Human melanoma progression in skin reconstructs : biological significance of bFGF. The American Journal of Pathology. 2000;156:193–200. doi: 10.1016/S0002-9440(10)64719-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hsu M, Meier F, Nesbit M, Hsu J, Van Belle P, Elder D, et al. E-cadherin expression in melanoma cells restores keratinocyte-mediated growth control and down-regulates expression of invasion-related adhesion receptors. The American Journal of Pathology. 2000;156:1515–25. doi: 10.1016/S0002-9440(10)65023-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Colombino M, Capone M, Lissia A, Cossu A, Rubino C, De Giorgi V, et al. BRAF/NRAS Mutation Frequencies Among Primary Tumors and Metastases in Patients With Melanoma. Journal of Clinical Oncology. 2012 doi: 10.1200/JCO.2011.41.2452. [DOI] [PubMed] [Google Scholar]
- 34.Smalley KS, Lioni M, Herlyn M. Life isn’t flat: taking cancer biology to the next dimension. In Vitro Cellular & Developmental Biology - Animal. 2006;42:242–7. doi: 10.1290/0604027.1. [DOI] [PubMed] [Google Scholar]
- 35.Smalley KS, Haass NK, Brafford PA, Lioni M, Flaherty KT, Herlyn M. Multiple signaling pathways must be targeted to overcome drug resistance in cell lines derived from melanoma metastases. Mol Cancer Ther. 2006;5:1136–44. doi: 10.1158/1535-7163.MCT-06-0084. [DOI] [PubMed] [Google Scholar]
- 36.Horning JL, Sahoo SK, Vijayaraghavalu S, Dimitrijevic S, Vasir JK, Jain TK, et al. 3-D tumor model for in vitro evaluation of anticancer drugs. Mol Pharm. 2008;5:849–62. doi: 10.1021/mp800047v. [DOI] [PubMed] [Google Scholar]
- 37.Oltval ZN, Milliman CL, Korsmeyer SJ. Bcl-2 heterodimerizes in vivo with a conserved homolog, Bax, that accelerates programed cell death. Cell. 1993;74:609–19. doi: 10.1016/0092-8674(93)90509-o. [DOI] [PubMed] [Google Scholar]
- 38.Engesater BO, Sathermugathevan M, Hellenes T, Engebraten O, Holm R, Florenes VA, et al. Targeting inhibitor of apoptosis proteins in combination with dacarbazine or TRAIL in melanoma cells. Cancer Biology & Therapy. 2011;12:47–58. doi: 10.4161/cbt.12.1.15714. [DOI] [PubMed] [Google Scholar]
- 39.Chawla-Sarkar M, Bae SI, Reu FJ, Jacobs BS, Lindner DJ, Borden EC. Downregulation of Bcl-2, FLIP or IAPs (XIAP and survivin) by siRNAs sensitizes resistant melanoma cells to Apo2L//TRAIL-induced apoptosis. Cell Death Differ. 2004;11:915–23. doi: 10.1038/sj.cdd.4401416. [DOI] [PubMed] [Google Scholar]
- 40.Storr SJ, Safuan S, Mitra A, Elliott F, Walker C, Vasko MJ, et al. Objective assessment of blood and lymphatic vessel invasion and association with macrophage infiltration in cutaneous melanoma. Mod Pathol. 2012;25:493–504. doi: 10.1038/modpathol.2011.182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Creagan ET, Kovach JS, Moertel CG, Frytak S, Kvols LK. A phase I clinical trial of recombinant human tumor necrosis factor. Cancer. 1988;62:2467–71. doi: 10.1002/1097-0142(19881215)62:12<2467::aid-cncr2820621202>3.0.co;2-5. [DOI] [PubMed] [Google Scholar]
- 42.Selby P, Hobbs S, Viner C, Jackson E, Jones A, Newell D, et al. Tumour necrosis factor in man: clinical and biological observations. Br J Cancer. 1987;56:803–8. doi: 10.1038/bjc.1987.294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Greco WR, Bravo G, Parsons JC. The search for synergy: a critical review from a response surface perspective. Pharmacol Rev. 1995;47:331–85. [PubMed] [Google Scholar]
- 44.Greco WR, Unkelbach HD, Poech G, Suehnel J, Kundi M, Boedeker W. Consensus on concepts and terminology for combined-action assessment: the Saariselka Agreement. Arch Complex Environmental Studies. 1992;4:65–9. [Google Scholar]
- 45.Oberoi-Khanuja TK, Karreman C, Larisch S, Rapp UR, Rajalingam K. Role of Melanoma Inhibitor of Apoptosis (ML-IAP) Protein, a Member of the Baculoviral IAP Repeat (BIR) Domain Family, in the Regulation of C-RAF Kinase and Cell Migration. Journal of Biological Chemistry. 2012;287:28445–55. doi: 10.1074/jbc.M112.341297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Oberoi TK, Dogan T, Hocking JC, Scholz R-P, Mooz J, Anderson CL, et al. IAPs regulate the plasticity of cell migration by directly targeting Rac1 for degradation. Embo J. 2012;31:14–28. doi: 10.1038/emboj.2011.423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Laukens B, Jennewein C, Schenk B, Vanlangenakker N, Schier A, Cristofanon S, et al. Smac mimetic bypasses apoptosis resistance in FADD- or caspase-8-deficient cells by priming for tumor necrosis factor α-induced necroptosis. Neoplasia (New York, NY) 2011;13:971–9. doi: 10.1593/neo.11610. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.